Buy ebook Neuro-oncology compendium for the boards and clinical practice [team-ira] (true pdf) macie
Neuro-Oncology Compendium for the Boards and Clinical Practice [Team-IRA] (True PDF) Maciej M. Mrugala
Visit to download the full and correct content document: https://ebookmass.com/product/neuro-oncology-compendium-for-the-boards-and-clini cal-practice-team-ira-true-pdf-maciej-m-mrugala/
More products digital (pdf, epub, mobi) instant download maybe you interests ...
Neuro-Oncology for the Clinical Neurologist 1st Edition
NEURO-ONCOLOGY COMPENDIUM FOR THE BOARDS AND CLINICAL PRACTICE
NEURO-ONCOLOGY
COMPENDIUM FOR THE BOARDS AND CLINICAL PRACTICE
EDITED BY Maciej M. Mrugala, MD, PhD, MPH, FAAN
MAYO CLINIC PHOENIX, AZ, USA
Na Tosha N. Gatson, MD, PhD, FAAN
BANNER MD ANDERSON CANCER CENTER
PHOENIX, AZ, USA
GEISINGER COMMONWEALTH SCHOOL OF MEDICINE
SCRANTON, PA, USA
Sylvia C. Kurz, MD, PhD
EBERHARD KARLS UNIVERSITY
TÜBINGEN, GERMANY
Kathryn S. Nevel, MD
INDIANA UNIVERSITY SCHOOL OF MEDICINE INDIANAPOLIS, IN, USA
Jennifer L. Clarke, MD, MPH
UNIVERSITY OF CALIFORNIA SAN FRANCISCO, CA, USA
Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and certain other countries.
Published in the United States of America by Oxford University Press 198 Madison Avenue, New York, NY 10016, United States of America.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by license, or under terms agreed with the appropriate reproduction rights organization. Inquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above.
You must not circulate this work in any other form and you must impose this same condition on any acquirer.
CIP data is on file at the Library of Congress
ISBN 978–0–19–757377–8
DOI: 10.1093/med/9780197573778.001.0001
This material is not intended to be, and should not be considered, a substitute for medical or other professional advice. Treatment for the conditions described in this material is highly dependent on the individual circumstances. And, while this material is designed to offer accurate information with respect to the subject matter covered and to be current as of the time it was written, research and knowledge about medical and health issues is constantly evolving and dose schedules for medications are being revised continually, with new side effects recognized and accounted for regularly. Readers must therefore always check the product information and clinical procedures with the most up-to-date published product information and data sheets provided by the manufacturers and the most recent codes of conduct and safety regulation. The publisher and the authors make no representations or warranties to readers, express or implied, as to the accuracy or completeness of this material. Without limiting the foregoing, the publisher and the authors make no representations or warranties as to the accuracy or efficacy of the drug dosages mentioned in the material. The authors and the publisher do not accept, and expressly disclaim, any responsibility for any liability, loss, or risk that may be claimed or incurred as a consequence of the use and/or application of any of the contents of this material.
Printed by Integrated Books International, United States of America
DEDICATIONS
The years 2020–2021 were particularly difficult for the medical community due to the global pandemic. Hats off to the providers who maintained excellent care of their patients and helped to usher innovations using telemedicine and distance learning. This book is dedicated to those on the frontline (physicians, advanced care providers, nurses,
aides, technicians, and others) and those acting behind the scenes (laboratory medicine, maintenance, housekeeping, food services, and more). Finally, to all the healthcare workers who continuously go to great lengths to take the best care of patients. Together, we elevate and educate in medicine.
IN MEMORIAM
Gordon A. Watson, MD, PhD, co-author of the Rare Tumors chapter, was a beloved Associate Professor and Vice Chair for Clinical Affairs for the Department of Radiation Oncology at Indiana University. He was among the most popular and busiest physicians in the department and the Indiana University Melvin and Bren Simon Comprehensive Cancer Center. Due to his vast wisdom and well-known desire to help, his input was always sought after for the most challenging of clinical cases. He was an outstanding physician in every way imaginable, who is dearly missed by his colleagues, patients, family, and friends.
CONTENTS
Foreword
Preface
Acknowledgments
Contributors
PART I NEURO-ONCOLOGY OVERVIEW
1. neURoPAtHoLoGY PRIMeR 3
Ibrahim Kulac and Melike Pekmezci
2. IMAGInG oF centRAL neRVoUs sYsteM tUMoRs 29
Shobhit Kumar Garg and Rajan Jain
PART II ADULT NEURO-ONCOLOGY
3. GLIoBLAstoMA
Rimas V. Lukas
4. DIFFUse GLIoMAs (WHo GRADe 2–3)
Haroon Ahmad and David Schiff
5. MenInGIoMA
Thomas J. Kaley
6. ePenDYMoMA
Iyad Alnahhas, Appaji Rayi, Wayne Slone, Peter Kobalka, Shirley Ong, Pierre Giglio, and Vinay K. Puduvalli
7. tUMoRs oF tHe PItUItARY GLAnD
Reginald Fong and Andrew R. Conger
8. GeRM ceLL tUMoRs
Hirokazu Takami
9. IntRAVentRIcULAR tUMoRs
Samanthalee C. S. Obiorah, Richard S. Dowd, and Steven A. Toms
10. RARe tUMoRs In ADULts, IncLUDInG MeDULLoBLAstoMA
Kevin Shiue, Logan S. DeWitt, John M. Kindler, and Gordon A. Watson†
11. PRIMARY centRAL neRVoUs sYsteM LYMPHoMA AnD HeMAtoPoIetIc neoPLAsMs AFFectInG tHe neRVoUs sYsteM 161
Mary Jane Lim-Fat and Lakshmi Nayak
12. MetAstAtIc DIseAse In tHe centRAL neRVoUs sYsteM 179
Jessica A. Wilcox and Adrienne A. Boire
13. tUMoRs AFFectInG tHe sPInAL coRD 199
Terrence Verla, Kara L. Curley, Matthew T. Neal, and Alexander Ropper
PART III PEDIATRIC NEURO-ONCOLOGY
14. ePIDeMIoLoGY AnD GeneRAL oVeRVIeW oF PeDIAtRIc centRAL neRVoUs sYsteM tUMoRs 219
Scott L. Coven
15. neURosURGIcAL MAnAGeMent oF PeDIAtRIc centRAL neRVoUs sYsteM tUMoRs 228
Nir Shimony, Mohammad Hassan A. Noureldine, Cameron Brimley, and George I. Jallo
16. PeDIAtRIc eMBRYonAL tUMoRs 240
Aaron Mochizuki and Sonia Partap
17. PeDIAtRIc LoW-GRADe GLIoMA 255
Sheetal Phadnis and Theodore Nicolaides
18. PeDIAtRIc HIGH-GRADe GLIoMA 267
Sameer Farouk Sait, Morgan Freret, and Matthias Karajannis
PART IV MANAGEMENT OF NEURO-ONCOLOGIC DISEASE
19. neURosURGIcAL MAnAGeMent oF ADULt centRAL neRVoUs sYsteM tUMoRs 289
Andrew J. Gogos, Ramin A. Morshed, and Shawn L. Hervey-Jumper
20. RADIotHeRAPY In neURo-oncoLoGY 309
Molly Havard Blau and Lia M. Halasz
21. PRIncIPLes oF sYsteMIc tHeRAPY In neURo-oncoLoGY 324
Nikolaos Andreatos and David M. Peereboom
22. DeVIce-BAseD tReAtMents In neURo-oncoLoGY 341
Ekokobe Fonkem, Amir Azadi, and Ramya Tadipatri
PART V COMPLICATIONS OF SYSTEMIC AND CENTRAL NERVOUS SYSTEM CANCERS
23. neURoLoGIc coMPLIcAtIons In cAnceR PAtIents 355
Katherine B. Peters
24. PARAneoPLAstIc neURoLoGIcAL sYnDRoMes 370
Cristina Valencia-Sanchez and Maciej M. Mrugala
25. neURo-oncoLoGIc eMeRGencIes 385
Maya Hrachova, David Gritsch, Simon Gritsch, and Maciej M. Mrugala
PART VI GENETICS AND NEURO-ONCOLOGIC DISEASE
26. FAMILIAL sYnDRoMes In neURo-oncoLoGY 407
Radhika Dhamija, Joseph M. Hoxworth, and Ashok R. Asthagiri
27. tHe neURoFIBRoMAtoses: neURoFIBRoMAtosIs 1, neURoFIBRoMAtosIs 2, AnD scHWAnnoMAtosIs 421
Kun-Wei Song and Scott R. Plotkin
PART VII SPECIAL CONSIDERATIONS IN NEURO-ONCOLOGY
28. DesIGnInG AnD DecIPHeRInG cLInIcAL tRIALs: MetHoDoLoGIcAL InGReDIents to noURIsH A HeALtHY stUDY 441
Michael Glantz and Alireza Mansouri
29. PRoceDURes AnD WeLL-cARe cLInIcs In neURo-oncoLoGY: RoLes FoR ADVAnceD PRActIce PRoVIDeRs 456
Erika N. Leese, Alyssa Callela, and Na Tosha N. Gatson
30. neURo-oncoLoGY In tHe eLDeRLY 468
Valeria Internò, Roberta Rudà, and Riccardo Soffietti
31. PALLIAtIVe cARe, ReHABILItAtIon, AnD sUPPoRtIVe cARe In neURo-oncoLoGY 480
Jamal Mohamud and Kathryn S. Nevel
32. tHe neURo-oncoLoGY oF WoMen (noW) 491
Na Tosha N. Gatson, Kerianne R. Taylor, Maria L. Boccia, and Terri L. Woodard
FOREWORD
This slim volume manages to be both comprehensive and succinct at the same time—a welcome combination. The editors compiled a list of stellar authors, and employed a standard structure for each chapter, making this book easy to read and the information highly accessible. The book fills a critical need for those seeking rapid and current information to care for patients with a neuro-oncologic problem. The focus is on primary intracranial tumors, but the authors also cover CNS metastases, and other neuro-oncologic topics such as paraneoplastic syndromes. They review the core diagnostic methods and summarize their clinical relevance; the vivid images of MRI scans and pathology throughout the book are especially informative. The latest therapeutic studies are summarized in a clear fashion along with the potential benefits and toxicities of each therapeutic modality. For those seeking information to prepare for an oncology or neurooncology Board certification examination, they will find the key points of each chapter highlighted throughout the text, followed by flash cards and multiple-choice questions for future referral and test practice. The editors and authors are to be congratulated on this highly practical compilation of data; it is a “must have” for anyone who cares for neuro-oncologic patients.
Lisa M. DeAngelis, MD Physician-in-Chief and Chief Medical Officer
Memorial Sloan Kettering Cancer Center
New York, NY, USA
Within the growing field of Neuro-Oncology, heavy-volume textbooks or detailed review articles do not meet the need for rapidly accessible and up-to-date practice-related information. This comprehensive, well-structured and good–to-read volume allows the newcomer to swiftly achieve an overview and the more advanced practitioner to recapitulate important facts, i.e. on recent trials, for which main efficacy outcomes and unwanted effects are summarized.
The present book by a group of experienced practitioners from all important subspecialties feeding into Neuro-Oncology covers many different aspects of modern Neuro-Oncology with the focus on primary Central Nervous System (CNS) tumors, CNS metastases, and neurological complications of cancer. Each chapter is structured in a standard way, allowing for recapitulating diagnostic challenges, clinical courses, and standard treatments including clinical trial updates. The book follows a high educational standard and fosters learning, self-testing as well as preparation for board certification in Neuro-Oncology for learners around the globe. The editors and authors are to be congratulated on this highly scholarly and useful volume, which certainly will help to inform care for patients with neuro-oncologic disease.
Wolfgang Wick, MD Chairman, Neurology Clinic, University of Heidelberg Neuro-Oncology Program Chair, National Center for Tumor Diseases, Heidelberg and German Cancer Research Center Heidelberg, Germany
PREFACE
tHe neURo-oncoLoGY coMPenDIUM
It is with great joy that we deliver this textbook to our readers. This work would not have been possible without the tremendous dedication of the contributing authors, the Oxford University Press staff, and our team of editors. We started this journey several years ago when the five of us identified gaps in the neuro-oncology curriculum for test takers and those who sought an up-to-date clinical reference guide. Our field changes rapidly, leaving gaps in the neuro-oncology library for books that would provide a high-yield overview of the various facets of our sub-specialty. This work is designed to not only address the board exam requirements but also provide practical knowledge for those who take care of patient population. We were fortunate that some of the top talents from across the world were enthusiastic to contribute to this project. A tremendous thanks is owed to the contributing authors, who spent countless hours making sure the content of their chapters was filled with relevant information and laid out to maximize
retention and recall of the information for the boards and daily clinical practice. We hope this textbook will become a staple educational offering and will be widely used by all who wish to enhance their knowledge in neuro-oncology.
On a personal note, working on this book brought us immense joy, allowed for excellent networking, and solidified our friendships for the years to come. We are very grateful for this opportunity! We wish you an enjoyable read, success on the board exams, and satisfaction in your clinical practices. We would appreciate your feedback and remain open to new and continued collaborations for future editions of the NeuroOncology Compendium.
The Editors Maciej M. Mrugala
Na Tosha N. Gatson
Sylvia C. Kurz
Kathryn S. Nevel
Jennifer L. Clarke
ACKNOWLEDGMENTS
We would like to acknowledge the patients and their caregivers who allowed us to share in their care and for their participation in clinical trials or other research that led to advancements in our field. Words of gratitude go to scientists and academicians who led the research projects described in this book. We would also like to acknowledge donors and funding agencies for their contribution to the science presented in this book. Finally, we would like to thank the Society for Neuro-Oncology for supporting our original idea for this work and Craig Panner and Oxford University Press team for taking our project on.
CONTRIBUTORS
Haroon Ahmad, MD University of Maryland, School of Medicine Baltimore, MD, USA
Iyad Alnahhas, MD
Thomas Jefferson University Philadelphia, PA, USA
Nikolaos Andreatos, MD Mayo Clinic Rochester, MN, USA
Ashok R. Asthagiri, MD University of Virginia Charlottesville, VA, USA
Amir Azadi, MD Honor Health Neuroscience Scottsdale, AZ, USA
Molly Havard Blau, MD, MS University of Washington Seattle, WA, USA
Maria L. Boccia, PhD, LM
Baylor University Robbins College of Health and Human Sciences Waco, TX, USA
Adrienne A. Boire, MD, PhD Memorial Sloan Kettering Cancer Center New York, NY, USA
Cameron Brimley, MD Geisinger Health Danville, PA, USA
Alyssa Callela, PA-C Geisinger Health Danville, PA, USA
Jennifer L. Clarke, MD, MPH University of California San Francisco, CA, USA
Andrew R. Conger, MD, MS Geisinger Health Danville, PA, USA
Scott L. Coven, DO, MPH
Riley Hospital for Children at IU Health Indianapolis, IN, USA
Kara L. Curley, PA-C Mayo Clinic Phoenix, AZ, USA
Logan S. DeWitt, DO Indiana University School of Medicine Indianapolis, IN, USA
Radhika Dhamija, MD Mayo Clinic Phoenix, AZ, USA
Richard S. Dowd, MD, MS Tufts Medical Center Boston, MA, USA
Reginald Fong, MD Geisinger Health Danville, PA, USA
Ekokobe Fonkem, DO
Medical College of Wisconsin Milwaukee, WC, USA
Morgan Freret, MD, PhD
Memorial Sloan Kettering Cancer Center New York, NY, USA
Shobhit Kumar Garg, MD, FRCR William Harvey Hospital, East Kent Hospitals University Foundation Trust Ashford, Kent, UK
Na Tosha N. Gatson, MD, PhD, FAAN Banner MD Anderson Cancer Center Phoenix, AZ, USA
Geisinger Commonwealth School of Medicine Scranton, PA, USA
Pierre Giglio, MD The Ohio State University Columbus, OH, USA
Michael Glantz, MD Penn State College of Medicine - Milton S. Hershey Medical Center Hershey, PA, USA
Andrew J. Gogos, MBBS, FRACS
St Vincents Hospital Melbourne Fitzroy, AU
David Gritsch, MD
Mass General Brigham Boston, MA, USA
Simon Gritsch, MD, PhD Mass General Brigham Boston, MA, USA
Lia M. Halasz, MD University of Washington Seattle, WA, USA
Shawn L. Hervey-Jumper, MD University of California San Francisco San Francisco, CA, USA
Joseph M. Hoxworth, MD Mayo Clinic Phoenix, AZ, USA
Maya Hrachova, DO University of Oklahoma Oklahoma City, OK, USA
Valeria Internò, MD University of Bari Bari, Italy
Rajan Jain, MD
New York University Grossman School of Medicine New York, NY, USA
George I. Jallo, MD
Johns Hopkins All Children’s Hospital St. Petersburg, FL, USA
Thomas J. Kaley, MD Memorial Sloan Kettering Cancer Center New York, NY, USA
Matthias Karajannis, MD, MS Memorial Sloan Kettering Cancer Center New York, NY, USA
John M. Kindler, MD
Indiana University School of Medicine Indianapolis, IN, USA
Peter Kobalka, MD Ohio State University Columbus, OH, USA
Ibrahim Kulac, MD Koç University School of Medicine Istanbul, Turkey
Sylvia C. Kurz, MD, PhD Eberhard Karls University Tübingen, Germany
Erika N. Leese, PA-C Geisinger Health Danville, PA, USA
Mary Jane Lim-Fat, MD, MSc Sunnybrook Health Sciences Toronto, ON, Canada
Rimas V. Lukas, MD Northwestern University Chicago, IL, USA
Alireza Mansouri, MD, MSc, FRCSC Penn State Health Hershey, PA, USA
Aaron Mochizuki, DO
Cincinnati Children’s Hospital Medical Center Cincinnati, OH, USA
Jamal Mohamud, DO Indiana University School of Medicine Indianapolis, IN, USA
Ramin A. Morshed, MD University of California San Francisco San Francisco, CA, USA
Maciej M. Mrugala, MD, PhD, MPH, FAAN Mayo Clinic Phoenix, AZ, USA
Lakshmi Nayak, MD
Dana-Farber Cancer Institute, Harvard Medical School Boston, MA, USA
Matthew T. Neal, MD, MBA Mayo Clinic Phoenix, AZ, USA
Kathryn S. Nevel, MD Indiana University School of Medicine Indianapolis, IN, USA
Theodore Nicolaides, MD Caris Life Sciences New York, NY, USA
Mohammad Hassan A. Noureldine, MD, MSc University of South Florida Tampa, FL, USA
Samanthalee C. S. Obiorah, BSc Warren Alpert Medical School of Brown University Providence, RI, USA
Shirley Ong, MD
The Ohio State University Wexner Medical Center Columbus, OH, USA
Sonia Partap, MD
Stanford University & Lucile Packard Children’s Hospital Palo Alto, CA, USA
David M. Peereboom, MD Cleveland Clinic Cleveland, OH, US
Melike Pekmezci, MD University of California San Francisco, CA, USA
Katherine B. Peters, MD, PhD, FAAN Duke University School of Medicine Durham, NC, USA
Sheetal Phadnis, MD University of Alabama Birmingham, AL, USA
Scott R. Plotkin, MD, PhD Massachusetts General Hospital Boston, MA, USA
Vinay K. Puduvalli, MD UT MD Anderson Cancer Center Houston, TX, USA
Appaji Rayi, MD Charleston Area Medical Center Health System Charleston, WV, USA
Alexander Ropper, MD Baylor College of Medicine Houston, TX, USA
Roberta Rudà, MD University of Turin Turin, Italy
Sameer Farouk Sait, MBBS Memorial Sloan Kettering Cancer Center New York, NY, USA
David Schiff, MD University of Virginia Health System Charlottesville, VA, USA
Nir Shimony, MD St. Jude Children’s Hospital Memphis, TN, USA Johns Hopkins University SoM Baltimore, MD, USA
Kevin Shiue, MD Indiana University School of Medicine Indianapolis, IN, USA
Wayne Slone, MD
Ohio State University Columbus, OH, USA
Riccardo Soffietti, MD, PhD University Hospital of Turin Turin, Italy
Kun-Wei Song, MD Massachusetts General Hospital / Dana-Farber Cancer Institute Boston, MA, USA
Ramya Tadipatri, MD Banner MD Anderson Cancer Center Gilbert, AZ, USA
Hirokazu Takami, MD, PhD University of Tokyo Tokyo, Japan
Kerianne R. Taylor, RN Banner MD Anderson Cancer Center Phoenix, AZ, USA
Steven A. Toms, MD, MPH Brown University / Lifespan Health System Providence, RI, USA
Cristina Valencia-Sanchez, MD, PhD Mayo Clinic Scottsdale, AZ, USA
Terrence Verla, MD
Baylor College of Medicine Houston, TX, USA
Gordon A. Watson† , MD, PhD Indiana University School of Medicine Indianapolis, IN, USA
Jessica A. Wilcox, MD Memorial Sloan Kettering Cancer Center New York, NY, USA
Terri L. Woodard, MD, MPH Baylor University College of Medicine Houston, TX, USA UT MD Anderson Cancer Center Houston, TX, USA
† Deceased
PART I. | neURo-oncoLoGY oVeRVIeW
1 | NEUROPATHOLOGY PRIMER
IBRAHIM KULAC AND MELIKE PEKMEZCI
IntRoDUctIon
Tumors of the central nervous system (CNS) have been traditionally classified based on their histomorphologic features and by presumed cell of origin. Histologic grading has been across tumor entities, and each tumor is assigned to a grade as part of the nomenclature. Histologic grading is only one of the prognostic criteria, and a tumor with low histologic grade may behave aggressively depending on the location, patient’s performance status, radiographic features, proliferation index, and, more recently, genetic changes.
The revised fourth edition (2016) of the World Health Organization (WHO) classification introduced a combined histologic and molecular classification (integrated diagnosis), which dramatically changed the nomenclature.2 But this updated classification and grading scheme has already become insufficient, given our rapidly growing understanding of molecular features and numerous new techniques including methylation profiling. For this purpose, a group of experts gathered to create cIMPACT-NOW (Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy—Not Official WHO), which has already published multiple update reports, and many of these updates are expected to be incorporated into the imminent fifth edition of the WHO classification (2021).3 Important changes in the upcoming classification include designation of WHO grade using Arabic numerals, rather than Roman numerals (which practice we have adopted for this volume) and reserving the term “glioblastoma” for isocitrate dehydrogenase (IDH)-wildtype diffuse astrocytomas with histologic or molecular features of grade 4, while all IDH-mutant astrocytomas will be diagnosed as such with accompanying histologic grade (2–4). In this chapter we cover the most common CNS tumors in a fairly detailed manner and briefly touch base with the relatively rare ones using the current diagnostic criteria and terminology, some of which may change in the near future.
GLIAL tUMoRs
CNS glial tumors are a diverse group of neoplasms originating from the supportive cellular elements of CNS. Glial tumors can be classified as diffuse (infiltrative) or solid glial tumors by their growth pattern.
Glial tumors can be classified as diffuse (infiltrative) or solid by their growth pattern.
DIFFUSE GLIAL TUMORS
The mainstay of the classification of adult diffuse gliomas is the presence of IDH1/IDH2 mutations, which is associated with concerted CpG island methylation at many gene loci (G-CIMP phenotype) and with better outcome.
Editors’ Note: This book is being published just as the new 2021 World Health Organization (WHO) classification of CNS tumors is being released.1 While this chapter is largely based on the 2016 classification, significant impending changes are noted where appropriate. 4 For the most up-to date information, please see: WHO Classification of Tumours; 5th Edition. Central Nervous System Tumours. Edited by the WHO Classification of Tumours Editorial Board. International Agency for Research on Cancer 2021.
Diffuse gliomas (Chapter 4) have an infiltrative growth pattern, in which neoplastic glial cells diffusely infiltrate into the existing neuropil structures of the gray and white matter without a sharp border between the tumor and the adjacent parenchyma. Earlier classifications of diffuse gliomas relied only on the morphologic features; however, the 2016 WHO classification of diffuse glial tumors has had a major update, including the addition of certain genetic alterations to the diagnostic criteria, thus providing an integrated diagnosis.2 Currently, the mainstay of the classification is the presence of IDH1/IDH2 mutations, which is associated with concerted CpG island methylation at many gene loci (G-CIMP phenotype) and with better outcome. Furthermore, regardless of the morphologic features, tumors with IDH mutation and 1p/19q co-deletion are categorized as oligodendroglioma, while the rest are diagnosed as astrocytoma. While this classification addresses many important issues and provides an increased level of objectivity, its application to pediatric diffuse gliomas has been somewhat limited. Pediatric-type diffuse gliomas rarely harbor IDH mutations, and their molecular features overlap significantly with solid glial and glioneuronal tumors. Those with oligodendroglial morphology often harbor FGFR1 alterations, and astrocytomas often show MYB/MYBL1 rearrangements.5 And, unlike adult counterparts, pediatric high-grade gliomas frequently harbor H3 G34 mutations, and infantile gliomas are frequently associated with NTRK, ALK, or ROS fusions.6–8 The upcoming WHO classification will incorporate molecular alterations of pediatric-type diffuse gliomas
and address the role of molecular alterations in grading of diffuse gliomas.
Pediatric type diffuse gliomas rarely harbor IDH mutations, and their molecular features overlap significantly with solid glial and glioneuronal tumors.
D IFFUSE A STROCYTOM A , IDH-M UTANT ,
WHO G RADE 2
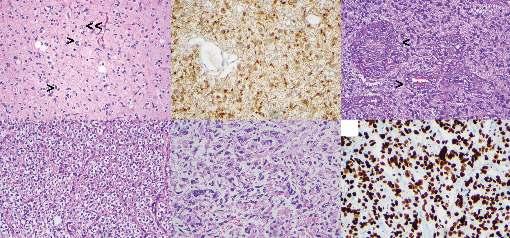
Diffuse astrocytomas are composed of moderately pleomorphic cells with hyperchromatic, oval to round nuclei with irregular nuclear contours interspersed in the parenchyma where neurons or axonal fibers are visible between tumor cells (see Figure 1.1A). Cellularity may vary but often it is moderately increased compared to normal parenchyma. Mitoses are absent or rare. Necrosis or microvascular proliferation is not seen. By definition, these tumors have a mutation in either IDH1 or IDH2, and the most common mutation is the R132H mutation in IDH1 9 Concurrent mutations in ATRX and TP53 are typical features of IDH-mutant diffuse astrocytomas.10 Immunohistochemistry (IHC) is almost always very helpful for diagnosis. Antibody against IDH1 R132H mutant protein shows diffuse cytoplasmic positivity in majority of the tumors, as seen in Figure 1.1B, although it is negative in tumors with non-canonical IDH mutations. Therefore, sequencing for other IDH1 and IDH2 mutations may be necessary. Loss of nuclear ATRX expression by IHC suggests presence of an ATRX mutation and can be used to strongly argue against an oligodendroglioma. While strong and diffuse p53 nuclear
staining is suggestive of a TP53 mutation, the sensitivity and specificityaswellastheidealcutoffvalueofp53stainingarestill debated. Tumor cells are variably positive with GFAP, OLIG2, SOX10, and MAP2, none of which is specific for the diagnosis. Neurofilament protein is useful to identify the entrapped axonal fibers, confirming the infiltrative growth pattern.
A NAPLA STIC
A STROCYTOM A , IDH-M UTANT , WHO G RADE 3
Anaplastic astrocytomas either arise from a grade 2 diffuse astrocytoma or, more frequently, are diagnosed de novo. Histomorphological features are somewhat similar to diffuse astrocytoma, IDH-mutant, WHO grade 2, but they tend to have more prominent cytologic atypia, increased cellularity, and elevated mitotic activity. Necrosis and microvascular proliferation are not seen. Increased number of mitoses is the key characteristic feature of anaplastic astrocytoma, IDH-mutant, WHO grade 3; however, no strict cutoff is set for the number of mitosis for the diagnosis, and mitotic activity should be assessed in the context of specimen size. Molecular characteristics of these tumors are also similar to their grade 2 counterpart, including mutations in IDH1 or IDH2, ATRX, and TP53 genes, but with more frequent copy number alterations. There is ongoing debate about the utility of the current grading system since some studies showed no significant difference in prognosis between histologic grade 2 and 3 astrocytomas while others did so.11–13 Future studies may refine mitotic thresholds and may identify additional genetic alterations associated with more aggressive clinical behavior to establish better grading criteria.
Figure 1.1 Infiltrating glial tumors. (A) Diffuse astrocytoma, WHO grade 2: Note atypical tumor cells with irregular nuclear borders (>) diffusely infiltrating to background cortex evidenced by entrapped cortical neurons (>>), Hematoxylin & eosin (H&E). (B) Immunohistochemistry for isocitrate dehydrogenase (IDH)-1 R132H mutant protein. (C) Glioblastoma, WHO grade 4: Note multilayered endothelial cells referred to as microvascular proliferation (>), H&E. (D) Oligodendroglioma, IDH-mutant and 1p/19q-co-deleted, WHO grade 2: Note fine, chicken-wire vasculature and perinuclear halos giving cells a fried-egg appearance, H&E. (E) Diffuse midline glioma, H3 K27M-mutant, WHO grade 4: Note that the absence of microvascular proliferation or necrosis does not preclude the designation of WHO grade 4, H&E. (F) Immunohistochemistry for H3 K27M mutant protein in diffuse midline glioma.
G LIOBL A STOM A , IDH-M UTANT , WHO G RADE 4
IDH mutant glioblastoma comprises approximately 10% of all glioblastomas and either arises from a preexisting low-grade astrocytoma or develops de novo. These tumors are composed of pleomorphic cells with increased mitotic activity and contain microvascular proliferation and/or areas of necrosis, oftenpalisading necrosis (see Figure 1.1C). Presence of an IDH mutation can be demonstrated by IDH1 R132H IHC in most cases.14 ATRX loss and p53 positivity are also common findings similar to other IDH-mutant astrocytomas. IDH1 and IDH2 sequencing should be performed for IDH1 R132H IHC-negative glioblastomas if there is a history of a lower grade glioma or if a patient is younger than 55 years at the time of diagnosis and additional IHC studies suggeststrongassociationwithanIDHmutation(i.e.,ATRXloss).
While the morphologic features of IDH-mutant and IDH-wildtype glioblastomas are essentially identical, their molecular features and clinical outcomes are significantly different.15,16 Recent studies demonstrated that homozygous deletion of CDKN2A/B in IDH-mutant diffuse gliomas without microvascular proliferation or necrosis is associated with a disease course at least as adverse as a histologically defined IDH-mutant glioblastoma.17 Based on a recent consensus paper, there is a consideration to change the grading scheme of IDH-mutant diffuse gliomas, including assigning grade 4 to tumors with CDKN2A/B loss regardless of presence of microvascular proliferation or necrosis, and limiting the term “glioblastoma” to IDH-wildtype tumors,18 so that these tumors will instead be named astrocytoma, IDH-mutant, WHO grade 4.
G LIOBL A STOM A , IDH-W ILD TYPE , WHO G RADE 4
Thevastmajorityofhigh-gradegliomasinolderadultsareIDHwildtype glioblastomas (Chapter 3). They are characterized by highly atypical, pleomorphic cells with frequent mitoses, necrosis, and/or microvascular proliferation, similar to their IDHmutant counterparts (see Figure 1.1C). Chromosome 7p gain in combination with 10q loss is the most frequent chromosomal alteration in glioblastoma. Frequent molecular alterations include TERT promoter mutation, CDKN2A/B homozygous deletion, EGFR amplification, PTEN truncating mutations, and other alterations in p53/MDM2, CDKN2A/RB1, and receptor tyrosine kinase pathways.19 MGMT (O6-methylguanine-DNA methyltransferase) promoter methylation, which is predictive of response to alkylating agents such as temozolomide, is seen in a subset of glioblastomas; however, it is more common in IDH-mutant glioblastomas that have a G-CIMP phenotype.20
IDH-wildtype glioblastomas are characterized by highly atypical, pleomorphic cells with frequent mitoses, necrosis, and/or microvascular proliferation.
There are a number of histomorphologic variants, some of which are associated with unique molecular alterations or clinical behavior. Giant cell glioblastoma is predominantly composed of highly atypical, multinucleated or mononuclear giant cells and harbors frequent TP53 mutations. Small cell glioblastoma is relatively monomorphic, with bland morphology mimicking a low-grade glioma but harbors frequent
EGFR amplification and other genetic features of glioblastoma. Epithelioid glioblastoma has a dominant population of closely packed epithelioid, sometimes rhabdoid cells; shows significant morphologic and molecular overlap with anaplastic pleomorphic xanthoastrocytoma (PXA); and nearly half of them harbor BRAF V600E mutation. Gliosarcoma is another wellrecognized morphological variant that has areas resembling various sarcomas, most often undifferentiated sarcoma, previously referred as fibrosarcoma. Similar to IDH-mutant glioblastoma, tumor cells are positive for glial markers (GFAP, OLIG2, SOX10). IHC for IDH1 R132H is negative. Nuclear ATRX expression is retained. Ki67 proliferation index is high.
Gliosarcoma is another well-recognized morphological variant that has areas resembling various sarcomas, most often undifferentiatedsarcoma,previouslyreferredas fibrosarcoma
O LIGODENDROG LIOM A , IDH-M UTANT , 1 P /19
The classic morphologic features of oligodendroglioma includes an infiltrative tumor composed of cells with round nuclei with a crisp, fine chromatin structure on a background of delicate, chicken-wire vasculature. Due to the processing steps in routine histopathology, the cytoplasm looks clear, which gives the cells a classic fried egg appearance on hematoxylin and eosin (H&E)-stained sections, as seen in Figure 1.1D. So-called secondary structures represent accentuation of the tumor cells around neurons (perineuronal satellitosis) and blood vessels and in subpial regions. Microcalcifications and myxoid degeneration is common. Tumor cells with eccentric, eosinophilic cytoplasm are referred to as minigemistocytes, which can be quite numerous. Occasional mitoses can be seen, but the tumors do not show a brisk mitotic activity.
The classic morphologic features of oligodendroglioma includes an infiltrative tumor composed of cells with round nuclei with a crisp, fine chromatin structure on a background of delicate, chicken-wire vasculature.
By definition, all oligodendrogliomas are IDH-mutant; therefore, IDH1 R132H stain is positive in almost all cases. Cases with histomorphologic features of an oligodendroglioma but negative IDH1 R132H stain should be further tested for non-canonical IDH mutations by sequencing. Similarly, all oligodendrogliomas harbor co-deletion of entire chromosome arms of 1p and 19q, which can be shown by fluorescence in-situ hybridization (FISH), array comparative genomic hybridization (aCGH) or next-generation sequencing, although sensitivity and specificity may vary.2 Frequent alterations include mutations in FUBP1 and CIC genes, located on chromosomes 1p and 19q, respectively. Oligodendrogliomas frequently harbor TERT promoter mutations and are ATRX-wildtype; therefore, they show retained ATRX nuclear staining by IHC.10 Likewise, diffuse strong p53 staining is not expected as TP53 mutations are not common in oligodendrogliomas. Tumor cells are usually positive with OLIG2, SOX10, and MAP2,
Q
C O -D ELETE D , WHO G RADE 2
none of which is specific for the diagnosis. GFAP staining is usually limited and more often seen in minigemistocytes or occasional tumors with astrocytic morphology.
A NAPLA STIC O LIGODENDROG LIOM A , IDH-M UTANT , 1 P /19 Q C O -D ELETE D , WHO G RADE 3
Anaplastic oligodendroglioma arises either de novo or by progression of a grade 2 oligodendroglioma. Anaplastic oligodendrogliomacanbedefinedasatumorwithhighcellularity, brisk mitotic activity (>6 mitosis per 10 high-power field [HPF}), and/or microvascular proliferation and/or necrosis that shows the classic molecular oligodendroglioma signature (IDH mutation and 1p/19q co-deletion). Anaplastic oligodendrogliomas are prominently more cellular than grade 2 oligodendrogliomas but may still show classic oligodendroglioma morphology with round nuclei, perinuclear halos, and chicken-wire vasculature. The highest grade assigned for a tumor of oligodendroglial lineage is grade 3, per WHO 2016.2 Molecular features of anaplastic oligodendroglioma are similar to grade 2 oligodendroglioma, with increased copy number alterations and more frequent CDKN2A loss.Inadditiontodefiningmolecularalterations(IDH mutation and 1p/19q codeletion), anaplastic oligodendrogliomas also typically harbor TERT promoter mutations.
D IFFUSE M ID LINE G LIOM A , H3 K27M-M UTANT , WHO G RADE 4
As the name specifies, diffuse midline glioma, H3 K27Mmutant (Chapter 18), is an infiltrative glioma, typically of astrocytic morphology, involving midline structures such as thalamus, basal ganglia, and spinal cord, which harbors a K27M mutation involving histone H3.3 (H3F3A) or H3.1 (HIST1H3B/C) genes. Tumors often consist of monomorphic, small glial cells and frequently exhibit features of high-grade glioma, as seen in Figure 1.1E.21 However, paucicellular tumors without mitoses, necrosis, or microvascular proliferation are not uncommon. Regardless of the histologic features, these tumors are assigned WHO grade 4. Additional molecular alterations include frequent mutations in TP53, PTEN, ATM, PPM1D, and CHEK2 22,23 ACVR1 mutations and FRGR1 rearrangements seem to correlate with the type of histone mutation and tumor location.24 These tumors are diffusely positive for H3K27M antibody that reacts with both mutant protein products of either H3.1 or H3.3 genes, as seen in Figure 1.1F.25 Mutation leads to inhibition of PRC2 complex, resulting decreased levels of trimethylation at the lysine 27 mark of histone 3 (H3K27me3), which can also be detected by IHC.
Regardless of the histologic features, diffuse midline glioma, H3 K27M mutant, is assigned WHO grade 4.
D IFFUSE H EMISP H ERIC G LIOM A , H3 G34-M UTANT
H3 G34-mutant gliomas (Chapter 18) are defined by their molecular alteration but show variable histology.26,27 It is not yet accepted as a distinct entity by the WHO 2016 classification, but it is often seen in cerebral hemispheres of young adults. Most commonly, they have a biphasic appearance, with a glial component characterized by a fibrillary background and atypical, pleomorphic cells, and areas of embryonal features
which sometimes may be the dominant feature. Because of the embryonal features, in the past, these tumors were diagnosed as glioblastoma, with primitive neuronal features, or as a CNS embryonal tumor. They are almost always high grade and often have morphological hallmarks of a high-grade glial tumor such as microvascular proliferation and/or palisading necrosis which is very helpful for the correct diagnosis. A GFAPpositive, OLIG2-negative hemispheric tumor with loss of nuclear ATRX staining and diffuse p53 staining in the absence of IDH mutations is highly suggestive of a H3 G34 mutation.
D IFFUSE A STROCYTIC G LIOM A , IDH-W ILD TYPE
Although diffuse astrocytoma, IDH-wildtype, WHO grade 2 and anaplastic astrocytoma, IDH-wildtype, WHO grade 3 have been includedinthe2016WHOclassification,theyhavebeenreferredto as “provisional entities.” It is well-documented that IDH-wildtype astrocytomas have significantly worse outcome than IDH-mutant astrocytomas, and some tumors show a disease course similar to IDH-wildtype glioblastomas. Multiple studies showed that this group may include diffuse gliomas with distinct molecular alterations other than IDH mutations, such as H3 K27M or H3 G34R mutations, and these tumors should now be diagnosed as such. Recent cIMPACT-NOW updates have recommended additional molecular testing to identify these tumors, which are expected to behave aggressively, and the group uses the terminology “diffuse astrocytic glioma, IDH-wildtype with molecular features of glioblastoma WHO grade 4.”19 Molecular features suggesting aggressive behavior in this setting includes EGFR amplification, TERT promoter mutation, and gain of chromosome 7q with concurrent loss of chromosome 10p.
Molecular features suggesting aggressive behavior in IDHwildtype astrocytoma include EGFR amplification, TERT promoter mutation, and gain of chromosome 7q with concurrent loss of chromosome 10p.
A STROCYTOM A , N OT O THERWISE S PECIFIE D (NOS) AND O LIGODENDROG LIOM A , NOS
These tumors have the morphological features of an astrocytoma or an oligodendroglioma but the molecular workup needed for definitive integrated diagnosis cannot be completed. In this setting, the WHO 2016 classification recommends that pathologists use these diagnostic categories. Although the diagnostic categories for oligoastrocytoma and anaplastic oligoastrocytoma, NOS, still exist in the WHO guidelines, these categories should only be used when further testing will not or cannot be completed.28 Histologic features, corresponding molecular alterations and integrated diagnosis pathways in diffuse adult and pediatric gliomas are summarized in Figures 1.2 and 1.3.
SOLID GLIAL TUMORS
P ILOCYTIC A STROCYTOM A , WHO G RADE 1
Pilocytic astrocytoma is the most common glioma in children but can also be seen in adults of all ages. Although clinical and radiological features of pilocytic astrocytomas are
Histologic features
IDH1/IDHZ status
1p/19q status
Frequent alterations
Histologic grade
Alterations associated with molecular grading
DIAGNOSIS
Other diagnoses to consider and rule out
oligodendroglioma
1p/19q-codeleted
CIC and/or FUBP1 mutation TERT promoter mutation
Grades 2 and 3
Diffuse glioma (adult type)
Astrocytoma
IDH-mutant
1p/19q-Intact
ATRX-mutant TP53-mutant
Grades 2 and 3
No CDKN2A/B homozygous deletion
(Anaplastic)
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted, WHO grade 2 (or 3)
CDKN2A/B homozygous deletion
(Anaplastic) Astrocytoma, IDH-mutant, WHO grade 2 (or 3)
Grade 4Grade 4
Astrocytoma, IDH-mutant, WHO grade 4*
Glioblastoma, IDH-wildtype, WHO grade 4
IDH-wildtype
1p/19q-Intact
Grades 2 and 3
TERT promoter mutation
EGFR amplification
Polysomy 7/Monosomy 10
Pediatric-type gliomas, especially in younger adults
None
(Anaplastic) Astrocytoma, IDH-wildtype, WHO grade 2 (or 3)
Non-infiltrating gliomas
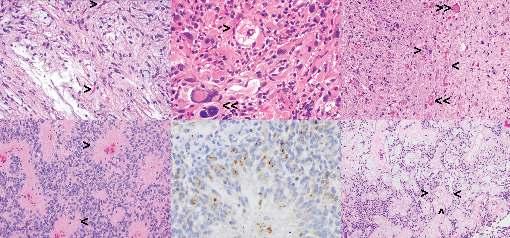
often pathognomonic for the diagnosis, histomorphology can be challenging since they have a wide range of tissue patterns. Most commonly pilocytic astrocytomas have a biphasic growth pattern with relatively compact areas composed of bipolar glial cells with long, hair-like (piloid) processes and loosely arranged microcystic areas composed of bland, glial cells with small nuclei and short, cobweb-like processes in a variably myxoid background (see Figure 1.4A). Rosenthal
Figure 1.2 Summary of classification of adult type diffuse gliomas. Location
H3 K27M status
Age group
H3 G34 status
Histologic grade
Additional mutations
Other alterations
DIAGNOSIS
H3 K27M-mutant
Diffuse glioma (pediatric type)
H3 K27M-wildtype
High-grade
ACVR1
FGFR1/NF1
TP53, ATRX
Diffuse midline gioma, H3 K27M-mutant, WHO grade 4
Other diagnoses to rule out and syndromic associations
Figure 1.4 Solid glial, glioneuronal, and ependymal tumors. (All except panel E are hematoxylin & eosin [H&E] stained sections.) (A) Pilocytic astrocytoma, WHO grade 1: Note bland bipolar cells, microcystic background, and Rosenthal fibers (>). (B) Pleomorphic xanthoastrocytoma, WHO grade 2: Note mixture of spindle cells, large atypical cells with smudgy irregular nuclei (>), and large cells with foamy (xanthomatous) cytoplasm (>>). (C) Ganglioglioma, WHO grade 1: Note large, dysmorphic ganglion cells (>) in the background of bland spindled glial cells and numerous eosinophilic granular bodies (>>). (D) Ependymoma, WHO grade 2: Note perivascular fibrillary anucleate zones, referred to as perivascular pseudorosettes (>). (E) Epithelial membrane antigen (EMA) immunohistochemical stain shows dot-like paranuclear staining in ependymomas. (F) Myxopapillary ependymoma, WHO grade 1: Note papillary structures containing a central vessel with mucoid degeneration surrounded by small ependymal cells (>) in the myxoid background.
fibers (intracytoplasmic, thick, and long eosinophilic fibers) are often seen in compact areas, and eosinophilic granular bodies (EGBs) are prominent in microcystic areas. Atypical cells with naked nuclei can be seen, especially at the periphery of the lesion, raising the differential diagnosis of a diffuse astrocytoma. Oligodendroglioma-like cytomorphology can be prominent in other cases. Tumors may show degenerative atypia as well as multinucleated cells.
Most commonly pilocytic astrocytomas have a biphasic growth pattern with relatively compact areas composed of bipolar glial cells with long, hair-like (piloid) processes and loosely arranged microcystic areas composed of bland, glial cells with small nuclei and short, cobweb-like processes in a variably myxoid background.
Pilocytic astrocytomas are highly vascular and may show complex glomeruloid vascular structures, especially lining the tumoral cyst walls. Necrosis can be focal or extensive and is usually an infarct type as opposed to a palisading type; therefore, this should not be interpreted as an alarming finding. Likewise, mitosis can be seen and may be quite frequent. While the clinical significance is not fully established, mitotic counts in excess of 4 mitoses per 10 HPFs associated with palisading necrosis may indicate a potential for more aggressive behavior, and these tumors are referred to as anaplastic pilocytic astrocytoma, without an officially designated WHO grade.
Pilocytic astrocytomas typically harbor alterations activating the mitogen-activated protein (MAP) kinase pathway, most frequent of which is a tandem duplication of BRAF resulting KIAA1549-BRAF fusion.29 Other typical alterations include BRAF V600E, KRAS, and NF1 mutations, and FGFR1 and RAF1 fusions. Presence of additional alterations such as CDKN2A/B homozygous deletion, ATRX mutation, or TERT promoter mutation seems to be associated with more aggressive behavior.30
Tumor cells are positive for GFAP, OLIG2, S100, and SOX10. Synaptophysin often shows weak cytoplasmic staining, and this should not be interpreted as infiltrative growth pattern or evidence for a neuronal component. Neurofilament is useful to demonstrate well-demarcated, solid growth pattern with scarce entrapped axons at the periphery. However, it should be kept in mind that rare examples, especially in cerebellum, may show an extensive infiltrative pattern. Because these tumors do not harbor IDH mutations, IDH1 R132H stain is expected to be negative. Although Ki67 proliferation index is typically low (<5%), some cases may reveal a quite high index.
PILOMYXOID ASTROCYTOMA
Pilomyxoid astrocytoma (PMA) is considered a variant of pilocytic astrocytoma, with additional, somewhat characteristic features. These tumors tend to localize in the thalamic or hypothalamic region and are seen in earlier ages compared to pilocytic astrocytomas.31,32 PMAs are solid tumors composed of monomorphous, bipolar cells with fusiform nuclei and long glial processes in a homogenous myxoid background. Tumor
cells are typically arranged around blood vessels, forming pseudorosettes. Immunohistochemical and molecular features are almost identical to pilocytic astrocytomas, with frequent MAP kinase pathway alterations.33 Current literature emphasizes a worse prognosis for PMAs compared to pilocytic astrocytomas; however, a definite grade has not been assigned.
P LEOMORP HIC X ANTHOA STROCYTOM A , WHO G RADE 2
Pleomorphic xanthoastrocytoma (Chapter 10) is a wellcircumscribed glial neoplasm with a frequent cystic component and is typically located superficially with leptomeningeal extension. It is characterized by large, often multinucleated cells with abundant foamy cytoplasm (referred as xanthomatous cells), which may show variable nuclear atypia, and a frequent spindle cell component with abundant pericellular reticulin (see Figure 1.4B). EGBs and perivascular lymphocytic infiltrate are frequent, but not specific for diagnosis. These tumors are categorized as WHO grade 2 with mitotic activity of less than 5 mitoses per 10 HPFs by definition. Necrosis is not a common finding, but its presence is not sufficient for a WHO grade 3 designation.
Typical molecular alterations include BRAF V600E mutation and concurrent CDKN2A homozygous deletion, which are present in more than half of the cases. Noncanonical BRAF mutations, BRAF and RAF1 fusions, and complex chromosomal copy number changes have been also reported.34 Tumor cells are positive for glial (GFAP, OLIG2) and occasional neuronal (MAP2, synaptophysin) markers. CD34 is also commonly positive in PXAs. BRAF V600E mutation can be demonstrated with a mutation-specific antibody using IHC. There is correlation with CDKN2A homozygous deletion and loss of p16 IHC expression, which can be useful in the diagnostic workup of these cases.
Typical molecular alterations in PXA include BRAF V600E mutation and concurrent CDKN2A homozygous deletion.
A NAPLA STIC P LEOMORP HIC X ANTHOA STROCYTOM A ,
WHO G RADE 3
Anaplastic PXA shares morphologic similarities with PXA, WHO grade 2, but, by definition, anaplastic PXA has a mitotic rate of 5 or more mitoses per 10 HPFs. While necrosis is common, its significance in the absence of mitotic activity is unclear. Molecular features also overlap with PXA and include combination of CDKN2A homozygous deletion with BRAF or RAF1 alterations. Recent studies suggested that anaplastic transformation may be due to accumulation of additional copy number changes and/or TERT gene alterations (amplification or promoter mutation).35
S UBEPEN DYMAL G IANT C ELL A STROCYTOM A ,
WHO G RADE 1
Subependymal giant cell astrocytoma (SEGA) is a benign tumor arising in the wall of the lateral ventricle, composed of large, ganglion/gemistocyte-like cells and variable amounts of spindle cells arranged in sweeping fascicles. Considerable pleomorphism and multinucleated giant cells are common and have no impact on prognosis. Vascular-rich stroma with
calcifications is also a common finding. Although a significant number of mitoses can be evident, this does not have an impact on the grade or outcome. Tumor cells are positive for GFAP and S100 and may show focal expression of neuronal markers such as NeuN. SEGAs are seen in the setting of tuberous sclerosis and show biallelic inactivation of TSC1 or TSC2 genes.
SEGAs are seen in the setting of tuberous sclerosis and show biallelic inactivation of TSC1 or TSC2 genes.
neURonAL AnD GLIoneURonAL tUMoRs
GANGLIOGLIOMA, WHO GRADE 1
Gangliogliomas (Chapter 10) are glioneuronal neoplasms composed of dysplastic ganglion cells (clustering, cytomegaly, binucleation, abnormal Nissl substance) intermixed with neoplastic glial cells with variable morphology resembling fibrillary astrocytomas, oligodendrogliomas, or pilocytic astrocytomas and are WHO grade 1 by definition (see Figure 1.4C). Anaplastic gangliogliomas (WHO grade 3) usually have increased cellularity and mitoses, necrosis, and microvascular proliferation.36 The glial component stains with glial markers (GFAP, OLIG2, SOX10). The dysplastic ganglion cells are positive with synaptophysin, show variable staining with NeuN, and show abnormal perikaryonic staining with neurofilament. They harbor solitary alterations activating MAP kinase pathways (BRAF p.V600E mutation and other mutations and fusions in BRAF, FGFR1, and FGFR2 genes).37,38 BRAF V600E mutationspecific stain can be helpful in the diagnosis.39 Additional genetic alterations (homozygous deletion of CDKN2A and DMBT1, gain/amplification of CDK4) are associated with anaplasia.40
DYSEMBRYOPLASTIC NEUROEPITHELIAL TUMOR, WHO GRADE 1
Dysembryoplastic neuroepithelial tumor (DNET) is a lowgrade glioneuronal tumor with nodular growth and is typically composed of axonal bundles perpendicular to pial surface (columns) lined by oligodendrocyte-like bland cells embedded in a mucoid matrix, which contains scattered “floating neurons.”41 Reported molecular alterations vary between studies and include FGFR1 kinase domain tandem duplications or missense mutations in more than 80% and BRAF p.V600E mutations in 30% of cases.38,42
OTHER NEURONAL AND GLIONEURONAL TUMORS
Multinodular and vacuolating neuronal tumor (MVNT) is a low-grade neuronal neoplasm with prominent nodules composed of small to medium-sized neuronal cells demonstrating prominent intracytoplasmic and stromal vacuolation.43 The tumor cells are positive with some glial and neuronal markers (OLIG2, synaptophysin) but negative with others (GFAP,