Immediate download The oxford handbook of neuronal ion channels arin bhattacharjee ebooks 2024
The Oxford Handbook of Neuronal Ion Channels Arin Bhattacharjee
Visit to download the full and correct content document: https://ebookmass.com/product/the-oxford-handbook-of-neuronal-ion-channels-arin-b hattacharjee/
More products digital (pdf, epub, mobi) instant download maybe you interests ...
Ion channels as therapeutic targets. Part A 1st Edition
Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and certain other countries.
Published in the United States of America by Oxford University Press 198 Madison Avenue, New York, NY 10016, United States of America.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by license, or under terms agreed with the appropriate reproduction rights organization. Inquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above.
You must not circulate this work in any other form and you must impose this same condition on any acquirer.
CIP data is on file at the Library of Congress
ISBN 978–0–19–066916–4
DOI: 10.1093/oxfordhb/9780190669164.001.0001
Printed by Integrated Books International, United States of America
Preface vii
Editor’s Bio ix
List of Contributors xi
SECTION 1 BASIC PRINCIPLES
1. Excitable Membrane Properties of Neurons 3
Leonard K. Kaczmarek
2. Ion Channel Permeation and Selectivity 33
Juan J. Nogueira and Ben Corry
3. Gating of Ion Channels 64
Rene Barro-Soria
SECTION 2 VOLTAGE- GATED CHANNELS
4. The Voltage-Dependent K+ Channel Family
Hanne B. Rasmussen and James S. Trimmer
5. Potassium Channel Mutations in Epilepsy
Elizabeth E. Palmer
6. The Voltage-Dependent Sodium Channel Family 198
Mariola Zaleska, Samantha C. Salvage, Andrew J. Thompson, Sivakumar Namadurai, Christopher L.-H. Huang, Trevor Wilkinson, Fiona S. Cusdin, and Antony P. Jackson
7. Specialized Sodium Channels in Pain Transmission 224
Yucheng Xiao, Zifan Pei, and Theodore R. Cummins
8. Sodium Channelopathies of the Central Nervous System 257
Paul G. DeCaen, Alfred L. George, Jr., and Christopher H. Thompson
SECTION 3 LIGAND- GATED CHANNELS
9. α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid and Kainate Receptors 291
G. Brent Dawe, Patricia M. G. E. Brown, and Derek Bowie
10. N-Methyl-D-Aspartate Receptors 343
Gary J. Iacobucci and Gabriela K. Popescu
11. Nicotinic Acetylcholine Receptors 374
Roger L. Papke
12. GABAA Receptor Physiology and Pharmacology 419
Martin Wallner, A. Kerstin Lindemeyer, and Richard W. Olsen
13. P2X Receptors 458
Annette Nicke, Thomas Grutter, and Terrance M. Egan
14. Large Conductance Potassium Channels in the Nervous System 486
Willy Carrasquel-Ursulaez, Yenisleidy Lorenzo, Felipe Echeverria, and Ramon Latorre
Alessio Masi, Maria Novella Romanelli, Guido Mannaioni, and Elisabetta Cerbai
SECTION 4 OTHER CHANNELS
16. Tandem Pore Domain Potassium Channels 571
Douglas A. Bayliss
17. TRPC Channels—Insight from the Drosophila Light Sensitive Channels 611
Ben Katz, William L. Pak, and Baruch Minke
18. Acid-Sensing Ion Channels 646
Stefan Gründer
19. TMEM16 Ca2+ Activated Cl– Channels and CLC Chloride Channels and Transporters 696
Anna Boccaccio and Michael Pusch
Preface
It was in 1939, just before the outbreak of World War II, when Alan Hodkin and Andrew Huxley first recorded an action potential from a squid giant axon. Yet, understanding why there are varied sizes, shapes, and numbers of action potentials elicited by different neurons and how these neurons transition from one mode of firing to another remains an important research pursuit of current-day neuroscience. With the advent of molecular cloning, we now know there are hundreds of ion channel genes particularly expressed in the brains of higher animal systems. The immense diversity of firing properties of neurons within the central and peripheral nervous systems can be in large part attributable to the vast number of ion channel genes. We now appreciate that stable neuronal network responses to diverse environmental stimuli are also likely due to the availability of multiple ion channel genes. With the ever-increasing number of pharmacological tools, including naturally occurring toxins and transgenic animal models, we are starting to understand the contributions different ion channels make to neuronal firing and network responses.
The purpose of this handbook is to provide the reader with an important reference to some of the identified functions of ion channels in neurons. The introductory chapters provide the reader with an overview of the major classes of ion channels and a comprehensive view of the mechanisms of ion channel selectivity and gating. The book is then divided into three categories: voltage-dependent ion channels, ligand-gated channels, and other channels. There are chapters focused on ionotropic neurotransmitter receptors and chapters devoted to understanding the relationship between channel dysfunction and neurological disease states. The plastic nature of neurons should become apparent as examples of channel regulation via phosphorylation, trafficking, and transcriptional changes will be described.
The great number of ion channel genes, many still with unknown function, cannot be encompassed by any one handbook. There are other online resources curating a list of all the identified ion channel genes and gene families, describing tissue distribution, and tabulating basic channel properties. This handbook, on the other hand, provides an in-depth, enriching look into ion channel biology and biophysics, emphasizing how ion channels contribute to neuronal function. It was challenging to bring this collection of chapters together, especially during these uncertain times; nonetheless, the handbook will provide the reader with an important, easy go-to reference on the fundamental roles of ion channels in neurons.
I thank Len Kaczmarek for considering me to spearhead this project. I thank him for his guidance and wisdom as well. I am indebted to all the chapter contributors, for the realization of this book could not have occurred without their immense efforts. I would like to especially thank Ada Brunstein, head of reference at Oxford University Press, who supported the project from the onset and worked hard to bring the production of the handbook together.
Editor’s Bio
Arin Bhattacharjee, PhD, obtained his bachelor’s degree and pharmacology certificate from the University of Alberta in 1992. He completed doctoral training at the University of South Alabama with Ming Li, PhD, in 1999, and completed postdoctoral training at Yale University with Leonard Kaczmarek, PhD. In 2005, Bhattacharjee was appointed assistant professor at the University at Buffalo in 2005, and currently, he is an associate professor of pharmacology and toxicology. Bhattacharjee investigates how sodiumactivated potassium channels contribute to neuronal firing in pain-sensing neurons, during inflammation and nerve injury. He has published 40 papers on ion channel biology and function.
List of Contributors
Rene Barro-Soria University of Miami, USA
Douglas A. Bayliss University of Virginia, USA
Arin Bhattacharjee
State University of New York at Buffalo, USA
Anna Boccaccio
Consiglio Nazionale delle Ricerche, Italy
Derek Bowie
McGill University, Canada
Patricia M. G. E. Brown McGill University, Canada
Willy Carrasquel-Ursulaez Universidad de Valparaíso, Chile
Elisabetta Cerbai University of Florence, Italy
Ben Corry Australian National University, Australia
Theodore R. Cummins
Indiana University, USA
Fiona S. Cusdin Granta Park, UK
G. Brent Dawe
McGill University, Canada
Paul G. DeCaen
Northwestern University, USA
Felipe Echeverria Universidad de Valparaíso, Chile
Terrance M. Egan
Saint Louis University School of Medicine, USA
Alfred L. George, Jr Northwestern University, USA
Stefan Gründer RWTH Aachen University, Germany
Thomas Grutter
Université de Strasbourg, France; Centre National de la Recherche Scientifique, France
Christopher L.-H. Huang University of Cambridge, UK
Gary J. Iacobucci
State University of New York at Buffalo, USA
Antony P. Jackson University of Cambridge, UK
Leonard K. Kaczmarek Yale University, USA
Ben Katz
Hebrew University, Israel
Ramon Latorre Universidad de Valparaíso, Chile
A. Kerstin Lindemeyer University of California, Los Angeles, USA
Yenisleidy Lorenzo Universidad de Valparaíso, Chile
Guido Mannaioni
University of Florence, Italy
Alessio Masi
University of Florence, Italy
Baruch Minke
Hebrew University, Israel
Sivakumar Namadurai University of Cambridge, UK
Annette Nicke
Ludwig-Maximilians-Universität München, Germany
Juan J. Nogueira
Australian National University, Australia
Richard W. Olsen
University of California, Los Angeles, USA
William L. Pak
Purdue University, USA
Elizabeth E. Palmer
University of New South Wales, Australia
Roger L. Papke
University of Florida, USA
Zifan Pei
Indiana University-Purdue University, USA
Gabriela K. Popescu
State University of New York at Buffalo, USA
Michael Pusch
Consiglio Nazionale delle Ricerche, Italy
Hanne B. Rasmussen University of Copenhagen, Denmark
Maria Novella Romanelli University of Florence, Italy
Samantha C. Salvage University of Cambridge, UK
Andrew J. Thompson University of Cambridge, UK
Christopher H. Thompson Northwestern University, USA
James S. Trimmer University of California, Davis, USA
Martin Wallner University of California, Los Angeles, USA
Trevor Wilkinson Granta Park, UK
Yucheng Xiao
Indiana University-Purdue University, USA
Mariola Zaleska University of Cambridge, USA
Section 1
BASIC PRINCIPLES
Chapter 1 Excitable Membrane
Properties of Neurons
Leonard K. Kaczmarek
Introduction
For the first couple of decades after the discovery of the ionic basis of neuronal action potentials using the giant axon of squid (Hodgkin & Huxley, 1952), a very common question among pioneers of cellular neurophysiology was “What is the molecular identity of the sodium (or calcium, or potassium) channel?” Clues slowly emerged that there might exist more than one type of each of these channels. For example, the pharmacology of sodium channels was found to vary in different types of neurons (Matsuda, Yoshida, & Yonezawa, 1978). Single-channel recordings revealed the existence of at least three types of unitary calcium channel (Nowycky, Fox, & Tsien, 1985). Rapidly inactivating potassium currents that differ from the traditional Hodgkin-Huxley current, termed the delayed rectifier, which repolarizes action potentials in squid axons were discovered in some types of neurons as well as in oocytes, suggesting that there may be two types of potassium channel (Connor & Stevens, 1971; Hagiwara, Kusano, & Saito, 1961). All such considerations were made irrelevant with the cloning of the genes for these classes of ion channels, nine for voltage-dependent sodium channels, 10 for voltage-dependent calcium channels, and over 70 for pore-forming subunits of potassium channels. Moreover, over 30 different genes have been found that encode nonselective cation channels, which are permeable to sodium, potassium, and calcium ions in different ratios. Add to this the array of chloride channels and ionotropic neurotransmitter receptors which are cation or anion channels gated by binding of a neurotransmitter, and the number of possible genetic ways of constructing an excitable cell becomes astronomical.
One potential explanation for the existence of a very large number of channels that apparently serve similar biological functions is that these are required for the diversity of firing patterns encountered in the nervous system. Different neurons have very different electrical properties (Figure 1.1). Some neurons never fire an action potential
unless activated by a strong excitatory input, while others are capable of firing in a regular pattern or in bursts, even in the absence of any synaptic stimulation. The duration of the action potential in different neurons ranges from hundreds of microseconds to tens of milliseconds. Some neurons fire at high rates for hours in response to maintained stimulation, whereas others cease to fire after only a few seconds of sustained excitatory input. Certain neurons will not fire in response to brief stimulation but will start to fire if their excitatory drive is maintained over a prolonged period. Still other neurons are triggered to fire spontaneously by synaptic stimulation and then continue to fire after the presynaptic input has ceased.
A further rationale for the large number of ion channel genes is that the diverse firing patterns illustrated in Figure 1.1 are not fixed but are known to be modulated by sensory stimuli and by the incoming patterns of synaptic stimuli to which a neuron is exposed (Kaczmarek & Levitan, 1987; Marder, 2012). Experiments first carried out with the nervous system of invertebrates and later confirmed in mammals demonstrated that the height and width of action potentials, the pattern of spontaneous firing, and the patterns evoked by an incoming stimulus can also be altered by synaptic and hormonal events. These changes are clearly linked to changes in animal behaviors. The onset of reproductive and feeding behaviors and simple forms of learning are known to be caused by such rapid changes in the intrinsic excitability of neurons that control these behaviors. Many of these changes can now be attributed to posttranslational modification of ion channels, such as phosphorylation of their regulatory domains. Because there are
Figure 1.1 Schematic diagram illustrating different types of neuronal firing patterns and ways in which they may be altered by modulation of ion channels.
myriad types of neurons in the nervous system and each type of neuron may respond in a different way to stimulation, this may require a very large set of ion channels, each of which is modulated in a slightly different way by posttranslational mechanisms.
Another potential explanation for the existence of the large assortment of ion channel genes has come from more recent findings that ion channels do more than simply conduct ions. The cytoplasmic and extracellular domains of ion channels bind other cell components such as signaling molecules, molecular motors, cell adhesion molecules, and the cytoskeleton (Kaczmarek, 2006; Lee, Fakler, Kaczmarek, & Isom, 2014). Some of these interactions allow channels to be trafficked to different locations in a cell. They also allow some channels, when activated, to propagate signals to the cell that may alter processes such as cell adhesion, transcription, and RNA translation. These activities have been termed nonconductive functions of ion channels.
This chapter will provide a bird’s-eye view of the multiple gene families that shape the electrical personality of neurons. Specifically, it will cover sodium, potassium, calcium, and nonselective channels. Of necessity, such an overview can give only a very brief outline of these channels, and other chapters in this volume will provide more specific details. Although ionotropic neurotransmitter receptors are bona fide ion channels, they are not normally considered to shape the intrinsic excitability of neurons; but they will also be covered in other chapters. Moreover, chloride channels can play important roles in shaping neuronal excitability, but their roles are covered elsewhere (Duran, Thompson, Xiao, & Hartzell, 2010; Ha & Cheong, 2017; Rahmati, Hoebeek, Peter, & De Zeeuw, 2018).
Sodium Channels
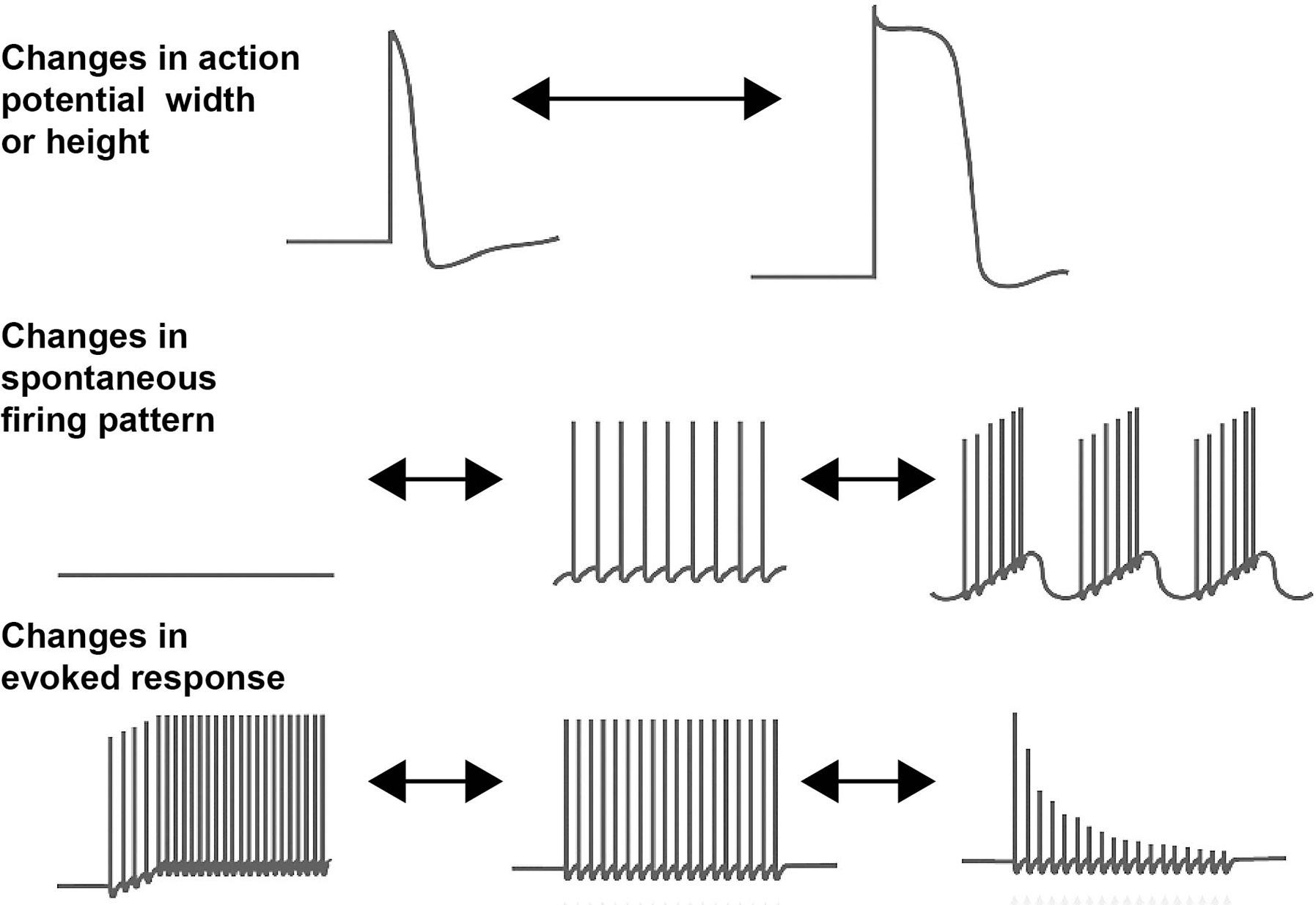
Voltage-dependent sodium channels shape the firing patterns of neurons in at least three different ways. Their classic role is to provide the upstroke of the action potential in mammalian neurons (Hodgkin & Huxley, 1952). They can also generate a persistent sodium current, sometimes termed INaP, that produces a persistent depolarization to drive repetitive firing (Crill, 1996). Finally, a type of sodium current termed resurgent current is activated during the repolarization of many types of neurons, enhancing their ability to fire at high rates (Lewis & Raman, 2014). There are nine genes for these channels, which are termed NaV1.1–NaV1.9. These encode the large pore-forming α subunits of the channels and have 24 transmembrane domains. These are organized into four tandem domains (domains I–IV), each of which has six transmembrane αhelical segments (termed S1–S6) (Catterall, Goldin, & Waxman, 2005) (Figure 1.2A). Segment S4 in each domain has a series of positively charged amino acids at three residue intervals, and it is the movement of these segments in response to changes in membrane potential across the membrane that renders the channels sensitive to transmembrane voltage. A loop between S5 and S6 in each domain provides the lining of the pore that conducts sodium ions.
Of the nine voltage-dependent sodium channels, NaV1.1, NaV1.2, NaV1.3, and NaV1.6 are expressed in neurons of the central nervous system (Table 1.1). Interestingly, the action potentials of peripheral neurons are driven primarily by a different set of channels, NaV1.7, NaV1.8, and NaV1.9. Because many of these peripheral neurons in dorsal root ganglia are nociceptors, they have been a promising target for pharmacological treatments to relieve pain. The neurotoxin tetrodotoxin (TTX), which is found in endosymbiotic bacteria within the gut of puffer fish, is a potent blocker of many of these sodium channels (Bane, Lehane, Dikshit, O’Riordan, & Furey, 2014); and sensitivity to TTX has become a conventional component of the way that individual NaV subunits, as well as sodium currents in neurons, are characterized (Table 1.1). In addition to the sodium channels listed in Table 1.1, there is a related channel termed NaX1, which is encoded by the SCN7A gene. This is insensitive to voltage changes across the plasma membrane but is regulated by changes in sodium concentrations outside the cell (Noda & Hiyama, 2015). It is expressed primarily in glial cells of sensory circumventricular organs.
While many older textbooks described action potentials as “all or none” events, it has become clear that the height and width of action potentials in neurons are subject to modulation by neurotransmitters and hormones. While much of this modulation reflects modifications in calcium or potassium channels, even voltage-dependent sodium channels can be modified by phosphorylation, leading to changes in the shape of action potentials and the ability to sustain repetitive firing (Scheuer, 2011). As one example, the sodium current in pyramidal cells of the prefrontal cortex is suppressed by activation of 5-hydroxytryptamine 2 (5-HT2) neurotransmitter receptors (Figure 1.2B). This is believed to be mediated by the phosphorylation of serine residues on the NaV1.2 α subunit by protein kinase C (Carr et al., 2003). The partial inactivation of Na+ current produced by 5-HT develops over many seconds (Figure 1.2C), increasing the threshold at which action potentials are generated in response to depolarization. This increase in threshold during exposure to 5-HT can be detected during repeated depolarizations that normally trigger repetitive firing and eventually prevents sustained firing during depolarization (Figure 1.2D,E) (Carr et al., 2003). Because sustained firing of pyramidal cells in the part of the brain is required for working memory in higher mammals (Constantinidis et al., 2018; Goldman-Rakic, 1995), neurotransmitter modulation of Na+ channels may influence which sensory events are subject to rapid recall.
Fully functioning sodium channels contain, in addition to their major pore-forming α subunits NaV1.1–NaV1.9, another set of intrinsic subunits, the sodium channel β subunits, which have only a single-membrane-spanning region (Namadurai et al., 2015; O’Malley & Isom, 2015). There are four of these, β1–β4, encoded by the genes Scn1b–Scn4b. All four β subunits are found in different types of neurons, and their binding to the α subunits controls insertion into the plasma membrane as well as the voltage dependence and the kinetics of activation and inactivation of the resultant channel complex. The extracellular region of the β subunits has an immunoglobulin domain, and the β1 subunit has been shown to function as a cell adhesion molecule that can regulate axonal fasciculation and neurite outgrowth (Figure 1.2F) (O’Malley & Isom, 2015). For example, the β1 subunits promote the outgrowth of neurites from cerebellar granule neurons, and this requires Na+ entry through its associated NaV1.6 α subunit (Brackenbury et al., 2010). Loss of the β1 subunit results in the disorganization of the axons of these neurons in the cerebellum (Figure 1.2G) (Brackenbury et al., 2008).
Figure 1.2 Voltage-dependent sodium channels. (A) Schematic diagram of the transmembrane topology of a sodium channel α subunit. (B–E) Stimulation of 5-hydroxytryptamine 2 (5-HT2) receptors reduces Na+ current in pyramidal cells of mouse prefrontal cortex. (B) An example of Na+ current measured by stepping the membrane potential from –100 to –20 mV before and after (red trace) application of 2,5-dimethoxy-4-iodoamphetamine (DOI), a 5-HT2 receptor agonist. (C) Time course of the decrease in current after application of DOI. (D) Examples of the effects of repeated depolarization of pyramidal cells on the firing of action potentials before and after application of DOI. (E) Plots of the membrane threshold for triggering an action potential with repeated stimulation as in (C) before and after DOI application. Data from Carr et al. (2003). (F) Sodium channels contribute to cell adhesion through their β subunits. Schematic diagram of Na+ channels on the processes of two different neurons. The associated β1 subunits can mediate cell–cell adhesion either directly by β1–β1 binding or indirectly through other associated cell adhesion molecules such as contactin (O’Malley & Isom, 2015). (G) Genetic deletion of the Na+ channel β1 subunit results in the disorganization of the axons of cerebellar granule neurons in the cerebellum. In this image, the growing axons have been labeled (green) with an antibody to a cell adhesion molecule, Transient Axonal Glycoprotein 1(TAG-1), that is restricted to these neurons (Brackenbury et al., 2008).
(b)
(f) (g)
Table 1.1
Voltage-Dependent Sodium Channels
Sodium Channel Gene for α Subunit Expression Pharmacology
NaV1.1 SCN1A Central neurons
TTX-sensitive
NaV1.2 SCN2A Central neurons TTX-sensitive
NaV1.3 SCN3A Central neurons TTX-sensitive
NaV1.4 SCN4A Skeletal muscle TTX-sensitive
NaV1.5 SCN5A Cardiac muscle TTX-resistant
NaV1.6 SCN8A Central neurons TTX-sensitive
NaV1.7 SCN9A Neurons of dorsal root ganglion TTX-sensitive
NaV1.8 SCN10A Neurons of dorsal root ganglion, cardiac muscle TTX-resistant
NaV1.9 SCN11A Neurons, nociceptive neuron in dorsal root ganglion and trigeminal ganglion TTX-resistant
Note. TTX = tetrodotoxin.
Calcium Channels
The opening of voltage-dependent calcium channels during neuronal firing allows calcium ions to enter the cytoplasm from the extracellular medium. This triggers numerous cellular events, including changes in gene transcription, adenosine triphosphate (ATP) production in mitochondria and neurotransmitter release in presynaptic terminals. In many neurons, calcium currents also contribute to determining the height and width of action potentials and/or to setting the membrane potential and patterns of spontaneous firing. There are 10 genes that encode these channels, which are grouped into three families: CaV1.1–CaV1.4, CaV2.1–CaV2.3, and CaV3.1–CaV3.3 (Table 1.2). As with sodium channels, these are the large pore-forming α subunits of the channels and have 24 transmembrane domains organized into four domains, comparable to those shown in Figure 1.2A for NaV subunits (Figure 1.3A) (Catterall, 2011; Simms & Zamponi, 2014). The members of the first family of calcium channels (CaV1.1–CaV1.4) are termed Ltype channels and are characterized by their sensitivity to block by dihydropyridines (Nanou & Catterall, 2018). Because they become activated at positive potentials, such as those encountered during an action potential, they are also described as highvoltage-activated channels. The individual members of the second class, CaV2.1, CaV2.2, and CaV2.3, have been termed P/Q-, N-, and R-type calcium channels, respectively. Like the L-type channels, they activate during action potentials but are insensitive to dihydropyridines. A general distinction between the biological roles of these first two classes is that calcium entry through L-type channels is typically linked to metabolic changes including changes in gene transcription, while neurotransmitter release at
Table 1.2
Voltage-Dependent Calcium Channels
Calcium Channel Gene for α Subunit Expression Characteristics
CaV1.1 CACNA1S
CaV1.2 CACNA1C
Skeletal muscle L-type
Heart/smooth muscle L-type
CaV1.3 CACNA1D Neurons L-type
CaV1.4 CACNA1F Photoreceptors L-type
CaV2.1 CACNA1A Neurons P/Q-type
CaV2.2 CACNA1B Neurons N-type
CaV2.3 CACNA1E Neurons R-type
CaV3.1 CACNA1G Neurons T-type
CaV3.2 CACNA1H Neurons T-type
CaV3.3 CACNA1I Neurons T-type
synaptic endings is typically triggered by P/Q-, N-, or R-type channels. This is not, however, a hard-and-fast rule because the CaV1.3 and CaV1.4 channels provide the major source of calcium entry for triggering release at some presynaptic terminals such as those of sensory hair cells in the auditory system and retinal photoreceptors, respectively (Catterall, 2011).
The members of the third class of calcium channels, CaV3.1–CaV3.3, are termed T-type for “transient” channels and are also described as low-voltage-activated channels. In contrast to the first two classes of calcium channels, they both activate and inactivate at negative membrane potentials close to or below the resting membrane potentials of most neurons. This allows them to shape the endogenous firing pattern of many neurons. For example, repeated bursts of action potentials followed by interburst hyperpolarizations can be shaped by a cycle of activation, inactivation, and then recovery from inactivation of T-type channels (Choi et al., 2010).
As with the voltage-dependent sodium channels, the voltage-activated calcium channels interact with auxiliary subunits that regulate their assembly, trafficking, and biophysical properties. The best characterized of these are the α2δ, β, and γ subunits that bind the pore-forming voltage-activated channel α subunits (Arikkath & Campbell, 2003; Dolphin, 2013). The CaV1 family also interacts with a variety of calcium-binding proteins including calmodulin (CaM) and a family of CaM-like calcium-binding proteins termed CaBPs (Hardie & Lee, 2016).
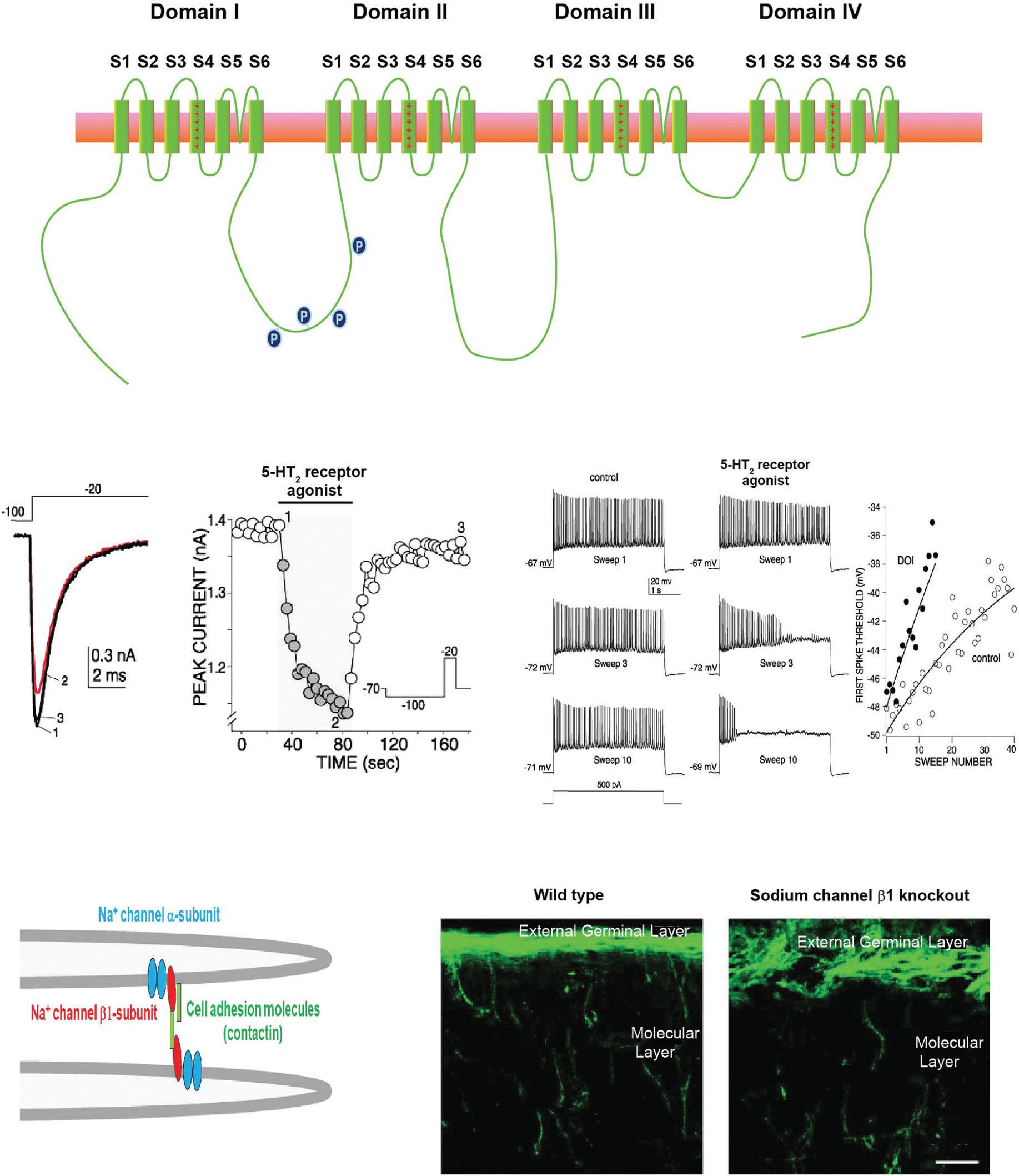
Voltage-gated calcium channels are potently modulated by a variety of second messenger pathways, including phosphorylation and the direct binding of the βγ subunits of G proteins (Figure 1.3A) (Catterall, 2011). In the CaV2.1 and CaV2.2 channels, which predominate at numerous presynaptic terminals, there exists a sequence, termed the synprint site, which allows these channels to interact directly with soluble Nethylmaleimide-sensitive factor activating protein receptor (SNARE) proteins that
Figure 1.3 Modulation of voltage-dependent calcium channels. (A) Schematic diagram of the transmembrane topology of a calcium channel α subunit, indicating sites of interaction with regulatory components in different families of calcium channels. (B) Cell-attached single-channel recordings of channels in neurosecretory bag cell neurons of Aplysia (Strong, Fox, Tsien, & Kaczmarek, 1987). Left panels show recordings under control conditions, where only a single species of L-type channels can be detected. Right panels show recordings after application of an activator of protein kinase C (PKC), which causes the insertion of a P/Q channel (White & Kaczmarek, 1997; Zhang et al., 2008). In this condition two sizes of unitary openings can be detected, those of the control L-type channel as well as those of the larger-conductance P/Q channel. (C) Pseudocolor images of levels of intracellular Ca2+ in the growth cone of a bag cell neuron after stimulation of a train of action potentials. Images show the same growth cone before and after application of the PKC activator. Insertion of vesicles containing the P/Q channel causes an increase in the size of the membrane and the appearance of new sites of calcium entry (Knox, Quattrocki, Connor, & Kaczmarek, 1992). (D) Voltage clamp traces (upper panel) and current–voltage relation (lower panel) of mammalian CaV2.2 channels expressed in a cell line (Su et al., 2012). The current of the wild-type channel (WT CaV2.20 is increased by coexpression with activated Cdk5 (Cdk5/p35), but currents of a CaV2.2 mutant in which eight Cdk5 phosphorylation sites were mutated are not affected by Cdk5. (E) Immunogold labeling for CaV2.2 in cultured hippocampal neurons transduced with control green fluorescent protein (GFP) or with the WT or phosphorylation site mutant CaV2.2 channels. Accumulation of the transduced channel at active zones occurs with the WT channel but not for the mutant. (F) Quantification of colocalization of CaV2.2 in the three conditions of (E) (Su et al., 2012).
control fusion of synaptic vesicles with the plasma membrane. Thus, modulation of these channels, through mechanisms such as phosphorylation, produces strong direct effects on neurotransmitter release, as well as shaping neuronal excitability (Catterall, 2011). In some cases, activation of a kinase results in the insertion of new calcium channels into the plasma membrane from an intracellular pool. An example of this is provided in Figure 1.3A, which shows single-channel recordings of channels in neurosecretory neurons of Aplysia. Under control conditions, only a single species of L-type (CaV1.3) can be detected, but activation of protein kinase C causes the insertion of a P/Q (CaV2.1) channel that contributes to increased release of neuropeptides resulting from enhanced calcium influx during action potentials (Figure 1.3B). Another example is shown in Figure 1.3C, which shows that currents of mammalian CaV2.2 (the N-type channel) are substantially enhanced by activated cyclin-dependent kinase 5 (Cdk5) (Su et al., 2012).
Phosphorylation of a serine residue in the distal cytoplasmic C-terminal tail of this channel by Cdk5 not only increases the probability of channel opening with depolarization but also serves to tether the channel to the presynaptic active zone, resulting in enhanced neurotransmitter release (Figure 1.3D,E).
Because Ca2+ ions impact a vast number of cellular functions, including neurotransmission, gene expression, and mitochondrial function, the opening of channels that are permeable to Ca2+ ions has consequences beyond mere regulation of excitability. Some of the protein–protein interactions of Ca2+ channels with cytoplasmic signaling molecules serve to promote these functions (D’Arco & Dolphin, 2012). For example, the close association of CaV1.3 channels and perhaps other L-type channels with CaM and clusters of CaM kinase II results in the rapid activation of this kinase when the channel opens. This triggers a preferred signaling pathway to the nucleus, resulting in the phosphorylation of nuclear CREB (cyclic adenosine monophosphate (AMP) response element binding protein) and changes in gene transcription (Wheeler et al., 2012). Some of the signaling functions of calcium channel proteins may, however, not require the entry of Ca2+ ions. For example, a fragment of the CaV1.2 channel, termed CCAT (the calcium channel–associated transcriptional regulator), can be transcribed independently of the full-length channel itself and acts as a transcription factor in the nucleus (GomezOspina et al., 2013). Perhaps the earliest example of how calcium channels play a key role that does not require calcium flux is found in excitation–contraction coupling in skeletal muscle, where the function of CaV1.1 is to act as a voltage sensor that is coupled to a different channel, the ryanodine receptor, to release Ca2+ ions from internal stores (Bannister & Beam, 2013; Calderon, Bolanos, & Caputo, 2014).
Potassium Channels
The first-described and most traditional role of a potassium channel is to open during depolarization, restoring the membrane potential to a resting negative value after an action potential. It may therefore be surprising that there are over 70 different genes that encode pore-forming α subunits of potassium channels, compared to only nine or 10 for the sodium and calcium channels. Moreover, in contrast to sodium and calcium channel
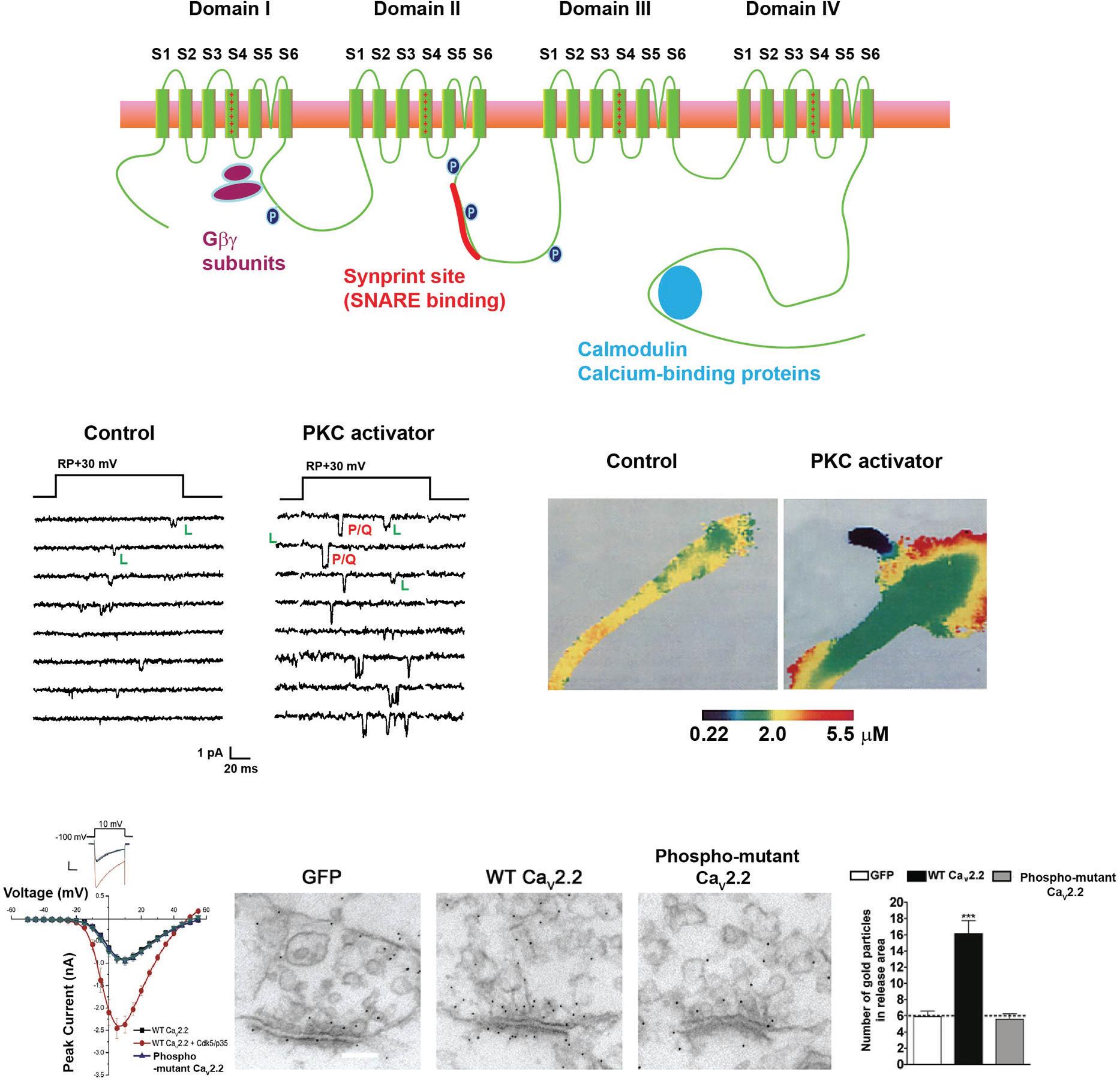
α subunits which are comprised of a single long polypeptide, the pore of most potassium channels is typically comprised of tetramers of α subunits, each of which corresponds to one of the four domains of a sodium channel (Figure 1.4A). A functional potassium channel is either a homomer of four identical α subunits or a heteromer containing different α subunits (Figure 1.4B). This, combined with the fact that there exist multiple splice variants for most of the α subunits, means that the number of potential potassium channel complexes is astronomical.
The potassium channel α subunits can be classified into five groups: (1) voltagedependent channels of the KV family, (2) calcium-activated channels (KCa channels), (3) sodium-activated channels (KNa channels), (4) inwardly rectifying channels (Kir channels), and (5) two pore domain channels (K2P channels). The KCa channels can be further divided into two types, large-conductance (KCa1.1, also called big-conductance [BK] channels) and small-conductance (KCa2 and KCa3 channels, also called smallconductance [SK] or intermediate-conductance [IK] channels). A diagram illustrating the transmembrane topology of each of these groups is shown in Figure 1.4A.
Voltage-Dependent Potassium Channels
Potassium channels that open selectively when the membrane potential of a cell membrane is depolarized constitute the largest subclass. There are 40 different genes that encode the pore-forming α subunits of these channels (Table 1.3). They all have six αhelical transmembrane segments (S1–S6), with a P-domain between S5 and S6. As with the voltage-dependent sodium and calcium channels, basic amino acids (lysine or arginine) are repeated every three residues in the fourth transmembrane segment (S4), and the movement of these charged residues in response to transmembrane changes in voltage triggers channel opening.
These Kv channels can be divided into 12 groups based simply on homology. Space precludes a detailed description of the channels in each group, but there are some general rules. Kv1 channels are typical delayed rectifiers in the Hodgkin-Huxley sense, and Kv1.1 and Kv1.2 are often clustered at the juxtaparanodes adjacent to the nodes of Ranvier (Rasband & Peles, 2016). Kv2 channels arrange themselves into distinct clusters that are coupled to underlying layers of endoplasmic reticulum (Kirmiz et al., 2018). Kv3 channels are commonly expressed in neurons capable of firing at high rates (Kaczmarek & Zhang, 2017). Kv4 channels inactivate rapidly during maintained depolarization and are expressed in neuronal dendrites (Birnbaum et al., 2004; Chen et al., 2006). Some of the groups, specifically Kv5, Kv6, Kv8, and Kv9 α subunits, are “silent” subunits in that they do not form functional channels by themselves when expressed in heterologous cells. They can, however, coassemble with Kv2.1 channels to generate heteromeric channels with properties that differ from those of Kv2.1 homomeric channels alone (Enyedi & Czirjak, 2010; Hugnot et al., 1996; Richardson & Kaczmarek, 2000).
Figure 1.4 Potassium channels. (A) Schematic diagrams of the transmembrane topology of each of the five classes of potassium channels. (B) Images of the structure of a KNa1.1 potassium channel obtained using cryo-electron microscopy (Hite et al., 2015). Each of the four identical α subunits that make this tetrameric channel is shown in a different color. Side and end-on views are shown.
Table 1.3 Voltage-Dependent Potassium Channels
Voltage-Dependent Potassium Channel Genes for α Subunit
KCNH2, KCNH6, KCNH7 Expressed in many tissues including neurons and cardiac cells; loss of Kv11.1 causes cardiac long QT syndrome
KV12.1, 12.2, 12.3
HERG channels (“human EAG-related gene ”)
KCNH8, KCNH3, KCNH4 Primarily expressed in neurons ELK channels (“EAG-like”)
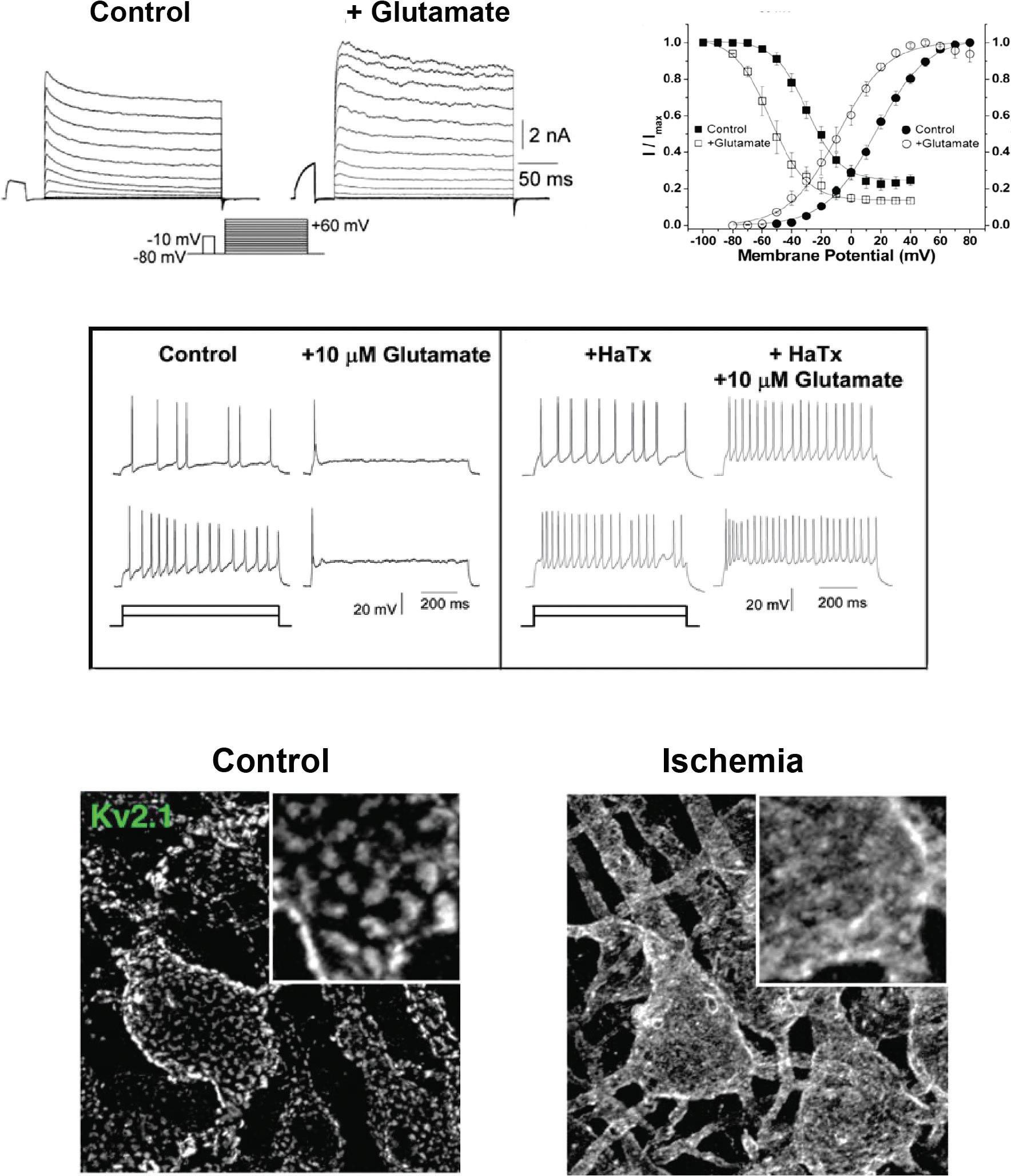
One example of how regulation of Kv channels influences neuronal firing patterns is provided by the Kv2.1 channel, which contributes the major component of K+ current in hippocampal neurons, as well as many other types of neurons. The amplitude and voltage dependence of this current are regulated by multiple phosphorylation sites on the Kv2.1 protein. Application of the neurotransmitter glutamate to hippocampal neurons shifts its voltage dependence of activation and inactivation and increases current amplitude (Mohapatra et al., 2009) (Figure 1.5A,B). This reduces neuronal excitability (Figure 1.5C). One of the interesting aspects of the Kv2.1 channel is that many of these channels aggregate to form discrete clusters on the soma and proximal dendrites of the neurons (Figure 1.5D). The channels in these clusters are thought to be inactive as K+ channels. Instead, the clustered channels serve to link the plasma membrane to layers of endoplasmic reticulum that lie immediately
Figure 1.5 Modulation of the Kv2.1 channel. (A) Application of a low concentration of glutamate increases K+ current in cultured hippocampal neurons. (B) Glutamate alters the voltage dependence of activation and inactivation of K+ current in these neurons. (C) Left panel shows that a low concentration of glutamate suppresses the firing rate of hippocampal neurons in response to two different levels of depolarizing current. Right panel shows that the effects of glutamate on firing are abolished in the presence of hanatoxin (HaTx), an inhibitor of Kv2.1 channels. (A–C) are from Mohapatra et al. (2009). (D) Immunostaining of clusters of Kv2.1 channels on cortical neurons under control conditions and after dissolution of the clusters during acute brain ischemia (Misonou et al., 2008).
under the surface. This is mediated by Kv2.1-binding proteins that couple the two types of membrane (Johnson, Leek, & Tamkun, 2019). The clustering of the channels and their coupling to the endoplasmic reticulum are rapidly modulated in response to many biological stimuli including neurotransmitters such as glutamate as well as
(a) (c) (d)
(b)
by conditions such as seizures or ischemia (Figure 1.5D) (Misonou, Thompson, & Cai, 2008). This is regulated by phosphorylation sites on Kv2.1 that are distinct from those that alter voltage dependence. The full biological significance of the dual biological function of these channels and the independent modulation of each function are active areas of investigation.
Calcium-Activated Potassium Channels
Calcium-activated potassium channels can be divided into two groups with quite distinct properties (Kaczmarek et al., 2017). The first group contains only one member, KCa1.1, which has also been termed the BK or maxi-K channel because of its large conductance measured in single-channel recordings (Table 1.4). Although it has one more transmembrane segment than KV subunits (Figure 1.4A), it is essentially a voltagedependent KV channel with a large cytoplasmic C-terminal domain that allows it to be activated by Ca2+ ions (Latorre et al., 2017). The central action of the binding of Ca2+ is to alter the voltage dependence of the channel, increasing open probability at more negative potentials. This channel is very widely expressed in the central nervous system as well as many non-neuronal tissues. These include vascular smooth muscle, where activation of these channels plays a central role in the regulation of blood pressure. Many different splice isoforms can be generated from KCNMA1, the gene that encodes KCa1.1, channels, such that the properties and regulation of these channels can differ from cell to cell. One splice isoform specifically targets KCa1.1 channels to the inner membrane of mitochondria (Balderas, Zhang, Stefani, & Toro, 2015).
The second group of Ca2+-activated K+ channels is comprised of four members (KCa2.1, KCa2.2, KCa2.3, and KCa3.1; Table 1.4) (Adelman, Maylie, & Sah, 2012). The first three in this group are usually termed SK channels because of the small conductance relative to that of the BK channels. The fourth channel, KCa3.1, is sometimes termed the
Table 1.4 Calcium-Activated Potassium Channels
Calcium-Dependent Potassium Channels Genes for α Subunit Characteristics Alternate Names
KCa1.1
KCa2.1
KCa2.2
KCa2.3
KCa3.1
KCNMA1
KCNN1
Voltage- and Ca2+-dependent BK, Slo1
Voltage-independent, contribute to slow afterhyperpolarization in neurons SK1
KCNN2 Voltage-independent, contribute to slow afterhyperpolarization in neurons SK2
KCNN3
Voltage-independent, contribute to slow afterhyperpolarization in neurons SK3
KCNN4 Primarily in non-neuronal cells; major role in immune cells and red blood cells SK4, IK, Gardos channel
IK channel because it has an “intermediate” conductance. These channels are not sensitive to changes in voltage. Moreover, in contrast to the BK channels, the pore-forming α subunits do not bind Ca2+ ions directly. Instead, the Ca2+-binding protein CaM is an intrinsic component of each channel complex. Because of their insensitivity to transmembrane voltage, they play a very different physiological role from BK channels in neuronal firing. Specifically, they hyperpolarize and slow neuronal firing as long as intracellular Ca2+ levels are elevated, as occurs during prolonged neuronal firing. This usually leads to adaptation of the firing rate during maintained stimulation. The activation of SK channels and their trafficking into and out of synaptic spines in neurons of the hippocampus have been shown to regulate the synaptic plasticity that underlies some forms of learning and memory (Adelman et al., 2012).
Sodium-Activated Potassium Channels
Potassium channels activated by elevations in cytoplasmic Na+ were first described in cardiac cells and were subsequently found to contribute to shaping the excitability of a variety of neurons (Dryer, 1994; Kaczmarek, 2013; Kaczmarek et al., 2017; Kameyama et al., 1984). There are two known pore-forming subunits of these channels, KNa1.1 and KNa1.2; and these have also been termed Slack and Slick, respectively (Table 1.5) (Bhattacharjee & Kaczmarek, 2005; Kaczmarek et al., 2017). The first of these, KNa1.1, is expressed primarily in the nervous system, while KNa1.2 is expressed much more widely. The pore-forming α subunit of KNa1.1 binds a variety of other proteins including FMRP (fragile X mental retardation protein), Phactr-1 (phosphatase and actin regulator protein-1), and TMEM16-C (transmembrane protein 16-C) (Brown et al., 2010; Fleming et al., 2016; Huang et al., 2013). Mutations in this subunit result in early-onset epilepsies and very severe intellectual disabilities (Kim & Kaczmarek, 2014).
Inward Rectifier Potassium Channels
Inwardly rectifier potassium channels (Kir channels) are simpler in structure than the channels we have considered thus far in that they are comprised of only two α-helical