ChemicalCatalystsforBiomassUpgrading
Editedby MarkCrocker
EduardoSantillan-Jimenez
Editors
MarkCrocker UniversityofKentucky DepartmentofChemistryandCenterfor AppliedEnergyResearch 2540ResearchParkDrive Lexington KY40511 UnitedStates
EduardoSantillan-Jimenez UniversityofKentucky CenterforAppliedEnergyResearch 2540ResearchParkDrive Lexington KY40511 UnitedStates
Allbookspublishedby Wiley-VCH arecarefullyproduced.Nevertheless, authors,editors,andpublisherdonot warranttheinformationcontainedin thesebooks,includingthisbook,to befreeoferrors.Readersareadvised tokeepinmindthatstatements,data, illustrations,proceduraldetailsorother itemsmayinadvertentlybeinaccurate.
LibraryofCongressCardNo.: appliedfor
BritishLibraryCataloguing-in-Publication Data
Acataloguerecordforthisbookis availablefromtheBritishLibrary.
Bibliographicinformationpublishedby theDeutscheNationalbibliothek TheDeutscheNationalbibliotheklists thispublicationintheDeutsche Nationalbibliografie;detailed bibliographicdataareavailableonthe Internetat <http://dnb.d-nb.de>.
©2020Wiley-VCHVerlagGmbH& Co.KGaA,Boschstr.12,69469 Weinheim,Germany
Allrightsreserved(includingthoseof translationintootherlanguages).No partofthisbookmaybereproducedin anyform–byphotoprinting, microfilm,oranyothermeans–nor transmittedortranslatedintoa machinelanguagewithoutwritten permissionfromthepublishers. Registerednames,trademarks,etc.used inthisbook,evenwhennotspecifically markedassuch,arenottobe consideredunprotectedbylaw.
PrintISBN: 978-3-527-34466-6
ePDFISBN: 978-3-527-81481-7
ePubISBN: 978-3-527-81480-0
oBookISBN: 978-3-527-81479-4
Typesetting SPiGlobal,Chennai,India PrintingandBinding
Printedonacid-freepaper
10987654321
Contents
Preface xiii
1UpgradingofBiomassviaCatalyticFastPyrolysis(CFP) 1 CharlesA.Mullen
1.1Introduction 1
1.1.1CatalyticPyrolysisOverZeolites 4
1.1.1.1CatalyticPyrolysisOverHZSM-5 4
1.1.1.2DeactivationofHZSM-5DuringCFP 9
1.1.1.3ModificationofZSM-5withMetals 13
1.1.1.4ModificationsofZSM-5PoreStructure 18
1.1.2CFPwithMetalOxideCatalysts 20
1.1.3CFPtoProduceFineChemicals 24
1.1.4OutlookandConclusions 26 References 27
2TheUpgradingofBio-OilviaHydrodeoxygenation 35 AdetoyeseO.Oyedun,MadhumitaPatel,MayankKumar,andAmitKumar
2.1Introduction 35
2.2Hydrodeoxygenation(HDO) 37
2.2.1HydrodeoxygenationofPhenolasaModelCompound 38
2.2.1.1HDOofPhenolic(Guaiacol)ModelCompounds 38
2.2.1.2HDOofPhenolic(Anisole)ModelCompounds 40
2.2.1.3HDOofPhenolic(Cresol)ModelCompounds 40
2.2.2HydrodeoxygenationofAldehydeModelCompounds 41
2.2.3HydrodeoxygenationofCarboxylicAcidModelCompounds 43
2.2.4HydrodeoxygenationofAlcoholModelCompounds 44
2.2.5HydrodeoxygenationofCarbohydrateModelCompounds 44
2.3ChemicalCatalystsfortheHDOReaction 45
2.3.1CatalystPromotersforHDO 48
2.3.2CatalystSupportsforHDO 49
2.3.3CatalystSelectivityforHDO 49
2.3.4CatalystDeactivationDuringHDO 50
2.4ResearchGaps 51
2.5Conclusions 52 Acknowledgments 52 References 53
3UpgradingofBio-oilviaFluidCatalyticCracking 61 IdoiaHita,JoseMariaArandes,andJavierBilbao
3.1Introduction 61
3.2Bio-oil 63
3.2.1Bio-oilProductionviaFastPyrolysis 63
3.2.2GeneralCharacteristics,Composition,andStabilizationofBio-oil 63
3.2.2.1AdjustmentofBio-oilCompositionThroughPyrolytic Strategies 65
3.2.2.2Bio-oilStabilization 66
3.2.3ValorizationRoutesforBio-oil 69
3.2.3.1Hydroprocessing 69
3.2.3.2SteamReforming 70
3.2.3.3ExtractionofValuableComponentsfromBio-oil 71
3.3CatalyticCrackingofBio-oil:FundamentalAspects 71
3.3.1TheFCCUnit 71
3.3.2CrackingReactionsandMechanisms 73
3.3.3CrackingofOxygenatedCompounds 74
3.3.4CrackingofBio-oil 76
3.4Bio-oilCrackingintheFCCUnit 78
3.4.1CrackingofModelOxygenates 78
3.4.2CoprocessingofOxygenatesandTheirMixtureswithVacuumGasOil (VGO) 78
3.4.3CrackingofBio-oilandItsMixtureswithVGO 79
3.5ConclusionsandCriticalDiscussion 86 References 88
4StabilizationofBio-oilviaEsterification 97 XunHu
4.1Introduction 97
4.2ReactionsoftheMainComponentsofBio-OilUnderEsterification Conditions 102
4.2.1Sugars 102
4.2.2CarboxylicAcids 109
4.2.3Furans 113
4.2.4AldehydesandKetones 114
4.2.5Phenolics 116
4.2.6OtherComponents 117
4.3ProcessesforEsterificationofBio-oil 121
4.3.1EsterificationofBio-oilUnderSubcriticalorSupercritical Conditions 121
4.3.2RemovaloftheWaterinBio-oiltoEnhanceConversionofCarboxylic Acids 121
4.3.3In-lineEsterificationofBio-oil 123
4.3.4EsterificationCoupledwithOxidation 123
4.3.5EsterificationCoupledwithHydrogenation 123
4.3.6StericHindranceinBio-oilEsterification 124
4.3.7CokinginEsterificationofBio-oil 125
4.3.8EffectsofBio-oilEsterificationontheSubsequent Hydrotreatment 129
4.4Catalysts 132
4.5SummaryandOutlook 136 Acknowledgments 137 References 137
5CatalyticUpgradingofHolocellulose-DerivedC5 andC6 Sugars 145 XingguangZhang,ZhijunTai,AminOsatiashtiani,LeeDurndell,AdamF.Lee, andKarenWilson
5.1Introduction 145
5.2CatalyticTransformationofC5–C6Sugars 146
5.2.1IsomerizationCatalysts 147
5.2.1.1Zeolites 149
5.2.1.2Hydrotalcites 151
5.2.1.3OtherSolidCatalysts 154
5.2.2DehydrationCatalysts 154
5.2.2.1ZeoliticandMesoporousBrønstedSolidAcids 156
5.2.2.2SulfonicAcidFunctionalizedHybridOrganic–InorganicSilicas 159
5.2.2.3Metal–OrganicFrameworks 163
5.2.2.4SupportedIonicLiquids 164
5.2.3CatalystsforTandemIsomerizationandDehydrationofC5 –C6 Sugars 165
5.2.3.1BifunctionalZeolitesandMesoporousSolidAcids 165
5.2.3.2MetalOxides,Sulfates,andPhosphates 167
5.2.3.3Metal–OrganicFrameworks 172
5.2.4CatalystsfortheHydrogenationofC5 –C6 Sugars 172
5.2.4.1NiCatalysts 173
5.2.4.2RuCatalysts 176
5.2.4.3PtCatalysts 178
5.2.4.4OtherHydrogenationCatalysts 178
5.2.5HydrogenolysisCatalysts 179
5.2.6OtherReactions 183
5.3ConclusionsandFuturePerspectives 184 References 186
6ChemistryofC—CBondFormationReactionsUsedinBiomass Upgrading:ReactionMechanisms,SiteRequirements,and CatalyticMaterials 207 TuongV.Bui,NhungDuong,FelipeAnaya,DuongNgo,GapWarakunwit, andDanielE.Resasco
6.1Introduction 207
6.2MechanismsandSiteRequirementsofC–CCouplingReactions 208
6.2.1AldolCondensation:MechanismandSiteRequirement 208
6.2.1.1Base-CatalyzedAldolCondensation 208
6.2.1.2Acid-CatalyzedAldolCondensation:MechanismandSite Requirement 214
6.2.2Alkylation:MechanismandSiteRequirement 219
6.2.2.1LewisAcid-CatalyzedAlkylationMechanism 219
6.2.2.2BrønstedAcid-CatalyzedAlkylationMechanism 220
6.2.2.3Base-CatalyzedAlkylation:MechanismandSiteRequirement 225
6.2.3Hydroxyalkylation:MechanismandSiteRequirement 225
6.2.3.1BrønstedAcid-CatalyzedMechanism 227
6.2.3.2SiteRequirement 228
6.2.4Acylation:MechanismandSiteRequirement 229
6.2.4.1MechanisticAspectsofAcylationReactions 230
6.2.4.2RoleofBrønstedvs.LewisAcidinAcylationOverZeolites 232
6.2.5Ketonization:MechanismandSiteRequirement 234
6.2.5.1MechanismofSurfaceKetonization 234
6.2.5.2SiteRequirement 238
6.3OptimizationandDesignofCatalyticMaterialsforC–CBond FormingReactions 239
6.3.1Oxides 239
6.3.1.1Magnesia(MgO) 239
6.3.1.2Zirconia(ZrO2 ) 245
6.3.2Zeolites 248
6.3.2.1ZSM-5 248
6.3.2.2HY 254
6.3.2.3HBEA 257 References 259
7DownstreamConversionofBiomass-DerivedOxygenatesto FineChemicals 299 MichèleBesson,StéphaneLoridant,NoémiePerret,andCatherinePinel
7.1Introduction 299
7.2SelectiveCatalyticOxidation 300
7.2.1Introduction 300
7.2.2CatalyticOxidationofGlycerol 301
7.2.2.1GlyceroltoGlycericAcid(GLYAC) 301
7.2.2.2GlyceroltoTartronicAcid(TARAC) 304
7.2.2.3GlyceroltoDihydroxyacetone(DHA) 305
7.2.2.4GlyceroltoMesoxalicAcid(MESAC) 305
7.2.2.5GlyceroltoGlycolicAcid(GLYCAC) 305
7.2.2.6GlyceroltoLacticAcid(LAC) 306
7.2.3Oxidationof5-Hydroxymethylfurfural(HMF) 307
7.2.3.1HMFto2,5-FurandicarboxylicAcid(FDCA) 307
7.2.3.2HMFto2,5-Diformylfuran(DFF) 309
7.2.3.3HMFto5-Hydroxymethyl-2-furancarboxylicAcid(HMFCA)or 5-Formyl-2-furancarboxylicAcid(FFCA) 310
7.3Hydrogenation/Hydrogenolysis 310
7.3.1Introduction 310
7.3.2HydrogenolysisofPolyols 310
7.3.2.1HydrodeoxygenationofPolyols 311
7.3.2.2C–CHydrogenolysisofPolyols 314
7.3.3HydrogenationofCarboxylicAcids 316
7.3.3.1LevulinicAcid 316
7.3.3.2SuccinicAcid 318
7.3.4SelectiveHydrogenationofFuranicCompounds 320
7.3.5ReductiveAminationofAcidsandFurans 323
7.4CatalystDesignfortheDehydrationofBiosourcedMolecules 324
7.4.1Introduction 324
7.4.2GlyceroltoAcrolein 325
7.4.3LacticAcidtoAcrylicAcid 328
7.4.4SorbitoltoIsosorbide 330
7.5ConclusionsandOutlook 331 References 331
8ConversionofLignintoValue-addedChemicalsviaOxidative Depolymerization 357 JustinK.Mobley
8.1Introduction 357
8.1.1CautionaryStatements 360
8.2CatalyticSystemsfortheOxidativeDepolymerizationofLignin 361
8.2.1EnzymesandBio-mimeticCatalysts 361
8.2.2CobaltSchiffBaseCatalysts 363
8.2.3VanadiumCatalysts 367
8.2.4Methyltrioxorhenium(MTO)Catalysts 368
8.3CommercialProductsfromLignin 369
8.4StepwiseDepolymerizationof β-O-4Linkages 369
8.4.1BenzylicOxidation 369
8.4.2SecondaryDepolymerization 376
8.5HeterogeneousCatalystsforLigninDepolymerization 382
8.6Outlook 386 Acknowledgments 386 References 386
9LigninValorizationviaReductiveDepolymerization 395 Yang(Vanessa)Song
9.1Introduction 395
9.2Late-stageReductiveLigninDepolymerization 396
9.2.1MildHydroprocessing 398
9.2.2HarshHydroprocessing 404
9.2.3BifunctionalHydroprocessing 407
9.2.4LiquidPhaseReforming 410
9.2.5ReductiveLigninDepolymerizationUsingHydrosilanes,Zinc,and Sodium 414
9.3ReductiveCatalyticFractionation(RCF) 416
9.3.1ReactionConditions 417
9.3.2LignocelluloseSource 417
x Contents
9.3.3AppliedCatalyst 427
9.4Outlook 428 Acknowledgment 429 References 429
10ConversionofLipidstoBiodieselviaEsterificationand Transesterification 439
AminTalebian-KiakalaiehandAminNorAishahSaidina
10.1Introduction 439
10.2DifferentFeedstocksforBiodieselProduction 441
10.3BiodieselProduction 441
10.3.1AlgalBiodieselProduction 442
10.3.1.1NutrientsforMicroalgaeGrowth 443
10.3.1.2MicroalgaeCultivationSystem 444
10.3.1.3Harvesting 444
10.3.1.4Drying 445
10.3.1.5LipidExtraction 446
10.4CatalyticTransesterification 446
10.4.1HomogeneousCatalysts 446
10.4.1.1AlkaliCatalysts 446
10.4.1.2AcidCatalysts 448
10.4.1.3Two-stepEsterification–TransesterificationReactions 448
10.4.2HeterogeneousCatalysts 450
10.4.2.1SolidAcidCatalysts 451
10.4.2.2SolidBaseCatalysts 451
10.4.3Enzyme-CatalyzedTransesterificationReactions 453
10.5SupercriticalTransesterificationProcesses 454
10.6AlternativeProcessesforBiodieselProduction 455
10.6.1UltrasonicProcesses 455
10.6.2Microwave-AssistedProcesses 456
10.7Summary 459 References 459
11UpgradingofLipidstoHydrocarbonFuelsvia (Hydro)deoxygenation 469
DavidKubi ˇ cka
11.1Introduction 469
11.2Feedstocks 471
11.3Chemistry 472
11.4Technologies 475
11.5Catalysts 477
11.5.1SulfidedCatalysts 477
11.5.2MetallicCatalysts 480
11.5.3MetalCarbide,Nitride,andPhosphideCatalysts 483
11.6ConclusionsandOutlook 489 References 490
12UpgradingofLipidstoFuel-likeHydrocarbonsandTerminal OlefinsviaDecarbonylation/Decarboxylation 497
RyanLoe,EduardoSantillan-Jimenez,andMarkCrocker
12.1Introduction 497
12.2LipidFeeds 500
12.3deCOx Catalysts:ActivePhases 502
12.4deCOx Catalysts:SupportMaterials 508
12.5ReactionConditions 509
12.6ReactionMechanism 511
12.7CatalystDeactivation 516
12.8ConclusionsandOutlook 518 References 518
13ConversionofTerpenestoChemicalsandRelated Products 529
AnneE.Harman-Ware
13.1Introduction 529
13.2TerpeneBiosynthesisandStructure 529
13.3SourcesofTerpenes 532
13.3.1ConifersandOtherTrees 532
13.3.2EssentialOilsandOtherExtracts 534
13.4IsolationofTerpenes 535
13.4.1TappingandExtraction 535
13.4.2TerpenesasaBy-productofPulpingProcesses 536
13.5HistoricalUsesofRawTerpenes 536
13.5.1AdhesivesandTurpentine 536
13.5.2Flavors,Fragrances,Therapeutics,andPharmaceutical Applications 537
13.6CatalyticMethodsforConversionofTerpenestoFineChemicalsand Materials 537
13.6.1HomogeneousProcesses 538
13.6.1.1HydrationandOxidationReactions 538
13.6.1.2HomogeneousCatalysisfortheEpoxidationofMonoterpenes 541
13.6.1.3Isomerizations 541
13.6.1.4ProductionofTerpeneCarbonatesfromCO2 andEpoxides 543
13.6.1.5PolymersandOtherMaterialsfromTerpenes 545
13.6.1.6“ClickChemistry”RoutesfortheProductionofMaterialsand MedicinalCompoundsfromTerpenes 548
13.6.2HeterogeneousProcesses 551
13.6.2.1IsomerizationandHydrationof α-Pinene 551
13.6.2.2HeterogeneousCatalystsfortheEpoxidationofMonoterpenes 553
13.6.2.3Isomerizationof α-PineneOxide 555
13.6.2.4VitaminsfromTerpenes 555
13.6.2.5DehydrogenationandHydrogenationReactionsofTerpenes 557
13.6.2.6ConversionofTerpenestoFuels 558 Acknowledgments 560 References 561
Contents
14ConversionofChitintoNitrogen-containingChemicals 569
XiChenandNingYan
14.1WasteShellBiorefinery 569
14.2ProductionofAminesandAmidesfromChitinBiomass 571
14.2.1SugarAmines/Amides 571
14.2.2FuranicAmines/Amides 574
14.2.3PolyolAmines/Amides 576
14.3ProductionofN-heterocyclicCompoundsfromChitinBiomass 579
14.4ProductionofCarbohydratesandAceticAcidfromChitin Biomass 581
14.5ProductionofAdvancedProductsfromChitinBiomass 584
14.6Conclusion 587 References 587
15Outlook 591
EduardoSantillan-JimenezandMarkCrocker
Index 599
Preface
Formostofhumanhistorybiomasshasbeentheprincipalenergysourcepoweringhumandevelopment.Indeed,biomasscanbeutilizedfortheproductionof heat,steam,motivepowerandelectricity,andcanbeconvertedviathermal,biological,orchemicalpathwaysintochemicalsandfuels.Althoughthetwentieth centurycanberegardedasthe“petroleum”century,i.e.thetimewhenpetroleum cametotheforeasthepredominantsourceoffuelsandchemicals,inrecent yearsinterestinbiomassconversiontochemicalsandfuelshasgrownsteadily, drivenbytheissuesofsustainabilityandenvironmentalprotection.Increasing concernssurroundingglobalwarmingandthecontributionoffossilfueluseto atmosphericCO2 levelsmeanthathumanityisonceagainlookingtobiomass resourcesfortheproductionofessentialcommodities.
Giventhecomplexityofbiomass,asreflectedinthewiderangeoffunctional groupspresent,alongwithitsrecalcitrance–asexemplifiedbythelignincomponentofbiomass–catalysishasamajorroletoplayintheconversionofbiomass tousefulmolecules.However,althoughcatalysisisintegraltomodernlife(it hasbeenestimatedthatcatalysisisinvolvedatsomestageintheproductionof ca.80%ofallmanufacturedgoods)theuseofcatalysisismuchlessdevelopedfor theproductionofchemicalsfrombiomasscomparedtopetroleum.
Againstthisbackdrop,thisbookaimstoprovidethereaderwithadetailed descriptionofthecatalystsandcatalyticprocessesemployedinthesynthesisof chemicalsandfuelsfrombiomass,theinformationbeingorganizedinawaythat coversthemostabundantandimportanttypesofbiomassfeedstock.Theissue ofcatalystdesignisemphasizedthroughout,bearinginmindthatcatalystsused forbiomassprocessingmustoftenfunctioninaqueousenvironmentsandinthe presenceofpotentialpoisonssuchasmineralcomponents.
Twogeneralapproachescanbediscernedfortheconversionofbiomass tochemicalsandfuels,involvingeither(i)conversionofthewholebiomass intoanintermediateproductsuchaspyrolysisoilorsyngasthatcanthenbe catalyticallyupgradedtousefulproducts,or(ii)fractionationofthebiomassinto itsmaincomponents,followedbytheupgradingofthesefractionsusingtailored conversionprocesses.Followingthisrationale,Chapters1–4ofthisbookfocus ontheapplicationofcatalyststothepyrolysisofwholebiomassandtothe upgradingofbio-oils.Subsequentchaptersfocusonthevalorizationofbiomass constituents.Chapters5–7discusscatalyticapproachestotheprocessingof biomass-derivedoxygenates–asexemplifiedbysugars–viareactionssuch
xiv Preface
asreforming,hydrogenation,oxidation,andcondensationreactions.Lignin isconsiderednext,Chapters8and9providinganoverviewofcatalystsfor ligninvalorizationviaoxidativeandreductivemethods.Afterthat,Chapters 10–12considertheconversionoffatsandoilstofuelsandterminalolefinsby meansofesterification/transesterification,hydrodeoxygenation,anddecarboxylation/decarbonylationprocesses.Chapters13and14provideanoverviewof conversionprocessesbasedonterpenesandchitin,twoemergingfeedstocks witharichchemistry.Finally,Chapter15summarizessomeoftheemerging trendsinthefieldofcatalysisforbiomassvalorizationandlooksaheadtofuture developments.
Theeditorsofthisbookaredeeplyindebtedtotheauthorsofthechapters. Withouttheirtime,effortandexpertisethisbookwouldnothavebeenpossible.WewouldalsoliketothankLeslieHughesattheCenterforAppliedEnergy Researchforherunflagginghelpinthepreparationofthemanuscript.
July2,2019 Lexington,Kentucky,U.S.A.
MarkCrocker EduardoSantillan-Jimenez
UpgradingofBiomassviaCatalyticFastPyrolysis(CFP)
CharlesA.Mullen
USDA-AgriculturalResearchService,EasternRegionalResearchCenter,600E.MermaidLane,Wyndmoor,PA, USA
1.1Introduction
Definedasheatingofanorganicmaterialinanonoxidativeenvironment,pyrolysishasbeenrecognizedfordecadesasthemostefficientprocessforconvertinglignocellulosicbiomassintoadenseliquid,commonlycalled pyrolysisoil or bio-oil [1–3].Themostcommonlyusedconditionsforconversionofbiomassto liquidhavebeenhighheatingratestotemperaturesofaround500 ∘ C,atatmosphericpressure,theso-calledfastpyrolysisprocess[1–3].Thefastpyrolysisprocessoffersmanyadvantagesthatmakeitattractiveforconversionofbiomassto bio-fuelintermediatesandproductionofrenewablechemicals.Theseadvantages includehighliquidyields(>60%insomecases)andproductionofapotentially valuablecoproductin bio-char .Thissolid,consistingoffixedcarbonandminerals,hasbeenshowntobeagoodsoilamenderandapotentialroutetosequester carbon[4–6].Withthepotentialutilizationofthecombustibleoffgases,andif neededsomeofthebio-char,pyrolysiscanbepoweredbyitsownenergy,making itanearlyself-sufficientprocessrequiringfewotherinputs[3].
Bio-oilcontainshundredsofoxygenatedcompoundsderivedfromthecellulose,hemicellulose,andligninthatcomprisethebiomass.Inrecentyears, muchhasbeenmadeofbio-oilasapotentialintermediatetotheproduction ofadvancedhydrocarbontransportationfuelsorasafeedstockfromwhich toisolaterenewablechemicals.However,commercialorevenprecommercial successforutilizationofthesebio-oilshasbeenlimitedtolowervalueapplicationssuchasuseasboiler-typefuelsforheatandpower[3,7]orutilization asanasphalt-likematerial[8].Thetechnicalreasonfortheselimitationsisthat thecompositionofthebio-oil,comprisinghighconcentrationsofreactiveoxygenatedfunctionalgroups,plusthepresenceofcatalyticmicrosolids,makesthe mixturethermallyunstable[9–11].Therefore,processingtechnologiesrequiring evenmoderateheatingofthebio-oilmixture,suchasdistillation,resultin productionofintractablematerials[12].Whilecatalytichydrodeoxygenation (HDO)hasbeenthepost-productionupgradingchoiceforrefiningofbio-oilto hydrocarbonstobeusedasfuels,theunstablenatureofthebio-oilalsomakes ChemicalCatalystsforBiomassUpgrading, FirstEdition.EditedbyMarkCrockerandEduardoSantillan-Jimenez. ©2020Wiley-VCHVerlagGmbH&Co.KGaA.Published2020byWiley-VCHVerlagGmbH&Co.KGaA.
1UpgradingofBiomassviaCatalyticFastPyrolysis(CFP)
thisHDOprocessdifficult.Themosteffectivepost-productiondeoxygenation processesdevelopedrequiremultiplecatalytic,highpressurehydrotreating steps,atsignificantcost,makingtheproductionofalowmarginfuelproductof questionableeconomicviability[13–15].
Becauseoftheselimitations,researchershavesoughttodevelopprocessesthat alterthechemicalpathwaysduringpyrolysistoproduceamorestablebio-oil productwithmorefavorablecompositionsforvariousend-useapplications, includingHDO.Utilizationofheterogeneouscatalystsduringthepyrolysis process,termed catalyticfastpyrolysis (CFP),hasreceivedthemostattention. Becauseoftheinterestinadvancedhydrocarbonbio-fuels,themostcommon goalofcatalyticpyrolysishasbeentoproduceapartiallydeoxygenated,thermallystablepyrolysisoilthatismoreamenabletofinalHDO-typeupgrading tofuel-rangehydrocarbons.However,alternativeprocesseshaveaimedtoconvergechemicalpathwaystowardproductionofvariousindividualcompounds orgroupsofcompoundsforpetrochemicalorfinechemicaluses.Inthischapter wewilldiscussCFPprocessesbothaimedatgeneraldeoxygenationandthose aimedattargetedclassesofmolecules.Forthepurposesofthischapter,wewill considercatalyticpyrolysisprocessesthatfallwithinthefollowingdefinitions: (i)heterogeneouscatalyticprocessingofbiomasspyrolysisvaporseither insitu (pyrolysisandcatalysisoccurinthesamereactorzone)or exsitu (pyrolysisand vaporphasecatalysisaredecoupled)and(ii)reactionstakingplaceininertor reactive(butnon-oxidative)atmospheresatnearatmosphericpressure.Therefore,catalytichydrothermalorsolventliquefaction[16]andothertechnologies suchaspressurizedhydropyrolysis[17]areoutsidethescopeofthischapter.
Asmentionedearlier,CFPencompassesprocesseswheresolidbiomasscontactsthecatalystandbothpyrolysisandvaporupgradingoccurinthesamereactor,theso-called insitu methods,andprocesseswherethepyrolysisandcatalytic vaporupgradingoccurinseparatereactors,called exsitu or vaporupgrading methods.SimplifiedschematicsofthetwoprocessesarepresentedinFigure1.1. Themainadvantageofthe insitu methodisitssimplicity–the“one-pot”reaction systemsavescapitalcostasitdoesnotrequireasecondreactor.However,there areseveraladvantagesthatdecouplingthetwostepsallowsfor[18].Whileitis wellknownthattemperaturesinthe500 ∘ Crangeproducethemaximumyieldof condensablerangespeciesfrompyrolysisofmostbiomass,theidealrangeforvariouscatalyticprocessesmaybesignificantlydifferent,dependingonthecatalyst andthedesiredendproducts.Furthermore,becausethe insitu processrequires contactofsolidbiomasswithsolidcatalyst,reactorsfortheunitoperationtendto belimitedtoonlyfluidizedbeds.Decouplingtheprocessallowsthecatalysisstep tooccurineitherafixedorfluidizedbedandalsoallowsforothervariousreactor typestoaccomplishthepyrolysis.Anotherveryimportantfactoristhatthe exsitu processallowsforremovalofbio-charanditsassociatedmetalcontentpriorto introductionofthecatalyst.Thishasimportantimplicationsforcatalystlifetimes asinorganicmaterialscontainedinthebiomass,particularlyGroup1andGroup 2metals,canpoisoncatalysts,leadingtomorefrequentcatalystreplacementor replenishmentandahighercatalystdemand.Thisisanespeciallyimportantconsiderationforzeolite-catalyzedprocessesbecausetheyarehighlysusceptibleto thistypeofdeactivation,asdescribedlater.
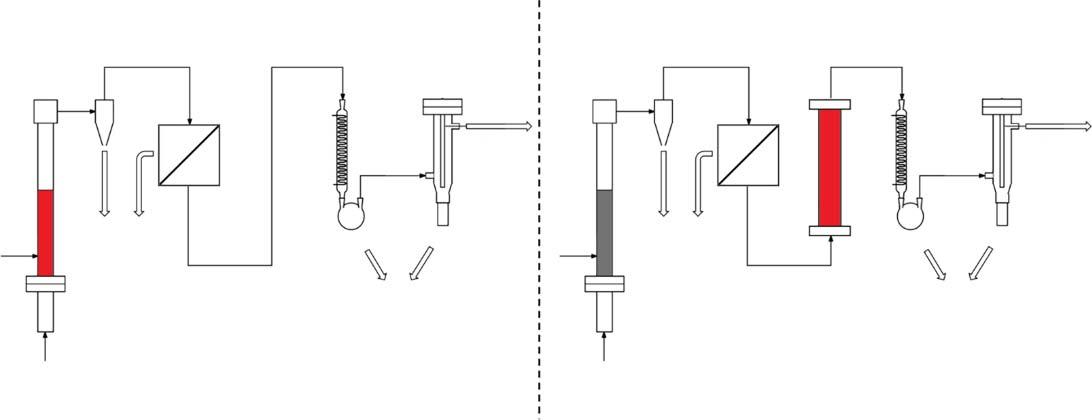
Figure1.1 Simplifiedprocessschematicscomparing(a) insitu and(b) exsitu catalyticpyrolysis.
Biomass inlet
(a) (b)
Biomass inlet
Bio-char
1.1.1CatalyticPyrolysisOverZeolites
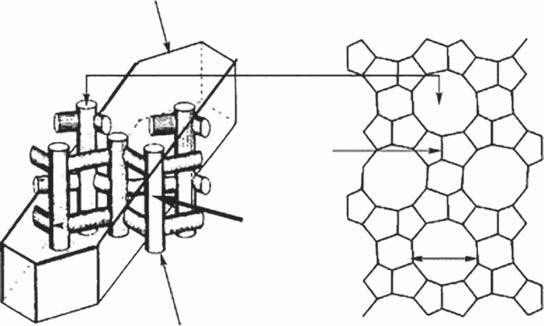
Zeolitesandsimilarmaterialshavebeenbyfarthemostcommoncatalysts employedinCFPprocesses.Zeolitesarecrystallinesubstanceswithastructure characterizedbyaframeworkoflinkedtetrahedra,eachconsistingoffour oxygenatomssurroundingacation[19–21].Zeolitesoccurnaturally,butthe adventofsyntheticzeolitesinthe1950s,freeofthedefectsandimpuritiesfound innature,iswhentheiruseinchemicalcatalysistookoff[20].Industrialscale catalyticuseofzeolitesstartedin1962,withtheuseofzeolitesXandYfor fluidcatalyticcracking(FCC)ofheavypetroleumfractions,aprocessthatisstill todayoneofthehighestvolumechemicalprocessesused.Otherpetrochemical usesincludehydrocracking,isomerization,disproportionation,andalkylation ofaromatics.Theframeworkofzeolitesisusuallysilicaandaluminabased.This frameworkcontainsopencavitiesintheformofchannelsandcages[19].These areusuallyoccupiedbyH2 Omoleculesandextra-frameworkcationsthatare commonlyexchangeable[19].Insilica–alumina-basedmaterials,theneedfor theextra-frameworkcationistobalancethechargeimbalancecreatedbythe fourcoordinateAl(III)sites,asopposedtotheneutralSi(IV)sites(Figure1.2) [19–21].Often,intheactivecatalysts,someoralloftheextra-frameworkcations areBrønstedacid(H+ )activesites,whichalongwiththeLewisacidityofother sitesareresponsibleforinitiatingthecatalyticreactions.Thechannelsallowthe passageofguestspeciestotheseactivesites[19–21].InbiomassCFPtheseguest speciesaremoleculesderivedfromthebreakdownofthebiopolymerscausedby theinitialpyrolysisreactions.Theporesizeandshape,Brønstedacidstrength andsitedensity,andthepresenceofothertypesofactivesitesareimportant factorsintheactivityandselectivityofthecatalysts.
1.1.1.1CatalyticPyrolysisOverHZSM-5
SeveraldifferenttypesofzeoliteshavebeentestedascatalystsforthedeoxygenationofpyrolysisvaporsincludingZeoliteSoconyMobil-5(ZSM-5),Y,beta, mordenite,andferrierite[22–26].Amongthesetypesofzeolites,theZSM-5 (alsocalledMFIformordeniteframeworkinverted)-basedmaterialshaveshown themostpromiseandreceivedthemostattentionandwillbethefocusofthis section.ZSM-5-typezeoliteshavebeenshowntoselectivelyconvertmolecules fromawidevarietyofsourcestoaromatichydrocarbons.Amongtheseprocesses aremethanoltoolefinsandgasolineandcrackingofwastehydrocarbonplastics [27,28].Aromatichydrocarbonsaregoodtargetproductmoleculesfrom biomass,because,likethebiomassstartingmaterials,theyhavelowH/Cratios, meaningthebiomasscarboncanbemoreefficientlyconvertedwithoutthe
Figure1.2 BrønstedandLewisacidsitesofzeolites.
Macroscopic shape of a crystal of ZSM-5
Pore opening
5.5 Å
Microscopic internal surface of a crystal of ZSM-5 External surface of ZSM-5
Figure1.3 StructureofZSM-5.Source:Leietal.2003[30].Reproducedwithpermissionof RoyalSocietyofChemistry.
additionofanexternalsourceofhydrogen[29].TheshapeselectivityofZSM-5is responsibleforconvergencetowardaromatics.ZSM-5hasathree-dimensional poresystemcomprisingstraight10-memberedring5.2 × 5.7Åchannelsconnectedbysinusoidal5.3 × 5.6Åchannels(Figure1.3)[25,30].Thisputsitinthe categoryofamediumporesizezeolite.Thisporestructurecreatesamasstransfer effect,limitingthemoleculesthatenterandinteractwiththechemicalfunctionalityoftheacidsitebasedonsize.Atthesametime,theconfinedspacelimitsthe geometrythatcanoccurinthereactiontransitionstates,forcingthechemistry throughcertainpathways,creatingtheobservedproductselectivity[24].
Biomasscatalyticpyrolysis,whetherthephysicalprocessisperformedinthe insitu or exsitu configuration,encompassestwodistinctchemicalprocesses. First,pyrolyticdecompositionofthebiopolymersproducesoxygenatedvapors, aerosols,andbio-char.Thecatalystisinvolvedinthesecondstepwherein someoftheoxygenatedgasphaseproductsinteractwiththecatalyst,initiating reactionsthatalterthecompositionofthegasstreamandhencethecondensed productthatiscollected.Thevariousoxygenatedpyrolysisproductsthatarethe substratesinthecatalyticreactionshavedifferinginteractionswiththecatalyst, dependingonseveralfactorsincludingwhethertheirmolecularsizeallowsentry intotheporesofthecatalyst,theirdiffusionratethroughthechannels,andthe interactionoftheirchemicalfunctionalitywiththecatalystactivesiteandother reactantspresent.DuringcatalyticpyrolysisoverHZSM-5,mostofthearomatic hydrocarbonsareultimatelyderivedfromthecarbohydrateportions(cellulose andhemicellulose)ofthebiomass,whilelignin-derivedvaporsareconsidered primarily,butnotentirely,asprecursorstocokeformation[30,31].Theportions oftheligninthatdocontributetothehydrocarbonpoolaremostlyderivedfrom thecarbonchainlinkersorsidechainsratherthanthephenolicunitsthemselves [32].Thechemicalpathwaysthatoccurtoproducearomatichydrocarbonsare summarizedinFigure1.4.Thekeystepafterproductionofoxygenatedvapors
Levoglucosan
Primary pyrolysis vapors (cellulosic)
Hydrocarbon pool
Primary pyrolysis vapors (lignin)
Oligomerization aromatization
Cracking
Diels–Alder
Hydrocarbon pool intermediates (minor) Furans Other intermediate oxygenates
Alkyl phenols catcheols
Figure1.4 MajorchemicalpathwaysfromprimarypyrolysisvaporstoaromatichydrocarbonsviaCFPoverHZSM-5.
Aromatic hydrocarbons Aromatic hydrocarbons
PAH, coke
1.1Introduction 7 viatheinitialpyrolyticdepolymerizationisthedehydrationofcompoundssuch asanhydrosugarstoformfurans[33].Thisstepcanoccuronthesurfaceof thecatalysts,producingsmallermoleculesthatcandiffuseintothemicropores [25,34,35].Here,twomainpathwaystowardaromaticsbecomeoperative. Thefirstpathwayinvolvesdecarbonylationtoformolefinswhichcangoonto oligomerizeandaromatize.Thismixtureofolefinsandproducthydrocarbons isreferredtoasthe hydrocarbonpool [33].Alternatively,Diels–Alder-type cycloadditionreactionsoffuranswitholefinscandirectlyproducearomatics upondehydrationofthebicycliccycloadditionadduct[36].Relatedreactionsof otherprimarypyrolysisproductscanalsoproduceCO2 alongwitholefinsand alkanes.
ManystudieshavebeenconductedontheeffectsofprocessconditionsforcatalyticpyrolysisofbiomassandtheircomponentsoverHZSM-5,focusedeither onthegoalofproducingpartiallydeoxygenatedbio-oil(fordirectbulkuseor furtherprocessing),orfortheproductionofaromatichydrocarbonsandlight olefins[22–26,29,37–49].AnestimatedcarbonbalanceforCFPofbiomassover HZSM-5ispresentedinFigure1.5[49],andTable1.1summarizesthedetailsof selectedstudieswhereproductsampleswereproduced(i.e.largerthananalytical ormicropyrolyzerstudies).Whiletherearemanyfactorsthatcontributetothe variationinthereportedresults,generallythelevelofdeoxygenationachieved followsaninversetrendwithbio-oilyield[50].Lowerbiomasstocatalystratios, correspondingtohighercatalystactivity,resultinmoredeoxygenationatthe expenseofbio-oilyield;potentialorganicliquidyieldislosttogasphaseproducts, water,andcoke[18,49].Whileoverallorganicliquidyielddecreasesduetoeliminationofoxygenatedspecies,yieldsofaromatichydrocarbonsgenerallyincrease withincreasingbiomasstocatalystratio.Residencetimeandspacevelocityconsiderationscanalsoaffecttheconversiontoaromatichydrocarbons.Inastudyon CFPofcelluloseoverHZSM-5inan insitu fluidizedbedprocess,atequalweight hourlyspacevelocity(WHSV),agasresidencetimeofabout8.6seconds(studied overarangeof5.6to ∼10secondsbychangingcarriergasflowrates)maximized aromaticyield[46].Selectivityforbenzeneandtoluenewerealsomaximizedat thisresidencetime,whileselectivitytoxylenesandnaphthaleneswasminimized.
Asmentionedearlier,thecellulosicportionofthebiomassismostefficiently convertedtoaromatichydrocarbons,whileonlyasmallportionoflignincanbe convertedtoaromatichydrocarbonsoverHZSM-5.Anumberofotherfactors relatedtothebiomasscompositioncanalsoaffecttheproductionofaromatics. Thisgivesrisetovariationinyieldsandqualityofliquidproductsandaromatic hydrocarbons.Biomasscomposition,alongwithprocessconditionsandcatalyst propertiesarealsoimportantvariablesincatalystdeactivationrates,whichare discussedindetaillater.Studiesofbiomasswithinasmallrangeofcompositions havefound,unsurprisingly,thatincreasedashcontent(particularlypotassium) correlateswithdecreasedconversionofcarbontoaromaticsduringCFP,due toanincreasedproductionofgases,andincreasedligninisalsocorrelatedwith decreasedaromaticsandincreasedcokeyield[51].
WithregardstothepropertiesofastandardHZSM-5zeolitecatalystthatinfluencebiomassvaporupgrading,thenumberofBrønstedacidsitesthebiomass isexposedto,controlledbytheacidsitedensity(inverselyproportionaltothe
Table1.1 SelectedresultsofbiomassCFPovertypicalHZSM-5catalysts.
BiomassScale(kg/h)ConfigurationSARa)
Cellulose ∼0.06 Insitu 305000.25—39.5[46]
Ligninmg Insitue) 2365015—8.2[32]
Pine ∼0.165 Insitu 3060060.315.5[43]
Pine2 Insitu —4750.33213.3/19.42[45]
Hybridpoplar2 Insitu —4750.33212.2/20.25[45]
Cornstover2 Insitu —4750.328.9/13.99[45]
Switchgrass2 Insitu —4750.33215.8/14.7[45]
Juniper2 Insitu —4750.33215.8/12.3[45]
Pinebark2 Insitu —4750.3328.9/18.9[45]
Switchgrass0.3 Insitu 30450–5000.51.29.4/23.5[41]
Corncob0.036 Insitu 48550525.5/14.69[38]
Pinewood20 Insitu 50550724/21.5[42]
Beechwood0.5 Insitu 505001127/19.5[47]
Beechwood0.5 Insitu 505002123/17.5[47]
Pine0.150 Exsitu 30500214.3/4.0[48]
Pine0.150 Exsitu 305000.6717.2/14.2[48]
Pine0.150 Exsitu 305000.4823.1/17.7[48]
a)SAR,silicaaluminaratio.
b)WHSV,weighthourlyspacevelocity. c)C%,carbonyield.
d)AHC,aromatichydrocarbons. e)Micropyrolyzer.
Light gases
Char + coke
Organic stream
Aqueous stream
Figure1.5 TypicalcarbondistributionfromCFPofbiomassoverHZSM-5andrelatedcatalysts. Source:Staraceetal.2017[49].ReproducedwithpermissionofAmericanChemicalSociety.
silica/aluminaratio)isthemostimportantvariablecontrollingactivity[40],along withthebiomasstocatalystratio.Intheabsenceofothervariations,thehigher theacidsitedensity,themoreactivethecatalyst.Thisgenerallyleadstohigher yieldsofaromatics;however,atveryhighacidsitedensities,thesmalldistance betweenactivesitescanpromotetheformationofcoke,decreasingtheinitial yieldofaromatichydrocarbonsandalsoleadingtomorerapidcatalystdeactivationasdiscussedinmoredetailinthesucceedingtext[52].Onestudylooking ataromatichydrocarbonyieldswhenusingHZSM-5withSiO2 /Al2 O3 = 23,30, 50,and80,found30astheoptimumtomaximizetheyieldofaromatichydrocarbons[40].LowerSiO2 /Al2 O3 alsocorrelatedwithhigherproductionofCO2 (but notCO),indicatingBrønstedaciddependenceforanydecarboxylationreactions, althoughthisisarelativelyminordeoxygenationpathwayinCFPoverzeolites. HighacidityalsoincreasedselectivityforbenzeneandtolueneoverC8+ aromaticsincludingxylenes,ethylbenzene,andindanes[40].
1.1.1.2DeactivationofHZSM-5DuringCFP
CatalystdeactivationisasignificantconcernforbiomassCFP.Zeolitesaresubjectedtothreemajordeactivationmechanismsduringtheprocess.Thefirstis formationofcokethatblocksaccesstothecatalystactivesite.Thisisthemost rapidformofcatalystdeactivationandcanhavenoticeableeffectsontheproduct distributionalmostimmediatelyatcumulativebiomass/catalystmassratiosof <1.Fortunately,thistypeofdeactivationisalsoreversible,viacombustionofthe coke.Asecondformofdeactivationresultsfrompoisoningofthecatalystactive siteswithinorganicspeciesfromthebiomass,particularlyalkalimetals.Thistype ofdeactivationis,forpracticalpurposes,irreversible.Athirdtypeofdeactivation isphysicaldegradationofthecatalyst,mostcommonlydealumination.Thiscan occurduetoexposuretohightemperaturesandsteamandcanalsooccurduring combustionprocessusedtoregeneratecatalystsfromcokedeposits.Thistypeof deactivationisalsoirreversible.
1UpgradingofBiomassviaCatalyticFastPyrolysis(CFP)
TherearetwomainmechanismsofcokeformationactiveduringbiomassCFP. Thefirstissimilartocokeformationthatoccursduringotherzeolitecatalyzed reactions(includingpetroleumprocessingandmethanoltoolefins),i.e.molecularweightgrowthofalreadyformedaromaticsbyreactionwithotheraromatics orolefinspresentinthehydrocarbonpool[50,53].Becausebiomasspyrolysis vaporsaredeficientinhydrogen,anydeoxygenationoccurringbydehydration reactionswillfurtherreducethehydrogencontentofthevaporsfavoringmolecularweightgrowthreactionsthatformlargearomaticmoleculeswithlowH/C ratiosthatcomprisecoke.Thisprocesscanbefurtherexacerbatedbyslowdiffusionofproductsoutofthezeolite[25].Anothersourceofcokeisdirectcondensationordepositionoflignin-derivedphenolics,whichhavepoorreactivity overZSM-5andtendtobetoobulkytonavigatetheporesofthecatalyst[54]. IfHZSM-5catalystsarenotregeneratedbycombustionofthecoke,complete deactivationofthecatalystandproductionofproductvaporswithsimilaroxygen contentandcompositiontononcatalyticpyrolysisareproducedatcumulative biomasstocatalystratiosofabout3or4to1forbiomasssuchaswoodandgrasses [50,55,56].Lignindeactivatescatalystsmuchquickerthancelluloseanduseof feedstockswithhighligninconcentrationmayresultinmorerapidcatalystdeactivationviacoking.Therapidcokingeffectcanaccountforasignificantportion ofthevariabilityinresultsandtheeffectofbiomasstocatalystratiosasseenin Table1.1,dependingonrunlengthsandregenerationprotocols.
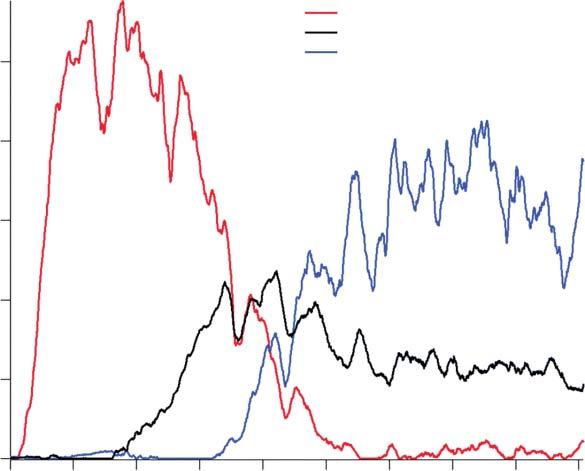
Theeffectofcokingontheproductvaporscanbeseenalmostimmediately, atverylowcumulativebiomasstocarbonratios.DuringCFPoverHZSM-5,the firstoxygenatestoappeararenotprimarypyrolysisvaporsbutpartiallydeoxygenatedspeciesincludingfurans,phenols,andalkylphenols(mostlycresols)[50]. Furansareintermediatesonthepathwayfromanhydrosugars(celluloseprimary pyrolysisproducts)toaromatics,andtheirappearanceisreflectiveofthelossof catalyticactivityforthedecarbonylationstepthatresultsintheirconversionto olefinsthatmakeuppartofthehydrocarbonpool(seeFigure1.4).Adecreasein COformationhasbeenobservedtooccuratlowcumulative/biomassratios[56].
Phenolshavealsobeenfoundasapartiallydeoxygenatedproductfrombothcelluloseandlignin[51,55–57].Unlikefurans,phenolsarenotintermediateson thepathwayfromcellulosicprimarypyrolysisvaporstoaromatichydrocarbons. Thereisevidencethattheyareformedviaareactionofdeoxygenatedspeciesstill boundtothecatalystwithwater,confirmedbyobservingtheincorporationof 17 O inphenolicproductsfromlabeledwateraddedinaCFPexperiment[57].These productsofpartiallydeactivatedcatalystsreachamaximumandthenbeginto becomelessconcentratedastheactivityforeventheinitialdehydrationstep becomesnullifiedbycokeblockingbothinternalandsurfaceacidsties[50].Primarypyrolysisproductsarethenobservedathighercumulativebiomass/catalyst ratios.Theproductvaporsconsistofamixtureofthethreegroupsuntilthe completedeactivationpointisreached.Theresultsofonestudymonitoringthe compositionofthevaporsoverthecourseofCFPuptoacumulativebiomassto catalystratioof4.5/1ispresentedinFigure1.6[50].
Cokingratescanvarywithcatalystproperties,particularlywithacidsite density.Asdiscussedearlier,theactivityoffreshcatalystsgenerallytrends withBrønstedacidsitedensity,butyieldsofaromatichydrocarbonswere
Figure1.6 Trendsinconcentrationofaromatichydrocarbons,intermediateproductsfrom pinepyrolysisoverHZSM-5(microreactor-MBMSsystem)vs.cumulativebiomasstocatalyst ratio.Source:Mukarakateetal.2014[50].ReproducedwithpermissionofRoyalSocietyof Chemistry.
optimizedatSiO2 /Al2 O3 = 30overanevenmoreacid-site-densecatalystwith SiO2 /Al2 O3 = 23[40].Thereasonforthisislikelyamorerapiddeactivationof themoreacidiccatalystduetoincreasedcokingovertheshortcourseofthe experiment.Onestudy,whichdecoupledactivityanddeactivationfromthe totalnumberofacidsites,foundthatHZSM-5withaSiO2 /Al2 O3 of40had comparableinitialactivityfortheproductionofaromatichydrocarbons,butthe deactivationratewassignificantlyslowerthancatalystswithhigheracidsite density(Figure1.7)[52].Somestudiesonmodelcompoundshaveshownthat closeproximityofacidsitescanfacilitatethecondensation(C—Cbondand H2 Oforming)reactionsbetweenoxygenatedintermediates,leadingtocoking [52,58].Achangeintheamountofextra-frameworkalumina,whichcanserve asnucleationsitesformolecularweightgrowth,canalsoleadtocoking.Inthe followingSections1.1.3and1.1.4thatdiscussmodificationofHZSM-5,the effectsofvariouschangestothecatalyststructurewithrespecttodeactivation areconsidered.
Alkalimetalspresentinthebiomassarealsoaproblematicsourceofcatalystdeactivation.Alkalimetalscanaccumulateoncatalystsrapidlyandcanion exchangewithBrønstedacidsites,deactivatingthesites.Intwostudiesof in situ CFPoverHZSM-5(wherecatalystswereregeneratedfromcoking,oneby useofacirculatingfluidizedbedandonebyintermittentregeneration)mineral
1UpgradingofBiomassviaCatalyticFastPyrolysis(CFP)
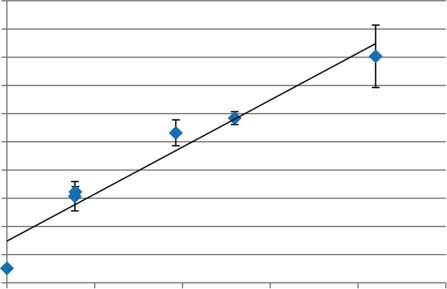
Figure1.7 Comparisonof alkylbenzenesproductiononHZSM-5 Si/Al = 11.5,25,and40fromoak pyrolysisvaporswithfixedtotalsitesat 500 ∘ C.Source:Wanetal.2015[52]. ReproducedwithpermissionofJohn WileyandSons.
Switchgrass/catalyst (w/w) Concentration selected impurties (ppm)
Figure1.8 ConcentrationoftotalCa,Cu,Fe,K,Mg,Na,andPonHZSM-5atdifferentlevelsof catalystexposuretoswitchgrassinafluidizedbedatpyrolysisconditions(500 ∘ C,N2 ).Values aretheaverageofthreeinductivelycoupledplasmaspectroscopy(ICP)measurements.Error barsareonestandarddeviation.Source:MullenandBoateng2013[41].Reproducedwith permissionofAmericanChemicalSociety.
accumulationincludingCa,K,Mg,Na,andPwasfoundtobelinearwithtime onstream(orcumulativebiomass/catalystratio)[41,42].Eachstudy,onewith switchgrass(∼2.6%ash)andonewithpinewood(∼0.4wt%ash),foundalkali concentrationonthecatalystwasaround1wt%atcumulativebiomasstocatalystratiosofaround20bymass(Figure1.8).Alinearcorrelationwasalsofound betweentheconcentrationoftheinorganicspeciesandtheBrønstedacidity, suggestingthatatleastsomeionexchangewithacidicsiteshadoccurred.This hadtheeffectofdecreasingthecatalystactivity,bothstudiesreportobserving increasesinbio-oiloxygencontentwithincreasingcumulativebiomasstocatalystratios.Becausethesecatalystsweresubjectedtohightemperaturesduring
regenerationfromcoking,physicaldamagemaybeanotherreasonforactivity loss,asdescribedinmoredetailhereafter.However,anotherstudyspecifically lookedattheeffectofKbypreparingionexchangedcatalysts(2.95wt%loading) andfoundadrasticreductionincatalystacidityandyieldofaromatichydrocarbonsviaCFPofcelluloseandswitchgrass[59].Thesamestudyalsofoundan increaseinfuransandphenolics,andthesameproductsarefoundwhencatalysts begintodeactivateasaresultofcoking.Useof exsitu CFPmethods,wherebythe largemajorityoftheinorganicspeciesareremovedfromthevaporstreamwith thebio-char,cangreatlyreducetheimpactofthistypeofdeactivation.
Ineither insitu or exsitu CFP,however,zeolitecatalystsneedtoberegeneratedfromcoking,likelycontinuously,ifmaximizedaromatichydrocarbon productionisthegoal.Regenerationusuallyconsistsofcombustionofthecoke depositsinanairatmosphere.Becausecombustionisanexothermicprocess,it ispossiblethathotspotsmayoccurduringtheregenerationprocesses,making itmorelikelyforcatalystdamagetooccur.Inonestudywhereacommercial spray-driedZSM-5catalystwasusedin30successivereaction/regeneration cycleswheretheregenerationwasperformedofflineat580 ∘ Coveralongperiod (15–20hours),noevidenceofdealuminationorlossofcrystallinitywasfound, onlymacroscopicphysicaldamage–somebreakingapartofthespraydried particles–wasobserved[43].Asmalllossinperformancewasattributedto alkalimetalpoisoning.However,intheaforementionedworkusingacirculating fluidizedbedwheretheregenerationconditionsweremuchmorerapidat measuredtemperaturesof650–670 ∘ C,evidenceofdealuminationwasevident andadecreaseinmicroporevolumeswasobserved[42].Thenegativeeffectson performancewerelimitedat100hourstimeonstream,butmaybecomemore significantwithadditionalexposuretothereaction/regenerationconditions. Onlyrecentlyhasworkbeendonetooptimizetheconditionsthatcontrol temperatureduringregenerationtopreventcatalystdamage.Arecentreport foundthatthemeasuredtemperaturejustaboveafixedbedregenerationreactor correlatedwellwiththeexcessofoxygenoverthecombustionstoichiometry intheatmosphere[60].Limitingtheoxygento15vol%duringcombustion significantlyincreasedthecatalystlifetimeforconversionoffurantoaromatics. Further,controllingthetemperatureviaadditionofsteamfurtherimproved thelifetimeofthecatalyst.Thisstudyalsoexploredtheideaofacontrolled regenerationwhereatargetedamountofcokeremainedonthecatalyst.Some otherprocessesusethistypeofpartialdeactivation(equilibriumstate)tocontrol productselectivity,anideathatwarrantsfurtherstudyforbiomassCFP.
1.1.1.3ModificationofZSM-5withMetals
TherehavebeenseveralattemptstomodifyZSM-5tofurtherimproveyields, changetheselectivityoftheproducts,and/ordecreasecatalystdeactivationrates. OnefacilewaytomodifythezeoliteisviaionexchangeoftheBrønstedacid siteswithanactivemetalcation.Thereareafewmethodstodothis,including ion-exchangeandincipientwetnessimpregnation.Somemetalscanbeincorporatedintotheframeworkofthezeoliteduringsynthesis.Metalscannotonly adjusttheacidityofZSM-5byreplacingtheprotonwithametalcationbutalso promotedesirablereactionsduringpyrolysis,potentiallyincreasingtheyieldof
1UpgradingofBiomassviaCatalyticFastPyrolysis(CFP)
desirablecompoundswhilereducingcokeformationonthecatalyst.Amongthe metalsthathavebeenstudiedforthispurposeareZn,Mo,Ga,Ni,Co,Cu,Mn, andFe[61–75].Somemetal-modifiedZSM-5catalystsarecurrentlybeingused commerciallyfortheconversionoflighthydrocarbonstoaromatics.Forexample, aGa-modifiedZSM-5catalysthasbeenusedforthearomatizationoflightalkanesintheCyclarprocessbyUOPandBP[76–78],whileZn/HZSM-5isused fortheproductionofaromaticsfromolefin-richhydrocarbonsintheAlphaprocessfromAsahiKaseiChemicalsCorp.[79].Modificationwithgalliumhasbeen amongthemoststudiedapproachesforbiomassCFP[43,56,68,70–73].Galliumincorporationbyionexchangeorimpregnationhasbeenshowntoeffect 21–50%increasesinyieldsofaromatichydrocarbonsoverparentZSM-5catalysts.CatalystssynthesizedwithGainthezeoliteframeworkwerelesssuccessful forproductionofaromaticsfromamodelcompound(furan)[68].Table1.2summarizesresultsfromvariousselectedCFPstudiesusingGaZSM-5.
ThepresenceofgalliumincreasesthenumberofLewisacidsitespresentin thecatalyst,increasingitsdehydrogenationactivity.Ionexchangemethodsfor preparationofGaZSM-5havebeenshowntoonlyeffectaminimalamountof actualionexchange,andmuchofthegalliumexistsasGa2 O3 onthesurfaceofthe catalyst[76].Thesmallamountofgalliumthatisexchangedislikelyresponsible fortheincreasedyields.Inasimilarprocess,conversionofethanoltoaromatics,aphysicalmixtureofGa2 O3 andHZSM-5showednoimprovement,butthe ionexchangedzeolitedid[77].Furthermore,reductivepretreatmentappearsto mobilizethegalliumtoionexchange,furtherincreasingactivity.Theexchanged Ga(III)sitesaremostimportantfortheobservedactivity.Thereductionthat allowsthismobilitymaynotactuallyperformareductiontoGa(I)butproduce low-coordinateGa(III)hydridespecies[77].Preservationofthesestatesmayfurtherenhancethearomaticyieldforalkanearomatizationreactions.
InCFP,gallium-modifiedZSM-5catalyststhathavebeenprereducedhave shownespeciallystrongdehydrogenationactivity(asmeasuredbyproduction ofH2 ),althoughthisrapidlydecreasesuponexposuretopyrolysisvaporsasa resultofeitheroxidationofGa(I)toGa(III)orconversionoflowcoordinate activeGa(III)speciestofullyligatedspecies.Ineithercase,theresultant gallium(III)-exchangedcatalystcontinuedtoshowsuperiorperformancefor productionofaromatichydrocarbons.Theincreaseddehydrogenationand/or decarbonylationactivityaffordedincreasesinolefinformationandintheirrate ofaromatization.ComparedwithparentHZSM-5catalysts,thosemodifiedwith galliumshowedmoreselectivitytowardlessalkylatedaromatics(benzeneand toluene)andawayfromxylenes[68,71].Intermsofproductionofdeoxygenated bio-oil,onestudyoftheCFPofpinewoodshowedaslightincreaseinyieldof organicphasebio-oil(from ∼25to ∼26–28C%)withGaZSM-5overHZSM-5 withadecreaseinoxygencontentoftheproduct(from18.4to17.1–17.7wt%) [73].Furthermore,GaZSM-5catalystshaveshownlongerlifetimeswithrespect todeactivationduetocokingthanstandardHZSM-5[56].Figure1.9depicts howGaZSM-5(SiO2 /Al2 O3 = 30)continuedtoexhibitsuperiordeoxygenation activityathighercumulativebiomasstocatalystratiosthandiditsparent HZSM-5(SiO2 /Al2 O3 = 30)orHZSM-5(SiO2 /Al2 O3 = 80)duringCFPof eucalyptuswood[56].