Cancer consult: expertise in clinical practice, volume 1: solid tumors & supportive care, 2nd editio
Visit to download the full and correct content document: https://ebookmass.com/product/cancer-consult-expertise-in-clinical-practice-volume-1 -solid-tumors-supportive-care-2nd-edition-syed-a-abutalib/
More products digital (pdf, epub, mobi) instant download maybe you interests ...
Prostate Cancer, Second Edition: Science and Clinical Practice Jack H. Mydlo Md Facs
Precision Cancer Therapies, Volume 1: Targeting Oncogenic Drivers and Signaling Pathways in Lymphoid Malignancies: From Concept to Practice Owen A. O'Connor
Cancer Consult: Expertise for Clinical Practice, John Wiley and Sons, Inc. (2014)
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by law. Advice on how to obtain permission to reuse material from this title is available at http://www.wiley.com/ go/permissions.
The right of Syed A. Abutalib, Maurie Markman, Al B. Benson III and Hope S. Rugo to be identified as the authors of the editorial material in this work has been asserted in accordance with law.
Registered Offices
John Wiley & Sons, Inc., 111 River Street, Hoboken, NJ 07030, USA
John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK
Editorial Office
The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK
For details of our global editorial offices, customer services, and more information about Wiley products visit us at www.wiley.com.
Wiley also publishes its books in a variety of electronic formats and by print-on-demand. Some content that appears in standard print versions of this book may not be available in other formats.
Trademarks: Wiley and the Wiley logo are trademarks or registered trademarks of John Wiley & Sons, Inc. and/or its affiliates in the United States and other countries and may not be used without written permission. All other trademarks are the property of their respective owners. John Wiley & Sons, Inc. is not associated with any product or vendor mentioned in this book.
Limit of Liability/Disclaimer of Warranty
The contents of this work are intended to further general scientific research, understanding, and discussion only and are not intended and should not be relied upon as recommending or promoting scientific method, diagnosis, or treatment by physicians for any particular patient. In view of ongoing research, equipment modifications, changes in governmental regulations, and the constant flow of information relating to the use of medicines, equipment, and devices, the reader is urged to review and evaluate the information provided in the package insert or instructions for each medicine, equipment, or device for, among other things, any changes in the instructions or indication of usage and for added warnings and precautions. While the publisher and authors have used their best efforts in preparing this work, they make no representations or warranties with respect to the accuracy or completeness of the contents of this work and specifically disclaim all warranties, including without limitation any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created or extended by sales representatives, written sales materials or promotional statements for this work. The fact that an organization, website, or product is referred to in this work as a citation and/or potential source of further information does not mean that the publisher and authors endorse the information or services the organization, website, or product may provide or recommendations it may make. This work is sold with the understanding that the publisher is not engaged in rendering professional services. The advice and strategies contained herein may not be suitable for your situation. You should consult with a specialist where appropriate. Further, readers should be aware that websites listed in this work may have changed or disappeared between when this work was written and when it is read. Neither the publisher nor authors shall be liable for any loss of profit or any other commercial damages, including but not limited to special, incidental, consequential, or other damages.
Library of Congress Cataloging-in-Publication Data
Names: Abutalib, Syed A., editor. | Markman, Maurie, editor.
Title: Cancer consult : expertise in clinical practice. Volume 1, Solid tumors & supportive care / edited by Syed A. Abutalib, Maurie Markman, Al B. Benson and Hope S. Rugo.
Description: Second edition. | Hokoben, NJ : John Wiley & Sons Ltd., 2023. | Includes bibliographical references.
Identifiers: LCCN 2023024465 | ISBN 9781119823735 (paperback) | ISBN 9781119823742 (pdf) | ISBN 9781119823759 (epub) | ISBN 9781119823766 (ebook)
Subjects: LCSH: Tumors--Treatment--Examinations, questions, etc.
Hematologic Malignancies and Transplantation and Cellular Therapy
Aurora St. Luke’s Medical Center
Advocate Aurora Health Care Milwaukee, Wisconsin
Maurie Markman, MD
Professor, Department of Medical Oncology and Therapeutics Research City of Hope
President, Medicine & Science, City of Hope, Atlanta, Chicago, and Phoenix
Kenneth C. Anderson, MD
Program Director, Jerome Lipper Multiple Myeloma Center and LeBow Institute for Myeloma Therapeutics
Kraft Family Professor of Medicine
Harvard Medical School
Dana Farber Cancer Institute, Boston, MA
James O. Armitage, MD
Joe Shapiro Professor of Medicine, University of Nebraska Medical Center Omaha, NE
Editor’s Biography
Syed Ali Abutalib, MD, is Medical Director, Hematologic Malignancies and Transplantation and Cellular Therapy, Aurora St. Luke’s Medical Center. He also served as active member of Executive Committee on Education for American Society of Transplantation and Cellular therapy (ASTCT®) and is one of the Associate Editors for The ASCO Post®. Dr. Abutalib is the Founding Editor of Advances in Cell and Gene Therapy, a Wiley Journal. In addition, he is Associate Professor of Medicine at Rosalind Franklin University of Medicine and Science. Dr. Abutalib has co-edited 16 Hematology/Oncology and Cellular/ Gene therapy books over a short span of 10 years and has published in a number of cancer magazines, journals, and textbooks. He continues to collaborate with most cancer experts worldwide in the area of Hematology/Oncology and stem cell transplantation to explore and develop innovative ideas and breakthroughs in Medical Education with a prime focus to improve patient care.
Maurie Markman, MD, is Professor of Oncology and Therapeutics Research, City of Hope and President of Medicine & Science, City of Hope Atlanta, Chicago, Phoenix. A nationally renowned oncologist, Dr. Markman has more than 20 years of experience in cancer treatment and gynecologic research at some of the country’s most recognized facilities. Dr. Markman has served as the Vice President for Clinical Research and Chairman of the Department of Gynecologic Medical Oncology at M.D. Anderson Cancer Center in Houston, Texas. Prior to that, Dr. Markman spent 11 years as Chairman of the Department of Hematology/ Oncology and Director of the Taussig Cancer Center at the Cleveland Clinic Foundation. He also spent five years as Vice Chairman of the Department of Medicine at Memorial Sloan-Kettering Cancer Center in New York.
Al B. Benson III, M.D FACP FASCO is a Professor of Medicine in the Division of Hematology/Oncology at Northwestern University’s Feinberg School of Medicine in Chicago, Illinois, and the Associate Director for Cooperative Groups at the Robert H. Lurie Comprehensive Cancer Center. He is a recipient of the American Society of Clinical Oncology (ASCO) Statesman Award (Fellow of ASCO) and has served on a number of ASCO committees, often as chair or co-chair. Dr Benson is currently Vice-Chair of the ECOGACRIN Cancer Research Group, past Chair of the ECOGACRIN Gastrointestinal Committee, Co-Chair of Cancer Care Delivery Research Committee, a member of the NCI Rectal/Anal Task Force and Co-Chair of the International Rare Cancers Initiative (IRCI), Anal Cancers Committee. In addition, he is a Past President of the Illinois Medical Oncology Society, Past President of the Association of Community Cancer Centers (ACCC), a board member and past-Chair of the Board of Directors of the National Comprehensive Cancer Network (NCCN) and the past President of the National Patient Advocate Foundation. Dr. Benson has published extensively in the areas of gastrointestinal cancer clinical trials, health services research and cancer treatment guidelines.
Hope S. Rugo, MD, FASCO, is Director of Breast Oncology and Clinical Trials Education and a Professor of Medicine at the University of California San Francisco Comprehensive Cancer Center. Dr. Rugo is an internationally well-known medical oncologist specializing in breast cancer research and treatment. She entered the field of breast cancer in order to incorporate novel therapies with excellent quality of care into the treatment of breast cancer and is a principal investigator of multiple clinical trials focusing on novel targeted therapeutics to improve the treatment of breast cancer. Her research interests include immunotherapy and combinations of targeted agents to treat breast cancer, and management of toxicity. She is co-chair of the Safety Committee for the multicenter adaptively randomized phase II I-SPY2 trial, co-chair of the Triple Negative Working Group of the Translational Breast Cancer Research Consortium (TBCRC) and a member of the Alliance breast committee. Dr. Rugo is an active clinician committed to education.
Preface
Over the past decade there have been tremendous advances in the oncology arena resulting in both improved survival and quality of life for individuals requiring management of a malignant disease. However, this surge in available therapeutic options has quite often resulted in increased complexity of care. As a result, patients frequently request a “second opinion.”
It is also commonplace for oncologists to discuss with a colleague a difficult or unusual case, or the management of a patient who presents with serious comorbidities, to insure that the individual is given the greatest opportunity to experience the benefits of therapy while minimizing the risks of possible treatment- related harm. Such discussions occur both within a particular specialty (e.g., surgery, radiation, or medical oncology) and between various specialties.
Further, as cancer management becomes more multimodal in nature, with an increasing focus on maximizing the opportunity for extended survival and at the same time optimizing quality of life, the requirement for essential communication between individual specialists with their own unique knowledge and experience of critically relevant components of care becomes ever more important.
It is with these thoughts in mind that the editors conceived of an oncology text that would focus on the “Expert Perspectives” of oncology professionals. The intent of this distinctive effort is to have each individual chapter be viewed as a “miniconsultation” provided by a specialist regarding a specific, highly clinically relevant issue in cancer management.
Considering the specific purpose and focus of the material presented, the book is written without detailed references (although “Recommended Readings” are included at the end of each chapter). However, many of the authors have prepared a more extensive reference list, and the editors will be happy to email any reader the more detailed reference lists for individual book chapters, if so requested.
The chapters that follow have been written by clinicians selected for their recognized clinical expertise and experience. It is hoped that those reading this book will find the material of value in their own interactions with their patients.
Syed A. Abutalib, Maurie Markman, Al B. Benson III, and Hope S. Rugo
Acknowledgment
Syed Ali Abutalib: I am immensely grateful to God for providing me with the inspiration, and strength throughout this endeavour. I would like to extend my heartfelt acknowledgment to the timeless wisdom of Ali Ibne Abutalib (PBUH), who once said, “Knowledge is a companion that will never betray you, an ornament that will enhance your worth, a friend that will be with you in solitude, and a guide that will lead you to righteousness.” Inspired by these profound words, I express my gratitude to the coeditors, and esteem contributors of this text. Their expertise, and dedication have significantly enhanced the breadth and depth of the content. Also, I would like to acknowledge my grandparents, Hamid, and Laila Jafry for their unwavering support, and my daughter, Sakeena Ali Abutalib. Lastly, I would like to extend my appreciation to the courageous cancer patients and their families for making me a better physician and a person.
Maurie Markman: To my grandsons, James, William, and Conrad.
Al B. Benson III: This text is dedicated to our patients who deserve the best we can offer and to the entire health care team that works tirelessly to help provide our patients the comprehensive cancer care that is so essential during the course of their illness.
Hope S. Rugo: To our dedicated colleagues who worked so hard to contribute excellent and updated manuscripts and who responded to revision requests and proof reviews with enthusiasm. To the editor, Dr. Abutalib, who has worked tirelessly and for untold hours to put this remarkable collection together, and of course to my coeditors and colleagues at Wiley as well. And of course, to my family, for your unwavering support and enthusiasm. This work is truly a tribute to the passion of oncologists to educate broadly, with the goal of improving patient care.
Part 1
Central Nervous System
1 Central Nervous System Tumors
Mark R. Gilbert and Edina Komlodi-Pasztor
Introduction
Central nervous system tumors can be divided into primary brain tumors and metastatic lesions. This chapter will focus on the most frequently occurring questions related to tumors originating from the brain, spinal cord, or associated tissue. Primary central nervous system lymphoma is discussed in a separate chapter. We will review practical questions related to epidemiology, classification, and treatment of primary brain tumors. We will discuss the incidence rates and the genetic and environmental predisposing factors related to brain cancers. A brief overview of the new fifth edition of the World Health Organization (WHO) classification system will be provided. We will summarize treatment recommendations of the most commonly occurring benign and malignant primary brain tumors, such as meningioma, oligodendroglioma, astrocytoma, and glioblastoma.
1. How do we classify brain tumors?
Expert Perspective: Brain tumors can be divided into two main groups: benign tumors and malignant cancers. Malignant cancers can be further subdivided into metastatic lesions and primary central nervous system cancers (PCNSC).
Instead of using the tumor, node, and metastasis (TNM) staging system as in systemic solid tumors and hematologic malignancies, central nervous system (CNS) cancers are classified based on the World Health Organization (WHO) grading system. This grading system has been undergoing dynamic changes in the setting of new advances in molecular genetics. Although traditionally this grading system was based on histological features, the revised WHO classification published in 2016 used molecular parameters for the first time in addition to histology to categorize tumor tissues. The role of molecular diagnostics in CNS tumor classification is being further solidified in the 2021 WHO classification, which follows the recommendations of the 2019 cIMPACT-NOW Utrecht meeting (Louis et al. 2021).
As described in the new fifth edition of the WHO classification system, using a layered reporting structure, including integrated diagnosis, histological diagnosis, CNS grading, followed by molecular information, is recommended. The integrated diagnosis incorporates important molecular features in addition to tissue-based histological findings, highlighting the importance of different diagnostic modalities in the accurate diagnosis in CNS tumors.
Brain and spinal cord tumors are graded from 1 to 4, where the higher numbers indicate faster growth and/or greater aggressiveness. Each tumor type has its own grading that represents its historically determined natural history but does not necessarily correlate with prognosis in this current era. For example, the five-year survival of a grade 3 meningioma is 64%, whereas almost all patients with a grade 4 WNT-activated medulloblastoma have long-term survival if treatment is provided. In recent years, molecular biomarkers became an important part of clinical care in neuro-oncology because they have promoted better categorization of CNS tumors. Consequently, the 2021 WHO classification enhances the use of molecular biomarkers to influence grading in many tumor types due to their powerful prognostic value. In addition, molecular biomarkers also have the potential to influence treatment in some cases. Imminent advances in molecular diagnostics are expected to further evolve the landscape of neuro-oncology soon.
2. What is the incidence of primary brain cancers?
Expert Perspective: Although CNS tumors (including malignant and non-malignant etiology) are the most common solid tumor types in children below age 14, they are less frequent in adults. Brain tumors are the 3rd most common tumor types in people between the ages of 15 and 39 with an average annual age-adjusted incidence rate of 11.54 per 100,000 population, and the 8th most common tumor type above age 40 with an average annual age-adjusted incidence rate of 42.85 per 100,000 population. In adults, 70.3% of brain tumors are non-malignant, and among them, meningioma is the most common tumor type (Table 1.1). Regarding adult malignant tumors, glioblastoma leads in frequency because it represents 14.5% of all brain tumors and 48.6% of malignant brain cancers. Malignant brain cancers are more prevalent in males than females (56% vs 44%, respectively). Gender differences are reversed in non-malignant cancers, where 36% of the cases occurred in males and 64% in females. Incidence rates for malignant primary brain tumors are highest in Whites (7.58 per 100,000), but non-malignant primary brain tumors are more common in Blacks (19.45 per 100,000) (Ostrom et al. 2020).
3. Do primary brain cancers have genetic predisposition?
Expert Perspective: Most adult primary brain tumors occur sporadically without an identifiable genetic predisposition. However, genetic susceptibility to brain cancers is suggested by tumor aggregation in families, genetic cancer syndromes, linkage analyses, and lymphocyte
Table 1.1 Molecular characteristics and grading of the most frequently occurring CNS tumors in adults.
mutagen sensitivity. Thorough medical history taking with close attention to the family cancer history can reveal family cancer clusters and/or raise suspicion for a genetic syndrome associated with brain cancers. The most common genetic conditions associated with primary CNS tumors include Lynch syndrome, neurofibromatosis type 1, Li-Fraumeni syndrome, tuberous sclerosis (Table 1.2). In addition to colorectal cancer, endometrial cancer, upper urinary tract cancer, and ovarian cancer, patients with Lynch syndrome are at high risk of developing gliomas in their fifth decade. Patients with neurofibromatosis type 1 can develop optic pathway tumors in childhood and/or glioblastoma, as well as malignant peripheral nerve sheath tumors in early adulthood. They are also more prone to be diagnosed with breast cancer, endocrine cancers, sarcoma, melanoma, ovarian cancer, and prostate cancer. Patients with Li-Fraumeni syndrome can also develop multiple cancers, including solid tumors (breast cancer, osteosarcoma, sarcoma, and adrenocortical cancer), hematologic malignancies (leukemia), and CNS tumors (choroid plexus tumor in infancy, medulloblastoma in childhood, and high-grade glioma in early adulthood). Patients with tuberous sclerosis may develop subependymal giant-cell astrocytomas before age 25 in addition to other solid tumors (like rhabdomyoma and kidney cancer). Recognizing a genetic syndrome based on clinical signs and genetic markers is crucial to provide appropriate medical management. The identification of a genetic syndrome may influence the treatment plan because targeted therapy has been increasingly accessible for specific mutations. In addition to that, it may also guide future cancer surveillance and the need for family cancer screening. (See Chapters 45–47.)
Table 1.2 Characteristics of the most common genetic conditions associated with primary central nervous system tumors. Name
Lynch syndrome Estimated 1 in 279 Multiple chromosomes, including chromosomes 2, 3, and 7
Expert Perspective: The question of environmental factors leading to brain tumors has been extensively investigated, but there are only a few environmental factors that have been confidently linked to tumorigenesis in the CNS. Among these, therapeutic ionizing radiation has been uniformly accepted as a risk factor for brain cancers including meningiomas, gliomas, and nerve sheet tumors. This risk is even higher when radiation exposure occurs at young age. Although the significance of diagnostic radiation exposure has not been fully clarified, some studies suggest increased risk of brain cancers in patients who receive head CT scans in childhood. In contrast, allergic and ectopic conditions seem to reduce the risk of developing CNS tumors (such as glioma and meningioma) based on large epidemiologic studies. The role of hormones and hormonal therapy in brain cancer development has been thoroughly investigated without firm conclusion. The fact that meningioma is three to six times more common in females than males provided the biological basis of this hypothesis. So far, there is no clear evidence that hormone treatment would increase the risk of brain cancers, including meningioma and glioma. Another area of active investigation aims to explore the possible connection between cell-phone use and brain tumors. Although no consensus has yet been reached on this issue, some studies suggest that long-term use of or childhood exposure to cell phones is potentially carcinogenic. Occupational exposures and environmental factors, including but not limited to pesticides, lead, dust, formaldehyde, and sulfur dioxide, have been also explored without finding a significant relationship to brain cancers.
5. Does a grade 1 meningioma require treatment?
Expert Perspective: Meningiomas are the most common primary brain tumors, with an incidence rate of 8.81 per 100,000 population. They represent 38.3% of all tumors and 53.9% of all non-malignant brain tumors. Meningiomas are usually slow-growing, extra-axial tumors. Most often they are located along the parasagittal line, and less frequently they are in the spinal region. Usually they are found incidentally in asymptomatic patients, but they can cause progressive focal neurologic symptoms relevant to their location. Their incidence increases with age. They are more common in women, in Blacks, and in people with a remote history of radiation therapy to the CNS, especially if the exposure occurred in childhood. Some genetic disorders, such as neurofibromatosis type 2 and MEN1 syndrome, have been associated with an increased risk of developing meningiomas (Table 1.2).
Unlike most brain tumors, meningiomas are graded from 1 to 3 by the 2021 WHO classification system. Within the grades, 15 subtypes are distinguished histologically. Grade 1 benign tumors represent about 80% of meningiomas. Grade 2 is found in approximately 15–20% of the cases. Grades 1 and 2 are also referred to as non-malignant meningiomas, although both can undergo malignant transformation to higher grade, which is more likely with the grade 2 tumors. Grade 3 meningiomas are malignant tumors that represent 2% of meningiomas. The 10-year relative survival is 87.4% for non-malignant meningiomas and 59.6% for malignant meningiomas.
Due to the heterogeneous nature of meningioma cases (including patient factors and histological features of the specific tumor), individualized treatment should be offered for patients after careful evaluation of the risk and benefits of available therapeutic options.
Slow-growing asymptomatic tumors that do not invade or compromise healthy tissue can be monitored with periodic MRIs. Imaging should be repeated at three, six and twelve months after the diagnosis, then every 6–12 months for five years, followed by every one to three years thereafter. To determine tumor growth rate, imaging should be always compared to the first MRI. For large and symptomatic meningiomas, surgery is preferred if the location is accessible. For patients with unresectable meningioma, radiation treatment can be an effective alternative. Surgery followed by radiation therapy can be considered for grade 2 meningiomas, but prospective randomized trials comparing observation with radiation after resection are not yet completed. In contrast, radiation treatment is required for grade 3 meningiomas due to their high recurrence rates.
Increasing evidence supports the value of molecular sequencing of meningiomas, too. In addition to the undebatable role of the NF2 gene mutation in the development of meningiomas, the prognostic significance of other genetic alternations has been recognized in recent years. Based on the new 2021 WHO classification, a histologically grade 1 meningioma can be upgraded to grade 3 in the integrated reporting system if it harbors a TERT promoter mutation and/or homozygous deletion of CDKN2A/B. Therefore, molecular characterization of a meningioma is critical for the accurate tissue diagnosis and for the selection of appropriate treatment. In addition, molecular advances not only contribute to better characterization of specific tumor samples but may also contribute to the development of targeted therapy and better patient outcome in the near future.
6. What is the standard of care treatment of oligodendroglioma?
Expert Perspective: The diagnostic criteria have changed in the last few decades, and the 2021 WHO classification guideline categorizes tumors with mutation in the isocitrate dehydrogenase gene (IDH mutation) and combined whole-arm losses of 1p and 19q (1p/19q codeletion) as oligodendroglioma. This clear separation from other gliomas were needed due to the distinct pathology, molecular pathogenesis, treatment response, and prognosis of oligodendrogliomas. They are grouped into two grades based on their histological and molecular characteristics.
Grade 2 oligodendrogliomas are low-grade tumors. They have low mitotic activity and grow slowly, often for years prior to diagnosis. Molecular sequencing reveals no ATRX mutation but shows TERT promoter mutations. Grade 3 oligodendrogliomas, used to be called anaplastic oligodendrogliomas, are malignant tumors with atypical features including high cell density, increased mitotic rate, nuclear atypia, microvascular proliferation, and necrosis, but they also lack ATRX mutation and they harbor TERT promoter mutations. Patients with grade 3 oligodendroglioma have worse prognosis compared with patients with grade 2 tumors. There is some evidence that oligodendrogliomas start at low grade and ultimately evolve into more aggressive tumors, although this progression can’t always be detected because some patients are diagnosed with high-grade tumors at presentation.
Oligodendroglioma treatment should include maximal safe resection followed by additional treatment, which can include radiotherapy and chemotherapy at some point in the course of disease. Because the clear classification of oligodendroglioma is recent, prior clinical studies provide data on the treatment of oligodendroglioma mixed with other tumor types, like astrocytoma (per current classification). The most notable studies include RTOG 9802 for grade 2 tumors and RTOG 9402 and EORTC 269521 for grade 3 tumors. In
general, it is accepted that patients under 40 years old who underwent complete resection of a grade 2 oligodendroglioma and with no evidence of CDNK2A/B homozygous deletion may be followed by regular imaging. MRI with and without contrast is recommended every three to six months in the first five years, followed by every six to nine months or as clinically indicated, before transitioning to annual imaging. In this age group, the delay of treatment may improve quality of life without worsening the disease outcome. For grade 2 oligodendrogliomas diagnosed above age 40 or with incomplete resection, and for grade 3 tumors, fractionated external beam radiation treatment followed by chemotherapy is recommended. Beside the acute toxicities (including fatigue, headaches, and hair loss), long-term side effects of radiation treatment include cognitive impairment, hearing loss, meningioma, and overt brain injury called radiation-induced necrosis. The choice of postradiation chemotherapy remains controversial. Most data support the use of procarbazine, lomustine, and vincristine (PCV) as a choice of chemotherapy, although some centers avoid the use of vincristine due to lack of evidence of blood-brain barrier penetration and the universal development of peripheral neuropathy. Furthermore, the inclusion of a nitrosourea raises concerns about prolonged hematologic toxicity. Temozolomide, commonly used for astrocytoma and glioblastoma, has not been compared prospectively with PCV in the post-radiation setting but has a much better toxicity profile. However, clinical trials are underway to assess the utility of temozolomide in the treatment of oligodendroglioma. The initial study reported inferiority of temozolomide compared to radiation treatment. The current phase of the study compares radiotherapy with temozolomide to radiotherapy with PCV treatment.
7. How does one treat a WHO grade 3 IDH mutant astrocytoma?
Expert Perspective: Traditionally, an anaplastic astrocytoma was classified as a grade 3 astrocytoma. With the current 2021 WHO grading system, all astrocytomas are IDH mutant by definition and range from grade 2 to grade 4. Because clinical studies were designed based on prior classification systems or even before some of the currently used biomarkers were identified, data interpretation can be challenging at times.
In case of a WHO grade 3 IDH mutant astrocytoma, the CATNON study can be used for treatment guidance. The CATNON study is a randomized, open-labeled, phase III study that, using an innovative 2 × 2 factorial design, randomized 751 patients into four groups: radiation treatment alone, radiation treatment with concomitant temozolomide, radiation with adjuvant temozolomide, and radiation treatment with concomitant as well as adjuvant temozolomide (1:1:1:1). During data analysis, patients were divided based on IDH mutations status. Ultimately, data from 660 patients (444 with IDH mutation) were used to determine whether concomitant temozolomide or adjuvant temozolomide improve survival. The study revealed clinical benefit from adjuvant temozolomide (for 12 months) in patients with IDH1 or IDH2 mutant tumors. The five-year overall survival was 64.8% in the patients who did not receive adjuvant temozolomide compared to 82.8% who did. On the other hand, no benefit was found from daily concomitant temozolomide during radiation treatment in this patient population. The five-year survival was 80.5% without concomitant chemotherapy compared with 82.8% with concomitant temozolomide (van den Bent et al. 2021).
Mutations in the IDH1 or IDH2 gene provides an exciting opportunity for molecularly targeted treatment. Multiple studies are investigating the utility of IDH targeting treatment in different cancers, including glioma. For example, IDH-mutant inhibitors, ivosidenib and vorasidenib, have been approved by the Food and Drug Administration as a therapeutic option for IDH-mutated AML. In gliomas, the efficacy of ivosidenib has been assessed in a multicenter, open-label, phase I, dose escalation and expansion study. This study revealed median progression free survival benefit from ivosidenib in non-enhancing gliomas (considered to be low grade gliomas) but not in enhancing gliomas (considered to be high grade gliomas). Vorasidenib, an oral dual inhibitor of mutant IDH1/2 enzymes, significantly improved progression-free survival in patients with residual or recurrent grade 2 gliomas. This treatment delayed disease progression. These findings from the INDIGO trial represent a significant step forward in the treatment of patients with grade 2 glioma with IDH mutations. Moreover, there is increasing evidence that poly(ADP-ribose) polymerase (PARP) inhibition may be another treatment option; clinical trials are undergoing to assess the benefit of pamiparib with temozolomide. Also, vorasidenib is under evaluation in combination with pembrolizumab in an ongoing phase I study in grade 2/3 glioma.
Key clinical trial: CATNON, INDIGO.
8. Does age matter for glioblastoma treatment?
Expert Perspective: In 2005, a paper reported survival benefit from adding concomitant and adjuvant temozolomide to radiotherapy in glioblastoma patients. Since then, the Stupp protocol described in this paper became the standard of care in the treatment of this aggressive brain tumor. The regimen includes irradiation for a total of 60 Gy, 5 days per week for 6 weeks along with daily temozolomide (7 days a week) at 75 mg/m2, followed by 6 cycles of adjuvant temozolomide for 5 days in a 28-day cycle at 150–200 mg/m2. The median age of patients participating in the original study and who received the combination treatment with temozolomide with radiotherapy was 56 (ranged between 19 and 70 years old). The most common side effects include fatigue, thrombocytopenia, neutropenia, leukopenia, and constipation. The median survival benefit of this regimen was an additional 2.5 months for a total of 14.6 months, significantly longer than the 12.1 months in the “radiotherapy alone” arm.
The incidence of glioblastoma is increasing with age, and the median age at diagnosis is 65 years (Figure 1.1). At the same time, older patients are usually underrepresented in clinical studies aimed to treat glioblastoma. In addition to limited data on optimal treatment in this age group, clinicians need to consider other factors associated with age, such as poor performance status, additional medical conditions, increased risk of toxicity, and social support system. A small number of studies were specifically designed to address these issues. Among them, a phase III clinical study assessed the benefit of a short-course radiation treatment along with temozolomide in patients ≥ 65 years of age. Patients were randomized to receive either radiotherapy alone or concomitant chemoradiation. A total of 40.5 Gy radiation treatment was administered in 15 fractions over three weeks. Concomitant temozolomide was given at 75 mg/m2 for 21 days followed by adjuvant temozolomide at 150–200 mg/m2 dose for 5 days in a 28-day long cycle, for up to 12 cycles. The results showed that this short course of radiation treatment with or without temozolomide was well tolerated in this age group. The median number of adjuvant cycles was 5. The overall survival rate increased from 7.6 months (of the radiation treatment alone arm) to 9.3
months with the combination treatment. The benefit of chemotherapy was even greater in patients with MGMT methylated glioblastoma. This study provided evidence that it is reasonable to consider a shorter course, hypofractionated radiation therapy with temozolomide for the treatment of older patients (based on chronological or biological age).
Key Clinical Trial: Perry et al. 2017.
9. How does one diagnose pseudo-progression and necrosis?
Expert Perspective: Clinicians are often puzzled by the etiology of the non-specific posttreatment changes seen on brain MRI following completion of treatment for CNS tumors. These changes may represent tumor progression, pseudo-progression, or necrosis. Radiologically, they all present with new or enlarging contrast enhancement at the area of radiation exposure.
Even with additional imaging techniques, the exact diagnosis can be rarely given with confidence. Special MRI sequences, such as perfusion and diffusion weighted imaging, are frequently used even though they provide suboptimal sensitivity and specificity to distinguish tumor progression from tumor necrosis. MR spectroscopy may increase accuracy of the assessment; however, the imaging data may misrepresent the true etiology of the changes. Needless to say, that the proper diagnosis is crucial in order to provide appropriate management for the patient.
Pseudo-progression typically presents within three months of completing radiation treatment. The hypothesis is that radiation treatment damages all cells in the exposure field and leads to cell death of the tumor cells as well as healthy cells, including neurons and vascular cells. This cell death will initiate inflammatory changes and some local edema, which ultimately leads to blood brain barrier impairment. As a result, post-radiation scans are always used as a new baseline scan but not for treatment decisions. In cases of suspected pseudo-progression, it is advised to continue treatment and reimage the patient in 2–3 months, or sooner if needed. Pseudo-progression is usually asymptomatic, but if symptoms arise, a course of steroid treatment may lead to clinical and radiological improvement. Eventually, the contrast enhancing area on the MRI is expected to stabilize
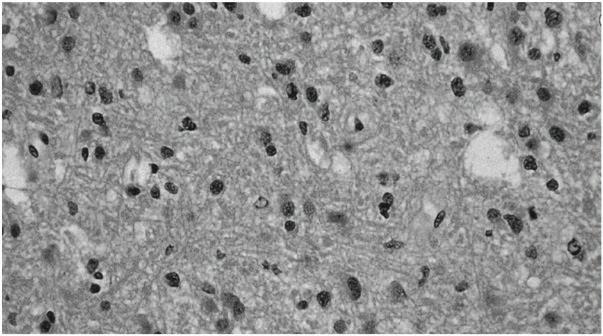
Figure 1.1 Histologic features of grade II astrocytoma, including increased astrocytic cellularity (“hypercellularity”). (Color Plate 1.1)
or regress if the changes are due to pseudo-progression. In questionable cases, surgery may be necessary for the proper diagnosis.
Treatment-related (radiation) necrosis presents as a delayed, severe degree of local tissue and vascular injury. It typically occurs 12–24 months following chemoradiation, but it may evolve several years later. There are multiple factors that play a role in the development of radiation necrosis, including the cumulative dose and fractionation of radiation treatment. Unlike pseudoprogression, which is often asymptomatic, radiation necrosis is often symptomatic and irreversible. It may even result in slowly progressive neurologic dysfunction. The risk of radiation necrosis needs to be discussed with patients who are expected to have a longer clinical course.
In many cases, time will help distinguish between progression, pseudo-progression, and necrosis. Yet time is of the essence when treatment is needed for true tumor progression. Until novel innovative methods become available for the precise diagnosis of these changes, clinicians need to exercise careful consideration when deciding between conservative management and active intervention.
10. What is the role of bevacizumab in neuro-oncology?
Expert Perspective: In solid tumors (such as renal cell carcinoma, metastatic colon cancer, and ovarian cancer), bevacizumab is used for its antitumor effect. In neuro-oncology, bevacizumab is used for its anti-angiogenic effect and often leads to clinical improvement as well as radiologic response evident by decreased contrast enhancement on MRI. Indeed, singleagent bevacizumab received accelerated FDA approval in 2009 followed by full approval in 2017 for the treatment of recurrent glioblastoma, based on demonstration of significantly increased progression-free survival based on the EORTC 26101 study, although there was no improvement in overall survival or time to neurologic deterioration. Subsequent studies also demonstrated no benefit on overall survival in patients with newly diagnosed or recurrent glioblastoma (AVAglio and RTOG 0825, EORTC 26101). In addition, some evidence suggests that patients who progress while undergoing treatment with bevacizumab do not respond well to further salvage therapy and, as a result, have limited eligibility for clinical trials. Therefore, it may be prudent to reserve bevacizumab as the final salvage option.
Bevacizumab is frequently used as a steroid-spearing agent. Dexamethasone is the drug of choice in patients who are symptomatic from brain edema. Some patients require short-term use of steroids, whereas others need it for longer time and may even become dependent on it. Long-term steroid use may significantly decrease patients’ quality of life and lead to multiple medical problems, especially in elderly patients or those who already have multiple comorbidities. Steroid-induced myopathy, insomnia, diabetes, weight gain, decrease in physical activity, mood changes, change in physical appearance, and fluid retention are among the most commonly experienced side effects. To avoid these problems, it is reasonable to consider bevacizumab after assessing the risks and benefits of this medications. The common side effects of bevacizumab include hypertension, headaches, epistaxis, and delayed would healing. The dangerous side effects include hemorrhage, thromboembolism, and spontaneous bowel perforation.
Bevacizumab has been shown in a randomized, placebo-controlled clinical trial to improve radiation-induced necrosis. A low dose and short course of treatment of bevacizumab is used for this purpose.