Visit to download the full and correct content document: https://ebookmass.com/product/protein-interactions-the-molecular-basis-of-interactom ics-1st-edition-volkhard-helms/
More products digital (pdf, epub, mobi) instant download maybe you interests ...
Charge and Energy Transfer Dynamics in Molecular Systems, 4th Edition Volkhard May
https://ebookmass.com/product/charge-and-energy-transferdynamics-in-molecular-systems-4th-edition-volkhard-may/
The Molecular and Cellular Basis of Neurodegenerative Diseases: Underlying Mechanisms 1st Edition Michael S. Wolfe
https://ebookmass.com/product/the-molecular-and-cellular-basisof-neurodegenerative-diseases-underlying-mechanisms-1st-editionmichael-s-wolfe/
Biochemistry, 7th ed: The Molecular basis of life 7th Edition James R. Mckee
https://ebookmass.com/product/biochemistry-7th-ed-the-molecularbasis-of-life-7th-edition-james-r-mckee/
eTextbook 978-0124105300 Streptococcus Pneumoniae: Molecular Mechanisms of Host-Pathogen Interactions
https://ebookmass.com/product/etextbook-978-0124105300streptococcus-pneumoniae-molecular-mechanisms-of-host-pathogeninteractions/
Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease 6th Edition Edition Roger N. Rosenberg
https://ebookmass.com/product/rosenbergs-molecular-and-geneticbasis-of-neurological-and-psychiatric-disease-6th-editionedition-roger-n-rosenberg/
The Phenomenal Basis of Intentionality 1st Edition Angela Mendelovici
https://ebookmass.com/product/the-phenomenal-basis-ofintentionality-1st-edition-angela-mendelovici/
Medicinal Plants in Asia and Pacific for Parasitic Infections: Botany, Ethnopharmacology, Molecular Basis, and Future Prospects Christophe Wiart
https://ebookmass.com/product/medicinal-plants-in-asia-andpacific-for-parasitic-infections-botany-ethnopharmacologymolecular-basis-and-future-prospects-christophe-wiart/
Immunosensing for Detection of Protein Biomarkers 1st Edition Edition Huangxian Ju
https://ebookmass.com/product/immunosensing-for-detection-ofprotein-biomarkers-1st-edition-edition-huangxian-ju/
Rights As Security: The Theoretical Basis Of Security Of Person 1st Edition Rhonda Powell
https://ebookmass.com/product/rights-as-security-the-theoreticalbasis-of-security-of-person-1st-edition-rhonda-powell/
ProteinInteractions ProteinInteractions TheMolecularBasisofInteractomics EditedbyVolkhardHelmsandOlgaV.Kalinina
Editors
Prof.VolkhardHelms SaarlandUniversity CenterforBioinformatics Saarbrücken Germany
Prof.OlgaV.Kalinina
HelmholtzInstituteforPharmaceutical ResearchSaarland(HIPS)/Helmholtz CentreforInfectionResearch(HZI); MedicalFaculty,SaarlandUniversity;and CenterforBioinformatics, SaarlandUniversity Saarbrücken Germany
CoverImage: Foregroundimage©VolkhardHelms Background©Shutterstock
Allbookspublishedby WILEY-VCH arecarefully produced.Nevertheless,authors,editors,and publisherdonotwarranttheinformation containedinthesebooks,includingthisbook, tobefreeoferrors.Readersareadvisedtokeep inmindthatstatements,data,illustrations, proceduraldetailsorotheritemsmay inadvertentlybeinaccurate.
LibraryofCongressCardNo.: appliedfor BritishLibraryCataloguing-in-PublicationData Acataloguerecordforthisbookisavailable fromtheBritishLibrary.
Bibliographicinformationpublishedby theDeutscheNationalbibliothek TheDeutscheNationalbibliotheklists thispublicationintheDeutsche Nationalbibliografie;detailedbibliographic dataareavailableontheInternetat <http://dnb.d-nb.de>
©2023WILEY-VCHGmbH,Boschstr.12, 69469Weinheim,Germany
Allrightsreserved(includingthoseof translationintootherlanguages).Nopartof thisbookmaybereproducedinanyform–by photoprinting,microfilm,oranyother means–nortransmittedortranslatedintoa machinelanguagewithoutwrittenpermission fromthepublishers.Registerednames, trademarks,etc.usedinthisbook,evenwhen notspecificallymarkedassuch,arenottobe consideredunprotectedbylaw.
PrintISBN: 978-3-527-34864-0
ePDFISBN: 978-3-527-83051-0
ePubISBN: 978-3-527-83052-7
oBookISBN: 978-3-527-83050-3
Typesetting Straive,Chennai,India
Contents Preface xv
1ProteinStructureandConformationalDynamics 1 VolkhardHelms
1.1StructuralandHierarchicalAspects 1
1.1.1SizeofProteins 1
1.1.2ProteinDomains 1
1.1.3ProteinComposition 2
1.1.4SecondaryStructureElements 3
1.1.5ActiveSites 3
1.1.6MembraneProteins 6
1.1.7FoldingofProteins 7
1.2ConformationalDynamics 8
1.2.1Large-ScaleDomainMotions 8
1.2.2DynamicsofN-TerminalandC-TerminalTails 9
1.2.3SurfaceDynamics 9
1.2.4DisorderedProteins 9
1.3FromStructuretoFunction 10
1.3.1EvolutionaryConservation 10
1.3.2BindingInterfaces 10
1.3.3SurfaceLoops 11
1.3.4PosttranslationalModifications 11
1.4Summary 11 References 12
2Protein–Protein-BindingInterfaces 15
ZeynepAbali,DamlaOvek,SimgeSenyuz,OzlemKeskin,andAttilaGursoy
2.1DefinitionandPropertiesofProtein–ProteinInterfaces 15
2.2GrowingNumberofKnownProtein–ProteinInterfaceStructures 18
2.3SurfaceAreasofProtein–ProteinInterfaces 21
2.4GapVolumeofProtein–ProteinInterfaces 22
2.5AminoAcidCompositionofInterfaces 22
2.6SecondaryStructureofInterfaces 23
2.7Protein–Protein-BindingEnergy 24
2.8InterfacesofHomo-andHetero-DimericComplexes 24
2.9InterfacesofNon-obligateandObligateComplexes 25
2.10InterfacesofTransientandPermanentComplexes 25
2.11Biologicalvs.CrystalInterfaces 26
2.12TypeI,TypeII,andTypeIIIInterfaces 27
2.13ConservedResiduesandHotSpotsinInterfaces 28
2.14ConclusionandFutureImplications 29 References 30
3CorrelatedCoevolvingMutationsatProtein–Protein Interfaces 39 AlexanderSchug
3.1Introduction 39
3.2AShortIntroductionintoBiomolecularModeling 41
3.3StatisticalInferenceofCoevolution 41
3.3.1LimitationsofLocalStatisticalInference 41
3.3.2Direct-CouplingAnalysis–APottsModelBasedonMultipleSequence Alignments 42
3.4SolvingtheInversePottsModel 43
3.5ContactGuidedProteinandRNAStructurePrediction 45
3.6Inter-MonomerInteractionandSignaling 45
3.7Summary 46 References 47
4ComputationalProtein–ProteinDocking 53 MartinZacharias
4.1Introduction 53
4.2RigidBodyProtein–ProteinDockingApproaches 56
4.3AccountingforConformationalChangesduringDocking 59
4.4IntegrationofBioinformaticsandExperimentalDataforProtein–Protein Docking 61
4.5Template-BasedProtein–ProteinDocking 62
4.6FlexibleRefinementofDockedComplexes 64
4.7ScoringofDockedComplexes 66
4.8ConclusionsandFutureDevelopments 67 Acknowledgments 68 References 68
5IdentificationofPutativeProteinComplexesin Protein–ProteinInteractionNetworks 77 SudharshiniThangamurugan,MarkusHollander,andVolkhardHelms
5.1Protein–ProteinInteractionNetworks 77
5.2IntegrationofVariousPPIResourcesinPublicDataRepositories 79
5.3Protein–ProteinInteractionNetworksofModelOrganisms 80
5.3.1PPINof Saccharomycescerevisiae80
5.3.2PPINofHuman 83
5.4AlgorithmstoIdentifyProteinComplexesinPPINetworks 84
5.4.1MolecularComplexDetection(MCODE) 84
5.4.1.1Definitions 85
5.4.1.2Algorithm 86
5.4.1.3Examples 88
5.4.2ClusteringwithOverlappingNeighborhoodExpansion (ClusterONE) 89
5.4.2.1Definitions 89
5.4.2.2Algorithm 90
5.4.3Domain-AwareCohesivenessOptimization(DACO) 92
5.5Summary 94 References 95
6Structure,Composition,andModelingofProtein Complexes 101 OlgaV.Kalinina
6.1ProteinComplexStructure 101
6.1.1ProteinQuaternaryStructure 101
6.1.2ClassificationofProtein–ProteinInteractionInterfaces 102
6.1.3ClassificationandEvolutionofProteinComplexes 105
6.2MethodsforAutomatedAssignmentofBiologicalAssemblies 106
6.2.1AssignmentfromCrystallographicData 107
6.2.2EmployingMachine-LearningMethods 108
6.2.3LeveragingEvolutionaryInformation 109
6.3ComputationalApproachestoPredicting3DStructureofProtein Complexes 110
6.3.1CombinatorialDocking 110
6.3.2Homology-BasedComplexReconstruction 114
6.3.3PredictionfromSequence 115
6.3.4AssistedDocking 116
6.4ConclusionandOutlook 117 Acknowledgments 118 References 118
7Live-CellStructuralBiologytoSolveMolecularMechanisms: StructuralDynamicsintheExocystFunction 127 AltairC.Hernandez,BaldoOliva,DamienP.Devos,andOriolGallego
7.1Introduction 127
7.2StructuralBiologyUsingLightMicroscopyMethods 129
7.3HybridMethods:IntegrativeStructuralBiology 131
7.4IntegrativeModeling:TheCaseoftheExocystComplex 133
7.5Comparingthe InSitu ArchitectureoftheExocystwitha High-ResolutionCryo-EMModel 136
7.6DiscussionandFuturePerspectives 138 Acknowledgements 139 References 140
8KineticsandThermodynamicsofProtein–Protein Encounter 143
NicolasKünzelandVolkhardHelms
8.1Introduction 143
8.2ThermodynamicEnsemblesandFreeEnergy 143
8.2.1TheIsothermal–IsobaricEnsembleandtheGibbsFreeEnergy 144
8.3OverviewofComputationalMethodstoDetermineBindingFree Energies 146
8.3.1CoarseGraining 147
8.3.1.1BrownianDynamics 147
8.3.2EndpointMethods 149
8.3.2.1MM/PBSA/MM/GBSA 149
8.3.3PotentialofMeanForce/PathwayMethods 150
8.3.3.1ThermodynamicIntegration 151
8.3.3.2UmbrellaSampling(US) 151
8.3.3.3SteeredMD(SMD) 153
8.3.3.4Metadynamics 153
8.3.3.5AdaptiveBiasingForce(ABF) 155
8.3.4Replica-ExchangeMethods 155
8.3.4.1ParallelTempering 155
8.3.4.2Generalized/HamiltonianReplica-ExchangeMethods 156
8.3.5AdditionalPathwayMethods 156
8.3.6RelativeBindingFreeEnergies 156 References 157
9MarkovStateModelsofProtein–ProteinEncounters 163 SimonOlsson Notation 163
9.1Introduction 163
9.2MolecularDynamicsandMarkovStateModels 164
9.2.1MarkovStateModels:TheoryandProperties 165
9.3StrategiesforMSMEstimation,Validation,andAnalysis 169
9.3.1VariationalApproachforConformationalDynamicsandMarkov Processes(VACandVAMP) 169
9.3.2FeatureSelection 170
9.3.3DimensionalityReduction 171
9.3.4Clustering 172
9.3.5ModelEstimationandValidation 173
9.3.6SpectralGapsandCoarse-Graining 174
9.3.7AdaptiveandEnhancedSamplingStrategies 175
9.3.8PracticalConsiderationforStudyingProtein–ProteinEncounters 176
9.3.9AnalysisoftheAssociation–DissociationPathEnsemble 177
9.4TheConnectiontoExperiments 178
9.4.1ExperimentalObservability,ForwardModels,andErrors 178
9.4.1.1SourcesofErrorsandUncertainty 179
9.4.2PredictingExperimentalObservablesUsingMSMs 180
9.4.3IntegratingExperimentalandSimulationDataintoAugmentedMarkov Models 181
9.5Protein–ProteinandProtein–PeptideEncounters 182
9.6EmergingTechnologies 184
Acknowledgments 186
References 186
10TranscriptionFactor–DNAComplexes 195 VolkhardHelms
10.1Introduction 195
10.2PrinciplesofSequenceRecognition 197
10.3DimerizationofEukaryoticTFs 198
10.4DetectionofEpigeneticModifications 199
10.5DetectionofDNACurvature/Bending 200
10.6ModificationsofTranscriptionFactors 200
10.7TranscriptionFactorBindingSites 201
10.8ExperimentalDetectionofTFBS 201
10.8.1Protein-BindingMicroarrays 202
10.8.2ChromatinImmunoprecipitationAssays 203
10.8.3DamIDProfilingofProtein–DNAInteractions 204
10.9Position-SpecificScoringMatrices 204
10.10MolecularModelingofTF–DNAComplexes 204
10.11Cis-RegulatoryModules 205
10.12RelatingGeneExpressiontoBindingofTranscriptionFactors 207
10.13Summary 208 References 208
11TheChromatinInteractionSystem 213
SarahKreuz,Stefan-SebastianDavid,LorenaViridianaCortesMedina,and WolfgangFischle
11.1ChromatinIsaSpecialInteractionPlatform 213
11.2InteractionofProteinswithHistonePosttranslational Modifications 215
11.2.1TheHistoryofHistonePosttranslationalModificationsand theHistoneCode 215
11.2.2PeptidesandNucleosomalTemplatesforStudyingHistonePTMs 222
11.2.3QualitativeAnalysisofHistonePTMReadout 224
11.2.3.1CharacterizingBindingSpecificitiesofKnownReaders 224
11.2.3.2IdentificationofNewReaderProteins 225
11.2.4MolecularParametersofHistonePTM–ReaderInteraction 226
11.2.5CellularAssaystoCharacterizeHistonePTM–ReaderInteractions 227
11.2.5.1VisualizingHistone–ReaderInteractions 227
11.2.5.2ChromatinImmunoprecipitation 229
11.2.5.3CellularLabelingandAffinityEnrichment 231
11.3InteractionofProteinswithModifiedNucleicAcids 231
11.3.1DiscoveryofDNAMethylationandtheFirstReaderProteins 231
11.3.2RNAModifications 234
11.3.3ModifiedDNAandRNATemplates 234
11.3.4 InVitro AssaysforIdentifyingReadersofNucleicAcidMethylation 235
11.3.4.1AffinityPurificationtoIdentifyNovelModificationReaders 235
11.3.4.2CharacterizingBindingSpecificitiesofKnownReaders 235
11.3.5CellularAssaysforIdentifyingReadersofNucleicAcid Modifications 236
11.4UHRF1asanExampleofaMultidomainReader/WriterProteinof HistoneandDNAModifications 239
11.5HistoneChaperonesandChromatinRemodelingComplexes 241
11.5.1ChromatinAssemblyandRemodeling 241
11.5.2DiscoveryofHistoneChaperonesandChromatinRemodelers 242
11.5.3MethodsforIdentifyingHistoneChaperonesandRemodeling Factors 244
11.5.3.1ImmunoprecipitationAssays 244
11.5.3.2ComputationalMethods 244
11.5.4AssaystoStudyChaperoneandRemodelerActivities 245
11.5.5CellularAssays 245
11.6ChallengesinChromatinInteractomics 247 References 248
12RNA–ProteinInteractomics 271
CorneliaKilchert
12.1Introduction 271
12.2InteractionsofProteinswithmRNAandncRNA 272
12.3TheBasicToolbox 273
12.3.1MetabolicRNALabelingwithModifiedNucleobases 273
12.3.2RNA–ProteinCrosslinking 274
12.4RNA–ProteinInteractomics 276
12.4.1WhatProteinsAreBoundtomyRNA(orRNAinGeneral)? 276
12.4.1.1CatalogingtheRBPome 276
12.4.1.2InteractomesofSpecificRNAs 278
12.4.2WhichRNASpeciesAreBoundbymyRBP? 280
12.4.2.1CopurificationMethods:CLIPandDerivatives 280
12.4.2.2Proximity-DependentLabelingMethods 280
12.5Outlook 282
Notes 283 References 283
13InteractionBetweenProteinsandBiologicalMembranes 293 LorantJanosiandAlemayehuA.Gorfe
13.1Introduction 293
13.2ThePlasmaMembrane:OverviewofItsStructure,Composition,and Function 294
13.3Lipid-BasedandProtein-BasedSortingofPlasmaMembrane Components 295
13.3.1Lipid-BasedSortingandDomainFormation 295
13.3.2Protein-BasedSortingandMembraneCurvature 296
13.3.3ProteolipidSortingandMembraneDomainStabilization 297
13.4InteractionofPeripheralMembraneProteinswithMembrane Lipids 297
13.4.1Protein-BasedMembrane-TargetingMotifs 298
13.4.2Lipid-BasedMembrane-TargetingMotifs 301
13.5InteractionsandConformationsofTransmembraneProteinsinLipid Membranes 303
13.5.1GlycophorinAandEGFRasExamplesofSingle-PassTransmembrane Proteins 303
13.5.2GPCRasanExampleofMulti-PassTMHelicalProteins 306
13.5.3AquaporinasanExampleofOligomericMulti-PassTMProteins 306
13.5.4AntimicrobialPeptides:PeripheralorIntegral? 307
13.6Summary 308
Acknowledgment 308 References 309
14InteractionsofProteinswithSmallMolecules,Allosteric Effects 315
MichaelC.Hutter
Abbreviations 315
14.1Introduction 315
14.2ModesofBindingtoProteins 316
14.3TypesofInteractionBetweenProteinandLigand 317
14.3.1SaltBridges 317
14.3.2CoordinationofIonsviaLonePairs 318
14.3.3HydrogenBonds 319
14.3.3.1Definition 319
14.3.3.2OccurrenceandFunctionalityofHydrogenBondsinBiological Systems 320
14.3.3.3ClassificationofHydrogenBonds 321
14.3.3.4WeakHydrogenBonds 321
14.3.3.5HydrogenBondstoFluorine 322
14.3.3.6Nitrogenvs.OxygenasCompetingHydrogenBondAcceptors 322
14.3.3.7BifurcatedHydrogenBonds 322
14.3.4HalogenBonds 323
Contents
14.3.5vanderWaalsInteractions 324
14.3.6MutualInteractionsofDelocalized π-ElectronSystems 325
14.3.7Cation–π Interaction 325
14.3.8Anion–π Interaction 325
14.3.9UnusualProtein–LigandContacts 326
14.4ModelingIntermolecularInteractionsbyForceFieldsandDocking Simulations 326
14.5EntropicAspects 327
14.6AllostericEffects:ConformationalChangesUponLigandBinding 327
14.7AspectsofLigandDesignBeyondProtein–LigandInteractions 329
14.8Conclusions 330 References 330
15EffectsofMutationsinProteinsonTheirInteractions 333
AlexanderGressandOlgaV.Kalinina
15.1Introduction 333
15.2StructuralAnnotationofMutationsinProteins 334
15.2.1DatabasesforStructuralAnnotationofMutations 335
15.2.2DynamicStructuralAnnotationPipelines 340
15.3MethodsforPredictingEffectofProteinMutations 342
15.3.1PredictionofPhenotypicEffect 343
15.3.2EstimationofMutationEffectsbyModelingBiophysicalPropertiesof Proteins 344
15.3.3PredictionofMechanisticEffectsofMutationsonInteractionsof Proteins 345
15.4Conclusion 348 Acknowledgments 349 References 349
16NotQuitetheSame:HowAlternativeSplicingAffectsProtein Interactions 359
ZakariaLouadi,OlgaTsoy,JanBaumbach,TimKacprowski,andMarkusList ListofAbbreviations 359
16.1Introduction 359
16.2EffectsofAlternativeSplicingonIndividualProteins 362
16.2.1AlternativeSplicingandProteinStructure 362
16.2.2AlternativeSplicingandIntrinsicallyDisorderedRegions 362
16.3EffectsofAlternativeSplicingonProtein–ProteinInteraction Networks 367
16.3.1AlternativeSplicingRewiresProtein–ProteinInteractions 367
16.3.2AlternativeSplicinginDiseases 368
16.3.3ResourcesforStudyingtheEffectofAlternativeSplicingon Protein–ProteinInteractions 369
16.4ConclusionandFutureWork 373 References 374
17Phosphorylation-BasedMolecularSwitches 381
AttilaReményi
17.1Introduction 381
17.1.1StructuralandFunctionalEffectsofProteinPhosphorylation 383
17.2ReversibleProteinPhosphorylationinCellularSignaling:Writers, Readers,andErasers 386
17.3ProteinKinasesasMolecularSwitchesandasComponentsofSignaling Cascades 388
17.4MechanismsofPhosphorylationSpecificity:theImportanceofShort LinearMotifs 390
17.5ExamplesofPhospho-Switch-BasedBiologicalRegulation 392
17.6Conclusion 395 Acknowledgments 397 References 397
18SummaryandOutlook 401 VolkhardHelmsandOlgaV.Kalinina
18.1TechnicalStateoftheArt 401
18.2RoleofMachineLearning 401
18.3Challenges 402
18.4WhatPicture(s)MayEvolve? 403 References 404
Index 405
Preface Proteinsarebiochemicalmachinesthatparticipateinvirtuallyallkeyprocesses ofbiologicalsystems.Forexample,enzymescatalyzebiochemicalreactions,and hencecontrolthemetabolicstateofthecell.TranscriptionfactorsguideRNApolymerasetoreadofftheneededpartsofthegenome.Initiationfactorstellribosomes whatmRNAstoprocess.Receptorsinthecellmembranesensesignalsfromthe outside.Transportersandchannelproteinsmediateexchangeofsubstancesacross thecellandorganellemembranes,etc.Intheseprocesses,proteinsexerttheir biologicalfunctionbyinteractingwithotherproteins,nucleicacids,membranes, low-molecular-weightligandssuchassubstratesanddrugs,andsoforth.Inessence, alltheseinteractionsaregovernedbynonbondedinteractionsbetweenprotein atoms(backboneaswellasside-chainatoms)andatomsofthecorresponding interactionpartners,wherebythescaleoftheseinteractionsdiffersfromahandful ofinvolvedatomsforlow-molecular-weightligandstothousandsofatomsinlarge proteincomplexes.Consequently,methodsthatallowstudyingandpredictingthese interactionsdifferinscale.
Theresearchfieldthatdiscoversandanalyzesthismyriadofinteractionsistermed interactomicsandbuildsoncontributionsfromexperimentsandcomputation.The technologiesusedinthisfieldareconstantlybeingrefined,butstilllagsomehow behindthelevelatwhichindividualpairwiseinteractionscanberesolvedandpredictedintermsofthree-dimensionalstructure,bindingthermodynamics,specificity, etc.Oneimportantaspectofinteractomicsistointegratedatafromdifferentsources. Practicallyignoredsofararetheeffectsofpost-translationalmodificationsandalternativesplicingoninteractomics(whichareaddressedindetailinthisbook,see below).Itistheaimofthisbooktocapturethestate-of-artofcellularinteractomics involvingproteinsandtodescribeexistingtechnicalandconceptualchallengesthat needtobeovercomeinthefuture.
Thisbookpresentsanoverviewofproteininteractions,experimentaltechniques andfindings,computationaltoolsandresourcesthathavebeendevelopedto studythem.Initsfirstpart,weintroducethemolecularbasicsofproteinstructure (Chapter1)andpropertiesofprotein–proteinbindinginterfaces(Chapter2). Recently,new-generationsequencingmethodsyieldedlargeamountsofprotein sequencedatathatcanalsobeleveragedtopredictproteininteractions,inparticular, pairwiseprotein–proteininteractions(Chapter3).Inparallel,classicalmethods ofprotein–proteindocking(reconstructionofpairwiseproteincomplexesfrom isolatedstructuresoftheircomponents)havematuredandnowadayssuccessfully
managetoincorporateadditionalexperimentalconstraintsandmayevenaccount forproteinconformationalchanges(Chapter4).Asystemicviewofproteinpairwise interactionsisprovidedbyproteininteractionnetworks,whichcancompriseboth experimentallyresolvedandcomputationallypredictedinteractions(Chapter5).
Largeproteincomplexespresentanadditionalchallengeduetonumerousways inwhichindividualprotomerscaninteractwitheachother.Suchcomplexes canbeextractedfromproteininteractionnetworks(Chapter5)orpredictedina combinatorialfashionorusingexperimentalconstraints(Chapter6).Systematic integrationofmanyexperimentalconstraintswithstructuraldatacanprovide excitinginsightsintothestructureandevolutionofverylargeandcomplexprotein assemblies(Chapter7).
Thephysicsofproteininteractionswithdifferentpartnerstakesplaceatdifferentscales,sincethesizeofthepartnersandhencethenumberofindividual non-covalentinteractionsdifferconsiderably.Thesecondpartofthisbookanalyzes thesedifferentinteractionsandwaystomodelthemindetail.Westartwithcomputationaltechniquestoexaminethekineticsandthermodynamicsofinteractions betweenpairsofproteins(Chapter8),followedbyachapteronMarkov-statemodels thatstatisticallyevaluatealltransitionsalongassociationanddissociationpathways (Chapter9).Wecontinuewithprotein–DNAinteractionsexemplifiedbytranscriptionfactorbindingtoDNA(Chapter10)andchromatin(Chapter11),followed byachapterontheemergingfieldofprotein–RNAinteractions,e.g.duringthe preprocessingstageofpre-mRNAandwithnoncodingRNAs(Chapter12).Asmany signalingandtransportprocessesinvolvecellularmembranes,protein–membrane interactionsarethencoveredinChapter13,followedbyadiscussionofhow proteinsinteractwithlow-molecular-weightligandssuchasdrugs(Chapter14).
Allthesedifferentkindsandinstancesofproteininteractionscruciallycontribute totheflowofmatterandinformationinlivingcells.Ifsuchinteractionsare modulated,thiscanobviouslyaltermanycellularprocesses.Inthethirdpartof thisbook,threeimportanttypesofmodulatingeffectsareaddressed,namelythe effectsofgeneticmutations(Chapter15),ofalternativesplicing(Chapter16), andthoseofposttranslationalmodifications(Chapter17).There,themainfocus isagainplacedonhowprotein–proteininteractionsareaffected.Theimpactof thesetypesofproteinalterationsonothertypesofinteractions(e.g.withsmall molecules)islesswellunderstood,althoughprominentexamples,suchasdrug resistance-associatedmutations,exist.Computationalmethodsforsystematic assessmentofsuchchangesarestilltobedeveloped.
Theindividualchapterswerewrittenbyexpertsintheirfields,andweare extremelygratefultothemfortheirtimeandefforttheyinvestedinthis.Wehope thatthisbookpaintsacomplex,butversatileandinstructivepictureofalldifferent kindsofinteractionsthatproteinsengagein.Interactomics,buildingoncombined experimentalandcomputationalwork,isanemergingdisciplinethatbearsgreat promisetobetterunderstandthemolecularmechanismsoflife.Inourview,protein interactionsholdthekeytoit. VolkhardHelms
ProteinStructureandConformationalDynamics VolkhardHelms
SaarlandUniversity,CenterforBioinformatics,SaarlandInformaticsCampus,Postfach151150, 66041Saarbrücken,Germany
1.1StructuralandHierarchicalAspects 1.1.1SizeofProteins Thesizeofproteinsrangesfromverysmallproteins,suchasthe20-aminoacid miniproteinTrpcage,tothelargestproteininthehumanbody,titin,whichconsists ofabout27000aminoacidsandhasamolecularweightof3millionDalton.Generally,whenspeakingoftypicalproteins,werefertocompactproteinsofabout80to 500aminoacids(residues)insize.Tiessenetal.reportedthatarchaealproteinshad thesmallestaveragesize(283aa),followedbybacterialproteins(320aa)andeukaryoticproteins(472aa)[1].Amongeukaryotes,plantproteins(392aa)hadasmaller size,whereasanimalproteins(486aa)andproteinsfromfungi(487aa)werelarger.
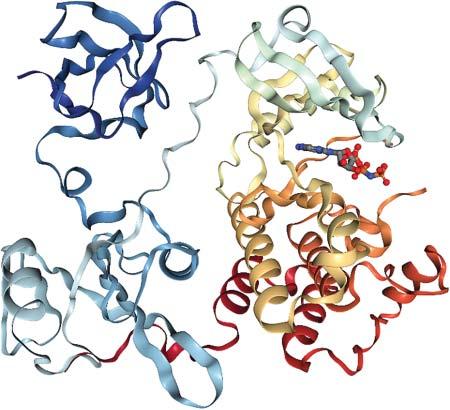
1.1.2ProteinDomains Thelargerasingleproteingets,thehigheristhechancethatitwillbecomposed ofmultiplestructurallydistinct“domains.”Thesearetypicallysequentialparts oftheproteinsequencewithacharacteristiclengthbetween100and200amino acids[2].Forexample,theproteinSrckinaseconsistsofanSH3domain(thatbinds toproline-richpeptides),anSH2domain(thatbindstophosphorylatedtyrosine residues),andthecatalytickinasedomain,seeFigure1.1.Intheinactivestate,the SH3domainwillholdontothelinkerconnectingSH2andcatalyticdomainthat containsseveralprolines,andtheSH2domainwillholdontoaphosphorylated tyrosineintheC-terminaltailofthecatalyticdomain.Thereby,allthreedomains arelockedinaconformationallyrestrictedstate.Onceactivatedbydephosphorylationofthetyrosine,thesecontactsarereleased,andthecatalyticdomaincan undergothecharacteristicPacman-typeopening/closingmotionofproteinkinases, enablingthebindingofadenosinetriphosphate(ATP).Intheclosedconformation, theactivesiteresiduescatalyzetransferoftheterminal γ-phosphateofATPtoa nearbytyrosineofasubstrateproteinboundontheSrckinasesurface.Thecatalytic ProteinInteractions:TheMolecularBasisofInteractomics,FirstEdition. EditedbyVolkhardHelmsandOlgaV.Kalinina. ©2023WILEY-VCHGmbH.Published2023byWILEY-VCHGmbH.
Figure1.1 X-raystructure (PDBcode1AD5)ofhuman Srckinase.Thepeptide sequencestartswithanSH3 domain(topleft),followed byanSH2domain(bottom left)andthenleadstothe catalytickinasedomain (right).ATPisbound betweensmall(top)and largelobe(bottom)ofthe kinasedomain.Source: FiguregeneratedwithNGL viewer.
domainofkinasesitselfconsistsoftwodomain-like“lobes,”asmallerN-terminal lobe(ofabout80aa)andalargerC-terminallobe(ofabout180aa).
Althoughmulti-domainproteinsexistinalllifeforms,morecomplexorganisms (havingalargernumberofuniquecelltypes)containmoreuniquedomainsanda largerfractionofmulti-domainproteins:eukaryoteshavemoremulti-domainproteinsthanprokaryotes,andanimalshavemoremulti-domainproteinsthanunicellulareukaryotes[3].
1.1.3ProteinComposition Thecompositionofaproteindependsonitsenvironmentanditsposttranslational modifications,suchasphosphorylationandsumoylation.Forexample,extracellular domainsofmostcellmembraneproteinsareoftenextensivelyglycosylated.Here, wewillfocusonthevaryingmixtureofthe20commonlyoccurringaminoacids thatmakeupmostofallexistingproteins.Water-solubleproteinspossessarather hydrophobiccoreandapolarsurfacethatisincontactwiththecytoplasm.This clearorganizationalprincipleprovidesthemaindrivingforceforthefoldingof water-solubledomainsviathe“hydrophobiceffect.”
Prokaryoticproteinscontainmorethan10%ofleucineandabout9%ofalanine residues,butratherfew(only1–2%)cysteine,tryptophan,histidine,andmethionineresidues[4].Brüneetal.comparedtheaminoacidcompositionofprokaryoticandeukaryoticproteins[5].Eukaryoteshavethehighestvariabilityforproline, cysteine,andasparagine.Aminoacidsshowinghighvariabilityacrossspeciesare lysine,alanine,andisoleucine,whereashistidine,tryptophan,andmethioninevary theleast.Cysteineismorecommonineukaryotesthaninarchaeaandbacteria, whereasisoleucineislessabundantineukaryotes.Theauthorsalsoanalyzedthe differentialusageofaminoacidsindomainsandlinkers.Prolineandglutamine, buttoasmallerextent,polarandchargedaminoacids,aremorecommoninlinkers
1.1StructuralandHierarchicalAspects 3 thatareratherexposedtosurroundingwater.Globulardomainscontainlargerfractionsofhydrophobicaminoacids,suchasleucineandvaline,andaromaticones, suchasphenylalanineandtyrosine.
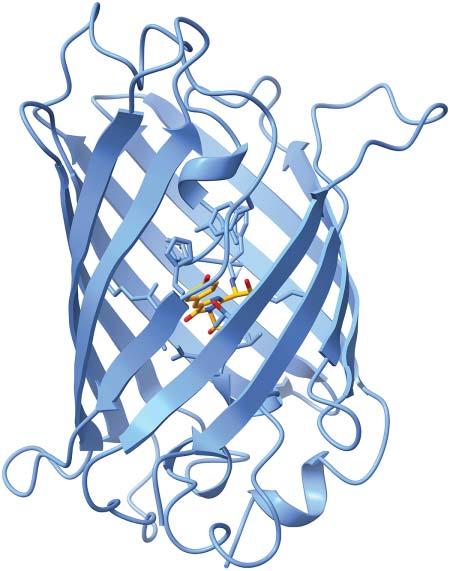
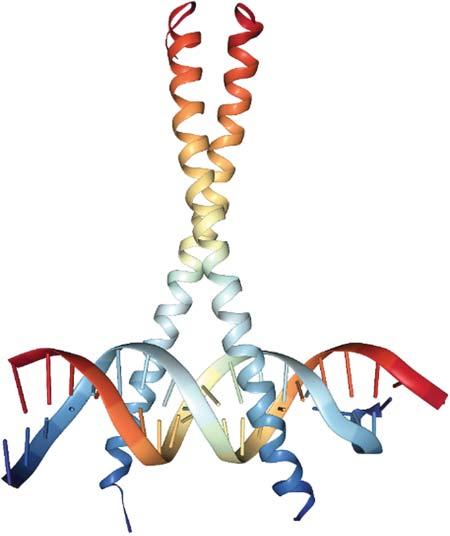
1.1.4SecondaryStructureElements Foldedproteinscontaintwotypesofsecondarystructureelements, α-helicesand β-sheets. α-Heliceshavelengthsbetween9and37residueswithapeakat11amino acids[6]. β-Sheetsareconsiderablyshorter,being2–17residueslongwithapeak at5residues[7].Thesecondarystructurecontentofproteinsrangesfrompurely helicalproteins,suchasmyoglobin,containingsix α-helices(seeFigure1.2)over mixed α/β proteinstoso-called β-barrels,suchasgreenfluorescentprotein(GFP), seeFigure1.3,orOmpmembraneporesintheoutermembranesofgram-negative bacteria.Secondarystructureelementsprovidestabilitytotheproteinstructureand serve,e.gtoanchorthecatalyticresiduesoftheactivesiteatprecisepositionsfrom eachother(seebelow). α-Helicesarealsothestructuralbasisofcoiledcoils,see Figure1.4,becausethehelicescannicelypackagainsteachother. α-Helicesarefrequentlyusedbytranscriptionfactors,suchasGCN4,attheDNA-bindinginterface, wherethe α-helicescanintercalateinthemajororminorgroovesoftheDNAdouble helix.
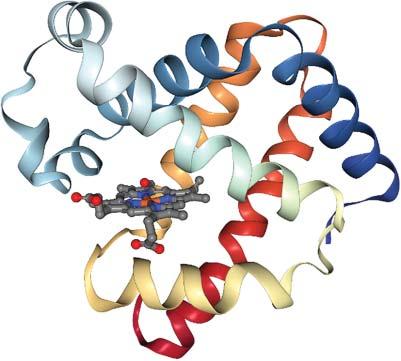
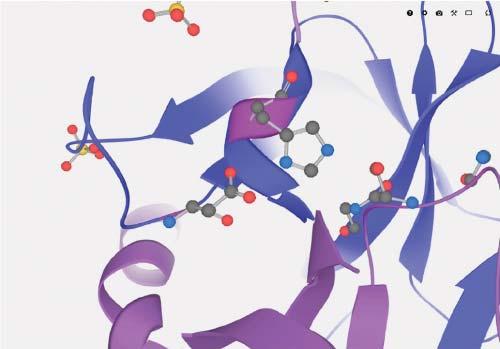
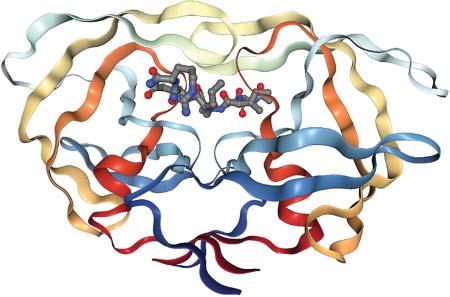
1.1.5ActiveSites Activesitesofenzymesarelocationswhereboundsubstratemoleculesundergo chemicalmodificationswhilebeingboundtotheenzyme.Figure1.5showsthe activesiteoftheserineproteasechymotrypsinogenAwiththecharacteristic catalyticresiduesserine,histidine,andasparticacid.Inprinciple,discussing enzymaticmechanismsisoutofscopeforthisbook,whichmostlydealswith interactionsthatproteinsengagein.Somemultienzymecomplexeshavingmultiple activesitesassembletoenabletheproductofonereactiontobepassedfrom
Figure1.2 X-raystructure(PDB code1MBN)ofmyoglobinfrom Physetercatodon.Theporphyrin cofactorisanchoredbetweensix alphahelices.Source:Figure generatedwithNGLviewer.
Figure1.3 X-raystructure ofthegreenfluorescent proteinfrom Aequorea victoria (PDBcode1EMA). Thebarrel-shapedstructure isformedby11beta-strands surroundingacentral alpha-helixholdingthe chromophore.Source: FiguregeneratedwithUCSF Chimera.
Figure1.4 X-raystructure ofGCN4dimerfrom S. cerevisiae forminga so-calledcoiledcoiland boundheretoDNA(PDB code1YSA).Source:Figure generatedwithNGLviewer.
Figure1.5 Catalytictriad–asparticacid,histidine,serine–intheactivesiteofaserine protease.Source:EuropeanMolecularBiologyLaboratory(EMBL).
oneactivesitetoother,whereitbecomesthesubstrateofafollow-upchemical reaction.Generally,accesstoactivesitesshouldnotbeprecludedbybindingto otherinteractionpartners,although,insomecases,bindingpatchesneedtobe closetotheactivesite,e.g.whenakinasebindsitssubstrateonapatchonthe surfaceofthelargelobesothataphosphategroupcanbetransferredfrombound ATPtoaserineresidueoftheboundsubstrateasmentionedbefore.
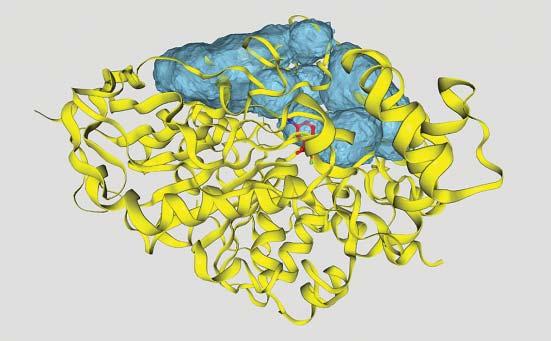
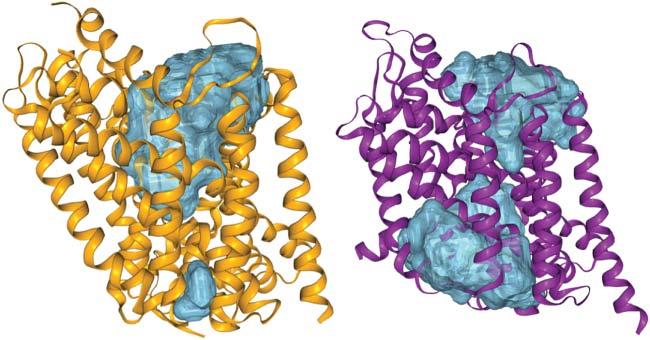
Often,theactivesitesofenzymesarelocatedontheproteinsurface,sothatsubstratescaneasilybindwhileremainingpartiallysolventexposed.Afrequentstructuralmotifisaflexibleproteinloopthatreachesovertheboundsubstrate,e.g.inHIV protease,seeFigure1.6.Inothercases,theactivesiteislocatedinsidetheprotein, suchasforcytochromeP450enzymesoracetylcholineesterase.There,substrates needtopassintotheproteinstructurethroughachannelthatmaybeuptoseveralnanometerslong,seeFigure1.7.Themainpurposeofsuchanarrangement istoplacethesubstrateinalow-dielectriccavitythatenablescomplicatedchemical reactionstotakeplace.Notethatthestrengthofelectrostaticinteractionsisinversely
Figure1.6 X-raystructure ofanHIVproteasedimer (PDBcode4HVP). Asubstratepeptideisbound intheactivesite.Accessto theactivesiteiscontrolled byopening/closing transitionsoftwoflexible loopsabovethepeptide (flaps).Figuregenerated withNGLviewer.
Figure1.7 Trimethylammoniotrifluoroacetophenoneligandboundintheactivesiteof acetylcholinesterasefrom tetronarcecalifornica (PDBcode1AMN).Thesurfacecontours illustrateseveralporesandcavitiesthatmakeuptunnelsleadingtotheinternalactivesite. Source:ThefigurewasgeneratedwiththeProPores2webserver(https://service .bioinformatik.uni-saarland.de/propores)[8].
proportionaltothedielectricconstantoftheenvironment.Inalowdielectricenvironment,chargedproteinresiduescanexertstrongerelectron-pullingorpushing effectsonthesubstrate.Enzymeactivesites,ligandbindingsites,ortranslocation poresofionchannelscaneitherresideinindividualproteinunitsorinbetweenthe interfacesofmultimers.
1.1.6MembraneProteins Integraltransmembraneproteinsareintegratedintocellularmembraneswhereby theiraminoacidchaincrossesthehydrophobicbilayeronceormultipletimes. Whiletheirsolubledomainshavethesamecompositionaswater-solubleproteins, themembrane-spanningpartshaveaso-called“inside-out”composition.These membraneregionsareveryhydrophobicontheoutsidethatisincontactwiththe aliphaticlipidchainsofthephospholipidbilayerandhaveapartiallypolarinterior thatoftencontainsawater-filledtranslocationchannelforsubstratemolecules. Whenthepeptidechaincrossesthebilayer,nohydrogenbondingispossiblewith thealiphaticlipidchainsthatareinstrongcontrasttothesituationinthewater phase.Tosatisfythehydrogenbondingcapacityofitsbackboneatoms,thechain thusadoptseitheran α-helicalconformationora β-sheetconformationinthe membrane.Betabarrelsconsistof8–22 β-sheets[9]butareonlyfoundintheouter membranesofgram-negativebacteria,mitochondria,andchloroplasts.Helical transmembraneproteinspossessbetween1andaround20alphahelices[10]that arebetween10and30residueslong.Themajorityofhelicalmembraneproteins possessonly1transmembranedomain(TMD),followedbythosehaving2TMDs andsmallerfractionswith3,4,7,and12TMDs[10].Oligomerizationisfrequently
1.1StructuralandHierarchicalAspects 7 foundamonghelicaltransmembraneproteins,wherebytheirbindinginterfaces consistofroughlyperpendicular α-helices.Manyreceptorsoncellsurfacesform functionaldimers.Ionchannelsformtetra-andhexamers,withtheion-conducting porebetweenthemonomers.Interactionsbetweenproteinsandmembranesare furtherdiscussedinChapter13.
1.1.7FoldingofProteins Predictingthefoldedstructureofaproteinfromitssequencehaslongbeenaholy grail.Inthemeantime,scientistshavebeenabletoputmanypiecesofthispuzzle together.Importantcontributionstothiswere,e.g.thephi-valueanalysisexperimentsbyFershtandcoworkersthatquantifythedegreeofnativefoldedstructure aroundmutatedresiduesinthefoldingtransitionstate[11]andthetheoretical workbyWolynes,Onuchic,andothers,whodrewananalogybetweenthefolding ofbiopolymersandrelaxationprocessesinspinglasses[12].Accordingtothis “newview”ofproteinfolding,apolypeptidechainfoldsonaruggedfunnel-shaped energylandscapewheretheentropyisplottedonthex-axisandtheenthalpy onthey-axis.Aproteinreachesthelowestfreeenergypoint,itsfoldedstate,by tradingentropyforenthalpy.Inthismodel,proteinchainsarenotabletofold properlyeitherabovethefoldingtemperature(whereadoptingacompactfolded structureisentropicallyunfavorable)orbelowtheglass-transitiontemperature (wheretheproteindynamicsessentiallyfreezebeforereachingthefoldedstate). TheDavidBakergrouphasbeenleadingtheproteinstructurepredictionfieldfor manyyearsusingtheirRosettasimulationmethodthatextensivelysamplesthe combinatorialstructuralmanifoldmadeupofsmallstructuralfragments[13]. Afurtherimportantadvancewasthebrute-forcemoleculardynamicssimulations bytheD.E.Shawgroup,whowereabletosimulatetherepeatedfoldingand unfoldingofsmallglobularproteinsatthefoldingtemperature[14].Recently, thecompanyDeepMindsuccessfullyapplieddeep-learningmethodstotacklethe problemofproteinstructureprediction[15,16].Theytrainedaneuralnetworkto makeaccuratepredictionsofthedistancesbetweenpairsofresidues.Inthelatest CriticalAssessmentofproteinStructurePrediction(CASP),theirmethodtermed AlphaFold2createdhighlyaccuratestructurepredictionswithamedianbackbone accuracyof0.96Årootmeansquaredeviation(RMSD)andall-atomaccuracyof 1.5ÅRMSD.
Proteinsaresynthesizedbyribosomeseitherinthecytosol,closetothemembrane oftheendoplasmicreticulum,orclosetothebacterialplasmamembrane[17].It isbecomingmoreandmoreclearthatportionsofthenascentpeptidechainsmay alreadystartadoptingalpha-helicalconformationswhilepassingthroughtheribosomalexittunnel.Allproteinsofthesecretorypathwayandallmembraneproteins arepassedfromtheribosometotheSectranslocon,anintegralmembranechannel intheendoplasmicreticulum(ER)membrane.Thepeptidesequencesofmembrane proteinsareabletoexittheSeccomplexsidewaysintothemembraneviaaso-called lateralgate.Proteinstargetedforthesecretorypathwayneedtotranslocateintothe ER,andoftengetglycosylatedbyanearbyoligosaccharyltransferaseenzyme.
1.2ConformationalDynamics Thermalmotionofatomsimpliesthatproteinsarenotrigidobjects.Yet,theycanstill befairlystiffandhaveapurescaffoldingfunction.Examplesofthisaretheproteins ofviruscapsidsorthecytoskeleton.Mostproteins,however,undergosometypeof conformationaltransitioneitherduringtheircatalyticcycle,whentheybindand unbindligands,oriftheyarepartofasignalingcascade.
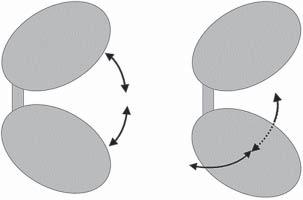
1.2.1Large-ScaleDomainMotions Proteinsconsistingofmultipledomainsorlobes(suchaskinases)canundergo large-scaleconformationaltransitionsbycharacteristicdomainmovements.Prototypesforthisarekinasesandlysozyme.Thefirstnormalmodetypicallydescribes aPacman-typeopening–closingtransitionofthetwodomainsrelativetoeach other,seeFigure1.8.Thesecondnormalmodewouldthenbeascissor-likemotion perpendiculartothefirstmode.Often,thesemovementsareconnectedtobiological functionsandfacilitateeitherligandbindingandunbindingorhelpincatalyzing theenzymaticreaction.Membranetransporters,suchastheleucinetransporter LeuT,undergoaconformationaltransitionbetweenaninward-facingconformation andanoutward-facingconformation,seeFigure1.9.
Figure1.8 Schematicillustrationofthefirst (lowestenergy)normalmodeofatwo-domain protein,suchasproteinkinases(left),andthe secondnormalmode(right).
Figure1.9 X-raystructuresofthebacterialleucinetransporterLeuTintheoutward-facing conformation(left,PDBcode3TT1)andintheinward-facingconformation(right,PDBcode 3TT3).ThefigureswereagaingeneratedwithProPores2(cf.Figure1.7).
Besidessuchlarge-scaledynamics,therestoftheproteinstructureisofcoursenot rigidbutundergoesconstantthermalmotionaswell.Sincethe1970s,time-resolved IRspectroscopywasusedtocharacterizethedynamicsoflaser-inducedCO dissociationfromtheinternalporphyrinringofmyoglobin[18].Theobserved multi-exponentialkineticsofthetimeneededforCOtorebindtotheporphyrin wasinterpretedtoreflecttheintrinsicdynamicsofthemyoglobinmatrix.Subsequently,Halleandcoworkersshowed,byNMR,thatwatermoleculesburiedinthe proteinbovinepancreatictrypsininhibitor(BPTI)exchangedwithbulksolventon timescalesofmilliseconds[19].Thisprovedthatevencompactglobularprotein structuresundergocontinuousconformationalbreathingtransitionsthatarelarge enoughtoallowthepassageofwatermoleculesinandoutofafoldedprotein.
1.2.2DynamicsofN-TerminalandC-TerminalTails N-terminusandC-terminusofaproteinchainaretypicallylocatedonitsproteinsurface,wheretheyoftenstretchoutintosolutionandhavesubstantial conformationalflexibility.Probably,thefunctionallymostimportantN-terminal tailsarethoseofhistoneproteins.Theyundergoposttranslationalmodificationsin manyways,andthisstronglyaffectstheirinteractionwithdouble-strandedDNA thatwindsaroundhistoneproteins.TheC-terminaltailsofproteinscanfunction, e.g.asrecognitionsitesforPDZadaptordomains.
1.2.3SurfaceDynamics
Aminoacidsidechainsonthesurfaceofproteinsoftenalsoshowconsiderableconformationaldynamics[20].Frequently,transientpocketsopenandcloseonproteinsurfacesonatimescaleoftensofpicoseconds.Thus,theproteinsurfacerather resemblesthesurfaceofasponge.Anothertypeoffunctionallyrelevantconformationalmotionsareloopmovementsontheproteinsurface,e.g.lipasespossessa looptermed“lid”thatcontrolsaccesstotheactivesitebeneath.Thesameisthe caseforHIVproteaseasmentionedbefore.Interestingly,ithasbeenarguedthat disease-associatedmutationsinproteinsoftenresultinflexibilitychangesevenat positionsdistalfrommutationalsites,particularlyinthemodulationofactive-site dynamics[21].
1.2.4DisorderedProteins X-raycrystallographyandCryo-EMareperfectstructuraltechniquestoresolve preciseconformationaldetailsofwell-orderedportionsofproteins.Obviously, N-terminus,C-terminus,andsurfaceloopsextendintothesolvent,andtheir conformationaldynamicsmaysometimesnotyieldpreciseelectrondensitythat canbedetectedagainstthebackground.Furthermore,itcameasasurprisewhen NMRexperimentsshowedinthemid1990sthatthereexistnumerous“disordered” proteinsthatdonotadoptawell-foldedconformationatall.Sometimes,they mayrefoldwhentheybindtootherproteins,orwhentheyundergoaphenotypic order-to-disordertransition,suchastheprionproteinthatismorefoldedinthe non-diseasestateandisthoughttobetheoriginofmadcowdisease.Allofus
containprionproteinsandweareusuallyjustfine.Accordingtothe“protein-only” hypothesis,thekeyeventinthepriondiseasepathogenesisoccurswhenthe cellularprionprotein(PrPC)undergoesaconformationaltransitionfroma mainly α-helix-richfoldedstructureintoaninfectiousandpathogenic β-sheet-rich conformer(PrPSc).PrPScpossessesabnormalphysiologicalproperties,suchas resistancetoproteolyticdegradation,relativeinsolubility,andthepropensityto polymerizeintoscrapieagents[22].
Monzonetal.distinguishedshortanddisorderedregions(between5and30 residueslong)thatareusuallyassociatedwithflexiblelinkersorloopsinfolded proteinsandso-calledlongdisorderregions(LDRs)thathaveatleast30consecutivedisorderedresidues.TheseLDRswerefoundtobeenrichedinchargedand hydrophilicaminoacidsanddepletedinhydrophobicones[23],suchasthelinker segmentsdiscussedbeforeinthecontextofproteindomains.Disorderedregions mayalsohaveimportantrolesinmediatingproteininteractions.Forexample, so-calledeukaryoticlinearmotifs(ELMs)arelocatedindisorderedregionsof proteinsandmediateinteractionsbetweenproteins[24].
1.3FromStructuretoFunction 1.3.1EvolutionaryConservation Oneimportantprincipleofevolutionarybiologyisthatfunctionallyimportant proteinregionstendtobeconservedbetweenrelatedorganismswhereasunimportantregionsaresubjecttoconsiderablevariation.Functionallyimportant regionsinclude,ofcourse,activesiteresidues.Mutationsofcatalyticresiduesmay renderenzymesnonfunctionalandare,therefore,rarelytolerated.Furthermore, conservationalsoextendstostructuralelements,suchasdisulfidebridgesand residuesinshortturns.
Ingeneral,structureisbetterconservedthansequence.Therefore,functionally relatedpairsofproteinsmaysometimesshowverylowsequencesimilarity,butfairly highstructuralsimilarity.Assumingthatbothproteinswerederivedfromadistant commonancestorprotein,itcameaboutthattheirstructureswereconservedduring evolution,buttheirsequenceswerenot,exceptforafewcrucialpositions.
1.3.2BindingInterfaces Manyproteinscarryouttheirfunctionbybindingtootherproteins,smallmolecules, membranes,ornucleicacids.Thisisactuallywhatallofthisbookisabout.Usually, thisinvolvesoneormorebindingpatchesonthesurfaceoftheproteins.Binding interfacesoftwoproteinshavesizesrangingfrom500to3000Å2 [25].Smallinterfacesarepreferredfortransientcontactsofsmallhydrophilicproteins,e.g.thoseof redoxproteinssuchastheelectroncarriercytochrome c.Incontrast,antibodiesbind totheirantigenswithratherlargeandhydrophobicinterfacesthatsupportpermanentoratleastlong-lastingcontacts.Also,permanentdimerstendtohaverather
1.4Summary 11 hydrophobicinterfaces.Howmuchoftheproteinsurfaceispartofaninterface dependsonthetotalsizeofthecomplex.Aninternalprotein,e.g.intheribosome mayevenbefullyshieldedfromsolventandallofitssurfacesareincontactwith otherbiomolecules.Protein–proteininteractionsandlargeproteincomplexesare discussedinChapters2–7.
DNAandRNAarestronglynegativelychargedduetotheirphosphatebackbones. Hence,proteinsneedtopossesscomplementary,positivelychargedsurfacepatches, tobeabletobindtoDNAorRNA.Suchpatchesaretypicallynotsuitableforbindingtootherproteins.However,therearecertainproteinsthatareabletomimic nucleotidepolymers.Oneexampleistheintracellularinhibitorproteinbarstarthat bindstotheRNAsebarnaseandpreventsitfromchewingupallmRNAandother RNAmoleculesinsidethecell.Thus,barnaseonlyactsextracellularly.Barstarhasa stronglynegativebindingpatchtomimicthenaturalsubstrateRNA.Chapters10–12 giveadeeperinsightintoproteininteractionswithnucleicacids.
Thetopologyandcompositionofbindinginterfaceswillbediscussedindetailin Chapter2.
1.3.3SurfaceLoops Surfaceloopsareused,forexamplebyantibodies,tobindtotheirantigensvia complementarity-determiningregions(CDRs).Asmentioned,surfaceloopscan alsoregulatetheaccesstotheactivesiteofproteins,andtheymaycontaincleavage sitesforrestrictionenzymes.Notethatcleavageisalmostasfrequentlyobserved in α-helicesasinregionswithoutsecondarystructure,suchasloops,butlessin β-strands[26].
1.3.4PosttranslationalModifications
Often,theactivityofproteinsisdeterminedbytheproperplacementofposttranslationalmodificationstosurfaceresidues.Forexample,about75%ofallhuman proteinsgetphosphorylated,oftenatmultiplepositions[27].Othermodifications areglycosylation,farnesylation(e.g.oftheRasprotein),etc.Ubiquitinationoften endsthelifeofproteinsbecausethismodificationtargetsthemfortransporttothe proteasomethatshredspeptidesequencesintosmallcomponents.Themodification sitesareusuallylocatedontheproteinsurfaceandthemodificationsareplacedby otherenzymes,againinvolvingproteininteractions.Posttranslationalmodifications areimportantmarkersforbindingpartnersandmayalsoaffectproteinconformation (seeChapter17forfurtherdiscussion).
1.4Summary Thecharacterizationofproteinstructurehasbecomefairlyroutinethesedays.For about70%ofallhumanproteins,thereexiststructuralmodelseitherfromexperimentaldeterminationorfromhomologymodeling[28].Infact,DeepMind,incooperationwithEuropeanBioinformaticsInstitute(EBI),recentlypublishedstructural
1ProteinStructureandConformationalDynamics
modelsproducedwithAlphaFoldforallhumanproteinsandproteinsofseveral othermodelorganisms[29].Somebelievethateventheproteinfoldingproblem hasbeen,atleastpartially,solved.Despitealltheaccumulatedknowledge,westill donotknowthefunctionofaconsiderablefractionofthehumanproteins,andit isveryhardtorationalizethefunctionaleffectsofposttranslationalmodifications ortoevenpredictthem.Wehavealimitedunderstandingofwhatdeterminesproteininteractions,andwearerarelyabletocorrectlypredictthestructuresofprotein assembliesfromscratch,withoutadditionalexperimentalevidence.
References 1 Tiessen,A.,Pérez-Rodríguez,P.,andDelaye-Arredondo,L.J.(2012).Mathematicalmodelingandcomparisonofproteinsizedistributionindifferentplant, animal,fungalandmicrobialspeciesrevealsanegativecorrelationbetween proteinsizeandproteinnumber,thusprovidinginsightintotheevolution ofproteomes. BMCRes.Notes 5:85.https://bmcresnotes.biomedcentral.com/ articles/10.1186/1756-0500-5-85.
2 Wheelan,S.J.etal.(2000).Domainsizedistributionscanpredictdomainboundaries. Bioinformatics 16:613–618.
3 Yu,L.,Tanwar,D.K.,Penha,E.D.S.etal.(ed.)(2019).Grammarofprotein domainarchitectures. Proc.Natl.Acad.Sci. 116:3636–3645.https://www.pnas .org/content/116/9/3636.
4 Hormoz,S.(2013).Aminoacidcompositionofproteinsreducesdeleterious impactofmutations. Sci.Rep. 3:2919.
5 Brüne,D.,Andrade-Navarro,M.A.,andMier,P.(2018).Proteome-widecomparisonbetweentheaminoacidcompositionofdomainsandlinkers. BMCRes.Notes 11:117.
6 Kumar,S.andBansal,M.(1998).Geometricalandsequencecharacteristicsof α-helicesinglobularproteins. Biophys.J. 75:1935–1944.
7 Penel,S.etal.(2003).Lengthpreferencesandperiodicityin β-strands.Antiparalleledge β-sheetsaremorelikelytofinishinnon-hydrogenbondedrings. Protein Eng.Des.Sel. 16:957–961.
8 Hollander,M.,Rasp,D.,Aziz,M.,andHelms,V.(2021).ProPores2:webservice andstand-alonetoolforidentifying,manipulatingandvisualizingporesin proteinstructures. J.Chem.Inf.Model. 61:1555–1559.
9 Tian,W.,Lin,M.,Tang,K.etal.(2018).High-resolutionstructurepredictionof β-barrelmembraneproteins. Proc.Natl.Acad.Sci. 115:1511–1516.
10 Reeb,J.,Kloppmann,E.,Bernhofer,M.,andRost,B.(2015).Evaluationoftransmembranehelixpredictionsin2014. Proteins 83(3):473–484.
11 Matouschek,A.,Kellis,J.T.Jr.,Serrano,L.,andFersht,A.R.(1989).Mappingthe transitionstateandpathwayofproteinfoldingbyproteinengineering. Nature 340:122–126.
12 Onuchic,J.N.andWolynes,P.G.(2004).Theoryofproteinfolding. Curr.Opin. Struct.Biol. 14:70–75.
13 Yang,J.,Anishchenko,I.,Park,H.etal.(2020).Improvedproteinstructure predictionusingpredictedinterresidueorientations. Proc.Natl.Acad.Sci. 117: 1496–1503.
14 Robustelli,P.,Piana,S.,andShaw,D.E.(2018).Developingamoleculardynamics forcefieldforbothfoldedanddisorderedproteinstates. Proc.Natl.Acad.Sci. 115:E4758–E4766.
15 Jumper,J.,Evans,R.,Pritzel,A.etal.(2021).Highlyaccurateproteinstructure predictionwithAlphaFold. Nature 596:583–589.
16 Senior,A.W.,Evans,R.,Jumper,J.etal.(2020).Improvedproteinstructure predictionusingpotentialsfromdeeplearning. Nature 577:706–710.
17 Bornemann,T.,Jöckel,J.,Rodnina,M.V.,andWintermeyer,W.(2008).Signalsequence–independentmembranetargetingofribosomescontainingshort nascentpeptideswithintheexittunnel. Nat.Struct.Mol.Biol. 15:494–499.
18 Austin,R.H.,Beeson,K.W.,Eisenstein,L.etal.(1975).Dynamicsofligandbindingtomyoglobin. Biochemistry 14:5355–5373.
19 Denisov,V.P.,Peters,J.,Hörlein,H.D.,andHalle,B.(1996).Usingburied watermoleculestoexploretheenergylandscapeofproteins. Nat.Struct.Biol. 3:505–509.
20 Helms,V.(2007).Proteindynamicstightlyconnectedtothedynamicsofsurroundingandinternalwatermolecules. ChemPhysChem 8:23–33.
21 Campitelli,P.,Modi,T.,Kumar,S.,andOzkan,S.B.(2020).Theroleofconformationaldynamicsandallosteryinmodulatingproteinevolution. Annu.Rev. Biophys. 49:267–288.
22 Baral,P.K.,Yin,J.,Aguzzi,A.,andJames,M.N.G.(2019).Transitionoftheprion proteinfromastructuredcellularform(PrPC)totheinfectiousscrapieagent (PrPSc). ProteinSci. 28:2055–2063.
23 Monzon,A.M.,Necci,M.,Quaglia,F.etal.(2020).Experimentallydetermined longintrinsicallydisorderedproteinregionsarenowabundantintheprotein databank. Int.J.Mol.Sci. 21:4496.
24 Tompa,P.,Davey,N.E.,Gibson,T.J.,andBabu,M.M.(2014).Amillionpeptide motifsforthemolecularbiologist. Mol.Cell 55:161–169.
25 Janin,J.,Bahadur,R.P.,andChakrabarti,P.(2008).Protein–proteininteraction andquaternarystructure. Q.Rev.Biophys. 41:133–180.
26 Timmer,J.C.,Zhu,W.,Pop,C.etal.(2009).Structuralandkineticdeterminants ofproteasesubstrates. Nat.Struct.Mol.Biol. 16:1101–1108.
27 Sharma,K.,D’Souza,R.C.J.,Tyanova,S.etal.(2014).UltradeephumanphosphoproteomerevealsadistinctregulatorynatureofTyrandSer/Thr-based signaling. CellRep. 8:1583–1594.
28 Somody,J.C.,MacKinnon,S.S.,andWindemuth,A.(2017).Structuralcoverage oftheproteomeforpharmaceuticalapplications. DrugDiscoveryToday 22: 1792–1799.
29 Varadi,M.,Anyango,S.,Deshpande,M.etal.(2022).AlphaFoldproteinstructuredatabase:massivelyexpandingthestructuralcoverageofprotein-sequence spacewithhigh-accuracymodels. NucleicAcidsRes. 50:D439–D444.
Protein–Protein-BindingInterfaces ZeynepAbali 1 ,DamlaOvek 2 ,SimgeSenyuz 1 ,OzlemKeskin 3 ,and AttilaGursoy 2
1 KocUniversity,ComputationalScienceandEngineeringProgram,Istanbul,34450,Turkey
2 KocUniversity,ComputerEngineering,Istanbul,34450,Turkey
3 KocUniversity,ChemicalandBiologicalEngineering,Istanbul,34450,Turkey
2.1DefinitionandPropertiesofProtein–Protein Interfaces Thesurfaceregionswhereproteinsinteractwithothermoleculesarecalled protein-bindingsites.Iftheinteractionoccursbetweentwoproteins,then interactingbindingsitesforma protein–proteininterface.Interfacesinvolve aminoacidsfromeachsideformingmainlynon-covalentbonds.Interfacesmight alsocontaincovalentbonds,suchasdisulfidebridges,butwithlowerfrequency.
Thephysicalproximityofresiduesfromtwoproteinchainsdeterminestheinterfaceresiduesineachprotein.Interfacescanbedescribedusingavarietyofcomputationalmethods[1].Thesemethodsusestructuresofprotein–proteincomplexesand variousmetrics,suchasdistancebetweentheatomsbelongingtodifferentsubunits (proteinchain),oraccessiblesurfacearea(ASA).Interfaceresiduesdonotneedto becontinuousinsequencebutshouldbeclosetoeachotherin3Dspace.Here,we presentsomeofthecommonlyusedmethods.Adistance-basedapproachisoneof them.Residuesofaninterfacecanbedefinedbythedistancebetweentheiratoms. Athresholddistanceisdefined,usuallyrangingbetween4and6Å.Iftworesiduesof opposingchainshaveheavyatoms(non-hydrogen)withinthedefinedthresholddistance,thentheseresiduesarecategorizedas interfaceresidues [2,3].Someother studiesconsideronlythedistancesbetweenCα atomstoidentifyinterfaceresidues. WhenCα atomsareused,thethresholddistanceisusuallygreaterthantheonesused withheavy-atomapproaches,rangingfrom8to12Å[4–6].Anotherdistance-based methoddefinesthedistancebetweentwoatomsusingthevanderWaals(VDW) radiioftheindividualatoms.Tworesiduesaredefinedasinterfaceresiduesifthey haveatomswithinadistancethatissmallerthanthesumoftheirVDWradiiplusa thresholddistance(usually0.5Å)[7,8].
Distance-basedmethodsarenottheonlyonesforidentifyinginterfaceregions inproteincomplexes.Alternatively,ASAorrelativeaccessiblesurfacearea(rASA) ProteinInteractions:TheMolecularBasisofInteractomics,FirstEdition. EditedbyVolkhardHelmsandOlgaV.Kalinina. ©2023WILEY-VCHGmbH.Published2023byWILEY-VCHGmbH.
2Protein–Protein-BindingInterfaces
ofindividualresiduescanbeusedtofindinterfaceresidues.ASAistheareaofa moleculethatisaccessibletoasolvent.InASAcalculations,usually,aspherewith theradiusofawatermolecule(1.4Å)isrolledaroundtheproteintoprobeitssurface. ThereareseveralavailabletoolsforcalculatingtheASAofresiduesinaprotein,such asNACCESS([9]),POPScomp[10],orFreeSASA[11].rASAiscalculatedbytaking theratiooftwostatesofaresidue:(i)whenitisinthemostsolvent-exposedstate (inAla-X-AlaorGly-X-GlytripeptidewhereXistheresidueofinterest)and(ii)when itisinthefoldedconformationoftheprotein.
InterfaceregionsoncomplexescanbeidentifiedbyconsideringthechangeinASA (ΔASA).TheresidueASAsarecalculatedwhentheproteinisinitsmonomericform andincomplexform.IfthedifferencebetweenmonomericASAandthecomplex ASAislargerthanathreshold,thentheresidueisidentifiedasaninterfaceresidue. Athresholdvalueof1Å2 isgenerallyused[12].SPPIDER[13]isoneoftheavailabletoolsthatusesrASAvaluesforidentificationofinterfaceresidues.Itusesa4% thresholdofrASAchangebetweenthemonomerandthecomplexand ΔASA > 5Å2 Anotherstudyusesathresholdof25%forrASAand ΔASA > 0Å2 todefineinterface residues[14].
Thereareothermethodstodefineinterfacesthatarenotascommonasthementionedones.Forexample,Voronoidiagramsareusedasageometricapproachfor identifyinginterfacesandspecifyingtheboundariesofagiveninterface[15].There arealsosomestudiesthatembracegraph-basedapproachestodefineinterface regions[16].
Methodsfordefiningprotein–proteininterfacescanbeusedontheirownasa singlemethod,orasacombinationofmultiplemethods.Forexample,Hadarovich etal.definedinterfaceresiduesbya12Åatom–atomdistancecutoffbetweenthe interactingmonomersandtheneliminatedsmallinterfacesthathaveburiedsurface area <200Å2 perchain[4].Sincedistance-basedcalculationsarecompute-intensive, Cukurogluetal.definedinterfaceregionsfirstusing ΔASA > 1Å2 andthenbydistancecriteria.Theydefinedinterfaceresiduesas contacting (Figure2.1a)ifthe distancebetweenanytwoatomsofthetworesiduesfromdifferentchainsisless thanthesumoftheircorrespondingVDWradiiplusathresholdof0.5Å[17].
Amorecontinuousinterfacestructureisusuallypreferable.Besidestheinterface residuesthatareincontact,thenearby(neighbor)residuescanalsobeincludedin theinterfaceregionstomakeitmorecontinuousandtopreservethesecondarystructures[7,17,18].Afteridentifyingcontactingresidues,nearbyresiduesaredefined basedonthecontactingresidues.IfaresiduehasaCα atomatmost6Åawayfrom theCα ofacontactingresidue,thenitisdefinedasanearbyresidue(Figure2.1b). Nearbyresiduesprovideasupportingscaffoldforcontactingresiduesininterface regions[7].
Interfaceregionscanbedividedinto core and rimareas similartoregionsin proteinglobularstructures.Interfacecoresaresimilartoproteincores,andinterface rimsaresimilartoproteinsurfaces.Coreresiduescontributemoretothebinding affinityandspecificity[14,19–21].Coreandrimregionsaredefinedbythechange ofASAofresiduesuponcomplexformation.Ifasurfaceresiduebecomessolvent inaccessibleaftercomplexformation,itispartoftheinterfacecore;ontheother