











This project focused on predicting the pathogenicity of genetic mutations associated with breast cancer susceptibility genes (BRCA1, BRCA2, PALB2, and CHEK2). Leveraging bioinformatics databases and advanced machine learning algorithms, we developed a high-precision variant analysis pipeline integrating conservation scores, amino acid substitution matrices, and functional impact predictions.

• Genomic variant data collected from gnomAD for BRCA1 (2630), BRCA2 (5077), PALB2 (1783), and CHEK2 (1012).


• Protein sequences from Ensembl, conservation scores from UCSC PhyloP.
• Filtered variants to retain only high-confidence coding mutations (e.g., excluding intronic and conflicting variants).
• HGVS-format mutation data parsed to extract:
⚬ Reference Amino Acid (Ref_aa)
⚬ Alternate Amino Acid (Alt_aa)
⚬ Mutation Position
• Frameshift and stop codon analysis performed for functional context.

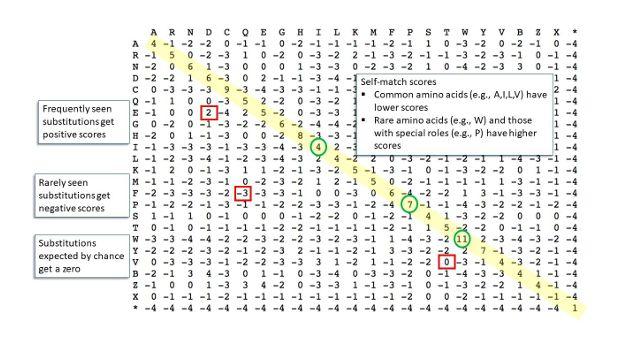
1: BLOSUM62 matrix showing amino acid substitution scores.
Used BLOSUM62 matrix to quantify evolutionary distance between wild-type and mutant amino acids.
Conservation Analysis with PhyloP
Integrated PhyloP conservation scores to determine evolutionary pressure at nucleotide positions.
5

Functional Impact Prediction
Used SIFT and PolyPhen2 (via Ensembl VEP API)
to evaluate whether the amino acid changes were likely benign or damaging.
Machine Learning Model Ensemble
• Models used:
⚬ Random Forest
⚬ Support Vector Machine (SVM)
⚬ K-Nearest Neighbors (KNN)
⚬ Logistic Regression
⚬ XGBoost
⚬ LightGBM
• Input: Feature set combining substitution scores, conservation scores, and functional impact.
• Output: Pathogenicity classification.
• Train-Test Split: 80:20

High accuracy and precision in variant classification.
Precision: 97% across benign and pathogenic classes.
Strong F1-score for binary classes; intermediate variants were less separable due to data imbalance.
The ensemble learning approach outperformed individual algorithms in prediction robustness and stability.

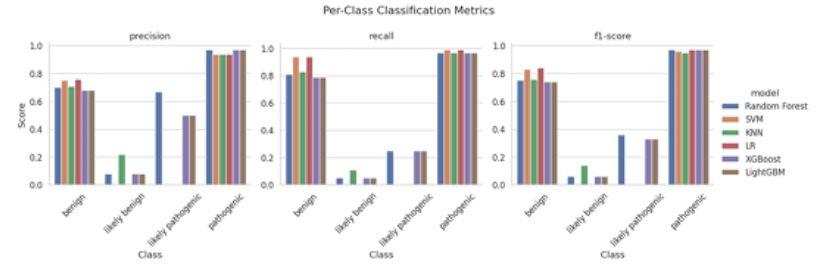
Figure 2 - Per-class classification metrics—precision, recall, and F1-score—across four variant categories (benign, likely benign, likely pathogenic, and pathogenic) for six machine learning models (RF, SVM, KNN, LR, XGBoost, and LightGBM). The bar charts highlight each model’s performance in correctly classifying variants within each pathogenicity class.
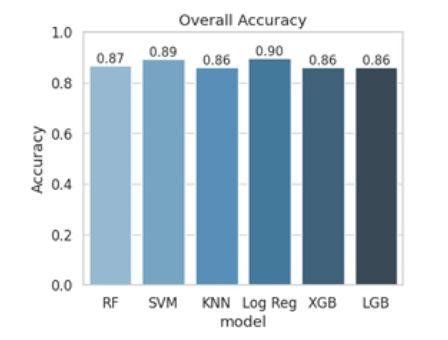
Figure 3 - Overall accuracy of six machine learning models Random Forest (RF), Support Vector Machine (SVM), K-Nearest Neighbors (KNN), Logistic Regression, XGBoost (XGB), and LightGBM (LGB)-in classifying variant pathogenicity. Logistic Regression achieved the highest accuracy (0.90), while all models exhibited performance above 0.85.
Figure 4 - Receiver operating characteristic (ROC) curves for the machine learning classifiers used in this study to predict mutation pathogenicity.
Shown are the ROC curves for the KNN, Logistic Regression, Naive Bayes, SVM, Random Forest, XGBoost, and LightGBM models. These

curves illustrate the trade-off between sensitivity and specificity across various decision thresholds. Higher AUC values, as observed for the SVM, LR, and RF models, indicate superior discriminative performance in clearly distinguishing benign from pathogenic variants, while the relatively lower AUCs for KNN and NB highlight challenges in classifying intermediate cases.
This project successfully delivered a state-of-the-art integrative framework combining evolutionary, structural, and functional bioinformatics features with machine learning algorithms for robust mutation classification. The approach offers a powerful strategy for early risk assessment in breast cancer and can be adapted to other genetic diseases.

We demonstrated:
• An advanced, reproducible bioinformatics pipeline.
• Superior classification power using ensemble learning.

• Scientific validation through conservation, substitution, and functional metrics
