
ENFERMEDADES NEUROLÓGICAS Y CONDUCTA
Dr. Joaquin Ortega Escobar
Profesor Titular Jubilado de la UniversidadAutónoma de Madrid
LECCION 5
Enfermedad deAlzheimer (EA)
• Historia:AloisAlzheimer yAuguste Deter.
• Anatomía patológica deAuguste D.
• Criterios diagnósticos de la EA.
• Epidemiología: Prevalencia, Incidencia, Mortalidad y Factores de Riesgo.
• Genética: PPA,Amiloide, Tau, Presenilina,ApoE.
• Prevención y Gestión
• Regiones cerebrales y EA. Etapas.
• Aproximación farmacológica
El 25 de noviembre de 1901, se admitió en elAsylum del Estado de Frankfurt a una mujer de 51 años que fue examinada por un prometedor psiquiatra,AloisAlzheimer.

Retrato deAloisAlzheimer (izquierda) y su pacienteAguste D (derecha).
Entre otros síntomas,Auguste Deter (oAuguste D.) mostraba los siguientes:
-Memoria gravemente deteriorada, -Afasia, -Comportamiento errático (impredecible), -Paranoia, -Alucinaciones auditivas.
Auguste D. murió el 8 de abril de 1906.
Una pequeña muestra de las alteraciones cognitivas deAuguste D es el siguiente interrogatorio deAlzheimer a la paciente:
“Si compras 6 huevos, a 70 centavos cada uno, ¿cuánto es? De modo diferente.
¿En qué calle vives? Puedo decirtelo, debo esperar un poco.
¿Qué te pregunté? Bueno, esto es Frankfurt am Main.
¿En qué calle vives? Calle Waldemar, no, no…
¿Cúando te casaste? Ahora no lo sé. La mujer vive en el mismo piso
¿Qué mujer? La mujer donde estamos viviendo. La paciente llama Mrs. G, Mrs.
G, aquí un paso más profundo, ella vive…
Le muestro una llave, un lapiz y un libro y los nombra correctamente. ¿Qué te mostré? No lo sé, no lo sé.
¿Es dificil, verdad? Tan ansiosa, tan ansiosa.
Le muestro 3 dedos; ¿cuántos dedos? 3.
¿Sigues ansiosa? Sí.
¿Cuántos dedos te mostré? Bien, esto es Frankfurt am Main.”
Sección de la corteza cerebral con numerosas placas amiloides y ovillos neurofibrilares (izqda) y ovillos neurofibrilares (dcha).

Tomado de M. B. Graeber (1998). Neurogenetics, 1, 223-228.

Tomado de A.Alzheimer (1911). Zeitschrift für die Gesamte Neurologie und Psychiatrie, 4, 356-385.
Macroscopicamente, existen diferencias entre un individuo sin demencia y un paciente con EA.
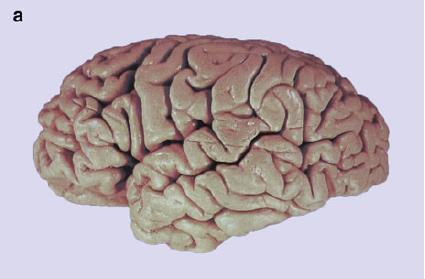

Obsérvese la atrofia severa del lóbulo temporal del paciente.
Diferentes
formas de placas amiloides.
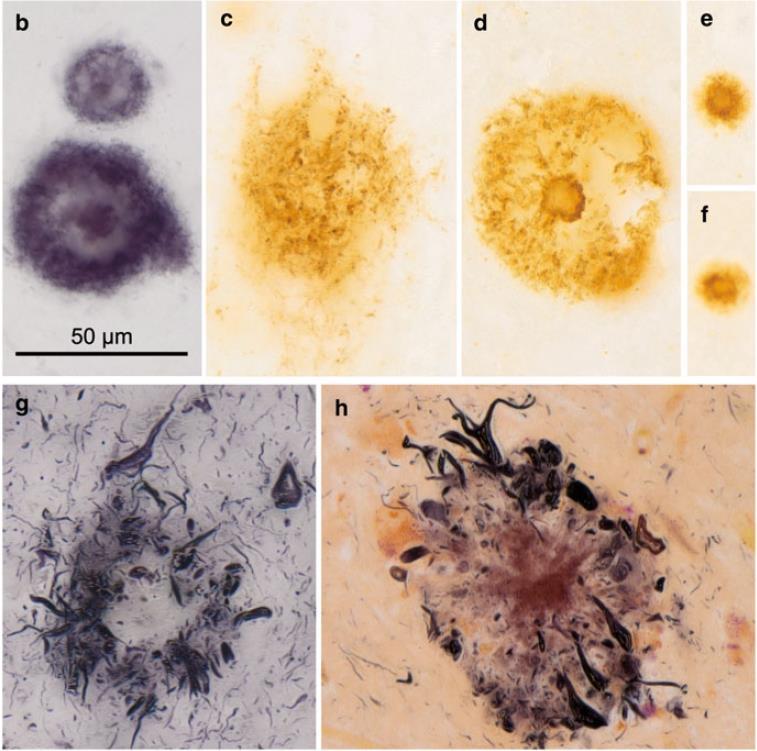
Diferentes formas de ovillos neurofibrilares (también se observan hilos neurofibrilares).

Criterios diagnósticos:
El síntoma inicial más frecuente notado por el paciente, y más particularmente por su pareja, son quejas de memoria.
No todos los aspectos de la memoria están afectados igualmente en la EA temprana. En particular, está afectada la memoria episódica anterógrada con pobre codificación y rápido olvido del material nuevo, por ejemplo una historia o una lista de palabras. También hay problemas con la memoria autobiográfica y semántica.
La demencia de Alzheimer en toda regla es precedida por una fase larga de declinar cognitivo lentamente progresiva que condujo al desarrollo del concepto de deterioro cognitivo leve como un estado amnésico con una alta tasa de progresión a la EA propiamente dicha. Hoy en día, la línea divisoria entre ambos se ha vuelto extremadamente borrosa.
Además de los problemas de memoria, en los enfermos de Alzheimer existen déficits en las habilidades visuoespaciales, en la velocidad mental, función ejecutiva y en la atención.
• La diagnosis ha pasado de una puramente patológica de los días de Alois Alzheimer, a una aproximación clínica excluyente en 1984*.
• Inicialmente, la diagnosis de la EA se restringió a la etapa de demencia (alteración cognitiva progresiva e importante que afecta a varios dominios); una persona con demencia no es capaz de vivir completamente independiente y en esto se diferencia del deterioro cognitivo leve. Posteriormente, con el desarrollo de los biomarcadores, la diagnosis de la EA se define por la presencia de ß-amiloide y de tau fosforilada.
El proceso diagnóstico comienza con la determinación de la presencia y severidad de la alteración cognitiva:
• Información de los familiares,
• Examen del estado mental por un clínico cualificado,
• Consideración de etiologías alternativas como enfermedad de los cuerpos de Lewy o demencia fronto-temporal,
• Confirmación mediante ligandos para beta-amiloide (florbetapir, florbetaben, flutemetamol y el componente Pittsburgh B [PiB]) y tau (18F-flortaucipir, p-tau181*), y la utilización de glucosa (hipometabolismo) que precede a la perdida de volumen cerebral.
• Biomarcadores basados en la sangre: ßA42/ßA40.
Los tres mejores biomarcadores de neuroimagen para la EA son: la atrofia del LTM en RMNe, el hipometabolismo en la CCP y temporoparietal en la TEP-18FDG y los depósitos de ß-Aen la TEP-amiloide.
¿Por qué son importantes los biomarcadores?
• Los estudios longitudinales mediante TEP-ßAdemuestran una muy baja acumulación de ßA, con un retraso temporal de 10-20 años entre el inicio de la acumulación y deterioro cognitivo sintomático y una desaceleración en dicho depósito a partir del deterioro cognitivo.
• Acumulación de ßAy edad: ~ 20% de personas a los 65 años y ~ 60% a los 85 años.
• ¿Por qué la elevada concentración de ßAno conduce a consecuencias clínicas? Tiene que suceder algo más: expansión de la tauopatía fuera del lóbulo temporal medio (Knopman et al., 2021; Long y Holtzman, 2019).

Comienzo y severidad de los síntomas clínicos pueden ser graduados mediante la escala “Clasificación
Clínica de la Demencia (CDR)”. 0=Sano; 0.5 cuestionable; 1 leve; 2 moderada; 3 grave
Los principales dominios cognitivos que son afectados en la EA son la memoria, el lenguaje, la función visuoespacial y la función ejecutiva. Las características amnésicas son más comunes con la edad de comienzo más tardía (>70 años), mientras que las no amnésicas son más comunes en personas más jóvenes.
Síntomas neuropsiquiátricos que co-ocurren con los déficits cognitivos: depresión, ansiedad, y aislamiento social en la demencia leve y, en etapas más avanzadas, delirios, alucinaciones, descontrol emocional o conductas físicamente agresivas.
Epidemiología:
• En 2018, Alzheimer’s Disease International estimó una prevalencia* de la demencia de aproximadamente 50 millones de personas en todo el mundo.
• La proyección al 2050 sería a 150 millones, con dos tercios viviendo en países de ingresos bajos o medios.
• La incidencia** de la EA está declinando en los países con ingresos económicos elevados (prevención de factores de riesgo cardiovasculares y/o mejora del nivel educativo).

Factores de riesgo:
• Edad avanzada ( >65 años, aunque esto no es una definición fija)
• Tener al menos un alelo APOE ε4.
• Ser mujer, especialmente después de los 80.
• Los factores de riesgo vascular no incrementan el riesgo de la patología de la EA, tal como se mide mediante marcadores del fluido cerebro-espinal o la TEP.
Genética:
• El riesgo de la EA depende en un 60-80% de factores heredables.
• El alelo APOE ε4 (ver más adelante) explica una parte sustancial de la heredabilidad pero no toda ella.
• Diversos estudios han incrementado el número de alelos de riesgo asociados a la EAa más de 40.
• Variantes genéticas protectoras: APOE ε2, mutación protectora Islandesa Ala673Thr, mutación Pro 522Arg en el gen PLCG2, mutación Christchurch en el alelo APOE ε3, mutación PSEN1E280A .
• Gen PSEN1: La proteína PSEN1es la subunidad catalítica de la γ-secretasa. Las mutaciones en las presenilinas desestabilizan las interacciones γ-secretasa-APP y hacen que se liberen prematuramente péptidos de ß-A más largos y propensos a la acumulación.
Evita la formación de ß-amiloide
Favorece la formación de ß-amiloide
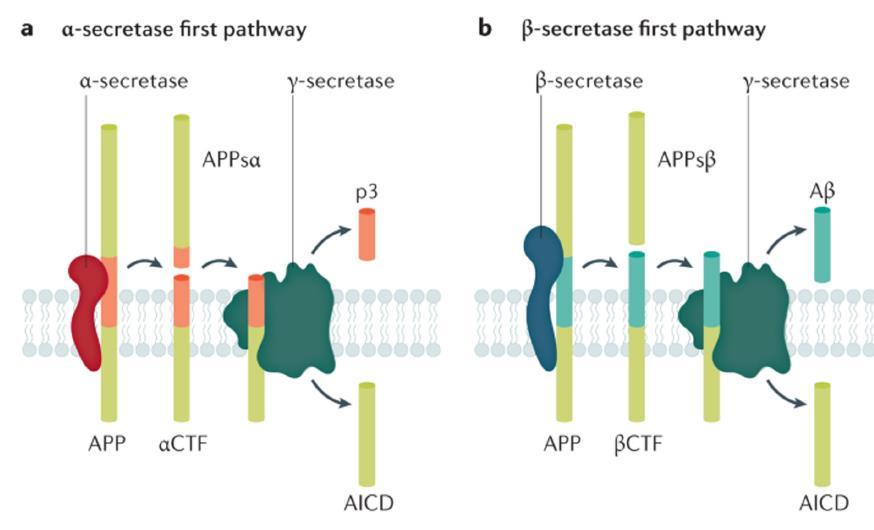
¿Cómo se forman las placas amiloides?
A partir de una proteína, la PPA, que es abundante en las sinapsis, tras la ruptura por ß- y γ-secretasas (vía amiloidogénica). Después de la producción, la ß-A es secretada al espacio extracelular como un monómero.
Evita la formación de ß-amiloide
Favorece la formación de ß-amiloide
La ß-amiloide:
• Interactúa con diversos receptores,
• Causa daños patológicos en espinas dendríticas y sinapsis.
Proteína tau (MAPT)
• Está normalmente presente en el axoplasma y en las zonas pre- y post-sinápticas.
• Su principal función es la estabilización de los microtúbulos del citoplasma.
• Tau puede ser propensa a modificaciones post-translacionales y agregarse. Cuando sucede esto, se acumula en una forma hiperfosforilada en el cuerpo neuronal y las dendritas (ovillos neurofibrilares e hilos de neuropilo).
• La tauopatía en el lóbulo temporal medial es el lugar inicial relevante para la cognición.
Apolipoproteina E (ApoE; gen ApoE)
• Producida predominantemente por astrocitos y microglia activada.
• ApoE se encuentra en las placas amiloides y en los ovillos neurofibrilares.
• Existe un alelo, el ApoE ε4, que va a afectar al comienzo de los síntomas clínicos de una manera dependiente de la dosis. Aproximadamente el 10% de los portadores de ApoE ε4 han incrementado la ß-A hacia los 57 años, mientras que los no portadores la incrementan aproximadamente 7 años más tarde.
Prevención:
• Ejercicio,
• Cambios en el estilo de vida,
• Estimulación cognitiva Gestión:
• Debe comenzar desde el momento de la interacción del clínico con el paciente y sus familiares,
• Compasión, paciencia y falta de superioridad,
• Ayuda a los cuidadores.




