Dissolution Testing to Support Clinical Bridging Studies Vivek S.Purohit





Dissolution and clinical performance.
Use of dissolution and clinical studies for clinical bridging using the BCS framework.
Clinical studies to support dissolution method development.
Design elements of biopharmaceutic clinical studies.
Jack Cook, Clinical Pharmacology, Global Product Development, Pfizer
Kyle Matschke, Biostatistics, Global Product Development, Pfizer
Dissolution research dates back to the 1890’s as branch of physical chemistry (Noyes-Whitney Equation, 1897)
• The realization that dissolution is important for bioavailability also dates back 1890’s.
• "... the composition of all compressed tablets should be such that they will readily undergo disintegration and solution in the stomach." C. Caspari, "A Treatise on Pharmacy", 1895, Lea Bros., Philad., 344.
• Serious attention to the relationship between dissolution and bioavailability only in 1950’s.
and Macheras,
of Pharmaceutics, 321(2006), 1
we using
we use
we have a framework to use
design
and set
we using our clinical studies to generate product understanding?
Can we use the clinical data more effectively to inform the development of dissolution test and product quality characteristics?

Each
Figure adapted from: Modelbased Drug Development, Clinical Pharmacology and Therapeutics, Vol 82(1), 2007
• Relative BA study with acceptance criteria specified based on the tolerance of deviation from target exposure considering safety and efficacy.
• Example Criteria Point estimate of the geometric mean ratio is contained within 80% 125%
• In vitro dissolution based bridging.
• Applicable to drugs with high solubility formulated as solution or very rapidly dissolving solid dosage forms.
• Approach is consistent with biowaiver considerations.
• May be applicable for drugs with low solubility conditional on comparable multimedia dissolution properties and inert excipients.
• Requires that the formulation excipients do not alter physiologic conditions affecting drug absorption.
• Population PK based bridging.
• Physiologic based pharmacokinetic (PBPK) modeling.
• Applicable to drug products where an adequately qualified PBPK biopharmaceutic model exists.
• Approach requires substantial amount of prior data necessary to establish and qualify a PBPK model. Will require prior planning.
Dissolution and/or PBPK based bridging
• Clinical studies to bridge formulations is a default option.
• Clinical bridging studies are typically run as estimation or relative BA studies during early development stages.
• E.g., solid dosage form Vs solution, relative BA of variant formulations etc.
• Clinical bridging studies that require confirmatory evidence of comparability and switchability are run as BE studies.
• E.g., clinical Vs commercial product, new formulation Vs commercial product.
• BA Vs BE Studies for bridging
• Any situation where the question takes the form of “What is the relative bioavailability of …..” would typically require BA study.
• BE studies are and should be reserved for situations requiring confirmatory evidence.
• BCS framework provides a useful risk-based bridging option*.
• BCS 1 drugs with very rapidly (≥85% for the mean percent dissolved in ≤ 15 minutes) or rapidly (≥85% for the mean percent dissolved in ≤ 30 minutes) dissolving drug products or similar in vitro dissolution characteristics (i.e., based on f2 comparison), under all of the defined conditions.
• BCS 3 drugs with very rapidly (≥85% for the mean percent dissolved in ≤ 15 minutes).
• In theory, for BCS 1 and 3 drug with drug products that meet above criteria, one can envision an entire program without a biopharmaceutic study.
• Population PK analysis from the patient studies can help check for bioavailability between different formulations.
• However, it is recommended that in initial stages, targeted biopharmaceutic studies should be planned to enable biopharmaceutic knowledge generation.
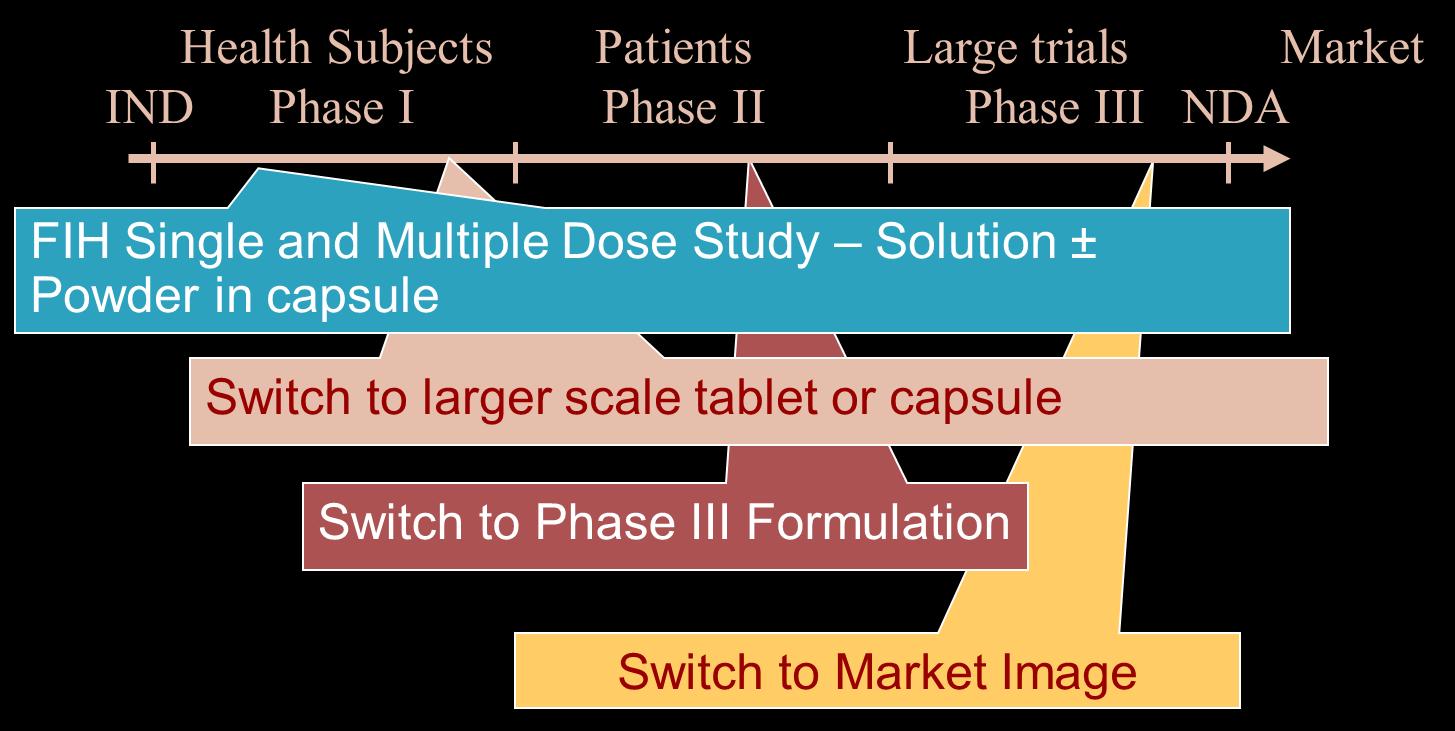
Clinical Bridging Studies and Dissolution Dissolution and/or PBPK based bridging• Can be implemented if there is confidence in the performance of the product.
• A Population PK model developed using the First in human data (FIH, single and multiple dose PK characterized).
• If FIH studes assess multiple prototype formulations, POPPK can assess relative BA.
• Each change of formulation would involve:
• Robust dissolution testing to assess comparability between the new and old drug product.
• Confirm exposures and relative bioavailability with POPPK.
• A clinical bridging study (BE) is likely to be required if the commercial product is different from clinical product used in pivotal trial.
• Except when eligible for biowaiver.
• Assume a level of risk with each introduction of an untested formulation.
• Risk higher for pivotal safety efficacy trials.
• Biopharmaceutic knowledge acquisition is limited.
• Does not allow testing of variant formulations.
• Scope of the dissolution test may be limited by the dissolution space tested.
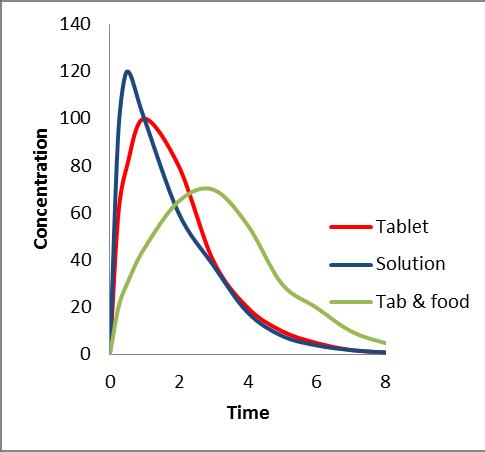
Dissolution and/or PBPK based bridging Clinical bridge• FIH/MD Study – PK profile from solution (Tmax < prototype tablet) and food effect (AUC() and Cmax()).
• BA study – Evaluated impact of relevant quality parameter (particle size, disintegration time etc.).
• AUC () and Cmax () at particle sizes greater 20 micron.
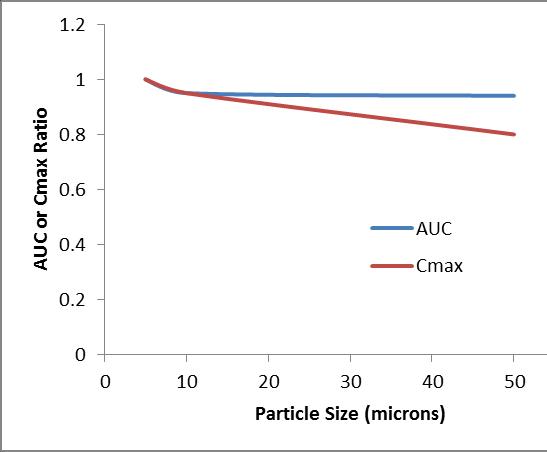
• If Cmax not critical for efficacy different interpretation
• All formulations found to be equivalent in a BA study with the Ph2 formulation.
• Useful for setting dissolution specification
• Final commercial product may be bridged using dissolution.
• If formulation excipients very different or a different dosage form may need a BE study.
• Subsequent to each study the dissolution test should be optimized to reflect the in vivo performance.
• Development of a PBPK model using incremental data from the BA studies is also encouraged.
• BCS 2 and 4 drugs will fall in this category
• Critical product attributes: DS Particle size, binder/granulation time, lubricant levels, disintegration time, coating thickness etc., can significantly affect product performance.
• Product design and dissolution test needs to evolve iteratively based on clinical data.
• Hence a more relevant dissolution method should be identified early.
• Optimized during the course of development.
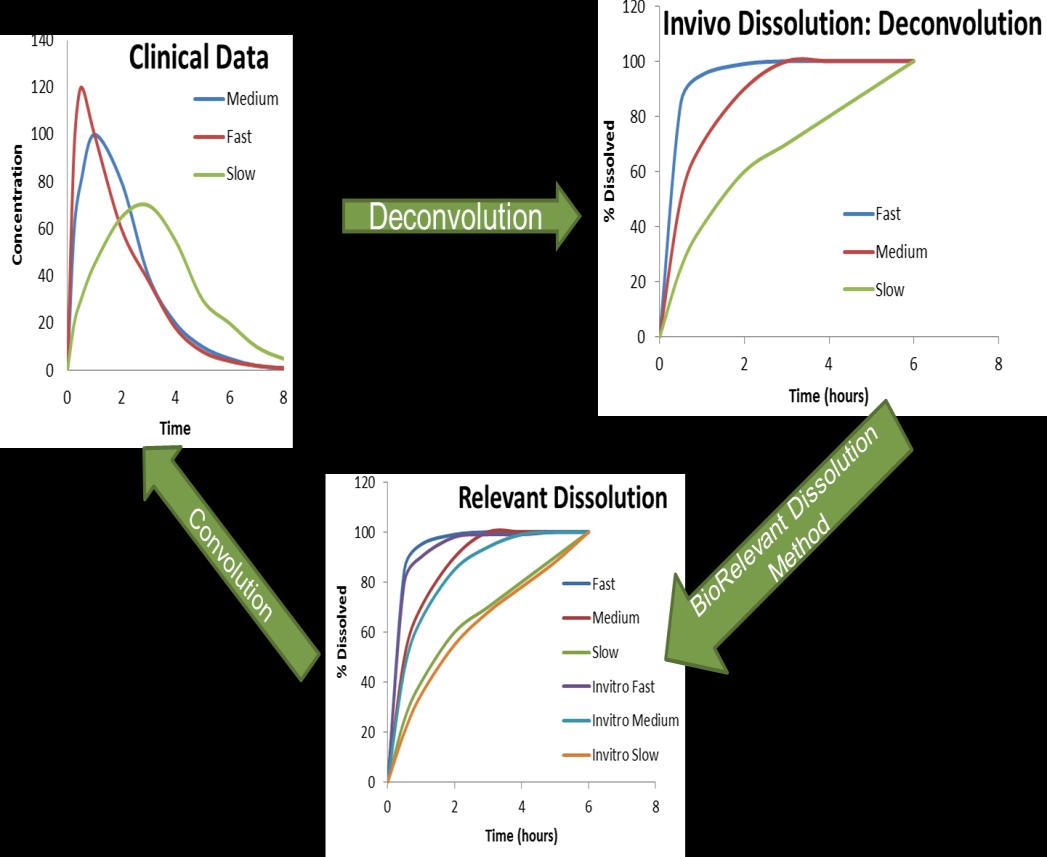
• Use of quantitative approaches (deconvolution convolution, modeling and simulation using Gastroplus or Simcyp) should be used for identifying a more biorelevant dissolution media.
• An evolving PBPK model development approach should be considered
• The PBPK model if adequately validated can be considered for justifying specifications and seek biowaivers.
• Ultimate goal could be to develop an IVIVC.
• Biorelevant dissolution = predictive of clinical performance.
• For BCS 1 and 3 drugs, multimedia dissolution represents a suite or battery of biorelevant tests which assess risk of failure.
• When using multimedia dissolution tests each condition is relevant.
• Multimedia dissolution tests taken together are predictive of low risk.
• For BCS 2 and 4 drugs developing a biorelevant dissolution test is a bit more involved.
• Establishing correlation between the dissolution testing conditions and result with clinical PK profiles is the key.
“BA for a given formulation provides an ESTIMATE of the relative fraction of the orally administered dose that is absorbed into the systemic circulation. BA for orally administered drug products can be documented by comparing a systemic exposure profile to that of a suitable reference product.”
relative
of
of
• Based on the estimated variability of the drug, precision is calculated for the relevant PK parameters (AUC and Cmax).
• Lower and Upper bounds of the 90% confidence intervals are considered for relevant geometric mean ratios to help illustrate the anticipated precision and operating characteristics one can expect for the given study design and sample size.
• Variability is based on the central tendency from past studies under similar conditions (ie. if study is given under fasted conditions, past fed studies should be excluded in the estimate).
• Sample size is based on the variability, geometric mean ratio (GMR) and desired power of the PK parameters being tested.
• GMR is assumed to be 1.05 (or 0.95) unless there is strong justification to use a higher value.
• The FDA requires ≥80% power.
• Variability is based on the central tendency from past studies under similar conditions.
• Criteria applied for declaring comparability and switchability:
• 90% confidence interval (CI) of geometric mean ratio (GMR) of test Vs reference for AUC and Cmax is contained with 80-125%.
• Typically employ variant formulations where a difference between the test and reference formulation can be expected.
• Hence, without an estimate of the magnitude of difference these studies are best designed as estimation BA studies.
• Can they be designed as confirmatory BE studies?
• Sure, under an assumption of no difference
• However, these studies will be large, expensive and the probability of success would be low especially when we employ variant formulations.
• Regulatory expectations are not clear or explicitly defined and not globally harmonized.
• A hybrid approach using BA studies with PBPK models or IVIVC models (when available) should be considered.
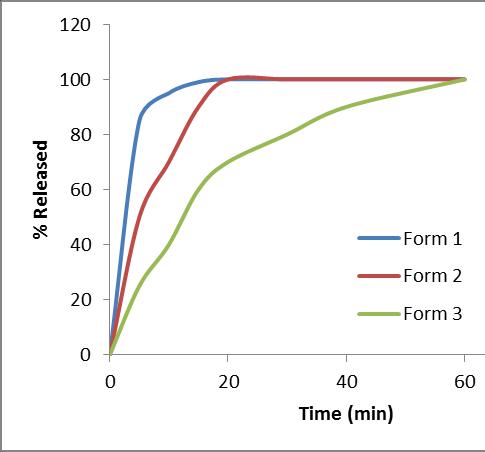
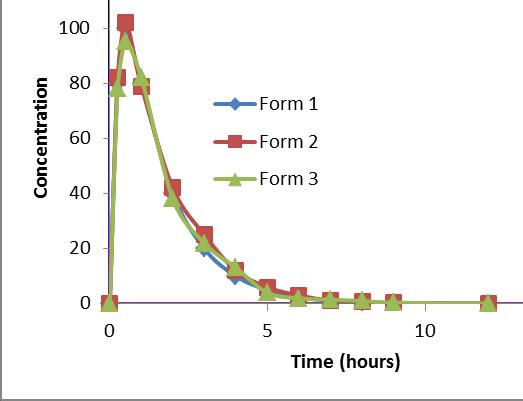
Most discriminating media
Form 2 – Reference
Form 1 GMR = 1.06 (Cmax) and 1.01 (AUC)
Form 2 GMR = 0.93 (Cmax) and 0.95 (AUC)
•
Bridging between formulations during development life cycle of drug products is a knowledge evolving process.
• Dissolution based bridging is a risk-based assessment for drug products of highly soluble drugs.
• Bridging formulations of low solubility drugs is more reliant on clinical studies with goal of developing bio-relevant dissolution test or IVIVC.
• Appropriately designed BA studies are adequate for supporting bridging in early stages and support dissolution development.
• GMR estimates of 0.9 to 1.10 are adequate to declare comparability.
• Development of robust dissolution tests can reduce reliance on clinical studies in healthy volunteers where participants do not receive any medical benefit.