All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IPI will be published in Summer 2024. ISSN No.International Pharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright. 2024 Senglobal Ltd./Volume
04 Editor’s Letter
TALKING POINTS
06 PCI Discusses Annex 1 Complaint Expansion Plans for Sterile Fill-Finish and Lyophilisation
IPI speaks with Jerome Detreille, Senior Director of Business Development at PCI, on updates following a $100 million investment into expanding on sterile fill-finish services. Jermone explores the expansion's impact on service offerings, operational advancements, and the benefits for clients in the biopharmaceutical industry.
08 AI & Automation: How Advanced is Adoption in Life Sciences R&D?
Life sciences organisations are adopting AI-powered automation to transform their R&D operations and improve patient safety. Emmanuel Belabe at ArisGlobal, concludes that the gap between ambition and reality suggests significant potential yet to be unlocked.
REGULATORY & MARKETPLACE
10 Alleviating Regulatory Burden Through Access Consortium
Access Consortium streamlines the regulatory approval process by allowing simultaneous assessment and approval across multiple jurisdictions. Piety Rocha at Pharmalex concludes that this pathway alleviates the burden on both health authorities and sponsors, enabling quicker access to essential medications.
12 Integrating Medical Devices into Pharmacovigilance Portfolios
Navigating the EU medical device regulatory landscape is essential for effectively integrating medical devices into pharmacovigilance portfolios. Humaira Qureshi at Qinecsa explores the practical challenges, regulatory obligations, and strategies for managing device surveillance, including compliance with the European Medical Device Regulation (MDR).
CLINICAL AND MEDICAL RESEARCH
16 The Oncology Crux – Asking the Smart Oncology Questions Before Shaping an Asset’s Journey to Market
Navigating the complex landscape of oncology clinical trials poses significant challenges for pharmaceutical companies aiming to differentiate their assets and maximise commercial potential. In this insightful article, Jason Gilbody, at Envision Pharma Group and of Envision Oncology Solutions, delves into the critical factors that shape successful product launches in oncology.
20 Engaging Minority Communities: Collaborative Practises and Long-Term Success
Exploring the challenges and imperatives in rare disease research, there is a critical need to engage marginalised communities in clinical trials and drug development. By fostering inclusivity and collaboration early on, researchers aim to advance understanding and treatment efficacy while promoting health equity. This article, authored by Neena Nizar at ICON and Deborah Requesens at JumpStart Program among others, emphasises that incorporating diverse perspectives and experiences is essential for shaping future advancements in rare disease therapies.
MANUFACTURING
22 The Importance of Polymorph Screenings: Risk Mitigation and Manufacturing Control
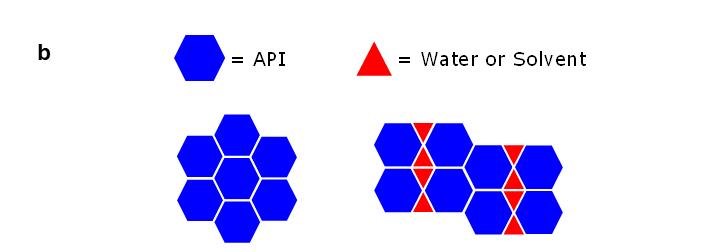
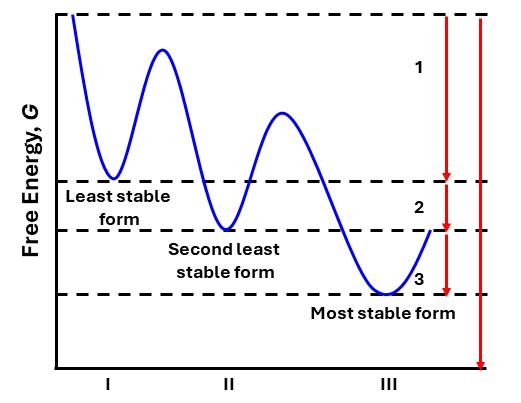
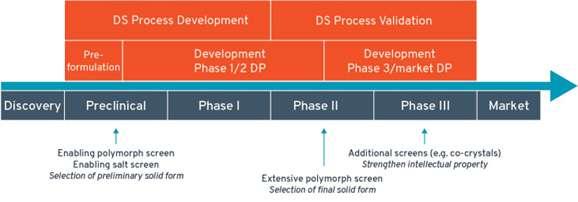
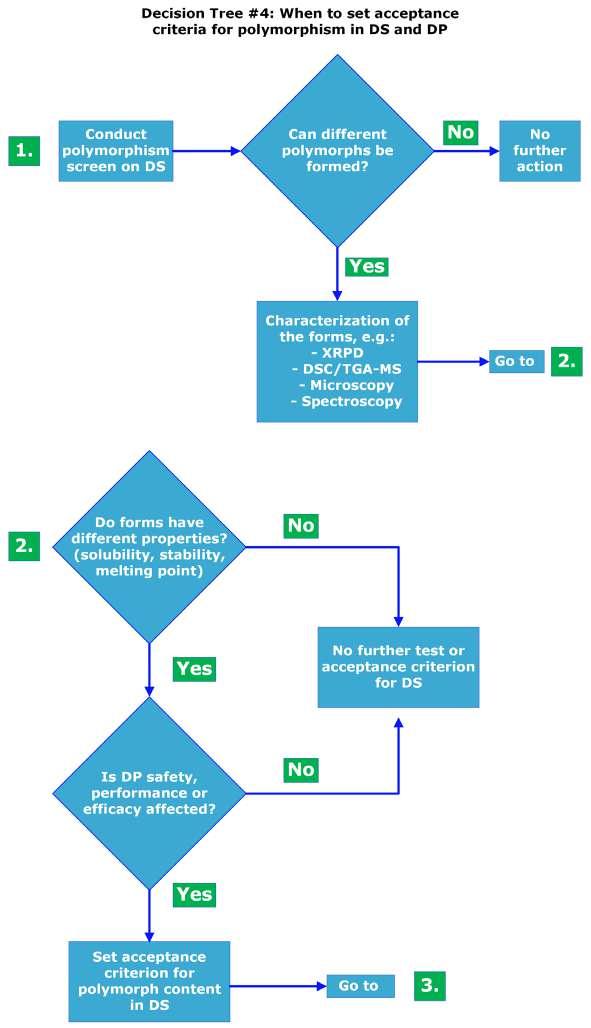
Polymorphism in pharmaceuticals may affect drug product (DP) development, clinical studies, manufacturing, quality and stability. Giovanna Brancatelli at Ardena Solid State Research explains that gaining knowledge of the polymorphic behaviour of active pharmaceutical ingredients (APIs) in the
early development phase is mandatory by performing polymorph screen programs.
28 Why X-ray Inspection is a Viable Quality Assurance Solution for Pharma Products
Efficient quality assurance is paramount in pharmaceuticals, ensuring product safety and compliance. Mike Pipe from Mettler-Toledo highlights x-ray inspection's pivotal role, detecting contaminants and verifying product integrity with minimal radiation exposure.
30 Adaptive, Innovative and Future-Ready: How CDMOs are Staying Sharp
As the pharmaceutical and biotech sectors evolve amidst shifting market demands, CDMOs play a pivotal role in driving innovation and sustainability. Thomas Otto of Vetter Pharma-Fertigung GmbH & Co. KG underscores the strategic investments in resilient supply chains and advanced technology that define the industry's future.
PACKAGING
32 Child Resistant Closures: Exploring New Paths Between Safety and Accessibility
Ensuring patient safety through child-resistant closures is crucial. Despite advancements and regulations, accidental ingestion remains a concern. Innovators are leading with solutions such as the ID-Cap, integrating biometric recognition for enhanced safety and accessibility. Anna Malori at Bormioli Pharma highlights the balancing of child safety with usability for all patients presents ongoing challenges.
34 Advancements in Pharmaceutical Packaging: Exploring Innovations in Sachet and Stick Pack Design
In packaging, research labs drive innovation. Focusing on laminates for single-dose applications, Andrea Grini at Universal Pack explores how these labs optimise packaging solutions through material studies, sustainability initiatives, and collaboration, ensuring enhanced product protection and environmental responsibility.
HEALTH OUTCOMES
36 Navigating the TGA’s Requirements for Combined Products
Combination products are integral to the healthcare system and play an increasingly important role in patient safety and medicinal usability, enhancing therapeutic benefits and improving outcomes. Today, combination products have gone well beyond simple drug-release delivery systems. Piety Rocha and Heyam Kalla at PharmaLex name a few of these, including wearable sensors, 3D-printed implantable modules, and digital drugs providing realtime monitoring.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
40 The End of Drug Shortages Begins with Data Transparency
Lack of data transparency across the supply chain is a core challenge for regulators and suppliers of all sizes. It prevents insights into supply fluctuations and their root causes. Vicki Cookson and Sofia Lange at Veeva Systems discuss that despite ongoing analysis and guidance, supply gaps continue, especially for generic drugs, which account for most of the medications prescribed in Europe and the United States.
44 Has AI Transformed Pharma’s Supply Chain? Pharmaceutical Packaging Expert Weighs In
Artificial intelligence (AI) is transforming pharmaceutical supply chain management by enhancing quality control, transparency through blockchain, cold chain management, inventory optimisation, drug discovery, and
personalised medicine. Steve Brownett-Gale from Origin highlights the integration of AI across these domains underscores its crucial role in advancing the industry.
46 Mastering Cold-Storage Management: Strategies to Minimise Pharmaceutical Losses
Effective temperature control in pharmaceutical logistics is crucial for safeguarding drug potency, mitigating financial losses, and reducing environmental impact from pharmaceutical waste. Mark Ross, Global Brand Manager for TITAN Containers ArcticStore, notes that robust cold-storage management is vital to achieving these objectives.
NASAL
& PULMONARY: DRUG DISCOVERY & DEVELOPMENT
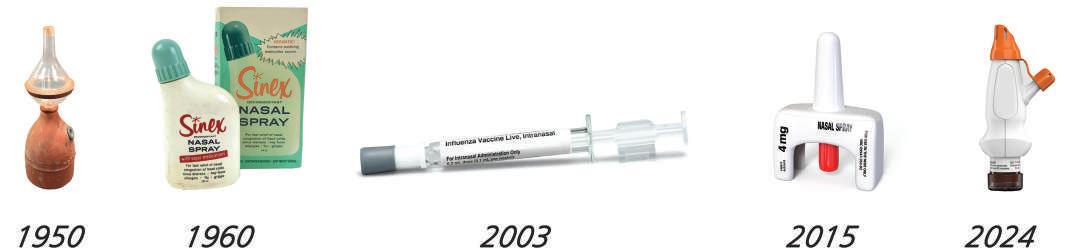
53 Development of Inhaled Products
The evolution of inhaled products over the decades has significantly improved the quality of life for asthma patients. Explores the development of inhalation devices like pressurised metered-dose inhalers (pMDIs) and dry powder inhalers (DPIs), highlighting advancements in drug formulations and environmental considerations. Lars Asking, CEO of MVIC (Medicon Valley Inhalation Consortium), underscores the substantial impact of these innovations in the field of respiratory care.
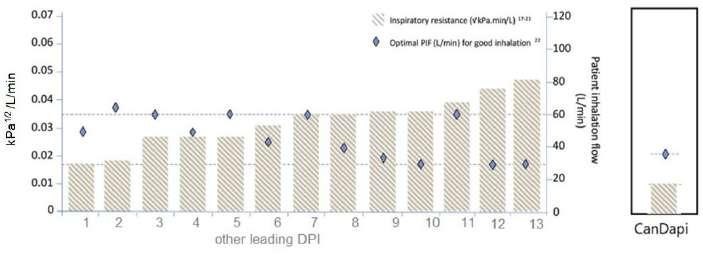
56 The Great Why Questions About Dry Powder Inhalers
Dry powder inhalers present significant challenges in managing asthma and COPD, including difficulties in use and high costs. Many patients struggle with poor inhalation techniques and high airflow resistance, limiting the effectiveness of these devices. Amnon Kritzman Kadron at CanDapi Ltd., concluding that adopting advanced, user-friendly DPI is essential for better patient outcomes.
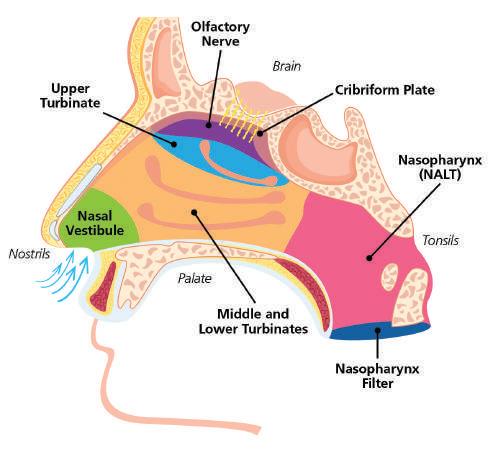
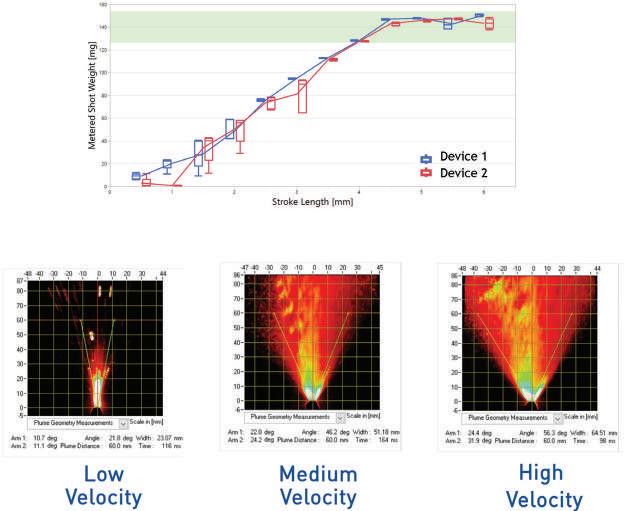
59 Emerging Trends in Nasal Spray Development: Tools for Future Success
Proveris Laboratories excels in developing Orally Inhaled and Nasal Drug Products (OINDPs), using advanced techniques that speed up drug development and approval. Grant Thursten and Alyssa Rubino from Proveris Laboratories highlight the growing trend of nasal spray therapies, emphasising their innovative tools that help pharmaceutical developers bring products to market more efficiently.
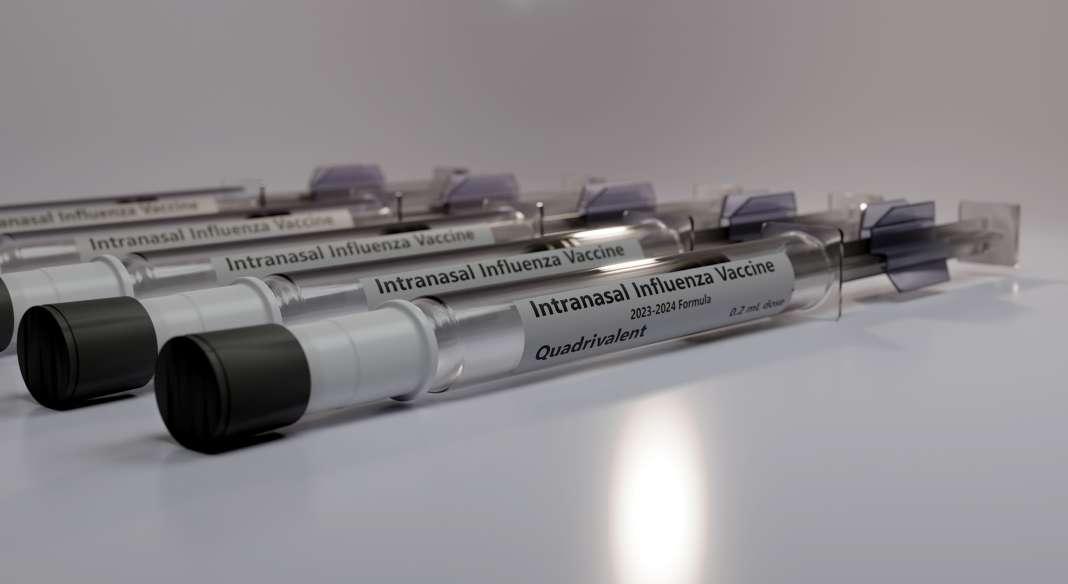
62 Advances in Intranasal Vaccine Delivery: A Promising Non-Invasive Route of Immunisation
The growing interest in nasal vaccines as a promising alternative to traditional injectable immunisation methods. Eleni Kehagia, et al. of the Department of Pharmacy, School of Health Sciences, National and Kapodistrian University of Athens reviews the latest advancements and evaluates the potential of intranasal vaccines for providing effective and non-invasive protection against diseases like influenza, pertussis, meningitis, and hepatitis.
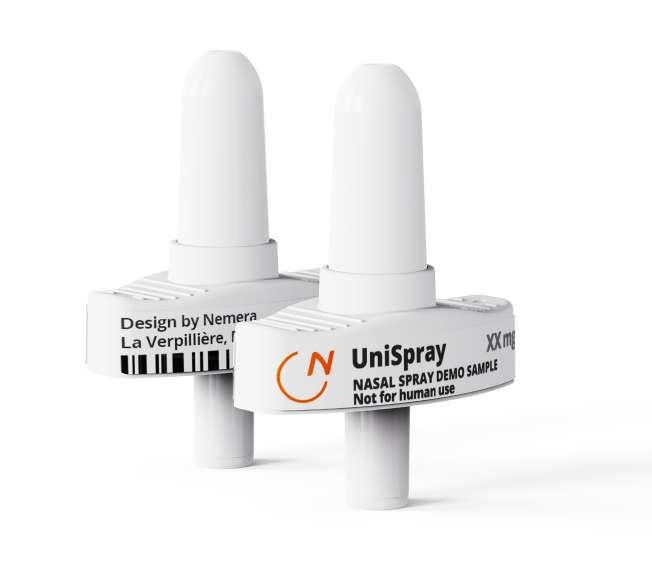
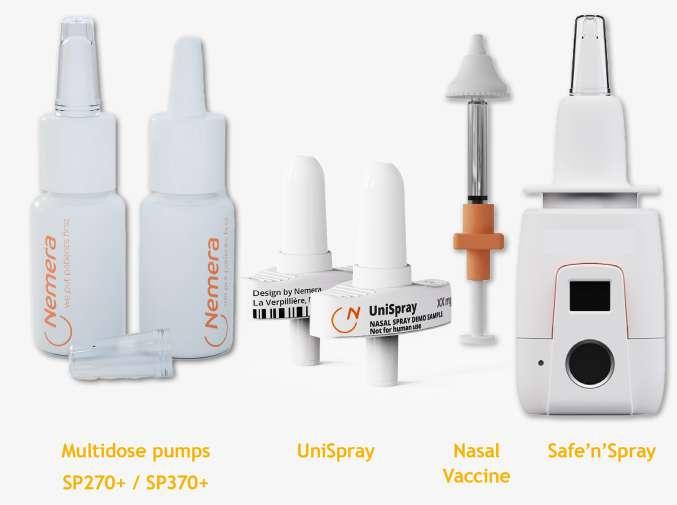
66 Accelerating the Go-to-market of Nasal Combination Products with Integrated Solutions
Explore the growing market for nasal drug delivery, highlighting its benefits for systemic and topical treatments. Learn how Nemera's innovative solutions streamline the development and launch of nasal combination products. Séverine Duband and Audrey Chandra from Nemera discuss their comprehensive approach to meeting market needs.
Versatile design intuitive delivery
Your fill volume may change, with Aidaptus® auto-adjust plunger technology your auto-injector doesn’t need to
Now in production
Accommodates both 1mL and 2.25mL glass syringes in the same device
See our auto-adjust plunger technology in action
Find out more by scanning the QR code or visiting ompharmaservices.com/ipi-june2024
In times like these, almost every stride we take is revolutionary. As the industry evolves, makes decisions, and grows, we do as well. In this summer edition of IPI, we are proud to present the steps this industry has taken forward – from piecing together the outcomes of $100 million dollar investments in sterile fill-finish services to delving into the latest advancements in intranasal vaccinations and powders. What is AI going to help us with? How do we remain mindful of the planet when approaching packaging and waste? Where do we stand with data transparency in a digitalheavy world?
In the Clinical and Medical Research section, a research paper that particularly resonates with me in today's societal context is Engaging Minority Communities, authored by Neena Nizar from ICON, among others; highlighting inclusivity, medical equity, and the implementation of collaborative mindsets to truly shape future advancements in rare disease therapies. This work is crucial, as we aren’t the machines; the machines are adapting on their own course, which we look into with a focus on automation and supply chain changes – and whether or not those changes are fruitful in the end. With some conflicting views on AI in different sectors of the pharmaceutical industry, we are extremely interested in the next waves of advancement and the research that is born from it.
As I’m sure you can see, this issue is packed with exciting developments – under our Manufacturing section, Mike Pipe from
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal Content Writer, Clarivate
Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
Mettler-Toledo highlights the stakes of X-Ray inspection's pivotal role in safety and compliance, detecting contaminants and verifying product integrity with minimal radiation exposure – another means to keep consumers safe and maintain efficiency at work. He’s joined in the section by Giovanna Brancatelli from Ardena Solid State Research, who discusses polymorphs, early screening programs, and managing risk in manufacturing – all to push forward.
This edition stands out with a dedicated sub-section for Nasal and Pulmonary under our Drug Discovery, Development & Delivery category; the rapid development of intranasal vaccinations following the COVID-19 outbreak highlights the trials and errors, step by step, of adjusting vaccines to be non-invasive and effective. Excitingly, Grant Thursten and Alyssa Rubino from Proveris Laboratories highlight the growing trend of nasal spray therapies, emphasising their innovative tools that help pharmaceutical developers bring products to market more efficiently.
Using dry powder inhalers for asthma and other things can be tough. They are hard to use, and they can be expensive. Many people do not breathe in the medicine the right way, and the inhalers themselves might make it hard to get the medicine into their lungs. Amnon Kritzman Kadron at CanDapi Ltd. looks into a new form of mesh inhaler that makes this process easier for those who need it – IPI aims to focus heavily on these new beginnings, and we are interested in constantly improving patient outcomes, innovative thinking, and in expanding the scope of respiratory care beyond traditional inhalers.
The commitment to this research is about putting our best foot forward. With our inhalation device section, the goal is to improve patient adherence and effectiveness of treatments. Similarly, in pharmaceutical packaging, innovations aim to ensure product safety and accessibility, particularly for vulnerable populations like children, while also taking the time to address environmental responsibility and sustainability concerns.
Safety, efficiency, and the path forward is what we want you to take away from this summer edition, and I hope you enjoy it!
Sara Shikooh, Editorial Manager – IPI
Georg Mathis Founder and Managing Director, Appletree AG
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey Litwin, M.D., F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Stanley Tam, General Manager, Eurofins MEDINET
(Singapore, Shanghai)
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Stefan Astrom, Founder and CEO of Astrom Research International HB
T S Jaishankar, Managing Director, QUEST Life Sciences
Pharmaceutical grade Benzalkonium Chloride
Minimize your risks by prioritizing high quality, consistency and reliable GMP manufactured raw materials.
Our Benzalkonium Chloride is manufactured following cGMP guide ICH Q7 for APIs, the highest available quality standard in the industry.
Get peace of mind with full traceability, high product purity, full pharmacopoeia compliance (PhEur, USP/NF, JP, ChP), audit access, and 30+ years of cGMP manufacturing experience.
novonordiskpharmatech.com
PCI Discusses Annex 1 Compliant, Large-scale Filling and Lyophilisation Expansion
And Plans to Increase Injectable Packaging Capabilities and Capacities
Q: In 2022 PCI Announced a $100 Million Investment into Expanding its Sterile FillFinish Service Offering – Can You Provide an Update on How This Is Progressing?
A: To enhance our sterile fill-finish capabilities and capacities we committed $100M to expand our sterile development and manufacturing campus of excellence in Bedford, NH with a new multi-product, 50,000 square foot facility. Over the past two years, we have meticulously planned and methodically executed the expansion, allowing us to grow our breadth of services as a global CDMO, providing best-in-class late stage clinical and commercial scale fully isolated vial filling and lyophilisation solutions for new and existing clients.
The new facility will provide increased capacity featuring Annex 1 compliant technology, including an aseptic fill-finish line within a fully isolated containment system, complete with twin lyophilizers (40 square meters / 430 square foot each) with automatic loading and unloading systems. The filling line can process batch sizes up to 300,000 vials at a rate of 400 vials per minute (vpm) providing much needed large-scale capacity for the filling of life changing small molecules and biologics, including highvalue APIs such as mABs, oligonucleotides, and peptide drug products.
With over 25 years’ experience in lyophilisation and sterile fill-finish manufacturing, PCI has developed industry leading technical expertise in the end-to-end processing of
often challenging and complex processes. Truly supporting full product lifecycle management from formulation and lyophilisation cycle development through clinical to commercial sterile manufacturing, this high-volume lyophilisation and liquid filling facility provides our clinical stage clients a seamless solution for scale-up as they prepare for commercialisation.
Q: How Does This New Technology Benefit Your Clients?
A: Our new large-scale vial filling, designed by Groninger and SKAN is among the industry’s most versatile isolator lines. This innovative technology streamlines the filling process and ensures the highest level of sterility assurance for the final product. The enclosed system safeguards drug integrity and operator safety, employing the latest SKANFOG® decontamination technology which has been proven to effectively eliminate any potential microorganisms. The SKANFOG system uses an advanced micronebulisation process to optimise particle size of the VHP droplets, this technology uses 80% less VHP than traditional vaporisation systems.
To meet the growing product demand of some of the largest commercial requirements from our clients, this highspeed line can fill 400 vpm, fulfilling batch sizes up to 300,000 vials. Additionally, the high-speed line’s smart fill technology minimises product loss, ensuring no wastage of precious drug substance through features
such as peristaltic pumps, defective vial identification prior to filling, re-stoppering, and under-fill correction.
While minimising any loss of API or Bulk Drug Substance, inline camera inspection systems check each vial for stopper placement and drug product. Our overarching philosophy at PCI, is to constantly strive for perfection, delivering a quality and sterility assured drug product to patients, while meeting regulatory standards including the recently revised Annex 1.
Q: When Will the New Site Be Operational, and What Additional Services Will PCI Offer?
A: The installation of the lyophilisation, filling and isolator equipment is underway with engineering and validation runs scheduled to take place in the autumn. The site will be ready for GMP production in the first quarter of 2025. We are currently facilitating client visits to the site, and providing the opportunity to reserve capacity to help secure their future sterile supply chains.
This site, is our third high throughput isolator based commercial sterile fill-finish facility that we have built across our global network in the last three years. Our expert technical, engineering and project management teams alongside our long-term contractor partners are experienced in delivering complex, time sensitive capital projects, and we have a high-level of confidence in meeting the 2025 operational timeline.
Q: What Efforts Does PCI Undertake to Build Robust Supply Chains and Mitigate Risk?
A: Recent events underscore the necessity for secure and nimble supply chains. Biopharmaceutical companies require service providers that offer robust integrated solutions, supporting drug products from early phase development and clinical studies through to commercialisation, mitigating the need to transfer between suppliers therefore reducing supply chain complexity and risk.
PCI ensures continuity of supply for life changing medicines by providing scalable sterile drug product development, manufacturing and lyophilisation solutions from development to commercialisation. Our integrated downstream custom assembly and packaging solutions for injectables, allows for ultimate knowledge sharing and communication between teams to ensure the drug product packaging is optimised for the product, patient and production. This integrated approach across global manufacturing and packaging teams not only streamlines supply chains but provides an accelerated delivery of life changing therapies to patients.
Q: What Investments Has PCI Made in Packaging Technology to Support the Growing Injectable Market Demand?
A: Complementing the continued growth and investment across our sterile manufacturing network, we continue to expand our Centres of Excellence for the clinical and commercial packaging of patient-centric delivery systems across Europe, North America and the UK. These facilities are equipped with advanced drug delivery packaging technologies for the final assembly, labelling and packaging of vials, cartridges, prefilled syringes, advanced safety syringes, autoinjector and pen devices, along with in-line serialisation. Combined with expanding global storage and distribution capabilities across a range of temperatures from controlled room temperature (15°C–25°C), through 2-8°C, -20°C, -40°C, -80°C to -196°C, ensures a seamless solution.
Recently we announced an investment of $150M in a new 310,000 square foot final assembly and packaging facility at our Rockford, IL, site to meet the growing market need of specialised assembly and packaging
for injectable drug-device combination products. Over 20 dedicated new suites will support the assembly and packaging of vials, prefilled syringes, autoinjectors, on-body injectors and pen-cartridges such as those for treating oncology, autoimmune diseases, diabetes and obesity. The investment also includes a 200,000 square foot warehouse for comprehensive storage solutions across various temperate ranges.
Q: How Does PCI Pharma Services Differentiate Itself From Other CDMOs in the Biologic Market?
A: PCI spans the entire drug product lifecycle, connecting development and commercialisation while de-risking supply chains to deliver true speed to market. Our expertise combined with innovative technologies means we deliver more than just a service; we are a trusted partner sharing an industry-leading depth and breadth of knowledge.
As part of the ongoing evolution of PCI we introduced Speed Solutions™ to the market combining value-added services and expertise for an integrated approach to every client project. Speed-solutions such as packaging design and artwork or quality and regulatory services, de-risks and reduces the complexity of our client partners supply chain by eliminating the need to transfer services to alternative suppliers.
Differentiating what we do, PCI Pharma Services is commitment to and invests in digital transformation utilising innovative technology. One such platform is PCI | Bridge™ which complements our project management capabilities by creating efficient and uncomplicated ways of working together. This industry-leading digital customer platform provides our client partners with real-time insights into their portfolio of work at our sites around the globe, unlocking productivity with access to real-time supply chain information and digital workflows.
Q: What Can We Expect in the Future From PCI as an Integrated Solutions Provider?
products. PCI are dedicated to fostering collaboration, creativity, and tailored solutions. Our purpose is that together, we bring lifechanging therapies to patients.
With over 50 years of experience, PCI is a respected provider of biopharmaceutical supply chain solutions worldwide. Our expertise enables us to deliver flexibility and excellence in all that we do. We will continue to invest in innovation, science, technology and digital transformation strategies to simplify processes and expedite drug product delivery to patients.
Central to our values and beliefs is our global commitment to environmental governance. Our ESG (Environmental, Social, and Governance) strategy, underscores our pledge to uphold environmentally sustainable practices, foster diversity and inclusivity, and positively impact our employees, partners, customers, patients, and communities in which we live and work. Our recently published inaugural Annual ESG Report outlines our achievements and ongoing commitment to set targets, aligning to the strategies of our customers as a trusted partner. We aim to act ethically, benefiting our employees, customers, communities, and the planet, while fulfilling a significant role in the pharmaceutical supply chain, enriching and extending lives.
Jerome Detreille
A: PCI remains committed to addressing dynamic market needs, whether in potent oral solid dose, sterile injectable process development, scale up, technology transfer, or custom packaging of specialised drug
Jerome Detreille, Senior Director Business Development EMEA, After graduating from Strasbourg University of Pharmacy, France, Jerome spent 13 years as European Director of Business Development in Catalent for sterile injectables, before moving to Penn Pharmaceutical Services as Senior Director New Business Development in 2012. Here, he was part of the executive team who built the potent Contained Manufacturing Facility (CMF), which won the ISPE Facility of the Year award for Facility Integration (2014). In 2014, PCI acquired Penn Pharmaceutical Services, and Jerome continues to act as Senior Director of New Business Development to support the growth of PCI, especially in the development and manufacturing of OSD and sterile drug products.
AI & Automation:
How Advanced is Adoption in Life Sciences R&D?
Forward-thinking life sciences organisations have switched on to the benefits of AI-powered automation, to transform their research and development operations and streamline their path to market while enhancing patient safety. But how far have they come, and where are investments currently focused to maximise the benefits? Here, ArisGlobal’s Emmanuel Belabe examines the progress leading pharma organisations are making and highlights the potential that has yet to be unlocked, drawing on new industry-specific research into evolving aspirations for AI and intelligent automation.
ArisGlobal’s new 2024 Industry Survey Report, Life Sciences R&D Transformation: Ambitions for Intelligent Automation & Today’s Reality, was conducted late last year with 80+ organisations, to understand more about the evolving attitudes toward it and the expectations of AI and intelligent regulatory and safety process automation. The international study, which spanned every patient treatment process domain, from CROs to young biotechs, sought to establish where life sciences companies are currently on the automation value spectrum, and where their ambitions lie; that is if rising data complexity necessitates a more ambitious technology use.
As our interview uncovers, the findings inform companies’ evolving use of intelligent automation, including the application of Generative AI; the growing role of real-world data in expediting the safe delivery of new drugs to market; and where companies plan to invest next.
Q: What was the Most Surprising Finding from the Research, in your View?
A: Emmanuel Belabe, ArisGlobal (EB): It’s probably the mismatch between aspiration and reality. At first sight it seems the Life Sciences industry is already quite mature in its use of next-generation automation, enabled by AI advancement. But a closer inspection
confirms that this is largely still an ambition rather than a reflection of companies’ current status. Although more than three-quarters of respondents (75%+ of surveyed organisations) say they already use some form of advanced automation within their processes today – up 13% from 2022 and just 5% in 2020, only 8% have applied advanced automation in “all” or “most” of their processes at this point. (By advanced automation, we mean the adoption of artificial intelligence (AI) and machine learning (ML).)
Q: What Does this Suggest to you?
A: It means that awareness of the opportunity for AI-based transformation of knowledge work and essential processes is high and growing, which is promising. Indeed, more than seven in ten respondents went on to express plans to expand business process automation over the next 18 months. The overarching trend is to move on from rudimentary, fairly crude process automation, toward something more material and a capability that is sophisticated, which tangibly alleviates the pressures on Safety and Regulatory professionals’ time.
Q: Can you Clarify the Difference?
A: Automation technology has come a long way from the early days of simple robotic process automation (RPA) solutions. These have harnessed optical character recognition and rules-based workflow to identify and manage standard documents and structured data in a fairly basic way. Today, thanks to intelligent, AI-powered automation, and the ability to identify and analyse all kinds of data, it’s become possible to distil all kinds of new actionable insights – irrespective of how colossal the volumes, or how diverse the range of sources. As AI and ‘deep learning’ solutions advance in line with the scale and sophistication of available data, life sciences R&D organisations are growing more ambitious in their ability to harness data and its insights in ever smarter ways. That could be to hone decision-making, expedite and remove cost from processes,
and deliver important treatments to patients more efficiently and affordably.
Q: What has Held Pharma Companies Back Until Now, with all of this?
A: The challenge previously has been how to assess, interpret and reliably harness vast amounts of unstructured data (the kind that exists in documents, in emails or on paper, rather than in a searchable database) which represent immense potential value if only Safety, Quality, Regulatory and Clinical teams had the resources to process it all.
This is where the latest knowledgebased intelligent automation technologies (including machine translation and a growing range of artificial intelligence capabilities, such as Generative AI and deep learning) come in. These next-generation capabilities make it possible to distil reliable, actionable insights from across that wealth of data in all its forms. It was the pace of companies’ transitions to intelligent automation that we were particularly keen to identify in the 2024 ArisGlobal Industry Survey Report.
Q: Has the Adoption of More ‘Basic’ Process Automation Slowed Down?
A: Not exactly. Our 2024 survey results, based on research conducted last year, showed that the adoption of basic automation by life sciences organisations had increased by 24% from 2022, and almost 30% from 2021. But the main focus now seems to be on the more advanced, smarter end of process automation. In the new research, 60% of organisational leaders confirmed that they are looking to explore new usage and/or increase usage of AI/Machine Learning within the next 18 months.
Q: What has Prompted this, Specifically, Do you Think?
A: As Life Sciences organisations are forced to do more with less, automation has been
Talking Point
increasingly applied to different processes and workflows. Certainly, companies seem to be realising the benefits of automation. In the survey, respondents reported the top five benefits of basic automation as being efficiency gains, increased quality, cost savings, increased speed, and reduced risk. But now they are craving more, which is reflected in that finding mentioned earlier, in which threequarters of companies point to at least some use of next-generation technologies. In other words, adoption of AI and machine learning is becoming widespread now.
Generally, respondents saw automation and AI as having the potential to increase positive outcomes and empower professionals across the drug development cycle. Increased adoption of these technologies is expected to benefit patients, through increased drug discovery, personalised medicine, clinical trial acceleration, enhanced care, and improved data analysis. Other anticipated business benefits include increased speed, superior decision-making, and more efficient processes – all critical success factors in a highly competitive industry.
Q: What About Challenges, or Barriers to Adoption of These More Advanced Capabilities?
A: Many organisations’ next-level AI ambitions are inhibited by integration challenges. While the most substantial blocker to AI adoption in life sciences is budget (cited by 53.6%), a notable 36% highlight lack of integration with existing technology. More than two-thirds (68%) of respondents went
on to say they considered or had found it “very difficult” or “somewhat difficult” to integrate automation technology with other systems and/or data.
This barrier to advanced automation goes some way to explaining some of the identified hesitancy around where the technology could take companies in the future. That’s particularly as new opportunities emerge to exploit real-world data (RWD) as part of critical but labor-intensive processes such as safety signal detection and validation.
Q: What Did the Survey Uncover in Terms of Companies’ Plans for More Ambitious Use of Data, Across Safety and Regulatory Operations?
A: There is an appetite to harness RWD where possible to unlock the next level of patient treatment innovation. In our research, just over half (51.2%) of respondents suggested that their organisation is already connected to some form of RWD, up from 31.5% in 2022; moreover, 54% of those currently connected to RWD said they are looking to increase the data’s usage.
For those not yet connected to RWD, 20.4% indicated that they are either in the process of connecting to such resources or have plans to within the next 18 months.
Access to high volumes of good-quality, current data will be important to build Large Language Models to drive Generative AI’s use in the industry, too. As it was for early automation, data is the key driver of
Generative AI. The more data an organisation can leverage, the stronger GenAI’s models (algorithms) can be accurately trained to help identify and qualify the next raft of industry breakthroughs.
According to respondents, the most important value from RWD in drug development is linked to clinical and patient safety activities. Respondents indicated that the ability to draw on and analyse additional data and insights could help de-risk clinical trials, expedite the trial process, speed up approval processes, and positively impact signal detection.
Q: Were There Any Additional Takeaways from the Research that Are Important to Mention?
A: The role of regulatory bodies/health authorities will be important in cementing the potential and the success of advanced automation in promoting safe new innovation in the industry. Without careful management, approaches, and implementation, RWD data use could become more of a point of contention in the interactions between regulatory agencies and organisations, several respondents indicated. RWD is being used within the US FDA’s Sentinel Initiative and by EMA in Europe, but there needs to be further willingness to partner with organisations in life sciences to pave the way to harnessing RWD in other use cases.
Emmanuel Belabe is Senior Vice President for Customer Success within the Global Customer Support and Solution Consulting organisation at ArisGlobal. ArisGlobal, an innovative life sciences technology company and creator of LifeSphere®, is transforming the way today’s most successful life sciences companies develop breakthroughs and bring new products to market. Headquartered in the United States, ArisGlobal has regional offices in Europe, India, Japan, and China. For more updates, follow ArisGlobal on LinkedIn.
Alleviating the Regulatory Burden Through the Access Consortium
While regulatory authorities are increasingly adopting reliance programs to assess applications, few offer the opportunity to take advantage of simultaneous assessment and approval timelines across borders and jurisdictions. This is what differentiates the Access Consortium, which provides joint scientific advice and work-sharing, reducing duplication and accelerating access to high-quality medicines for patients in need.1 The collaborative pathway alleviates the burden for both the health authority and the sponsor or marketing authorisation holder (MAH), enabling the submission of one dossier for all participating markets simultaneously.2 As part of a push for greater harmo-nisation of regulatory policy, Australia’s Therapeutic Goods Administration (TGA), Health Canada (HC), Singapore’s Health Sciences Authority (HSA) and Swissmedic formed the Access Consortium in 2007.3
Thirteen years later, following Brexit, the UK’s Medicines and Healthcare products Regulatory Agency (MHRA) has joined the fold.
How Work-sharing Works
Unlike reliance programs, where one national authority will consider assessments by another,4 Access is a work-sharing program, where member countries involved in an application agree to divide the modules according to their capability, experience and capacity.
All participating authorities agree to set evaluation times before the process begins, which gives sponsors a clearer timetable and enables teams to plan and prepare their organisational strategy.5
Each agency then delivers their modulespecific assessment report and list of questions to the other partner agencies. The consolidated assessment report and questions reflecting concerns regarding Modules 2 to 5 are then sent to the MAH, which is requested to provide responses within a defined time frame.
The country responsible for a specific module evaluates the responses and, if required, the other agencies may submit an additional set of questions for clarification.6
While differences between the modules are accepted, MAHs are asked to outline those in the information provided to each authority. Participating members discuss the differences to determine whether the application is suitable for a work-sharing arrangement.
Module 1 is treated differently because of the country-specific content and is reviewed separately by each participating agency.
At the end of the review period, each authority issues its decision on marketing authorisation independently.
Market authorisation or refusal of market authorisation by one regulator will not affect the decision or the timing of the decision by the remaining participating regulators.6
While indications and final product labelling could differ slightly in each country, the core evaluation and conclusion is common, which is quite similar to Europe’s Decentralised Procedure (DCP), where one agency acts as a reference regulatory agency (RRA) and evaluates Modules 2 to 5.
Strengthening Processes
The Access Consortium has in the past few years taken steps to strengthen its processes. There are now several working groups that support a range of activities across active substances, generics and IT architecture, as well as the more recent addition of a group dedicated to advanced therapy medicinal products (ATMPs).7
Interest in the Access pathway appears to be gaining momentum. According to the TGA, the first two submissions to all five agencies were approved through the New Active Substances Work-Sharing Initiative (NASWSI) in 2022–2023. Asciminib (Scemblix®) was approved by all five markets for the treatment of Philadelphia chromosomepositive chronic myeloid leukaemia, and Faricimab (Vabysmo®) was approved to treat macular degeneration.8
In 2023, the TGA approved seven new medicines through NASWSI. As of June 2023, NASWSI had approved 25 NAS work-sharing applications.9
The Access Consortium has also set up a pilot to find better ways to assess products that treat a serious, life-threatening, or severely debilitating condition, or one where no other treatment is registered in the participating regions. Established in late 2023, the Promise Pilot Pathway aims to find a common approach to the criteria for priority review. The assessment of an application is carried out by one agency with peer review by all participating agencies. Once the pilot ends, the pathway will undergo further review.10
Streamlining Approval
When an MAH submits applications in each country rather than through the Access Consortium, there can be significant delays in approvals in some markets.
For example, a pharmaceutical company has obtained registration from one of the Access regulators with a post-approval commitment to submit impurity-related information once it became available. However, another regulator had put a stop clock on the submission for the same product until the data was generated, resulting in an 18-month delay to the approval.
Experience suggests that, had the application been submitted through the Access Consortium, the health authorities would have likely come to an agreement, thereby reducing the approval delay in one Access market.
While, overall, the process can reduce the burden on global teams, companies considering the Access pathway should allow time to prepare for the administration involved.
Potential applicants will first need to seek permission to begin the process through an Expression of Interest (EOI) application to participate in work-sharing. This form will need to be sent to at least two Access members at the same time and requires the approval of each member before they can adopt the Access pathway.11
Companies interested in the Access pathway are also advised to take advantage of technical and logistical pre-submission meetings with the relevant health authorities to clarify expectaions.6 Both meetings are held after the EOI form has been submitted and before submitting the application.
Although modules 2 to 5 might be harmonised, MAHs may also need to meet country-specific requirements before initiating the process. The dossiers submitted should comprehensively address the requirements of all jurisdictions proposed for work-sharing. In Switzerland, for example, they will need to obtain an establishment license from Swissmedic and, in Australia, a Good Manufacturing Practice (GMP) clearance from the TGA.12
The Access pathway also allows companies to consider submission to the five markets concurrently with a submission to the United States and the European Union (EU).This could relieve the pressure on global pharmaceutical teams that are struggling to prioritise under the demands of often competing deadlines.
However, companies should first carefully consider what is required, as Singapore’s HSA and the TGA, for example, have rejected applications through Access for products approved in the EU, opting for reliance or comparable overseas pathways. Additionally, since all participating countries must agree on the timelines, it can add time to the process and therefore might not be the most appropriate path in all circumstances.13
The Impact on Sponsors
For MAHs or sponsors, the Access pathway also reduces the burden on global teams. If applying separately, the CMC, clinical and pre-clinical teams will face different sets of questions from each jurisdiction. This could result in endless requests for information dragging out the process, depending on the submission plan.
In one instance, a large pharmaceutical company that took a combination product through the Access pathway found that the consolidated list of questions enabled them to turn their attention to other priority products, rather than be burdened with a constant stream of requests for more information. This provided predictability for internal resourcing.
For small companies without the same human resources to plug the gaps, being able to answer one set of questions can be hugely advantageous.
Regulatory & Marketplace
A recent survey of MAHs that used the Access pathway found they were largely satisfied with the experience.9 They cited several benefits, including:
• Experience with a work-sharing pathway (76%)
• A near simultaneous approval in multiple countries (73%)
• A shorter review process compared with national timelines (61%)
• Fewer overall questions from health authorities (61%)
Busy regulatory affairs teams that would otherwise have had to adapt countryspecific dossiers, a process that experience shows can take several months, are freed up to focus on other priorities.
Data from the Centre for Innovation in Regulatory Science also shows that the median submission gap and median approval time for NAS approved through Access were faster than those approved by individual health authorities.14
Benefits for Authorities and Patients
The Access pathway clearly benefits health authorities too, with work-sharing helping to plug critical gaps in resources and skill sets.
Products are also becoming more complex, with specific expertise required to handle innovative biologics and ATMPs.
Dividing up the modules according to skill sets and capacity enables each authority to leverage the expertise of other agencies, while also making the most of their own.
What cannot be underestimated is the impact on patients. By streamlining the process to approval, Access paves the way for patients to gain earlier access to medicines that might otherwise take longer to get to the relatively smaller markets of the Access Consortium.
REFERENCES
1. Access Consortium, Strategic Plan 2021-2024. https://www.tga.gov.au/sites/default/files/ 2022-09/access-consortium-strategic-plan2021-2024.pdf
8. International work-sharing – First medicines approved by all five Access Consortium regulators, TGA. https://www.tga.gov.au/news/ media-releases/international-work-sharingfirst-medicines-approved-all-five-accessconsortium-regulators
9. Industry Perceptions and Experiences with the Access Consortium New Active Substance WorkSharing Initiative (NASWSI): Survey Results and Recommendations, Therapeutic Innovation and Regulatory Science, March 2024. https://link. springer.com/article/10.1007/s43441-024-00624-7
10. Industry Perceptions and Experiences with the Access Consortium New Active Substance WorkSharing Initiative (NASWSI): Survey Results and Recommendations, Therapeutic Innovation and Regulatory Science, March 2024. https://link. springer.com/article/10.1007/s43441-024-00624-7
11. Access Consortium New Active Substance (NAS) work-sharing initiative, TGA. https://www. tga.gov.au/access-consortium-new-activesubstance-nas-work-sharing-initiative
13. Why Novartis Rejected Access Consortium In Favor Of Direct UK Radioligand Filing, Pink Sheet, Sept 2022. https://pink.citeline.com/PS146972/ Why-Novartis-Rejected-Access-Consortium-InFavor-Of-Direct-UK-Radioligand-Filing
14. New drug approvals in six major authorities 2013-2022: Focus on orphan designation and facilitated regulatory pathways. https://cirsci. org/wp-content/uploads/dlm_uploads/2023/07/ CIRS-RD-Briefing-88-6-agencies-v.1.2.pdf
The information provided in this article does not constitute legal advice. PharmaLex and its parent Cencora, Inc., strongly encourage readers to review available information related to the topics discussed herein and to rely on their own experience and expertise in making decisions related thereto.
Piety Rocha is the Director, Head of Regulatory Affairs & Country VDC Head at Pharmalex, Australia. She has more than 20 years of experience in the Australian and New Zealand pharmaceutical industry. Piety is a seasoned regulatory affairs professional, with extensive knowledge and expertise covering innovative and generic prescription medicines across multiple therapeutic areas.
Piety
Rocha
Integrating Medical Devices into Pharmacovigilance Portfolios How to Navigate the Latest EU Medical Device Regulatory Landscape
to Successfully Adapt your Pharma Portfolio
Understanding medical device regulations is essential for pharmacovigilance professionals seeking to master device vigilance compliance and operational excellence. This article delves into the essentials necessary to navigate the complex EU device vigilance regulatory landscape and considers differences in drug and device surveillance requirements to help pharma partners confidently adapt departments to the changes.
It discusses the practical challenges of integrating medical devices into pharmacovigilance portfolios, the obligations of device manufacturers, how to navigate the European Medical Device Regulation, maintaining effective post-market surveillance and managing the life cycle of a medical device.
Implementing Medical Devices in Pharmacovigilance Portfolios
When addressing the practical challenges of implementing medical devices in pharmacovigilance portfolios, it is important to bear in mind the components that are specific for devices or covered geographies, including definitions. These include:
• all device risks well understood by the pharmacovigilance department
• specific risk management documentation for each device to understand reportable events in advance
• a procedure for contacting the Quality Assurance (QA) department of the manufacturer for clarification
Appropriate regulatory information, such as device class, signal reference number (SRN) and regulatory agency details should be readily available. This might sound trivial, but it is a common cause of unnecessary errors. A good way to solve this problem is to cite a device in a database and have all these attributes that do not change. It is also helpful to have any previous field safety corrective action documentation in hand.
It is not possible to have one template and use it for everything because every device is different and has specific risks.
Obligations and Responsibilities in the Device Sector
The general obligations of device manufacturers include implementing and keeping up-to-date a post-market surveillance system, maintaining a quality management system (QMS), and having a system for recording and reporting incidents and field safety corrective actions. The vigilance obligations are the manufacturers. A mandate must be in place which includes a responsibility to immediately inform the manufacturer about complaints and reports of suspected incidents.
Another key difference between drugs and devices concerns the obligations of economic operators in the device sector. In drugs, we have the marketing authorisation holder (MAH). In devices, we have got the manufacturer. If the manufacturer is not established in the EU, they must designate an authorised representative (AR). Pharmacovigilance departments may also be working closely with distributors as they should collect complaints and incidents and forward them to the manufacturer.
The Qualified Person for Pharmacovigilance (QPPV) is responsible for establishing and maintaining the pharmaceutical company's pharmacovigilance system, having an overview of the safety profiles and emerging safety concerns for the company's drugs, acting as a single contact point for Health Authorities on a 24-hour basis, monitoring product safety and risk-benefit balance, and ensuring the quality of the pharmacovigilance system. Device manufacturers must designate a Person Responsible for Regulatory Compliance (PRRC). While the roles are somewhat similar, the obligations differ and there should be close communication between them.
PRRC’s responsibilities, as set out in EU MDR (34), (35) and Article 15, include the QMS, regulatory documentation, postmarket surveillance and vigilance reporting, and devices used for clinical investigation.
The medical device systems audit approach (MDSAP) is a popular way to improve the penetration of new markets through mutual recognition of audits. The
important parts for pharmacovigilance departments handling device products are the agreements described in Annex 4. These include:
• Clause 8.2.2 which covers complaint handling
• Clause 8.2.3 on documented procedures for notifying regulatory authorities
• Clause 8.3.3 which covers procedures for issuing advisory notices in accordance with applicable regulatory requirements
Overview of Medical Device Regulation Requirements
European Medical Device Regulation (MDR) is not a standalone regulation. It must be taken in the context of other standards and industry guidelines and there are several which are particularly important for pharmacovigilance departments to note.
The most relevant ISO Standards are ISO 13485, ISO 14971 and ISO 10993. These cover quality management systems (QMS), risk management and biological evaluation respectively. Post-market surveillance is a central component of QMS in ISO 13485. This ISO standard includes a clause on reporting applicable complaints to the concerned regulatory authorities and emphasises the need for manufacturers to have documented procedures. Key points include:
• Broad definition of a complaint to cover quality and vigilance in all regions
• A record of the awareness date to meet different vigilance timelines across the globe
• A mandate for company employees to report or forward complaints
• A complaint form that enables vigilance reporting
• Appropriate contact methods
• A procedure for obtaining products back to enable root cause investigation
• A process for complaint investigation to enable regulatory reporting
• A process for issue escalation, for example an FSCA
Many legacy guidelines from MEDDEV – MEDical DEVices Guidance Documents created on behalf of the European
Commission – are also still valid. In particular, MEDDEV 2.12-1 rev.8, which covers vigilance, and MEDDEV 2.12/2 rev.2 which covers postmarket clinical follow-up (PMCF). MEDDEV 2.7/1 rev.4, covering clinical evaluation may also be relevant if the vigilance department is also completing literature searches.
The Medical Device Coordination Group (MDCG) guidelines specifically tie to EU MDR regulation. MDCG 2020-7 and MDCG 2020-8, cover Post Market Clinical Follow Up (PMCF) plan and report templates respectively, while MDCG 2023-3 covers vigilance terms and concepts outlined in the Regulation (EU) 2019/745 on medical devices. MDCG 2011-21 also has useful guidance on Periodic Safety Update Report (PSUR) according to EU MDR. The final big group of guidelines comes from The International Medical Device Regulators Forum (IMDRF) and the most important part of those for vigilance departments covers terminology. Without the terminology, it is impossible to complete the manufacture incident report form. All major database solutions currently on the market contain a device module including the IMDRF dictionary. Some concepts are specific for devices, for example, medical device
Regulatory & Marketplace
problem information. Others, like patient information, are compatible with clinically validated international medical terminology, known as MedDRA. Close cooperation between pharmacovigilance and quality assurance teams is vital to ensure proper investigation of device incidents which is also part of a device vigilance report.
Reporting Essentials and Timelines
There are a couple of essential forms in medical device vigilance. The first is a Manufacturer Incident Report (MIR) for Serious Incidents (MDR/IDVR) and Incidents (AIMDD/MDD/IVDD). This is used to report incidents involving devices made available on the EU market. The exception is expected side effects which are clearly documented in the product information, quantified in the technical documentation and subject to trend reporting.
Someone who has only previously worked in pharmacovigilance, opening an MIR form for the first time will likely find the form quite confusing because the concepts are very different. Each incident needs to be investigated. This investigation requires close coordination with the quality
assurance department because in every case, it is necessary to find the root cause of the event. In the initial form you need to explain what happened, and in the subsequent ones to include an analysis of why it happened. Were any corrective actions taken before? How much of this product was sold in the past? Unlike drugs, the number of units sold is known, making it possible to accurately estimate patient exposure and calculate rates of occurrence.
If it is necessary to take some sort of safety action, it will typically be done by circulating a field safety notice. Any field safety corrective action (FSCA) in respect of devices made available on the Union market must be recorded. This includes any FSCA undertaken in a third country if the reason for the action is not limited to the device made available in the third country.
Another key difference in reporting between pharmacovigilance and device vigilance is the Manufacturer’s Trend Report. In pharmaceuticals, you typically would not know how many patients were exposed to your products. In medical devices, this is known. Every device has a unique device
Regulatory & Marketplace
identification (UDI). Every manufacturer knows how many units of the device were sold and the estimate of patients treated by the device is part of each report. So, if there is a statistically significant increase in the frequency or severity of incidents, it would be a reportable event.
Post-market vigilance reporting is the responsibility of the manufacturer. Until the vigilance module of the European Database on Medical Devices (EUDAMED) is available, national vigilance reporting procedures will remain in place, this is covered by MEDDEV 2.12/1 Rev 8.
Reporting timelines are much shorter for devices than for drugs. A serious public health threat must be reported no later than two days after becoming aware of it. Serious incidents must be reported no later than 10 days after becoming aware and other reportable incidents no later than 15 days.
The two-day timeline is challenging because it is first necessary to learn about the incident and then decide if it is a serious public health threat. This can be particularly tricky when it comes to the interface between a pharmacovigilance department and medical device manufacturers.
Reporting Adverse Events
PMCF is a very popular way of obtaining clinical evidence for recertification of medical devices. Post-market vigilance covers any unexpected serious incidents, trend reports and any FSCA, as outlined above.
The National Competent Authority (NCA) must be notified and, in the case of FSCA, the member state where the manufacturer is registered. This follows MDR Article 87,88, 89 and 90. It is very important, before diving into reporting, to understand which section
applies and ask the NCA if they want to be informed about other reportable events.
In investigational studies, serious adverse events with a reasonably possible causal relationship with the device or procedure must be reported. There is an exception in the UK where all serious adverse events must be reported, not just the ones with a causal relationship.
The sponsor must also report any device deficiency that might have led to a serious adverse event, new findings in relation to any event mentioned above and reportable adverse events from third countries in which a clinical investigation is performed under the same investigation plan.
In investigational studies, MDCG 202010/1 classifies serious adverse events into four levels which are different to those found in drugs:
• Not related
• Possible: weak relation but cannot be ruled out completely
• Probable: relevant relation that cannot be reasonably explained by another cause
• Causal relationship: relation beyond a reasonable doubt
It is important to have the database properly set up to make sure the correct scale is used for devices.
Medical Device Product Life Cycle
If we look at the product lifecycle of a device, the crucial step for a vigilance department is to follow the post-market surveillance plan as defined by the manufacturer. These plans are very specific, there is no one-size-fits-all, so it is vital to communicate effectively with quality assurance teams on the device side. There is also an obligation to write a Periodic Safety Update Report (PSUR), which details all the safety information that occurred within a monitoring period. While this has the same name as the one for drugs, it is a completely different document and follows specific rules. There is a specific MDCG guideline – MDCG 2022–21 – for PSURs in devices.
Any outputs from this post-market surveillance life cycle become part of the risk management file.
Final Thought
Understanding the differences between drug and device requirements, valuing continuous education, and identifying strategies to
embrace and integrate products subject to different regulations is crucial. To support combined drug and device portfolios within primarily pharma-focused organisations, consider these actionable steps:
• Determine reportability: Establish clear criteria for what constitutes a reportable event.
• Adhere to timelines: Strictly follow the specified reporting timelines.
• Document reporting rationale: Clearly document the rationale for reporting decisions.
• Complete reports accurately: Provide detailed instructions for completing reports.
• Ensure medical input: Incorporate medical expertise in the reporting process.
• Manage translations: Ensure accurate translations for international submissions.
• Follow-up process: Implement a robust process for follow-up on reported events.
• Submission instructions: Provide clear instructions on submission procedures.
• Oversight responsibility: Designate responsibility for oversight of the reporting
• process.
• Trend and summary reporting: Establish processes for trend analysis and periodic summary reporting.
Humaira
Qureshi
Humaira has over 20 years’ strategic and operational experience in clinical and post-marketing drug safety solutions. As President of Qinecsa, Humaira is spearheading a movement to bring together scientific expertise with technology and create world-leading solutions that address the challenges faced by the pharmacovigilance industry and advance drug safety solutions to the next level of innovation, expertise and customer excellence. Prior to 2022 Humaira led a number of global pharmacovigilance businesses, delivering growth through customer excellence she has implemented sustainable operating models to support and manage PV operational solutions for many of our diverse clients.
talkfuture@pci.com
Driving development and connecting commercialization of sterile and lyophilized drug products, we are dedicated to your success in bringing life-changing therapies to patients.
Our end to end biologic solutions include:
• Sterile Formulation & Lyophilization Cycle Development
• Lyophilization and Sterile Fill-Finish Manufacturing
• Aseptic Robotic Technologies
• Analytical Support
• Clinical & Commercial Labeling & Packaging
• Refrigerated/Frozen Storage & Distribution
The Oncology Crux – Asking the Smart Oncology Questions before Shaping an Asset’s Journey to Market
With over 2,000 oncology clinical trials currently ongoing,1 the speed at which the market is evolving makes it increasingly difficult for pharmaceutical companies to differentiate their assets and maximise their value proposition, and thus their commercial potential. Asking the right questions at the outset of an asset's journey to market is essential to make informed decisions and develop a successful commercialisation strategy and product launch.
In this article, Jason Gilbody, Principal, Strategic Consulting at Envision Pharma Group and co-lead of Envision Oncology Solutions explores the unique challenges in the oncology market and the key factors that pharmaceutical companies should consider for a successful product launch.
What Makes Oncology Product Launches Different?
Launching an oncology drug differs significantly from launching drugs in other therapeutic areas due to the unique characteristics of cancer as a disease, the variability amongst cancers, the healthcare environment around treating cancers, and the specific needs and challenges faced by patients, healthcare providers, and payers in the oncology space. These differences shape the strategy, execution, and followup activities associated with oncology drug launches. Pharmaceutical companies can better prepare for oncology product launches early in development by asking questions, such as:
• Where does the product fit in complex oncology treatment landscapes?
• How does the regulatory approval path impact an oncology product’s launch strategy?
• How can patient support and engagement be used to support a successful oncology product launch?
• What special considerations are necessary when determining sales force size and structure when launching an oncology drug?
• What special considerations exist around pricing and reimbursement for oncology products?
Addressing these questions early in a product’s commercialisation journey allows developers to gain a deeper and more accurate perspective of the patients they serve and the markets they live in. The insights gained enable drug developers to make informed decisions, providing a solid foundation for an oncology product launch.
Where Does the Product Fit in Complex Oncology Treatment Landscapes?
The oncology treatment landscapes are characterised by rapid scientific advancements, evolving standards of care, and crowded development pipelines. New therapies are constantly being brought to market, leading to changes in the treatment guidelines. This makes it difficult for products to stand out and can be confusing for healthcare providers (HCPs) trying to understand where a drug fits into the current treatment landscape. Developers can address these challenges by continually evaluating the positioning of their products using competitive intelligence.
A deep understanding of the current and future market landscape, including competitor pipelines, product launches, and drug data forms the basis of successful launches. Given the competitiveness of oncology markets, having a well-informed, comprehensive, and highly agile launch strategy is a significant advantage. By understanding where the drug fits in the patient journey, educational materials and messaging can be targeted to the appropriate patient population.
The increased targeting of specific molecular pathways or biomarkers is a clear trend in oncology drug development. In 2023, all 14 new molecular entities approved for an oncology indication targeted a molecular pathway or biomarker.
2 Although this increase in precision medicine is promising for cancer patients, it also contributes to the challenging oncology landscape. For example, drugs targeting a specific molecular pathway or biomarker will likely require concurrent development and approval of a
companion diagnostic (CDx) to identify that specific target.
Managing the approval and launch of the CDx adds a layer of complexity and coordination when bringing an oncology drug to market. When a drug requires a CDx, it is imperative to educate HCPs on patient selection, testing, and treatment protocols that typically exist when dealing with precision medicines. A plan for educating the market on your drug’s CDx is critical to a successful launch. Even if a CDx is not necessary for your product, understanding how biomarker assays and genetic tests will be relied upon by HCPs to prescribe your product is critical when educating the market, and for the successful adoption of your product.
How Does the Regulatory Approval Path Impact an Oncology Product’s Launch Strategy?
New oncology drugs often qualify for expedited review pathways because of the high unmet needs in cancer treatment. Accelerated approvals in the US include Fast Track, Breakthrough Therapy, Priority Review, Accelerated Approval, and Real-time Oncology Review (RTOR). Oncology drugs can also be approved based on surrogate endpoints through conditional or accelerated pathways. For example, from 2006 to 2022, it was found that 71% of drug approvals for treatments of solid tumours were based on surrogate endpoints. 3 Accelerated approvals are designed to benefit patients but navigating them comes with uncertainty for oncology drug developers. Accelerated approval means developers need to get launch-ready faster, condensing time frames for corporate spending and launch execution. However, as this forces decisions to be made before all data is available, developers need to balance the risks and rewards associated with launch spending.
Being among the first-to-market for a certain patient population can be a huge competitive advantage. Therefore, developers must take advantage of accelerated approval opportunities whenever possible while also mitigating the associated risks. Additional post-marketing studies to confirm clinical benefits post-launch can help to reduce
some of the scepticism associated with products that have been through accelerated approval. Providing definitive evidence of clinical benefits, such as confirmed tumour shrinkage, allows pharmaceutical companies to address concerns about their products and anticipate any post-marketing changes to regulatory requirements.
How Can Patient Support and Engagement be Used to Support a Successful Oncology Product Launch?
Cancer diagnoses and subsequent treatment often come with relatively short treatment windows, high rates of morbidity, meaningful side effects, and a need to educate patients and get them on treatment quickly. The complexity of cancer diagnoses, the highly evolving standard of care in oncology treatments, and the use of increasingly personalised treatments make oncology treatments difficult for patients to understand. A clear and targeted approach to support and engage with patients can help them better navigate the labyrinth of these complexities.
Developers should consider engaging patient advocacy organisations geared towards oncology and specific types of
Clinical and Medical Research
cancer. Patient advocacy organisations play a prominent role in educating on the disease, the therapeutic options, the challenges with diagnosis and treatment, and provide patients with a much-needed support system that can speak to real-world experience with treatments. Given their high profile and reach within the patient community, partnering with advocacy groups can help boost awareness, disseminate education, and support patient access to new oncology treatments.
Coming to market with comprehensive patient support services can be another valuable asset when launching an oncology drug. Financial assistance programs and adherence support, along with additional training for HCPs, can alleviate administrative and monitoring requirements. Strategic considerations such as these can make a huge difference, dramatically increasing the chances of success at product launch.
What Special Considerations are Necessary When Determining Sales Force Size and Structure When Launching an Oncology Drug? The differences in cancer and oncology markets influence the optimal size and
structure of a sales force. Again, key factors include the complexity of treatments, specialized HCPs, and the importance of rapid patient care. Oncology drugs are prescribed by a small group of specialists made up of oncologists and haematologists. Oncology treatments are also usually administered in a hospital setting or specialised cancer centres. Smaller patient populations, higher drug costs, and complex selling processes necessitate a tailored approach to sales team structure.
Concentrated clusters of HCPs and facilities in urban centres can potentially expand the reach of each sales representative, enabling them to effectively engage with a larger number of HCPs. However, the relatively limited patient population can create a comparatively smaller target market for pharmaceutical sales, requiring a nuanced approach to resource allocation and sales strategy.
The limited number of patients and the high cost of each prescription within oncology must also be considered. Since cancer profoundly affects patients and their families, patient outcomes are an integral
STEAMING SOLUTIONS FOR ALL INDUSTRIES
Pressure regulators Control valves
Pipeline ancillaries
Steam traps
Special equipment
Clinical and Medical Research
consideration and activities that contribute to patient support and education should be considered. Sales representatives can also assist in navigating insurance approvals and reimbursement processes, addressing the substantial challenges patients may encounter in accessing oncology treatments. This approach aligns with the overall goal of delivering optimal care and improving patient outcomes in the field of oncology.
What Special Considerations Exist Around Pricing and Reimbursement for Oncology Products?
Pricing is the single most important factor to get right when launching any new product. This is particularly true in oncology where drugs are typically more expensive than drugs in many other areas, reflecting the complexities of development and their life-changing potential. High drug costs in oncology translate to increased scrutiny from payers and health technology assessment bodies (HTA) about cost-effectiveness and value.
This scrutiny means that securing reimbursement in oncology can also be more complex and contentious than in other therapeutic areas, making product pricing a real challenge. Setting an optimal price requires a thorough understanding of the drug’s unique value proposition, including its clinical benefits compared to existing treatments, its potential to address unmet needs, and its anticipated impact on patient outcomes. Given these considerations, it is important to bolster your product launch with early planning, including strategic, wellthought-out trial design and generation of relevant strong outcomes data.
Clearly demonstrating the value of a product and engaging in risk-sharing agreements or outcomes-based contracts are strategies that can overcome pricing and reimbursement challenges. Collecting and disseminating real-world evidence reinforces the value proposition of a drug, which is particularly relevant postlaunch. By providing strong evidence for the real-world effectiveness and safety of their products, oncology drug developers can unlock broader reimbursement and encourage adoption, ultimately leading to more patients being treated.
How Can Answering These Questions
Ensure a Successful Product Launch?
Entering the oncology drug market presents challenges due to its dynamic and intricate nature. A successful launch strategy requires
specialised knowledge, strategic flexibility, and a deep understanding of the needs of patients, healthcare providers, and payers.
By asking smart questions, pharmaceutical companies can shape an asset’s journey to market by developing a tailored commercialisation strategy and corresponding evidence-generated plan that considers real-world data needs. This comprehensive approach enables companies to move faster and more efficiently. Sound early decisions mean less back-tracking later on. This can also translate to better-targeted marketing messages, better relationships with key opinion leaders, and more effective patient support programs.
Strategic consulting partners can help navigate the complexities of an oncology drug launch. By leveraging experienced partners that have worked on launch plans and launch excellence frameworks across many biopharmaceutical products, oncology drug developers can quickly and effectively build a robust launch plan. Ultimately, this means that pharmaceutical companies can increase their understanding of industry benchmarks and best practices to help set up their oncology assets for a successful launch.
3. Chitkara, Akshit, Manoj P. Ria, Rajat Thawani, and Emerson Yu-sheng Chen, “Recent analysis of frequency of surrogate end points used in oncology clinical trials 2006–2022,” Journal of Clinical Oncology, 41:16, May 31, 2023.
Jason Gilbody
Jason has spent over 17 years in the consulting industry, with 7 years focused on life sciences. Jason’s expertise is supporting oncology companies with their commercial strategy and planning, including brand strategy, competitive response, launch planning and management, market research (primary and secondary), and data analytics. Jason has provided support to companies across numerous hematological and solid tumor types. Before joining Envision Pharma Group, Jason was with the Commercial Advisory Group at Syneos Health, focused on commercial strategy. He also served as a Vice President at the Analysis Group, where he led consulting engagements related to commercial operations for life sciences companies engaged in corporate litigation. Jason has also worked at Deloitte & Touche with a focus on healthcarerelated data analytics projects. He holds an MS in Finance from Brandeis University’s International Business School and a BA in Economics from Stonehill College.
Recombinant Protein-Reagent
PYROSTARTM Neo+ is based on a genetically engineered approach to produce reactive factors required for endotoxin detection at pharmaceutical and medical equipment manufacturing sites. With greater stability of the negative control and better endotoxin recovery in heparin and heparin-based compounds, Neo+ facilitates the same sensitivity of endotoxin detection as our traditional limulus amebocyte lysate (LAL) reagent, through a more sustainable and environmentally friendly method.
• Colorimetric method, can be used with an absorbance plate reader
• 3-Factor system mimics the same cascade reaction as traditional LAL
• Endotoxin-specific reagent eliminates the risk of false positives from (1-->3) ß-D-Glucan
• 100% free of horseshoe crab blood
• Quantitative range: 0.001 to 50EU/mL
• High sensitivity with less lot-to-lot variation
• Stable storage after dissolution (4 hrs. at 2-8°C and 2 weeks at -30°C)
Clinical and Medical Research
Engaging Minority Communities: Collaborative Practices and Long-term Success
The quest to understand and treat rare diseases poses unique challenges. Rare diseases, often referred to as orphan diseases, affect a small percentage of the population and are often neglected, however, within this complexity lies a crucial imperative: the need to engage marginalised communities in rare disease research and drug development processes.
We know that people experience the same disease differently therefore It’s essential that clinical trials include people with a variety of lived experiences and living conditions, as well as characteristics like race and ethnicity, age, sex, and sexual orientation. By fostering inclusivity, diversity, and collaboration, researchers and drug developers can gain deeper insights into the underlying causes of rare diseases, develop more effective treatments, and advance the cause of health equity for all.
But how do we engage with minority communities, empower them, and create lasting relationships long before we think about enrolling patients in a clinical trial?
Understanding the Minority Experience in Rare Disease
The first step is to understand that being a minority and facing a rare disease can be complex and challenging, often compounded by systemic barriers and disparities in healthcare access. Victoria Arteaga shares the isolation and stigma she faced when her threeyear-old daughter was diagnosed in 2018 with SYNGPA,1 a rare genetic disease. As a Latina with a daughter with a rare genetic disease, Victoria quickly understood the unique challenges of navigating the healthcare system by other Hispanic families. “Language, cultural and georgraphical barriers.” From the moment of her daughter’s diagnosis, Victoria and her family have felt it was important to reach out to the Latino and Hispanic communities to promote awareness and support and seek a cure for this condition.
Today, Victoria is the Latin America Director of the SynGAP Research Fund (SRF)
and co-founder of the Sociedad Hispana de Enfermedades Raras (SHER) (Hispanic Society for Rare Diseases). In these roles, Victoria advocates to raise awareness of rare diseases and improve access to resources for this under-represented community. Victoria states: “Creating opportunities for dialogue, listening to concerns, and incorporating feedback into decision-making processes can help ensure that different initiatives are responsive to community needs."
Empowering Advocacy and Self-advocacy
Education and awareness empower individuals within marginalised communities to advocate for their health needs and rights. By providing information about patient rights, available support services, and avenues for seeking assistance, these initiatives encourage self-advocacy and empower individuals to assert their voices in healthcare decision-making processes.
Deborah Requesens serves as the director of The Orphan Disease Centre's JumpStart program at the University of Pennsylvania’s School of Medicine State University. The program serves to establish and progress research agendas in emerging and neglected rare diseases. Deborah states: “The JumpStart program works closely with patient groups and foundations, pharma and biotech, and the academic community to drive therapeutic development for rare diseases.”
Deborah, who is also the co-founder of SHER, shares the importance of engaging with minority communities to build capacity and trust. “It is crucial to engage with underserved populations, such as the Hispanic and Latino communities, early and often, in the drug development process. It takes time to build trust and these interactions will ensure the development of therapies with the entire population in mind.”
Lack of cultural and linguistic competency among healthcare providers can impact the quality of care received by Hispanic and Latinx individuals with rare diseases. “Effective communication, patient education, and culturally sensitive approaches to care are essential for ensuring understanding, trust, and adherence to treatment plans,” says Deborah. “We need to also empower
and equip the healthcare providers, who may be from under-represented backgrounds themselves, with the information to share with patients and their families. Representation in all areas of healthcare is key.”
Listening to Underserved Voices
The Foundation for Sarcoidosis Research (FSR), an international nonprofit dedicated to finding a cure for sarcoidosis recently launched the Coalition for Clinical Trial Equity as part of their latest phase of the Ignore No More Initiative. This initiative aims to increase the representation of Black and African American patients in clinical trials, thereby improving health outcomes for marginalised communities.
FSR created an IRB-approved, anonymous survey for Black and African American patients to share their experience with sarcoidosis, as well as their knowledge, perspective and barriers faced in enrolling in clinical trials and the role it plays in advancing treatment for sarcoidosis patients. Mary McGowan, the CEO of FSR states: “The survey highlighted that the number one reason the black community is not enrolling in sarcoidosis clinical trials is because they are not being asked. They are not having conversations with their clinicians. In fact, the survey showed that only 6% of those that were asked did not participate.”
By engaging these communities in research efforts, we will gain valuable insights into the genetic, environmental, and socio-economic factors that contribute to the development and progression of sarcoidosis. “We must ensure that we are including a high percentage of those that have the highest prevalence and unfortunately the worst outcomes of the disease in clinical trials, as no doubt, we will gain a better understanding of the disease and improved therapies in a timelier fashion, leading to better patient outcomes for all communities.”
Homogeneity can limit the generalisability of clinical research and often findings may overlook important differences in how diseases manifest across diverse populations. By actively involving marginalised commu-
nities in clinical research, researchers can obtain a more comprehensive understanding of disease mechanisms, treatment responses, and potential adverse effects across different demographic groups. For instance, Mary points out: “Mental health is also a key component to consider as rare diseases, such as sarcoidosis, can have serious mental health ramifications.”
Celebrating Minority Populations
When Connie Lee learned that New Mexico has the highest prevalence of cavernous malformation (CCM) in the world because of a founder mutation that began in the early 1600s in the original Hispanic families, Common Hispanic Mutation, she launched what has become the Baca Family Historical Project in New Mexico.
Connie, the President and CEO of Alliance to Cure Cavernous Malformations, explains how the 4-year campaign connects the descendants of Cristóbal Baca and Ana Maria Pacheco Ortiz, one of the original Spanish settlers in northern New Mexico. “We facilitated connection through genealogy on Facebook and community gatherings, and by training the state’s extensive network of community health workers.” Connie shares how the work celebrates heritage, offers disease education and genetic testing, and creates opportunities for this extended family to meet and get to know each other. “We have created a book to preserve the connections and history.”
Clinical and Medical Research
Connie also found that the largest clinical and research databases of CCM patients in the US contained a disproportionately small number of Black patients. To address this disparity, the nonprofit organisation launched Breaking Barriers for Black CCM Health. “We need to listen to Black patients to help us understand their diagnostic and care challenges and build a community of Black patients to optimise support and disease knowledge”.
Increasing Access to a Fundamental Human Right
Access to healthcare is a fundamental human right, yet systemic inequalities persist, limiting the ability of marginalised individuals to benefit from advances in science and drug development. By prioritising inclusivity and diversity in engagement initiatives, stakeholders can work towards dismantling barriers and ensuring that all individuals, regardless of their background, have equitable access to the benefits of scientific progress.
Connie Lee
Connie Lee is the founder and CEO of the Alliance to Cure Cavernous Malformation patient research organisation. Among its many activities, the organisation created and supports two engagement initiatives: The Baca Family Historical Project in New Mexico and the national Breaking Barriers for Black Health project.
Mary McGowan
Mary McGowan is the CEO of the Foundation for Sarcoidosis Research the leading international organisation dedicated to finding a cure for sarcoidosis and improving care for sarcoidosis patients through research, education, and support. Mary has led an awareness campaign on African American Women and Sarcoidosis that has reach over 500,000 individuals through the awareness campaign, Ignore No More, and its second phase, Accelerate Clinical Trial Equity in Sarcoidosis (ACTe NOW!), to improve clinical trial engagement with Black Americans.
Neena Nizar
Neena Nizar, Ed.D is Director of Patient Advocacy Strategy at ICON’s Center for Rare Diseases. She has 20 years of rare disease patient advocacy strategy experience focused on development of nano-rare therapies and patient-centered outcomes. She has deep expertise in diversity, equity and inclusion strategy with experience across multiple healthcare settings and academia. Neena has strong expertise in the Middle Eastern/Asian patient advocacy landscape and is a highly regarded leader in the rare skeletal disease area. She is founder of The Jansen's Foundation and was named a WebMD Healthcare Hero in 2023.
Victoria Arteaga
Victoria Arteaga, MBA, is the Latin America Director of the Syngap Research Fund (SRF). Victoria’s journey with SRF started shortly after her daughter Amelia was diagnosed with a variant of the SYNGAP1 gene in 2018. As an SRF board member, Victoria has advocated for families in the US and Latin America to promote knowledge and collaboration. In addition, Victoria co-founded the Hispanic Society for Rare Diseases where she advocates to raise awareness of rare diseases and improve access to resources for this underrepresented community.
Deborah Requesens
Deborah Requesens, PhD, is the Director of the Orphan Disease Center’s JumpStart Program. This program serves to establish and progress research agendas in emerging and neglected diseases. Deborah is the cofounder of SHER (Hispanic Society for Rare Diseases), an organisation that promotes awareness about rare diseases in the Hispanic population and aims to increase the participation of this community in rare disease research.
The Importance of Polymorph Screenings: Risk Mitigation and Manufacturing Control
Polymorphism in pharmaceuticals may affect drug product (DP) development, clinical studies, manufacturing, quality and stability. Therefore, it is mandatory to gain knowledge of the polymorphic behaviour of active pharmaceutical ingredients (APIs) in the early development phase by performing polymorph screen programs.
The Importance of Polymorphism in Drug Development
Crystalline APIs often exhibit the “polymorphism” phenomenon, meaning that different crystalline arrangements of an API are possible in the solid state. The API’s polymorphic behaviour can significantly affect its physical properties.
Polymorphic transformations can be prompted in DP manufacturing by physical processes, including compression, grinding, milling, processes involving solvents and moisture, or under storage conditions. Unexpected changes in polymorphic form may affect the quality and efficacy of the final DP and dramatically impact patients.
The most notorious example of polymorphic conversion affecting DP quality and efficacy is the Ritonavir case.1 Form I, the only solid form of Ritonavir known in 1996, was marketed as a liquid formulation in hydroalcoholic mixtures. In 1998, precipitation from the DP solutions was noticed. This precipitation affected the API bioavailability as a lower amount of drug was present in the solution. Due to this inconsistency in the DP, Abbott had to remove the oral capsule formulation from the market. Investigations revealed that the emerged solid was a more stable polymorph, hence less soluble, known now as Form II. Eventually, the issue was solved, and the product returned to the market as a gel cap formulation. The discovery and reformulation of Ritonavir cost $250 million.2
This article will cover the issues related to polymorphism in drug development and the benefits of experimental polymorph screen studies.
Understanding Polymorphism and its Challenges
Polymorphism is defined as the ability of a solid substance to exist in various crystal forms (i.e. polymorphs). In other words, the same chemical entity can exhibit different molecular arrangements (Figure 1a).
Polymorphs demonstrate differences in molecular packing, including exposure of different functional groups to the crystal surface, crystal packing, intermolecular forces, crystal density and the presence of tunnels, voids, or channels. These structural features impact the physicochemical properties of the polymorph, including solubility, melting point, particle morphology, density, dissolution rate and hygroscopicity. Therefore, polymorphs can exhibit significant differences in their physicochemical properties despite the identical chemical composition.
The definition of “polymorphism” includes anhydrous, solvated and hydrated crystalline solids (Figure 1b). However, true polymorphs should exhibit the same chemical composition. For this reason,
solvates and hydrates are generally defined as pseudo-polymorphs.
Under particular conditions (e.g. temperature, pressure and humidity), the various polymorphs of an API have different free energies. The polymorph with the lowest free energy is considered the most thermodynamically stable form. All other forms are referred to as metastable.3
Metastable forms might be sufficiently stable under particular temperature, pressure and humidity combinations. However, they tend to evolve toward the thermodynamic equilibrium state, according to Ostwald’s rule of stages (Figure 24). Therefore, the metastable phase will eventually transform into the most thermodynamically stable form. Conversion rate will depend on kinetics. Sometimes, the conversion could be extraordinarily slow unless mediated by a solvent.
DP manufacturing processes (compression, grinding, milling) may induce polymorphic transformations caused by solvents, water, pressure and temperature.
Figure 1. a) Schematic representation of how the same API shapes into different molecular arrangements I, II and III. b) Schematic representation of compositions of anhydrous, and hydrated or solvated crystalline solids.
This might lead to the formation of undesired polymorphs. For instance, anhydrous forms might convert to solvates in processes involving solvents, or hydrates when exposed to uncontrolled humidity levels.
Unexpected polymorphic changes may affect the physicochemical properties of the drug, and ultimately the quality and efficacy of the final DP.
Innovative Approaches to Address Polymorphism in Drug Development
One of the major benefits of screening for polymorphs is the mitigation of potential risks a solid form change could pose. Simultaneously, the benefits associated with a solid form are assessed. The risk/benefit assessment will drive the selection of the final form optimal for development. This knowledge guarantees greater control over the selected solid form.
An additional important benefit of investigating the polymorphic behaviour of an API is that polymorphs are patentable. This knowledge strengthens the intellectual property (IP) of a certain drug.
Despite the considerable advances in crystal structure prediction in recent years,5 experimental screening is required to generate and analyse a certain polymorph.
A polymorph screen evaluates whether multiple polymorphs can exist for an API, and identifies thermodynamic and kinetic polymorphs, as well as pseudo-polymorphs (hydrates and solvates). The ultimate goal is to choose the optimal polymorph for development and understand its stability
and physicochemical properties. Usually, the preferred form for development is the thermodynamically stable polymorph, as it guarantees stability throughout the manufacturing process.
Polymorph screening studies provide information regarding desired polymorph solubility, preferred crystallisation solvent, optimised concentrations in mixed solvent systems, crystallisation conditions and other processing parameters.
In a typical polymorph screen, the API is recrystallised under various conditions promoting nucleation and crystal growth.6 Usually, different crystallisation methods, solvents and temperatures are tested.
1. Crystallisation Methods
Table 1 provides a brief (non-exhaustive) list of
Crystallisation method
Solvent equilibration
Thermocycling
Evaporation
Fast and slow cooling
Grinding
the crystallisation methods most commonly applied in polymorph screens. Depending on the time scale of the experiments, different crystallisation methods favour the nucleation of thermodynamically and metastable polymorphs.
2. Effect of Solvent
Most polymorph screen experiments are solvent-mediated. Therefore, the selection of solvents has a particular relevance in the study design.
Solvents with different polarities that belong to different classes (alcohols, ketones, amide, nitriles, aromatic and aliphatic hydrocarbons, acyclic and cyclic ethers) are usually included. Solvents differing in polarity and functional groups typically interact differently with the API molecule and as such favour different molecular arrangements in solution that translate into the solid state.7 Sufficient variability of solvents in the screen increases the chances of finding novel polymorphs.
When the size of the polymorph screen is limited, the focus should be on pharmaceutically accepted solvents (ICH Classes 3 and 2, Table 2). When the scope of the study can be broadened in a comprehensive polymorphism study, less common solvents can be tested. In the late phase of development, it is preferable to conduct polymorph screen programs in process-relevant solvents to verify that the solvents selected for manufacturing will guarantee a robust crystallisation process and that the process is not jeopardised by the appearance of undesired forms.
Diffusion of antisolvent vapours into API solution
Diffusion of solvent vapours onto solid API
Crystallisation method
Precipitation by antisolvent addition (reverse and forward mode)
Freeze-drying
Crystallisation templated by polymers
Table 1. Most commonly applied crystallisation methods in polymorph screens
Figure 2. Schematic diagram of energy states of polymorphs according to Ostwald’s rule of stages.
Manufacturing
3. Effect of Temperature and Relative Thermodynamic Relationship
As the free energy of a polymorph also depends on the temperature, the effect of temperature on the nucleation of polymorphs should be investigated in a polymorph screen.
For this reason, solvent equilibration experiments are usually performed both at room temperature (RT) and high temperatures.
Investigation of the effect of temperature on the polymorphs’ stability can give insight into their relative thermodynamic stability: two polymorphs can be monotropically or enantiotropically related.
When a single polymorph is always the most stable form regardless of temperature, the two polymorphs are monotropically related. When a different polymorph is the most stable polymorph at a higher temperature, the two polymorphs are
enantiotropically related. In enantiotropic systems, there is a temperature where the free energy of the two polymorphs is equal. This temperature is called transition temperature. One polymorph will be more stable below the transition temperature, and the other polymorph will be more stable above the transition temperature.
Investigating the relative thermodynamic stability of the two polymorphs is important to know the stability temperature range of the desired polymorph. This information is key to manufacturing the desired form consistently.
4. Characterisation of Polymorphs
X-ray powder diffraction (XRPD) is the foremost solid-state characterisation technique used to distinguish polymorphs. High-throughput XRPD analysis is used as a qualitative analytical tool for screening samples. High-resolution XRPD analysis is
used for quantitative analysis of polymorph mixtures (phase quantification).
Single crystal X-ray diffraction analysis is the definitive tool to obtain the crystal structure of polymorphs and can be used for obtaining calculated powder patterns.
Thermal analysis techniques such as differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), coupled with mass spectrometry or infrared spectroscopy (TGA/MS or TGA/ IR), play a particularly relevant role in the characterisation of polymorphs. Through TGA/MS or TGA/IR, it is possible to assess the solvation state of a polymorph (anhydrous, hydrate, solvate). Polymorph melting points are determined by DSC analysis, which also reveals other thermal events, such as recrystallisation, solid-solid phase transitions, desolvation and thermal decomposition.
Complementary analytical methods to characterise polymorphs include solidstate NMR, Raman, FT IR, and hot-stage microscopy.
5. Manufacture the Optimal Polymorph
Once the optimal crystalline phase has been selected, the following step is developing a robust crystallisation process. The most common crystallisation methods for manufacturing are cooling crystallisation and antisolvent precipitation.
In both cases, the addition of seeds of the selected crystal form to a supersaturated solution of API may help in initiating the crystallisation process of the desired form while preventing formation of unwanted forms. Crystal seeds serve, in fact, as nuclei where API molecules can attach themselves from the solution phase onto the surface of crystals, thereby continuing the crystal growth of the target solid form.
A seeding protocol therefore directs the crystallisation to the desired solid form and offers high control over the manufacturing process. This technique is commonly used in bulk drug synthesis.
Navigating Polymorphs from Early Development
The selection of the appropriate poly-morph with the desired stability and physicochemical properties must be realised in the early phase of the drug development process.
Conducting a comprehensive polymorph screening is costly and time-consuming.
A phase-appropriate approach to solid form screening is recommended as this carefully balances the importance of finding a stable solid form at the current stage of development against the cost of such a screen (Figure 3).
For compounds entering preclinical development, an Enabling Polymorph Screen is recommended. The key deliverable of this screening program is to identify a stable solid form that will generate reproducible dissolution and bioavailability, and, as such, minimise variability in pharmacokinetic and toxicology studies. During the program, solubility in 20 solvent systems is determined. The solubility information is fundamental for the design of the polymorph screen experiments, and followup crystallisation process development work. The Enabling Polymorph Screen explores crystallisation behaviour in 30 different solvents (or solvent combinations) in four crystallisation modes: solvent equilibration (at RT and 50°C), thermocycling, cooling and evaporative crystallisation. This screen is typically conducted at a stage where the drug substance (DS) is low in supply. This miniaturised approach generates sufficient data while consuming less than 2 g of API.
When the synthesis route of the API has been established and Phase II studies are approaching, it is the appropriate time to confirm the choice of the solid form with an Extensive Polymorph Screen. Building on the solubility data collected in the Enabling Polymorph Screen, the Extensive Polymorph Screen involves 10 different crystallisation modes (including solvent equilibration at 5, RT and 50°C, thermocycling, grinding, antisolvent crystallisation and vapour diffusion onto solids and solution, freezedrying, crystallisation templated by polymers, fast and slow cooling, evaporation) and approximately 300–400 experiments. The entire polymorph screen program consumes less than 10 g of API. The Extensive Polymorph Screen provides a near-complete picture of potential polymorphs and pseudopolymorphs and will guide the optimal solid form to progress into the final phases of development. The knowledge obtained from this screen also reinforces the IP position of the API. Additional screens may be conducted before product launch (e.g. salt/cocrystal screens) to identify and patent as many solid forms as possible.
Regulatory Requirements and Documents
Addressing Polymorphism
To guarantee the quality, safety and efficacy
of the DP, regulatory authorities now focus much attention on the physicochemical properties of APIs and require that the manufacturer has a thorough understanding of the drug polymorphism throughout the
product life cycle. Two main regulatory documents address polymorphism in DSs:
• FDA ANDAs: Pharmaceutical Solid Polymorphism9
Figure 3. Schematic representation of the phase-appropriate approach to solid form screening
Figure 4. Decision trees 4(1) and 4(2) adapted from ICH Q6A
DSs
Manufacturing
REFERENCES
1. Bauer, J., Spanton, S., Henry, R., Quick, J., Dziki, W., Porter, W., Morris, J., et al., Ritonavir: an extraordinary example of conformational polymorphism, Pharm. Res., 18, 6, 859-866, (2001).
2. V. Atkinson, Chemistryworld.com, Could mechanochemistry have saved Abbott Laboratories $250 million?
3. Lee, E. H., A practical guide to pharmaceutical polymorph screening & selection, AJPS, 9, 4, 163-175, (2014).
4. Ostwald, Von W., Studien über die Bildung und Umwandlung fester Körper. 1. Abhandlung: Übersättigung und Überkaltung. Zeitschrift f. Physik. Chemie, XXII, 289-330, (1897).
5. Cruz-Cabeza, A. J., Crystal structure prediction: are we there yet?, Acta Cryst. B, Structural Science, Crystal Engineering and Materials, 72, 4, 437-438, (2016); Price, S. L., Computed crystal energy landscapes for understanding and predicting organic crystal structures and polymorphism, Acc. Chem. Res., 42, 1, 117–126, (2009).
6. Newman, A., Specialised Solid Form Screening Techniques, Org. Process Res. Dev., 17, 457-471, (2013).
7. Gu C.H., Li H., Gandhi R. B., Raghavan K., Grouping Solvents by Statistical Analysis of Solvent Property Parameters: Implication to Polymorph Screening, Int. J. Pharm., 283, 117–125, (2004).
8. FDA, ANDAs: Pharmaceutical Solid Polymorphism, “Chemistry, Manufacturing, and Controls Information,” July 2007.
9. International Conference on Harmonisation (ICH) Q6A. “Specifications: Test procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances,” Step 5, May 2000.
• ICH Q6A Specifications: “Test procedures and Acceptance Criteria for New DSs and New DPs: Chemical Substances”9
The ICH guidelines provide a series of decision trees to evaluate the polymorphism and its impact on the physicochemical properties of the DS.
These decision trees should be followed sequentially. Initially, whether the DS exhibits polymorphism must be verified by performing a polymorph screen (Figure 4, decision tree 41). If so, it must be investigated whether the different polymorphic forms can affect the performance of the DP (difference in solubility and dissolution rate, for
instance) (Figure 4, decision tree 42). When polymorphism has been demonstrated for the DS and shown to affect these properties, the potential for change in polymorphic forms in the DP and whether such a change has any effect on product performance must be investigated (Figure 5, decision tree 43).
The questions of the decision trees can be answered by the information gathered in polymorph screen studies. The intent of decision trees is to either eliminate concerns about potential solid form changes (i.e. potential changes in polymorphic form have no effect on the DP) or demonstrate that the manufacturer has control of the solid form throughout the process.
Giovanna Brancatelli is Principal Scientist – Scientific Director at Ardena Solid State Research. She supervises R&D projects on polymorph, salt, cocrystal screens and preformulation studies. Giovanna joined Ardena in 2015 from Trieste University's Chemistry Department, where she conducted post-doctoral research on molecular recognition of biogenic amines in solid state. Her expertise includes polymorphism, crystallization, organic, inorganic and supramolecular chemistry, and X-ray crystallography. She earned her M.Sc. degree and Ph.D. in Chemical Sciences at Messina University.
Giovanna Brancatelli
Figure 5. Decision tree 4(3) adapted from ICH Q6A Specifications: “Test procedures and Acceptance Criteria for New DSs and New DPs: Chemical Substances.”
Why X-ray Inspection is a Viable Quality Assurance Solution for Pharma Products
We are all aware that the pharmaceutical industry operates within a highly regulated environment, where upholding product safety and quality is paramount. Manufacturers invest significant resources in validation processes to meet regulatory requirements. One technology solution to complete quality assurance checks in the pharmaceutical sector is product inspection. While many manufacturers rely on systems like vision inspection and metal detection, the use of x-ray inspection is gaining recognition as an additional tool for quality assurance. To understand the benefits of x-ray inspection, we must first look at its functionalities, then address the misconceptions. In this article, we will explore why x-ray inspection should be an invaluable asset in the pharmaceutical industry, providing manufacturers with comprehensive and accurate quality checks while dispelling concerns about its impact on products.
More than Contaminant Detection
The primary function of x-ray inspection is contaminant detection of a wide range of foreign bodies including metal, glass, mineral stone and high-density plastics. Simultaneously, x-ray systems perform valuable in-line quality checks including counting components, identify missing or broken products, monitoring fill levels, checking for damaged packaging and detecting agglomerates such as flavour and powder lumps.
Addressing the Misconceptions
To understand the benefits of x-ray inspection, we must first address the misconceptions. Pharmaceutical manufacturers often express concerns about the potential impact of x-ray inspection on their products. However, studies have consistently shown that the quantity and energy of x-rays used during product inspection are minimal, and the duration of exposure is extremely brief. For example, tablets undergoing x-ray inspection are typically exposed to low-energy x-rays for less than 0.2 seconds. In comparison, the dose of background radiation that pharmaceutical products receive while on the shelf, in transit
or during consumer ownership is significantly higher than the levels delivered by an endof-production line x-ray inspection system.
The US Food and Drug Administration (FDA) acknowledges that the dose levels received by objects passing through an x-ray inspection system are lower than the dose of background radiation received in a single day. Furthermore, and perhaps most crucially, the FDA asserts that there is no known danger in consuming medicines that have undergone x-ray inspection.1
Scientific studies have also been conducted to evaluate the effects of x-ray inspection on pharmaceutical products. For instance, researchers at the Department of Drug Delivery and Nano Pharmaceutics, Graduate School of Pharmaceutical Sciences, Nagoya City University in Japan conducted a study on the effect of x-rays on the pharmaceutical quality of drug tablets. The study exposed acetaminophen, ioxoprofen and mefenamic acid tablets to varying x-ray doses, ranging from 0.34mGy to 300Gy. These doses were significantly higher than those delivered by typical x-ray inspection systems. The evaluation of samples through formulation tests revealed that exposure to x-rays did not affect the pharmaceutical quality of the drug tablets. The exposed samples exhibited similar characteristics in dissolution, disintegration and hardness tests as control samples that were not exposed to x-rays. Additionally, when combined with accelerated temperature and humidity tests equivalent to six months of exposure, x-ray exposure did not affect the pharmaceutical quality of the samples. This study concluded that x-ray exposure at levels much higher than those encountered during product inspection had no significant effect on the efficacy or other properties of the drug tablets.2
Another study, conducted by Robert Bosch Packaging Technology and the PHAST Society for Pharmaceutical Quality Standards, exposed model pharmaceutical substances, tramadol HCl and nifedipine, to x-ray radiation for a period of 2 hours. This extended exposure time was significantly longer than the fraction of a second typically used during industrial x-ray inspection. However, no degradation was observed in
either substance after the 2-hour exposure period.3
These scientific studies collectively demonstrate that x-ray inspection at the levels used in pharmaceutical manufacturing does not significantly affect the quality or efficacy of the products. While not all formulations have been studied, the low doses of x-ray radiation involved in x-ray inspection systems are well below the levels of naturally occurring background radiation and do not pose a risk to product integrity.
Dispelling the Concern
Despite the availability of such comprehensive studies, pharmaceutical manufacturers often prefer to conduct their own tests. This cautious approach can be attributed to the stringent regulatory environment in which they operate. Regulatory bodies worldwide have established guidelines and requirements for pharmaceutical manufacturing including the Good Manufacturing Practice (GMP), the Good Distribution Practice (GDP), and manufacturers who want to comply with these regulations while maintaining the highest level of quality assurance.
Some manufacturers may have concerns specific to their products or processes that they believe require additional scrutiny. These concerns may stem from the complexity of the formulation, sensitivity to radiation or unique packaging requirements. Conducting their own tests allows manufacturers to address these specific concerns and gain confidence in the integrity of their products.
However, it is important to recognise that the extensive scientific studies and regulatory guidelines surrounding x-ray inspection provide a strong foundation for manufacturers to trust this inspection method. The studies have consistently demonstrated the safety and efficacy of x-ray inspection, and regulatory bodies acknowledge its effectiveness in ensuring product quality and safety.
Utilising the technology of x-ray inspection systems, manufacturers can start to see significant benefits on their production line. It eliminates the need for redundant testing, which can be costly and time-consuming, allowing manufacturers to streamline their
processes and focus on other critical aspects of production. By trusting in the proven capabilities of x-ray inspection systems, manufacturers can benefit from the expertise and experience of the producers of these systems, who have a deep understanding of the technology and its application in the pharmaceutical industry.
Expanding the Scope of X-ray Inspection
So where should x-ray inspection technology be used within the production process and what are its benefits? Traditionally, x-ray inspection in the pharmaceutical industry has been primarily used for specific applications, such as verifying the presence of tablets or needles. However, the potential of x-ray inspection extends far beyond these limited applications. When employed at the end of the production line, x-ray inspection becomes one of the most versatile and comprehensive inspection tools available to manufacturers. Unlike other inspection methods, x-ray inspection has the capability to inspect a diverse range of products, regardless of their shape or packaging.
One of the significant advantages of x-ray inspection is its ability to penetrate various materials, including metals, glass, high-density plastics and rubber. This capability enables the detection of dense foreign bodies that may contaminate pharmaceutical products. Metal fragments, glass shards or other foreign objects that could compromise product quality and safety can be reliably identified and removed from the production line, preventing them from reaching the consumers.
As mentioned at the start of this article, in addition to contaminant detection, x-ray inspection allows for other critical quality checks. It can verify the completeness and integrity of packaged products, detecting missing or damaged items within the packaging. This level of scrutiny is essential in the pharmaceutical industry, where missing components or damaged products can have severe consequences for consumers. For instance, checking the presence of needles in glucose monitoring systems, verifying their functionality is crucial, as a faulty device could have fatal implications for diabetic patients. By detecting and preventing the distribution of defective or compromised products, x-ray inspection plays a crucial role in upholding a company's brand image and reputation.
One of the notable advantages of x-ray inspection is its versatility. Unlike other
inspection methods that may be limited to specific product types or packaging formats, x-ray inspection can be applied to a wide range of pharmaceutical products, regardless of their shape, size or packaging materials. It is particularly beneficial for pharmaceutical manufacturers who commonly use metalised film or aluminiumbacked blisters as packaging methods. X-ray inspection systems can effectively inspect products contained within these packaging materials and check their integrity and quality.
By leveraging x-ray inspection technology at the end of the production line, pharmaceutical manufacturers can enhance their quality control processes and minimise the risk of contaminated or defective products reaching the marketplace. The comprehensive nature of x-ray inspection, coupled with its ability to inspect a variety of products and packaging formats, makes it an invaluable tool in upholding product safety and maintaining consumer trust.
Compliance and Traceability
Beyond its ability to detect defects, x-ray inspection systems also contribute to compliance with regulations and provide traceability. The data generated by x-ray inspection systems can facilitate compliance with regulatory requirements. Manufacturers can access comprehensive quality data and export it as needed, providing transparency and accountability. X-ray inspection systems enable the creation of individual images of each product, complete with date and time stamps, providing information to support due diligence that quality checks have been completed. The information is easily retrieval thereby facilitating easy tracking. Additionally, the reporting capabilities of x-ray inspection systems allow for detailed shift and production reports, including the number of rejected items. This level of documentation aids the validation process and ensures that approved settings remain unchanged during production, minimising the risk of errors or deviations.
Conclusion
X-ray inspection plays a crucial role in completing a large range of quality assurance checks without significantly impacting the efficacy or physical properties of pharmaceutical products. The technology offers comprehensive contaminant detection, quality checks, compliance facilitation and traceability, all of which contribute to brand protection and consumer safety. By embracing x-ray inspection as an additional tool in
Manufacturing
their quality assurance processes, pharmaceutical manufacturers can elevate their standards, mitigate risks, and have confidence that high quality products are entering the market. The time has come for the industry to recognise the immense value x-ray inspection brings and embrace its potential for a safer and more reliable pharmaceutical supply chain.
REFERENCES
1. Based on an airport security X-ray system, typically of higher dose than an inspection system for pharmaceutical products. From the FDA website: “There are no known adverse effects from eating food, drinking beverages, using medicine, or applying cosmetics that have been irradiated by a cabinet x ray system used for security screening. The radiation dose typically received by objects scanned by a cabinet x-ray system is 1 millirad or less. The average dose rate from background radiation is 360 millirad per year.” https://www.fda.gov/radiation-emittingproducts/security-systems/frequently-askedquestions-cabinet-x-ray-systems
2. Kazuaki Uehara, Tatsuaki Tagami, Itaru Miyazaki, Norikazu Murata, Yoshifumi Takahashi, Hiroshi Ohkubo & Tetsuya Ozeki (2015) Effect of X-ray exposure on the pharmaceutical quality of drug tablets using X-ray inspection equipment, Drug Development and Industrial Pharmacy, 41:6, 953-958, DOI: 10.3109/03639045.2014.917093
3. Martin Vogt, Elke Sternberger-Rutzel, Manuel Birke & Christoph Jacobs (2012) Influence of X-ray radiation as PAT method on the model substances tramadol HCl and nifedipine compared to the influence of UV-Vis radiation, TechnoPharm 2, Nr. 3, 1–12
Mike Pipe is a Head of Global Sales & Product Management. During his 15+ years at Mettler-Toledo, Mike has worked in both commercial and technical roles helping food and pharma manufacturers and brand owners achieve compliance, maximise productivity and drive consumer safety. In this international strategic role, Mike works closely with local and global customers to understand their business challenges and applications in order to identify the correct x-ray technology solutions. His experience includes helping manufacturers to reduce the risk of product recalls, increase productivity and lower total cost of ownership.
Web: www.mt.com/xraypharma
Mike Pipe
Adaptive, Innovative and Future-Ready: How CDMOs are Staying Sharp
As the pharma and biotech sector navigates economic uncertainties and evolving customer demands, the contract development and manufacturing industry is undergoing a significant transformation. To enhance their supply chain strategies, drug owners are increasingly relying on companies that can adapt to shifting requirements and maintain a competitive edge.
Leading CDMOs are demonstrating a heightened commitment to customer partnerships through strategic investments designed to adjust with evolving market demands. This is reflected in a forward-thinking approach, which aligns with the broader industry trend of a compound annual growth rate (CAGR) of 5–8% in global spending and demand for medicines through 2028, which raised by 2 percentage points compared to previous forecasts, according to the IQVIA Institute for Human Data Science Analysis.1
Despite rising costs, strategic decisions are necessary to maintain a competitive advantage and uphold the highest-possible quality standards in the development, aseptic fill and finish and packaging services. Outsourcing partners that invest throughout the value chain are well-positioned for future success. This includes contributing to reliable supply chains, providing advanced infrastructure, efficient processes and sustainable practices to expedite the market delivery of drug products.
To meet rapidly evolving customer needs, CDMOs should focus on the following areas:
Investing in Resilient Supply Chains
In an industry where speed is critical, supply chain disruptions can have severe consequences. Natural disasters, international trade tensions, cyberattacks and global pandemics are just a few of the shocks that can immobilise life science companies. Customers are recognizing the importance of real-time tracking, monitoring and traceability of products, from development, manufacturing to distribution and patient delivery. By
understanding that every step of the supply chain is visible and accountable, companies can enhance their operational efficiency, build trust with consumers and adapt more effectively to unforeseen challenges.
CDMOs are well positioned to manage resilient supply chains due to their specialised expertise, resource allocation and advanced technological capabilities. The foresight of purchasing, sales, and operations planning processes are vital in balancing cost containment and resource availability. These teams play a critical role in supplier negotiation, enabling strategies, internal collaborations, and navigating changing market demands. Established relationships with a comprehensive network of suppliers and logistics providers strengthens their ability to coordinate effectively and maintain supply chain integrity. By leveraging these strengths, outsourcing partners maintain the reliable and efficient delivery of pharmaceutical and biotech products, meeting the industry's high standards for transparency and accountability.
Revolutionising Advanced Infrastructure
CDMOs, by their specialisation, are wellpositioned to concentrate their resources
on staying at the forefront of technological advancements in their niche and highly specific development, manufacturing and packaging processes. They are also wellequipped to offer these capabilities to a wide customer base.
Technologies such as artificial intelligence (AI), virtual reality (VR) and autonomous, collaborative robots are actively driving this transformation. In fact, the global market for cleanroom robots is estimated to grow from $6.4 billion USD in 2022 to $25.6 billion in 2030, according to a report by Research & Markets.2 As a result, it is expected that there will be a continued and even heightened use of collaborative robots that work alongside human counterparts. For example, we operate robots called YuMi (short for you and me), which can be trained by employees to perform tasks independently, allowing operators to focus on more specialised projects. This digitalization reduces workloads and increases overall output, while also facilitating more sophisticated data analysis, driving innovation in drug development and manufacturing processes.
Forward-thinking service providers are leveraging technology to enhance customer
service through operational efficiencies. This approach is crucial for timely delivery of life-saving therapeutics by providing a costeffective solution. Moreover, the strategic focus on innovation means that companies remain relevant in a dynamic market and can meet the evolving needs of authorities, consumers and the patients.
Optimising Strategies for Speed and Efficiency
Efficiency in developing and manufacturing life-saving medicines is driven by time to market. CDMOs streamline manufacturing processes with proven methodologies, enabling efficient development while maintaining high-quality standards. Drug developers face intense competition and pressure as the trend towards simultaneous blockbuster and small-batch manufacturing is growing.
Leading outsourcing partners are setting an example for the industry by meeting emerging demands and contributing to more targeted and specialised drug development and manufacturing efforts to fulfil these needs in a timely manner. To do this, they enable biopharma companies to expedite the transition from preclinical development to commercial manufacturing and packaging by leveraging their expertise. CDMOs invest in facilities and versatile manufacturing platforms that enable scale-up/down capabilities, allowing adaptation to changing market dynamics and customer and patient needs. We are making this commitment to our customers through a 1.5 billion Euro investment over the next decade in expanding existing, cutting-edge aseptic production sites that will house several new cleanrooms, lab space for analytical services, new cool storage and warehousing, and more. Besides this, we are further expanding our footprint in both the U.S. and Germany.
Direct investment in new capacity, capabilities and technologies from the CDMO itself is another revealing sign that your outsourcing partner is in it for the long haul – as this is a tangible indicator that the CDMO is invested in their partners’ success.
Building a Sustainable Future
The shift towards sustainable and ecofriendly practices is no longer just an ethical decision; it has become a fundamental expectation shaping the future of aseptic fill and finish. The industry is acknowledging the importance of reducing its environmental footprint and addressing issues like resource depletion, pollution and climate change. This
Manufacturing
focus responds to societal expectations and is a strategic move to maintain the longevity and resilience of operations.
In addition to their dedication to reducing their carbon footprint, customers are now examining the sustainability initiatives of their partners more closely. As an outsourcing partner, being in agreement with these values and visions allows customers to gain an advantage and differentiate themselves from competitors who are slower to adopt sustainable practices.
In a highly competitive industry where customers are evaluating potential partners based on their sustainability practices, prioritising this can be the key factor in securing new customer partnerships, making it essential for CDMOs to focus on sustainability in order to stay competitive.
Looking Ahead to the Future
Biopharmaceutical companies seek partners that offer strategic, value-added services, flexible solutions and efficient operations across the value chain. New partnerships between manufacturers with complementary specialties are being pursued to synchronise expertise, shorten time-to-market, and deliver long-term value to customers.
Outsourcing partners must look ahead to the future of contract manufacturing through investing in a reliable supply chain, revolutionising technology, optimising strategies and building a sustainable future to stay ahead. This investment will attract more
drug owners seeking competitive advantages through collaboration with CDMOs.
REFERENCE
1. The global use of medicines 2024: Outlook to 2028. IQVIA Institute for Human Data Science Analysis. (2024, January 16). https://www.iqvia. com/insights/the-iqvia-institute/reportsand-publications/reports/the-global-use-ofmedicines-2024-outlook-to-2028
2. Global clean room robot market by type (articulated robots, Cartesian robots, Delta Robots), component (controllers, drives, end effectors), end user - forecast 2024-2030. Research and Markets. (2024, June). https://www. researchandmarkets.com/reports/5639896/ global-clean-room-robot-market-by-type
Thomas Otto has been Managing Director of Vetter Pharma-Fertigung GmbH & Co. KG since December 2002. He joined the company as a project engineer in 1990, after graduating from the Technical College in Stuttgart with an engineering degree in packaging technology and print processing. Prior to becoming Managing Director, Mr. Otto managed Vetter’s department of packaging materials development (1995–1999) and served as Vice President of R&D (2000–2002).
Thomas Otto
Child-Resistant Closures: Exploring New Paths Between Safety and Accessibility
In the intricate and highly regulated landscape of the pharmaceutical industry, patient safety is an aspect that unequivocally stands out in its importance. A cornerstone of this safety lies in the design and implementation of product packaging, with a particular emphasis on child-resistant closures. These solutions are meticulously designed and rigorously tested to deter accidental opening by children, significantly mitigating the risk of poisoning incidents resulting from unintentional ingestion.
Despite considerable advancements in packaging design, the introduction of stringent regulatory measures, and increased public awareness, cases of poisoning from accidental ingestion by children persist as a significant health concern. A report by the U.S. Consumer Product Safety Commission1 alarmingly indicates that pediatric poisoning continues to be a leading cause of injury and fatality among children. This ongoing issue underscores the urgent need for further innovation, vigilance, and education in the realm of child-resistant packaging.
The regulatory landscape for childresistant packaging is diverse and varies across different regions globally. In the United States, the landmark Poison Prevention Packaging Act of 1970 laid down specific requirements for the packaging of a wide array of household and pharmaceutical products. This was a significant step towards ensuring the safety of children from potentially harmful substances. Across the Atlantic, in Europe, UNI regulations establish standards for child-resistant packaging, including ISO 13127:2012, UNI EN ISO 8317:2016, UNI EN 14375:2016, and UNI CEN/TR 16353:2013. These standards provide a robust framework for manufacturers, guiding them in the creation of packaging that ensures both the safety and efficacy of child-resistant mechanisms.
Historically, the concept of childresistance has been primarily applied to primary packaging closure systems, such as bottle caps. The most prevalent systems
on the market include "Turn and Lift", "Push and Turn", and "Squeeze and Turn" features. These designs require a sequence of movements that are difficult for a young child to perform, thereby preventing accidental opening. However, the evolving needs of the pharmaceutical industry, coupled with increasing awareness about child safety, have led to the emergence of child-resistant solutions for blister packs and other types of secondary packaging. This expansion reflects the industry's commitment to safety across all forms of pharmaceutical packaging.
Designing a child-resistant capsule involves careful consideration of multiple factors. It's a complex process that requires a deep understanding of the needs of the patient, the operational requirements of the pharmaceutical industry, and the regulatory landscape. Bormioli Pharma, a pioneer in this field, was among the first players to introduce child-resistant capsules in the market. The company restlessly continues to innovate and evolve its offerings to meet the dynamic needs of patients and industry, demonstrating an unwavering commitment to safety.
While engineering a child-resistant capsule, the industrial needs of our customers should also be considered. Indeed, pharma companies require a robust product suitable to be repeatable in industrial processes, that 100% of production has already been checked with automatic vision systems confirming
improved capping performance to avoid inline failures.
Moreover, achieving a balance between safety and accessibility presents unique challenges. While child safety is a paramount concern, several studies have highlighted how child-resistant closures can inadvertently become a barrier for elderly patients or those suffering from certain diseases. Therefore, a significant challenge for the future of child-resistant capsules lies in finding alternative methods that ensure the safety of the youngest without creating barriers for the elderly or disabled.
Packaging
recently held together with some of the major Italian pharmaceutical companies and patients. The main objective of the workshop was having a clearer understanding of emerging and latent needs of people when using pharmaceutical products and identifying potential solutions to overcome them.
One potential solution could involve replacing the sequence of physical gestures needed to open the capsule with another opening mechanism. This could be cognitive-based, requiring a certain level of understanding to open the package, or technology-based, leveraging Internet of Things (IoT) technologies. Such an approach could offer a balance between preventing accidental access by children and ensuring accessibility for all patients, regardless of age, physical ability, or other parameters.
For example, Bormioli Pharma has recently showcased a new prototype representing a significant advancement in its pharmaceutical packaging technology.
This product concept, ID-Cap, is the result of a call4ideas launched in collaboration
with the Italian company Desall.com, and envisages a capsule with integrated biometric recognition which, by reading the fingerprint and a simple rotation of the cap, allows the patient to easily open the medicine. Biometric recognition can also take place via smartphones. The item is in fact designed both to be used independently and to be connected to a dedicated app, which also enables effective monitoring of treatment.
ID-Cap provides an advanced alternative to traditional CRC systems, while elevating pharmaceutical administration safety to unprecedented levels, extending the boundaries of pharmaceutical packaging, and meeting the emerging trend of personalised healthcare, perfectly tailored to patients’ needs.
In conclusion, child-resistant closures represent a critical component in pharmaceutical packaging. Their importance cannot be overstated in terms of safeguarding the health and well-being of the most vulnerable. However, it's equally crucial to continue innovating and researching solutions that effectively strike a balance between safety and accessibility. The goal is to ensure the protection of the most vulnerable without compromising usability for all patients. In this pursuit, the pharmaceutical industry must continue to evolve, innovate, and above all, prioritize patient safety. As we look towards the future, the continued development and refinement of child-resistant closures will play a pivotal role.
Anna Malori, PhD, received a Chemistry and Packaging Technology degree from the University of Parma, Italy. As Head of Product Management at Bormioli Pharma, she works to detect unmet market needs from which to then define new product strategies and create decision proposals. Anna Malori is responsible for full portfolio management, developing visions to build on the success of the company’s product range on the market, in close co-operation with all company departments.
That was one of the main takeaways of a Co-Innovation Lab that Bormioli Pharma
Anna Malori
Advancements in Pharmaceutical Packaging: Exploring Innovations in Sachet and Stick Pack Design
In the dynamic field of pharmaceutical packaging, research laboratories play a crucial role in advancing technology and optimising solutions for various packaging formats. One such laboratory focuses on the in-depth study of laminates and their application in sachets and stick packs, contributing significantly to the evolution of singledose packaging technologies.
These Labs are dedicated to conducting thorough analyses of laminates and customer products intended for use in sachets and stick packs. These studies are pivotal in understanding the complex interactions between packaging materials and pharmaceutical formulations, aiming to enhance product protection, usability, and compliance with regulatory requirements.
The primary goal of a laboratory is to advance packaging technologies by exploring a wide range of laminates suitable for singledose applications. Researchers delve into the properties of these laminates, evaluating their barrier capabilities, compatibility with different product formulations, and environmental sustainability aspects. This systematic approach enables the laboratory to identify optimal packaging solutions that meet the diverse needs of pharmaceutical and healthcare industries.
Sustainability is a core focus in these laboratories' research and development efforts. They are committed to exploring environmentally friendly packaging materials and processes that minimise environmental impact throughout the product lifecycle.
By incorporating recyclable materials, reducing packaging waste, and optimising energy efficiency in packaging production, researchers strive to contribute positively to global sustainability goals while meeting the stringent requirements of the pharmaceutical industry.
Moreover, a research laboratory emphasises the importance of optimising machine design to complement the characteristics of chosen laminates. Engineers collaborate closely with researchers to develop efficient packaging machinery that enhances productivity, reduces material waste, and ensures consistent quality in production.
Beyond material analysis and machine optimisation, the laboratory serves as a collaborative platform for industry stakeholders. Through partnerships with pharmaceutical companies, packaging suppliers, and regulatory bodies, researchers facilitate knowledge exchange and contribute to the advancement of best practices in pharmaceutical packaging.
In addition to its core activities, the laboratory explores emerging trends in active and intelligent packaging technologies. Researchers investigate innovative materials and design concepts aimed at improving product stability, shelf-life extension, and user experience. These efforts are driven by a commitment to continuous improvement and the application of cutting-edge technologies in pharmaceutical packaging.
Furthermore, the laboratory prioritises rigorous quality assurance and compliance with global regulatory standards. Researchers conduct thorough testing and validation to ensure that packaging solutions meet stringent criteria for safety, efficacy, and product integrity. This commitment underscores the laboratory's dedication to maintaining high standards of quality and reliability in pharmaceutical packaging.
Looking ahead, the laboratory remains dedicated to pushing the boundaries of innovation in single-dose packaging. By leveraging scientific research, technological expertise, and collaborative partnerships, researchers aim to address current
challenges and anticipate future needs in pharmaceutical packaging solutions.
In conclusion, the contributions of laboratories specialising in pharmaceutical packaging underscore their role as drivers of innovation and advancement. Through rigorous research, collaborative partnerships, and a commitment to excellence, these laboratories continue to shape the future of single-dose packaging technologies, benefiting the healthcare industry and consumers worldwide.
Andrea Grini graduated in 2023 with a MSc in Marketing and Communication from Urbino University, achieving a 110/110 cum laude. With over two years of experience at Universal Pack, Andrea became Marketing Executive at 24, known for innovative strategies and leadership. Their dedication to excellence, collaborative spirit, and problem-solving skills have established Andrea as a respected figure in marketing, setting new benchmarks in the field.
Andrea Grini
Vials with Exceptional Inner Surface Durability
For Biotech
Less interactions of drug molecules and formulations with the inner glass surface
• Optimized lyophilization process (less fogging)
• Reduced risk of glass delamination
For Diluents
Water for injection | Aqueous NaCl solution Lower pH shift
• Reduced risk of glass delamination
Health Outcomes
Navigating the TGA’s Requirements for Combination Products
Combination products are integral to the healthcare system and play an increasingly important role in patient safety and medicinal usability, enhancing therapeutic benefits and improving outcomes.
According to analysis, the drug-device combination market was valued at US$127.8 billion in 2022 and it is predicted to grow at a compound annual growth rate of 8.9% between 2023 and 2030.1
While combination products have been available for many years, advances in technologies, as well as the need for ‘user-friendly’ delivery of medicines, have led to an increase of these types of products globally and to a new generation of combination products.
Today, combination products have gone well beyond simple drug-release delivery systems, such as drug-eluting stents, to now offering intelligence and analytical capabilities that have the potential to reshape medicine.2 These include wearable sensors, 3D-printed implantable modules and digital drugs offering real-time monitoring.
Australia’s Combination Pathway
Australia’s Therapeutic Goods Administration (TGA) recently published its updated guidance on combination products and what it refers to as “boundary” products as a means to give sponsors and manufacturers greater clarity on the category and pathway for their therapeutic goods.3
The 2023 guidance updates the 2005 guidance, providing more context around what combination products are as well as additional considerations, given that much has changed in the market in the past 18 years.
In its guidance, the TGA defines combination products as those with components that have more than one therapeutic effect, for example, medicine-medical device combinations and medical devices that incorporate or are used
to administer a medicine. Boundary products are defined as those that have characteristics from two or more categories and for which the appropriate regulatory pathway is not necessarily obvious.
In addition to the guidance, in April 2024 the TGA provided a further update with examples of boundary and combination products and their product category.4 This provides manufacturers and sponsors with information on how the agency is likely to regulate common boundaries and combination products. As the TGA makes further determinations on these products, it is likely that examples will continue to be updated.
How the agency regulates boundary products will depend on their principal therapeutic effect, therapeutic claims and stated intended use. Boundary products can include products that contain medicinal substances that work in an ancillary way, which, depending on their therapeutic effect, may be regulated as medicines. Examples include alcohol swabs and disinfectants with antiseptic claims that may be classified as medicinal products, and nasal decongestion products and eye lubricants that, depending on the mode of action, may be a medical device or a medicinal product.3
In Australia, the way combination products are regulated depends on the primary mode of action (PMOA) for achieving their therapeutic effect, as well as the primary intended purpose. Drug device combinations may be regulated as either medicines or medical devices and would therefore be registered as such. Examples include:
• Devices that administer medicine, such as syringes and droppers – are regulated as medical devices
• Co-packaged medicines with devices where the medicine is the main component and the device is used to measure or administer the medicine –regulated as a medicinal product
• Integral combination products, where the device and medicine form a single, non-reusable unit, such as pre-filled inhalers – registered as a medicine
• Devices incorporating a medicine where the medicine’s action is considered
secondary, such as a heparin-coated catheter – registered as a medical device
Medical device companies must have their product in the Australian Register of Therapeutic Goods (ARTG) database unless “the product is exempt from ARTG inclusion, excluded from regulation by TGA or otherwise approved”. Where a device is a component of a medicinal product, it follows the Essential Principles from the Therapeutic Goods (Medical Devices) Regulations 2002 and, as such, does not need a separate ARTG entry, unless supplied separately.5,6 The Essential Principles are safety and performance requirements that determine whether the product has been designed according to safety principles and risk mitigation, that solutions are best practices, and that the benefits outweigh the risks.7
The medicinal component of a combination product is evaluated against appropriate TGA regulatory requirements, to assess the product’s safety, quality, and efficacy.
There are, however, innovative combination products that are harder to classify if there is no clearly defined PMOA, for example, customised 3D-printed scaffolds with incorporated medicinal products and bioprinting material that is classified as part of a medical device.8,9
Streamlining the Combination Product Pathway
In the devices space, there have been recent changes which now allow abridged pathways for drug-device combinations, permitting overseas manufacturers to avoid the expensive and extensive TGA full conformity assessment process under certain circumstances.
This paves the way for more innovative combination products that have been approved in some major markets to be brought into Australia more expeditiously.
For example, devices that incorporate a medicine can leverage certification through the European Union’s Medical Device Directive or Medical Device Regulation (MDR) for ARTG
Health Outcomes
application.10 If this is not available, they must undergo a TGA conformity assessment first, then subsequent ARTG application.
The abridged pathway can be a huge time saver since these are Class III medical devices in Australia and, without appropriate overseas evidence, can face a lengthy conformity assessment process with the TGA. This process includes in-depth assessment of the manufacturer's compliance with the Quality Management System requirements, as well as review of the technical documentation to establish compliance of the device with the applicable Essential Principles.
The regulator requires companies to supply a significant amount of documentation including device verification and validation information, supplier agreements, storage and packaging, sterilisation and other device documents. In addition, manufacturers may be subject to an onsite audit by the TGA prior to certification.11 Similarly, once certification is received, the TGA conformity assessment requires ongoing maintenance. For example, manufacturers are required to notify the TGA of any 'substantial changes’ to the QMS, manufacturing processes or
device design, and such changes must be assessed and approved by the TGA prior to implementation. This process is also lengthy and expensive.
In comparison, most changes to ARTGs supported by overseas evidence will not require notification to, or assessment by, the TGA, and can be implemented once the overseas body has assessed and approved the change.
It is important to note that while TGA does recognise overseas evidence from a number of countries for medical devices, that recognition is restricted to the EU when it comes to devices incorporating a medicine.9 That is because the conformity assessment process is similar to that carried out by a notified body in the EU – a requirement for the assessment and certification (or recertification) of most medical devices and in vitro diagnostic devices placed on the EU market.12
Similarly, when it comes to medicinal products, the TGA will, where possible, use assessments from comparable overseas regulators (COR). This will depend on whether
there is enough similarity between the TGA and a COR, as is the case with the European Medicines Agency, and whether other key criteria are met, such as that the indication proposed is equivalent.13
Patient Benefits of Abridged Pathways
Australia faces the same health challenges as many other markets, specifically a rising prevalence of chronic diseases such as cardiovascular disorders, diabetes, respiratory illnesses and cancer.
To address these issues, patients and physicians are looking to products that are designed to provide a more integrated and effective approach to patient care by combining drugs and medical devices, thereby controlling and targeting the release of the drug product and delivering minimally invasive treatment.1 The benefits of such an approach include improved treatment effectiveness, enhanced patient compliance, and simplified healthcare delivery.
In all major markets, streamlined regulatory pathways and the addition of clearer guidelines for combination products have facilitated faster approval processes.14
Health Outcomes
And the marketplace for combination products continues to evolve, as more innovative technologies emerge, paving the way for improved patient care.15 Regulatory authorities, including the TGA, are responding to these cutting-edge technologies through more streamlined pathways. Ultimately, the goal is to provide patients with products, including combination products, that can more effectively treat and diagnose lifethreatening or debilitating diseases, while continuing to demand manufacturers and sponsors meet safety, quality and efficacy requirements.
The information provided during this presentation does not constitute legal advice. PharmaLex Pty Ltd and its parent Cencora, Inc., strongly encourage readers to review available information related to the topics discussed herein and to rely on their own experience and expertise in making decisions related thereto.
REFERENCES
1. Drug Device Combination Products Market Size, Share & Trends Analysis Report By Product (Transdermal Patches, Infusion Pumps, Inhalers, Drug Eluting Stents), By Region, And Segment Forecasts, 2023 – 2030, Grand View Research. https://www.grandviewresearch.com/industryanalysis/drug-device-combination-market
2. Emerging trends in drug-device combination for advanced disease diagnosis and therapy, Nano Today, 2023. https://www.
3. Guidance on boundary and combination products, TGA, Dec 2023. https://www.tga.gov.au/ sites/default/files/2023-12/guidance-boundarycombination-products.pdf
4. Examples of boundary and combination products and their product category, TGA, April 2024. https://www.tga.gov.au/sites/ default/files/2024-05/examples-boundarycombination-products-and-product-category. pdf
5. Therapeutic Goods (Medical Devices) Regulations 2002, Australian Government, Federal Register of Legislation. https://www. legislation.gov.au/F2002B00237/latest/versions
6. Australian Regulatory Guidelines for Medical Devices (ARGMD), TGA, Nov 2023. https://www. tga.gov.au/resources/resource/guidance/ australian-regulatory-guidelines-medicaldevices-argmd
7. Quality, safety, and performance requirements for medical devices, TGA. https://www.tga. gov.au/how-we-regulate/manufacturing/ manufacture-medical-device/quality-safetyand-performance-requirements-medicaldevices
8. The regulatory challenges of innovative customized combination products, Sec. Regulatory Science, 2022. https:// www.frontiersin.org/articles/10.3389/ fmed.2022.821094/full?trk=public_post_mainfeed-card_reshare_feed-article-content
9. The Right Materials for Success in Bioprinting, Pharma’s Almanac, Feb 2023. https://www.
10. Use of market authorisation evidence from comparable overseas regulators / assessment bodies for medical devices (including IVDs), TGA, June 2023. https://www.tga.gov.au/sites/default/ files/use-market-authorisation-evidencecomparable-overseas-regulators-assessmentbodies-medical-devices-including-ivds.pdf
15. Early Development Considerations for Innovative Combination Products, FDA, 2006.
Piety Rocha
Piety Rocha is the Director, Head of Regulatory Affairs & Country VDC Head at Pharmalex, Australia. She has more than 20 years of experience in the Australian and New Zealand pharmaceutical industry. Piety is a seasoned regulatory affairs professional, with extensive knowledge and expertise covering innovative and generic prescription medicines across multiple therapeutic areas.
Heyam Kalla
Heyam Kalla is Associate Director, Team Lead Medical Devices / IVD at Cencora Pharmalex, Australia. As a regulatory affairs and quality assurance professional in the medical devices industry, Heyam has extensive experience in pre-market activities and post-market surveillance for medical devices in Australia and New Zealand.
Precision Revolution
Your quest for perfection ends here. Let’s revolutionize your manufacturing process, redefine precision, and gear up for excellence together. When you partner with KAHLE perfection isn’t a destination—it’s the journey we craft, gear by gear.
Kahle® is dedicated to providing custom automation machinery solutions for the Medical Device, Pharmaceutical, and Healthcare Industries around the world.
The End of Drug Shortages Begins with Data Transparency
Connecting Quality, Regulatory, and Manufacturing Data is Key to Improving Supply Chain Agility
For more than 15 years, shortages of critical and non-critical drugs [SL1] [MOU2] – from oncology treatments, emergency room and surgical suite essentials to antibiotics and flu medicine – have burdened the healthcare systems and compromised patient care and outcomes. “The issue has become uncomfortably real for me recently, as I searched for a pharmacy that could fill my daughter’s amoxicillin prescription,” said Vicki Cookson, strategy director, Vault RIM, Enterprise at Veeva.” “How would I feel, I wondered, if I`d needed immediate access not to a common antibiotic, but a treatment for a lifethreatening illness?”
Despite ongoing analysis and guidance, supply gaps continue, especially for generic drugs, which account for most of the medications prescribed in Europe and the United States. “For some generics manufacturers, margins in the low single digits can make consistent quality and compliance a challenge,” said Sofia Lange, strategy director, quality & manufacturing at Veeva. “The costs of not addressing quality can drive a small supplier out of business, further shrinking supplies.”
Lack of Data Transparency, on The Macro and Micro Level
Lack of data transparency across the supply chain is a core challenge for regulators and suppliers of all sizes. It prevents insights into supply fluctuations and their root causes. Decreased access to global compliance and quality data for the active pharmaceutical ingredients (APIs) and finished drugs manufactured offshore has only compounded supply risks. Between 2020 and 2022, FDA’s five-year backlogs for offshore API facility inspections have increased from 30% to 80%, and the agency is pivoting to remote and other inspection formats.
“Regulators, governments, patient advocates, and industry groups are actively working to solve today’s supply problems. By listing the generics most vulnerable to shortages, the European Medicine Agency has taken an essential first step, which
should help guide future efforts,” said Cookson. So far, discussions of next steps have emphasised the need for economic incentives, to help generics manufacturers update manufacturing, and to speed better approaches to supply chain data collection, monitoring, and analytics.
Meanwhile, at the “micro” level, at the individual facility and company, there is a new focus on establishing data transparency and connecting data across functions. Data-driven approaches are helping more companies reduce the risk of shortages by improving the efficiency of compliance and quality operations.
Connecting Cross-functional Data for More Agile Change Control
One area of focus is improving post-approval process change control, a time and labour intensive behind-the-scenes process that often leads to supply delays. Using traditional approaches, with disconnected data, separate IT systems, and manual processes, a single change control process can take from six months to two years to complete. Depending on the regulatory agencies and regional requirements involved, the work required can delay a drug’s availability by up to five years. Today, a typical large biopharma company manages 40,000 of these applications each year, with up to 200 for a single product.
Imagine the EMA has approved one manufacturer’s new therapy two years ago. Since then, the company has developed a safer manufacturing process that reduces product costs. Its leaders also plan to use a more sustainable packaging to reduce carbon footprint, and to shift from laboratory-based quality control to real-time batch release. Each of these continuous improvements may require separate regulatory agency approval.
Gathering the data required for each change takes months. First, regulatory teams must determine the impact of each change, and which countries and internal documents will be affected. Supply chain teams must then do the same for individual product lots.
According to Lange, the quality department must then ensure all change impact
assessments are completed, identify, and make sure affected documents and processes are up-to-date, incorporate changes into new training programs, and even qualify potential new suppliers. The team will also have to manage and keep track of these actions and estimate the potential risks of making each change.
Currently, at many companies, regulatory and quality teams use different electronic systems for each of these steps and communicate by email and phone. Delayed communication and errors can result in noncompliance and regulatory warning letters.
And this is only the beginning. After compiling, publishing, and submitting the applications, regulatory teams must optimise ongoing communication with regulatory agencies. In the end, regulators from each affected country can still decide that they need to reinspect the facility to re-approve the new and improved product, thus triggering additional delays in product availability.
Connecting Manual, Disconnected Processes to Gain Speed and Cut Costs
Unified approaches to quality and regulatory data management bring different software together onto a single platform, which can help streamline and simplify change control. They make it easier for users to meet regulatory requirements, and to spot and address problems faster.
Integrating quality, regulatory, and supply chain data, documents, and processes can enable even greater agility. It is now possible, for example, to connect regulatory and quality data and content, particularly product documentation, with a corporate enterprise resource planning (ERP) system.
A growing number of companies of all shapes and sizes are unifying quality and/ or regulatory data management. Some are connecting regulatory and quality operations to facilitate cross-functional collaboration, while others are connecting regulatory and quality data with their ERPs, which can potentially reduce batch-release timelines by up to 30%. Functional and cross-functional
QUALIFICATIONS & VALIDATIONS QUALIFICATIONS & VALIDATIONS AVAILABLE WORLDWIDE AVAILABLE WORLDWIDE AT YOUR FREIGHT FORWARDER AT YOUR FREIGHT FORWARDER
TEMPERATURE TEMPERATURE
Temperature protection of pharmaceutical and healthcare products in airfreight (+15°C + 25°C°) and (+2°C + 30°C) (+15°C + 25°C°) and (+2°C + 30°C)
Multilayer thermal blanket for PMC-ULD - Euro and Block pallets
Stress-tested in summer (+46°C) and winter (-15°C) profiles
Airfield Tarmac tested on solar power and greenhouse effects
Tel. (Belgium): +32-11.26.24.20
E-mail: info@krautz.org
Website: www.krautz.org
Logistics & Supply Chain Management
teams expect greater data transparency to make compliance and real time information exchange easier, which would mitigate the risk of drug supply gaps.
Assessing the Costs of Disconnected Systems
Every day that a drug isn't available costs a manufacturer from hundreds of thousands to millions of dollars, whether for highly specialised drugs or everyday over-thecounter medicines. Unified approaches that improve data visibility, centralise access to real-time information, and automate workflows are already proving that they can speed patient access to the treatments they need.
Tracking the time and cost of using traditional approaches and technologies can reveal surprising insights into the total cost of ownership and operation. Consider the savings and improvements from the following:
• Reorienting highly trained and qualified people away from manual, administrative tasks to focus on priority efforts, such as interaction with regulators
• Reducing the time spent on disconnected, one-off email and telephone communications, measured by the number of hours each employee spends on these efforts each day, within teams and across functions
• Minimising risk of errors and duplication of tasks resulting from disconnected, manual information exchange
• Strengthening patient and healthcare providers’ trust in access to critical treatments. Although this cannot be measured, quantifying missed product release deadlines over time could offer insight into performance gaps and trends.
• Avoiding the intangible, but significant reputational costs of having a drug supply problem.
Disconnected data not visible across systems can affect name-brand, generic, and biopharma manufacturers alike. Change control is only one of several behind-the-scenes operations that drain time and resources and delay patient
access to treatments. CMC submissions and submissions publishing are two other examples, both of which are undergoing significant change.
As the current drug supply situation has reminded us, we are all patients, and the industry’s supply chain issues affect us all. Solutions already exist to help automate more behind-the-scenes processes and maximise access to connected, real-time data. However, they can only work from a foundation of connected and transparent data, at the individual plant level and beyond.
Sofia Lange is Director of Strategy for Vault Quality, focusing on the European market, including but not limited to providing leadership to ensure customer success during implementation and postimplementation of Veeva Quality Systems. Sofía is a biochemist by profession, with more than 15 years of experience in pharmaceutical quality management and manufacturing, focusing on process improvement, mainly through computerised system implementation. Sofia has a broad range of experience in handling GMP licenses with MoH across the globe (Argentina, Brazil, Chile, Germany, Russia, Singapore, USA-FDA/ DEA), and is a certified ISO 9001, ISO14000 and IRCA GMP Pharmaceutical Lead auditor.
Vicki Cookson is Director of Strategy for Vault RIM at Veeva Europe. In her role, she is responsible for shaping the future of Veeva's Vault RIM offering, providing leadership to ensure customer success and helping life science companies to streamline global regulatory processes on a single, cloud-based platform to drive productivity, data quality and agility. Vicki has over two decades of experience in regulatory operations, having previously held roles at Liquent, Ipsen, AstraZeneca, and as a regulatory consultant at Parexel.
Sofia Lange
Vicki Cookson
Expand your horizons
CPHI Milan connects you to the global pharma community to level up your business at the heart of our industry.
Learn more
8-10 October 2024
Fira Milano, Italy
SCAN ME
Has AI Transformed Pharma’s Supply Chain? Pharmaceutical Packaging Expert Weighs in
The pharmaceutical industry is consistently seeking to improve, and now, with the rise of advanced artificial intelligence (AI), the industry has pursued leveraging this technology to revolutionise supply chain management by increasing efficiency, improving quality and accelerating innovative supply chain solutions.
If successful, the global economic impact of AI could boost GDP by $7–10 trillion by enhancing productivity and streamlining processes. This could translate to an estimated $60–$110 billion in annual economic value in pharmaceuticals.
While the concept of AI has been around for over a decade, AI with human-like intelligence has transformed industries worldwide, providing endless possibilities.
In this article, I explore whether or not AI has achieved the promising potential it offers to the pharmaceutical industry in its management of complex global supply chains, and how this could be enhanced.
Sophisticated Quality Control
Pharmaceutical packaging plays a crucial role in safeguarding critical medicines making quality control a vital component of the supply chain. These regulations vary from country to country, adding a layer of complexity to a critical global supply chain.
In the UK, regulations require that all packaging includes clear, understandable patient informational leaflets (PILs) and detailed information including the product name, dosage, chemical composition and safety information. Ensuring packaging adheres to these requirements is a timeconsuming and important task that can be optimised significantly through the use of advanced AI tools.
Innovations such as natural language processing (NLP) and machine vision, exemplified by technologies like the Cognex In-Sight system, are transforming quality control by ensuring accuracy and clarity while
identifying even the smallest but essential packaging errors.
This system also verifies compliance with safety standards, checking for unique identifiers to prevent counterfeiting and antitampering devices to prevent contamination and misuse, especially by children.
AI has notably improved quality control for advanced packaging types like pre-filled syringes and auto-injectors. For example, Amgen’s integration of AI in their quality control procedures detects air bubbles in viscous injectables and has led to a 70% increase in particle detection and a 60% decrease in false positives.
Overall, the integration of AI technologies like NLP and machine vision is significantly reinforcing the quality control processes in pharmaceutical packaging, thereby improving efficiency and productivity. But at the same time, patient safety is being enhanced.
Responsiveness and Transparency
In a global market, transparency substantially enhances supply chain risk management and improves access to safe, quality healthcare. In reality, this is often difficult to achieve, but advanced technological innovation has removed common pitfalls and barriers to achieving this transparency.
Blockchain technology provides a secure, immutable ledger that tracks every transaction of pharmaceutical products, boosting transparency and safeguarding against counterfeit medicines. Meanwhile, AI enables real-time monitoring of the supply chain, helping to prevent disruptions, predict market shifts, and detect counterfeit products through smart packaging and anomaly detection.
Combining blockchain with AI amplifies these benefits. Blockchain records transactions securely, while AI analyses this data to identify risks, forecast trends, and enhance decision-making. This integration not only maintains supply chain integrity but also helps stakeholders address inefficiencies, prevent drug shortages, comply with regulations, and ensure consistent quality control.
Lastly, blockchain and AI-integrated tools easily and meticulously store crucial information in microscopic detail, removing any chance of human error or negligence. This is particularly important should an incident arise, where every stage of the supply chain must be analysed against stringent industry regulations. This not only secures the supply chain but also builds trust and loyalty among stakeholders, including consumers.
Although this integration is a relatively novel approach and faces challenges like data silos and interoperability issues, continuous learning and improvement are key to overcoming these obstacles and achieving comprehensive supply chain transparency.
Cold Chain Management
In pharma, cold chain management is crucial to ensure the correct transportation and storage of temperature-sensitive products. This is essential to safeguard the safety, efficacy and quality of pharmaceutical products such as biopharmaceuticals, vaccinations and other conventional medicines.
This complex task requires efficient temperature control, specialised equipment and packaging solutions such as cold storage warehouses and thermal or insulated packaging, compliance with stringent regulations, meticulously planned logistics and distribution and technological innovation as discussed above.
Through predictive analysis and the proactive management of logistical disruptions, AI tools have transformed cold chain management procedures within the industry by optimising efficiency, enhancing safety and ensuring compliance with regulations.
For example, AI tools can forecast demand for cold chain products, allowing pharmaceutical companies to accurately respond to market demands, minimise waste and improve supply chain responsiveness to changing geo-political circumstances and other environmental factors which could disrupt the supply chain flow.
Furthermore, thanks to AI’s integration with Internet of Things (IoT) sensors, real-
Logistics & Supply Chain Management
time monitoring and immediate alerts for changes in temperature and humidity allow for efficient automated control systems which will adjust necessary environmental conditions autonomously, minimising the potential for human error and maintaining optimal conditions during transportation and storage of critical medicines.
Inventory Management
Inventory management is a pivotal component of the pharmaceutical supply chain, primarily due to the unique sensitivities of certain medications, including those with strict expiry dates and specific storage requirements.
Ensuring these medicines are stored under optimal conditions to prevent degradation and contamination is a critical priority, reflecting the complex nature of pharmaceutical logistics.
However, innovative use of AI technologies can minimise this complexity, streamlining the pharmaceutical supply chain through enhanced demand forecasting and optimising stock levels to minimise waste and cost.
AI’s advanced algorithms and machine learning capabilities allow it to analyse vast historical and real-time datasets to identify seasonal trends (such as seasonal viruses) and market dynamics. These capabilities are helping pharmaceutical companies to optimise their production schedules, reduce stock management issues and ensure timely availability of medicines when it matters.
As ever, the lessons learned from the Covid-19 pandemic have highlighted the importance of predictive analysis, demand forecasting and ensuring the abundant and timely delivery of crucial medicines in the face of a pandemic.
But drug shortages are an omnipresent and universal issue affecting all regions
around the globe, they are not simply an issue for third-world countries facing war and economic challenges. In the UK, pharmacists have described current medicine shortages as “beyond critical” with patients at risk of “imminent harm or death”.
Ultimately, AI’s optimisation of inventory management is not only creating resilient and robust pharmaceutical supply chains, but also ensuring the preservation of public health in the face of illness, through readily available and continuous supplies of medicines.
Drug Discovery and Innovation
The COVID-19 pandemic epitomises the crucial need for swift and ongoing drug discovery and development. Over the past decade, the digitisation of vast global medical data, including molecular screening profiles and health records, has fuelled AI-driven advancements in drug research, accelerating the creation of new treatments.
AI methods like de novo molecular design, structure-based design, and deep learning are revolutionising the drug discovery process, enabling the rapid analysis of biomedical data to speed up clinical developments. A notable case in point is Pfizer's use of AI in developing PAXLOVID, which cut computation times by 80–90%, significantly hastening research timelines.
In the UK, the MHRA's introduction of the AI-Airlock reflects a commitment to integrating AI in healthcare regulation, allowing for the testing and refinement of medical technologies in NHS settings safely and effectively.
With the looming threat of future pandemics, AI's ability to optimise logistics, predict demand, and ensure the timely distribution of essential medicines is becoming increasingly vital. AI-enhanced supply chains are essential for quickly delivering new, effective treatments during health crises, and ensuring global access to lifesaving drugs.
Personalisation
Scientific discovery has found that genetic differences significantly influence how individuals respond to medications, from common antibiotics to complex chemotherapy treatments. For example, recent news of a gene therapy trial restoring hearing to a young child otherwise born completely deaf showcases the impressive possibilities of personalised medicine in resolving unique medical issues.
But it is also because of these differences that adverse reactions (ADRs), which may be severe or life-threatening, can occur. For example, certain people lack the genes necessary to activate medications like codeine, rendering that medicine unhelpful in treating them.
To combat ADRs, the NHS is prioritising pharmacogenomics (personalised medicine) aiming to fully implement it by the end of the decade to improve drug and medical device safety.
The vast amount of complex genetic data presents challenges but AI is poised to address this by decoding, analysing, and organising large datasets, making it particularly effective in identifying genetic markers that predict treatment efficacy and safety as well as identifying anomalies.
These capabilities extend to optimising logistics in personalised medicine by forecasting demand, managing inventory, and ensuring timely delivery.
AI-enhanced supply chain management is vital for delivering personalised treatments efficiently, thereby improving patient outcomes and minimising the risk of adverse reactions.
Steve Brownett-Gale is a marketing professional with a career spanning both communications and products in B2B and B2C markets across Manufacturing and Services sectors. At Origin, in his role as Marketing Lead, Steve is responsible for positioning the company as a worldleading supplier of innovative and groundbreaking pharmaceutical packaging devices, as well as offering a unique and disruptive supply chain model. Established 60 years ago, Origin offers customers a remarkable range of versatile packaging solutions that respond to the unique needs of the global pharmaceutical marketplace. Origin engages in the design, manufacture, and consolidated supply of pharmaceutical packaging, partnering with licence holders and CMOs.
Web: www.originltd.com
Steve Brownett-Gale
Mastering Cold-Storage Management: Strategies to Minimise Pharmaceutical Losses
In an era where medical advancements are revolutionising healthcare, ensuring the potency and efficacy of pharmaceuticals is essential.
However, amidst the complex logistics of transporting pharmaceuticals, one critical factor often overlooked is temperature control. As temperatures fluctuate, so does the efficacy and safety of medicinal products. One study1 found that the pharmaceutical industry loses $35 billion each year to inefficient temperature-controlled logistics.
The losses associated with pharmaceutical waste are not only financial. These can delay the delivery of essential drugs and result in environmental damage from the disposal of drugs. So, how can effective cold storage management prevent further losses and reduce the harm caused by ineffective medicines?
Impact of Temperature Fluctuations
Many medications, particularly antibiotics and vaccines, are sensitive to temperature fluctuations. Some products may also be impacted by exposure to adverse humidity levels.
When exposed to temperatures2 outside the recommended range, these medications can become compromised or lose their potency.
Temperature fluctuations not only compromise the efficacy of the medications but also pose significant health risks when people do not get access to vaccines. This was particularly evident during the Covid-19 pandemic. The vaccines that were developed to protect against the Covid-19 virus had strict temperature requirements, raising concerns3 about whether they could be adequately stored, especially in developing countries.
Other medications such as monoclonal antibodies, often used for the treatment of cancer, are stored4 at temperatures between −20°C to −80°C. When kept in warm temperatures outside the manufacturer's
recommendations, their efficacy5 dramatically decreases.
There are multiple stages at which these temperature fluctuations can occur – from the transport of raw materials to the shipping of the finished products internationally. Of course, there are also many instances in which temperature fluctuations cannot be controlled, like when someone has to transport a medication from the pharmacy to their home.
One report estimates that around half of the vaccines6 distributed around the world go to waste because of poor temperature management. Another report by the United Nations Environment Programme7 has assessed that at this spoilage rate, a billion vaccines could be wasted. And it highlighted that even if valued at a non-profit cost of around $10 a vaccine, would still represent a massive loss.
Beyond the financial losses, there are also broader economic impacts that come into play when it comes to drug waste. They include increased healthcare costs due to ineffective treatments and the need for additional medical interventions. The NHS spendingvii on drugs has risen to nearly £19 billion in 2022/23, over 8 percent more than in the previous year. Hence, preventing wastage from storing drugs incorrectly is essential to ensure that costs don't spiral.
Additionally, drugs manufacturers have to comply with the strict regulations under which medicines are required to be transported. Failures in the temperature-controlled supply chain may prompt manufacturers to deny responsibility for the wastage of products. In some instances, logistics businesses may even be held responsible for the loss and suffer reputational harm as a result.
Ineffective temperature control can lead to increased pharmaceutical waste, contributing to environmental pollution and requiring complex disposal procedures. The NHS estimates that over 156,000 tonnes9 of medical waste is produced annually, which requires high-temperature incineration (HTI) or alternative treatment. It has been predicted that by implementing a comprehensive
waste strategy, over £11 million can be saved in costs, whilst dramatically reducing CO2 emissions.
Effective temperature control in the pharmaceutical supply chain is essential to minimise drug wastage, reduce environmental pollution and avoid the complex procedures associated with dealing with pharmaceutical waste.
Identify Weaknesses in the Cold Storage Chain
It's essential to evaluate the accuracy and reliability of temperature monitoring devices and data logging systems to mitigate the risks associated with temperature fluctuations in the cold storage chain.
These systems must be capable of providing real-time data and generating automatic alerts when deviations occur. Ensuring their accuracy involves regular calibration and maintenance.
Failures in cold storage may be visible through conditions such as condensation or mould growth. These are likely to occur when there is poor ventilation, fluctuations in temperature or poorly stored materials. Effective tracking systems are crucial in ensuring that these problems are dealt with before risking a loss of product.
Ensuring remote access to tracking systems and real-time notifications is key in the cold-storage management for addressing issues promptly.
Human error can also contribute to failures in the cold storage chain. Regular training for the staff involved in the handling and transporting of temperature-sensitive pharmaceuticals ensures that strict protocols are followed. Any deviations from the protocol demands an immediate response to prevent damage or loss.
Redundant temperature control systems10 are also beneficial in ensuring that there is adequate power and cooling capabilities in the event of a failure. Backup generators or alternative cooling units will maintain temperatures and warrant that nothing goes to waste.
Logistics & Supply Chain Management
Prioritise Compliance with Regulatory Standards
Regulatory bodies impose strict guidelines11 on temperature control during the storage and transportation of pharmaceuticals. Adhering to these standards not only upholds quality assurance but also mitigates the risk of non-compliance penalties and reputational damage.
Regulatory standards vary globally, and multinational pharmaceutical companies must navigate different compliance landscapes. Adopting the most stringent standards can help ensure universal compliance and quality.
In the UK, best practice recommends that the temperature monitoring should take place in all refrigerated transport and especially within shipments of high-risk products. Temperatures are required to be strictly controlled and monitored to provide temperature data for the entire journey. Daily monitoring that records minimum and maximum temperatures should take place at all storage locations and any recording devices should be calibrated.
Using electronic tracking systems built into cold-storage units allows for easy monitoring
and ensures that all records are kept in one place. This data is also required to be retained for five years.
In the UK, The Medicines and Healthcare Products Regulatory Agency (MHRA) carries out inspections12 to check that distribution sites comply with Good Distribution Practice (GDP). During these inspections, GDP inspectors will analyse several areas, including the equipment used to distribute medicine – this can include temperature monitoring and transportation arrangements.
If an inspector finds critical deficiencies or that improvements have not been made from previous inspections, consequences may include increased inspections or even a suspended licence, which will severely impact distribution plans.
While these regulations may seem onerous to comply with, maintaining comprehensive documentation of temperature control measures can pre-empt regulatory scrutiny and identify areas for improvement before external inspections.
Maintain Risk Mitigation Strategies
There are several risks to consider in the
logistics industry, from cybersecurity threats to transportation and equipment breakdowns.
As supply chains become more complex, pharmaceutical and logistics industries are more at risk of being exploited by criminals. In 2021, hackers targeted13 COVID-19 vaccine developers. The logistics industry is at a large risk of being hacked. One UK-based logistics company entered administration following a ransomware attack,14 which had caused major disruption.
This highlights the real-world threats that can occur as a result of cyber-attacks. Protecting against such threats requires robust cybersecurity measures, including data encryption and regular security audits.
Transportation delays,15 which have recently been experienced in the Red Sea, have posed some significant challenges for global trade. As a result of unrest in the area, shipping companies have been forced to divert from their usual trade routes and opt for more time-consuming routes.
This poses challenges for logistics companies that need to ensure cold storage maintains the required temperatures for longer periods.
Logistics & Supply Chain Management
Contingency plans and mitigation strategies are essential in addressing identified weaknesses, minimising the impact of disruptions on temperaturesensitive products. These plans should deal with potential disruptions such as equipment failure, transportation delays, and temperature deviations.
Regular drills and scenario planning can help ensure that teams are prepared to respond effectively to real-life incidents, minimising the impact on temperaturesensitive products. Established crisis management teams, trained to handle specific types of disruptions, can ensure that problems are met with a swift response.
Ensure Visibility within the Partner Network Integrated tracking systems provide transparency throughout the supply chain, empowering stakeholders to identify and address potential temperature deviations promptly. With many stakeholders involved in the logistics industry, each handoff in the supply chain introduces a potential risk for temperature deviations, mishandling, or delays.
Reliable tracking systems can mitigate these risks by providing real-time data and alerts. In the event of a temperature deviation, stakeholders can take immediate action to prevent damage to the product.
As technology rapidly develops, maintaining digital records of temperature data becomes more efficient, ensuring transparency and accountability. Advanced
sensors can continuously monitor environmental conditions and log this data to one platform that is easily accessible to multiple stakeholders. By automating this process, this ensures that data is up-to-date and accurate. This also reduces the risk of human error that may occur from manual record keeping.
This level of record-keeping also facilitates compliance with regulatory requirements, as detailed records can be readily accessed and provided to authorities when needed.
Additionally, the sharing of data with relevant stakeholders can enhance trust and collaboration, so that everyone is aware of the current status of shipments. This collaborative approach can both improve operational efficiency and strengthen relationships among supply chain partners.
Mark Ross, Global Brand Manager at ArcticStore, a TITAN Containers brand, providing flexible cold storage solutions for pharmaceuticals, supermarkets, and retail chains worldwide.
Dive into the Heart of Pharma at CPHI Milan 2024, October 8–10th
Join us at CPHI Milan 2024, the premier global event for pharmaceutical professionals, from October 8-10 at Fiera Milano. Celebrating 35 years of excellence, this event promises unparalleled opportunities for networking, innovation, and professional growth. With over 62,000 attendees and 2,500 exhibitors, CPHI Milan offers a unique platform to connect with industry leaders, explore cutting-edge exhibitions, and discover new awards such as ‘Woman of the Year’ and ‘Future Leader’. It’s where suppliers, innovators, and distributors converge, promising unmatched opportunities for networking and partnership.
Event Overview
CPHI Milan 2024 returns with expanded opportunities for networking, innovation, and collaboration in the pharmaceutical industry. This three-day exhibition immerses attendees in every facet of pharmaceuticals, emphasising supply chain resilience and sustainable practices. It’s a pivotal opportunity to connect with industry professionals and forge lasting partnerships.
“This year is particularly significant for CPHI as we celebrate our 35th anniversary. It will be our largest event yet, featuring new awards and enhanced bioproduction zones and content. As we return to Lombardy, a hub for pharmaceutical manufacturing and API production, I eagerly await welcoming the global pharmaceutical supply chain, its partners, and drug innovators to Italy. The partnerships forged here will drive future drug development and ultimately improve patient access and treatments," remarked Orhan Caglayan, Group Director, CPHI Milan.
With over 2,500 exhibitors and 62,000+ industry professionals, CPHI Milan provides unparalleled opportunities for networking and partnership-building essential for enhancing supply chain resilience in an increasingly dynamic market. Attendees can expect a dynamic agenda designed to inspire collaboration and facilitate discussions on the latest trends, innovations, and sustainable practices shaping the industry today.
What’s New at CPHI Milan 2024
Celebrating its 35th anniversary, CPHI Milan introduces several enhancements to elevate the attendee and exhibitor experience:
Supply Chain Resilience
Stay ahead with insights into new technologies and strategies essential for diversifying and stabilising the supply chain. CPHI Milan 2024 showcases the latest innovations and practical solutions to navigate the complexities of the pharmaceutical landscape. The event will focus on strategies for mitigating risks, ensuring continuity, and optimising operations within the supply chain, crucial in today's volatile market environment.
Awards Ceremony at CPHI 2024
On October 8th at Alcatraz Milano, the CPHI Awards Night will commence with a glamorous drinks reception, celebrating excellence and innovation in the pharmaceutical industry across 14 categories. Among the highlights is the new "Woman of the Year" award, honouring women leaders who inspire colleagues, champion diversity and inclusion, and advocate for industry progression. Nominees are evaluated on their leadership, collaboration, and ability to create opportunities for women while challenging the status quo.
Equally anticipated is the novel "Future Leader" award, recognising rising stars under 35 with at least five years of experience in pharma, biopharma, or academic institutions.
This award celebrates those who bring fresh perspectives, manage teams effectively, and introduce innovative ideas, marking them as the future leaders set to shape the pharmaceutical industry with their dedication and vision.
Key Highlights of CPHI Milan 2024 Unmatched Networking Opportunities
At CPHI Milan 2024, anticipate connecting with over 62,000 pharmaceutical professionals and 2,500 exhibitors, including industry giants like Lonza, Samsung Biologics, Thermo Fisher, and Pfizer CentreOne. It's expected to be the largest gathering yet, offering unprecedented networking possibilities. The event serves as a unique platform to establish new partnerships, strengthen existing relationships, and explore potential business ventures in a dynamic setting.
Conference Program Highlights
The CPHI Milan 2024 conference program is meticulously curated to inspire and educate attendees. Industry thought leaders lead insightful sessions on critical topics such as regulatory trends, technological innovations, and sustainable practices. From visionary keynote addresses on pharmaceutical R&D to interactive panel discussions on emerging therapies and market dynamics, the agenda promises a wealth of learning opportunities. Engage with over 150 expert speakers across 5 content tracks over 3 days at CPHI Milan,
ensuring a comprehensive exploration of the latest advancements and trends in the pharmaceutical industry.
Snapshot Overview of Some Key Sessions:
• Keynote: Advancing Immunisation with Next-Gen mRNA Vaccines
• When: Tuesday, October 8, 2024, from 11:00 AM to 11:30 AM
• Navigating Regulatory Landscapes: Ensuring Quality and Compliance in Pharma Ingredients
• When: Tuesday, October 8, 2024, from 1:15 PM to 1:40 PM
• Panel Discussion: Why the Middle East Can Be the New Frontier for NextGeneration Medicine
• When: Tuesday, October 8, 2024, from 4:15 PM to 5:00 PM
• Panel: Excipient Excellence: The Power of Excipient Grade Selection
• When: Wednesday, October 9, 2024, from 11:35 AM to 12:45 PM
Final Thoughts
CPHI Milan 2024 is more than an exhibition – it's a catalyst for change and advancement within the pharmaceutical industry. With a renewed focus on supply chain resilience, attendees gain actionable insights into navigating challenges, optimising operations, and ensuring supply continuity. By embracing these
trends and focusing on strategic imperatives, the pharmaceutical industry can achieve greater efficiency, resilience, and success in the years ahead. The future of pharmaceutical outsourcing is increasingly shaped by strategic, integrated, and collaborative partnerships. As the industry evolves, CDMOs and pharmaceutical companies must collaborate to navigate regulatory challenges, optimise supply chains, and deliver innovative therapies worldwide. CPHI Milan 2024 offers the perfect platform to explore these opportunities and
stay at the forefront of industry trends. Don't miss your chance to be part of the conversation and drive the future of pharmaceuticals forward.
Join Us
Join us at Fiera Milano, Italy, and immerse yourself in a dynamic environment where ideas flourish and partnerships thrive. Let's shape the future of pharma together. Visit CPHI Milan 2024 to learn more and register today!
Subsection: Nasal & Pulmonary Drug Development & Delivery
MVIC SYMPOSIUM 2024
ADVANCING THE DEVELOPMENT OF INHALED MEDICINES
16–17 October, Lund, Sweden
• Sharp, interesting, and relevant speeches within the field of inhalation
• Sponsor exhibition
• Scientific poster exhibition
• Symposium dinner
MVIC Symposium is a yearly event at Medicon Village in Lund, Sweden, covering the field of inhaled medicines. Join us and connect with industry inhalation experts, academic inhalation experts, company leaders, business developers, and students.
For more information and registration, visit: mvic.se/mvis-inhalation-symposium/
If you have questions, please contact us at info@mvic.se
Regulatory & Marketplace Nasal & Pulmonary
The Development of Inhaled Products
Today, we have decades of development of inhaled products behind us. This development has led to the availability of very efficient products that have remarkably improved the quality of life for patients with lung diseases, and particularly the asthmatic patients. In the early days, they had to put up with a life-threatening disease – now they are practically able to live a normal life. The development of these products is a combination of effective molecules and efficient delivery devices.
The development of these inhalation products can be split into the development of the actual device and that of the formulation incorporating the drug. Two types of devices are available at the moment, the pressurised metered dose inhaler (pMDI) and the dry powder inhaler (DPI). The pMDI development started in 1956 and was based on chlorofluorocarbons (CFCs) to create the liquid formulation and the high pressure, around 6 bars. It turned out to be very efficient but as the CFCs prompted the depletion of the ozone layer, these CFCs had to be phased out. This led to the development of hydrofluoroalkanes (HFAs) and the transition started in the 1990s. The increased global warming awareness highlighted that the HFAs had a high global warming potential (GWP), hence the urgency to find and register products that have a driving gas with low or no GWP. At the moment, the transition from HFAs to those with low GWP is well underway. Further to the environmental issues mentioned above with pMDIs, there has also been concerns about inhaler handling and the coordination between the inhalation manoeuvre and the releasing of the dose. This coordination issue is a general problem for pMDIs. Solutions exists, such as breath-activated mechanisms, but these are costly and are therefore not always included in the product development.
Dry powder development started alongside the need for high doses, higher than what can be delivered via a pMDI. For example, Fisons developed the Spinhaler, capable of delivering 20 mg of active pharmaceutical ingredient (API). Also, low
dose APIs were considered, which led to the development of capsule inhalers. Examples of such inhalers are the Rotahaler from GlaxoSmithKline (GSK) and HandiHaler from Boehringer Ingelheim. Likewise, the early developed Diskhaler from GSK is rather similar but has four or eight cavity blisters with each dose of the formulation individually packaged. These inhalers were technically simple, and the formulations were based on the use of large sugar particles, such as lactose or glucose. Two key benefits were identified with the capsule inhalers: 1. No need for hand-breath coordination as these DPIs were breath activated and 2. The API was kept dry in the capsules, resulting in a very beneficial stability.
DPIs were further refined with a focus on having many doses in each non-reusable inhaler. This led to the development of the Diskus (Accuhaler) from GSK and the Turbuhaler from AstraZeneca. The Diskus inhaler has up to 60 pre-filled doses separated on a blister strip, while the Turbuhaler has the powder in a reservoir from which the patient can measure a dose at use. The formulations also differ between these two inhalers. The Diskus has the traditional carrier-based lactose, in total 12.5 mg of powder, while the Turbuhaler has micronised particles only. Initially, the formulation in Turbuhaler was pure drug, e.g. budesonide. However, the low doses, e.g. 200 µg which is not much powder, made it difficult to meter a consistent dose. Later development increased the powder dose by mixing the API with micronised lactose resulting in a total amount of about 1 mg. Both these inhalers should be regarded as very modern, holding many doses, being portable, looking smart and having dose counters/indicators. These two inhalers were very successful, particularly the Diskus as it was registered and marketed in the USA. In 2011, the Advair Diskus was the bestselling pharmaceutical product globally with a total sale of $8.7 billion. Some additional development has taken place mainly with the new GSK device Ellipta. Ellipta is just a refinement of the Diskus that includes three different APIs.
Room for More Inhaled Products
Despite the accomplishments of very successful devices, not much has happened
in recent years. One can probably say that asthma and COPD are at the moment well treated diseases even though COPD is the third highest global cause of death, responsible for more than 5% of total deaths according to the WHO. Some generics inhalation products have reached the market and have mainly resulted in a lower cost for patients/payers.
The framework described above shows that inhaled products can indeed be successful and should bolster confidence that other products can be developed based on the inhalation route. It can also be concluded that patients are comfortable with these devices and able to use them correctly. One can even argue that inhaling a dose is both more convenient, quicker, and easier than to be administered, for example, as a subcutaneous dose. We see three areas of development for inhaled products: 1. Even better products for asthma and COPD, 2. Products against other respiratory diseases and 3. Products for systemic action.
Better products for asthma and COPD can integrate both improved devices and more efficient APIs. Inhaler improvements require means to increase the fraction of fine particles. For example, the dose released from Diskus only contains about 20% fine particles, i.e. particles small enough to have a chance to reach the lung. An increase in the fine particle fraction will be beneficial for several reasons, including a more efficient use of the API, resulting in the possibility to reduce the dose and thereby the cost of API. A lower dose will also reduce the side effects. Furthermore, if the fine particle fraction were to be increased, the relative variability of the dose to the lung would be reduced resulting in a more predictive dose within and between patients.
Building on the history of successful products against asthma and COPD, the development of products that target other respiratory diseases are underway. This includes products against pulmonary arterial hypertension (PAH), cystic fibrosis (CF), idiopathic pulmonary fibrosis (IPF), cough, etc. A successful development of products against these diseases will be of great value to many under-treated and suffering patients. The technical development of such products can
Nasal & Pulmonary
be more or less straightforward depending on how similar these products can be to the today existing ones. However, for example, if doses need to be high, currently available inhalers might be out of scope.
The lung is also a good route to reach the systemic circulation. Inhaling the API means that degradation in the gut and first pass metabolism are avoided. Unpleasant needle-sticks, intravenous or subcutaneous, are also avoided. Furthermore, many APIs dissolve and are taken up quickly in the lung resulting in a fast access to the systemic circulation. This principle can be used for example for getting a fast pain relief. Many diseases currently treated via dosing to the systemic circulation, mainly via the oral route, can be given via inhalation. Such examples exist already, for diabetes and Parkinson’s disease.
Before ending this summary of inhalation product development, it should be mentioned that an inhaled product may benefit from having a good patent protection as it is generally more difficult to develop inhaled generics than any other propositions. Additionally, as inhaled products include a device, there is an opportunity to make the product unique. There is also a possibility to include electronics to for example register at what time doses are taken and perhaps
the precise inhalation flow and inhaled volume.
Device Complexity
The types of different inhalation devices can be split into four groups: pMDIs, DPIs, soft mist inhalers (SMIs) and nebulisers. And within each group, one can find several different versions. Even if SMIs is the most recent group, we can already see a range of inhaler versions including the Respimat, but there are also many other under development. For nebulisers there is, strangely enough, no standardisation, even though the requirements, as having input from compressed air, a nozzle, and a reservoir for the medication, are quite simple. It is therefore surprising to find more than 100 different brands of nebulisers being marketed, many different in each country. The number of different standards for nebulisers makes the situation complex for all, including prescribing doctors, patients, and drug product developers. In pharmacies, this range of nebulisers is difficult to handle, both from a logistic perspective but also from a learning perspective. For example, does this particular nebuliser require a specific compressor and does this specific nebuliser fit a certain drug product?
Today, pMDIs are rather standardised with cans, valves, driving gas and adapters being
available off the shelf and all very similar. You just add the API. However, for dry powders, the situation is quite different. There are several different DPI devices both marketed and in development, but not off the shelf (more than single dose capsule inhalers), and these are different both from a technical perspective as well as from a patient`s view. If you want to develop a dry powder product that is both portable and holds many doses, the situation is difficult. For example, the Diskus and the Turbuhaler devices belong to the companies that had developed them and not to others. One could wish for a situation where there was an available DPI device platform, with many doses, being small and portable, intuitive to use, resulting in a high fine particle fraction and coming at a low cost. If this device was also reusable, beneficial from a cost and environmental perspective, it would be even better.
Conclusions
The availability of inhaled products, particularly for asthmatic patients, has transformed asthma from a life-threatening disease to a disease that patients can live with, feeling more or less healthy. Despite the current good situation, there is still room for cheaper, more convenient, and efficient products for this patient group as well. There is also room for using the inhaled route for other diseases, both local and systemic. This development should include both new drugs as well as a technical development within the field of inhaled drug delivery. Thus, there are many ways to develop inhaled products in the years to come. If needs are met and challenges are taken on by developers, the future of new inhaled products is set to be bright.
Lars Asking
With a background in physics, Lars has mainly worked with inhalation product development for more than 30 years in both small and large pharma including AstraZeneca and Novo Nordisk. In the 1990s, Lars was involved in the development of the modern way of testing inhalers including impactor development and calibration. Lars has now a broad general pharma understanding and has since 2016 been the CEO of MVIC (Medicon Valley Inhalation Consortium).
30+ YEARS OF EXPERIENCE IN INHALED & NASAL PRODUCT DEVELOPMENT
Support for small and large molecules across all respiratory delivery technology platforms from our GMP compliant laboratories.
• Formulation development
- Including particle engineering approaches
• Specialist support for biologics and DNA based medicines
- Dedicated suite for testing and characterisation
• Clinical manufacturing
• Independent device screening and selection
• CMC package support
• ICH stability study testing and program management
• QC and GMP batch release testing
• In-vitro bioequivalence (IV-BE) studies
• Support for formulation and testing of low GWP propellant pMDIs
• GMP IV-IVC tools
- MDRS
- Inhaled dissolution
- Idealised inlet models and realistic breathing profiles for impaction testing
+44 1763 261 648
intertek.com/inhalation
inhalation@intertek.com
3rd
ANNUAL INHALED & NASAL BIOLOGICS/DNA FORUM 2024
26-27 SEPT | CAMBRIDGE, UK
Join us for a ground-breaking and insightful forum on inhaled and nasal biologics development.
Don’t miss the opportunity to hear from world-renowned industry leaders sharing the latest learnings on inhaled & nasal biologics and DNA medicine development, including our keynote speaker, Dr Jenny Lam, Associate Professor in Pharmaceutics at the UCL School of Pharmacy, UK. Find out more online
CONFERENCE PARTNER
Nasal & Pulmonary
The Great Why Questions About Dry Powder Inhalers
The research pertains to the economic burden of asthma and chronic obstructive pulmonary disease and the impact of poor inhalation techniques with commonly prescribed dry powder inhalers as illustrated in three European countries. Currently, the direct cost burden of managing asthma and COPD for people using DPIs is €813 million, €560 million, and €774 million in Spain, Sweden, and the UK, respectively. Poor inhalation techniques comprised 2.2–7.7% of direct costs, totalling €105 million in these three countries alone. When lost productivity costs were included, total expenditure increased to €1.4 billion, €1.7 billion, and €3.3 billion in Spain, Sweden, and the UK, respectively, with €782 million attributable to poor inhalation technique across the three countries.1,2
Airflow resistance
A major issue is current airflow resistance: “Current guidance is in the line with these observations, suggesting that passive DPIs are all flow-rate dependent, and that young children and elderly patients are at risk of not being able to achieve the flow rates necessary to effectively disperse the powder. The underlying factors controlling inspiratory pressures, flow rates and dispensing, and dispersion characteristics of the various DPIs explain why this is the case. While it is also clear then that some patients at the extremes of the population, with poor muscle strength, may not be able to achieve the inspiratory flow rates to utilise a given DPI”.3 “In particular, the inspiratory airflow generated by the patient represents the only active force (a passive force for the device) able to produce the micro-dispersion (even if differently sized for each device) of the powdered drug to inhale. On the other hand, the extent of the patient’s inspiratory airflow depends on the patient’s airway and lung conditions, and, partially, on the intrinsic resistive regimen of the device”. “While low resistance DPIs are still regarded as the easiest and the most comfortable devices for the patient, they instead require a high inhalation airflow rate to the patient, not always
achievable. The reason is that the role of the other possible force involved in drug deagglomeration”.4
De-agglomeration
De-agglomeration is a major parameter in DPI design. “Dry powders designed for inhalation are very fine and can easily form agglomeration due to cohesion between individual particles and are hard to aerosolize. Despite the inhaler and formulation designs, patients are required to generate a forceful and deep inhalation through the DPI to de-agglomerate the powder formulation into respirable particles (with an aerodynamic size ≤ 5 µm) for efficient delivery to the lungs”.5
“In the inhaler, de-agglomeration is one of the most important particle behaviours since it determines the final particle size distribution and hence the FPF. For CapsuleBased DPI, the presence, dimension and movement of the capsule could greatly influence the flow field in the inhaler, as well as the de-agglomeration and dispersion of particles. The capsule is initially placed in the capsule chamber of the DPI and pierced in a certain way before inhalation to allow particles to be fluidised and move out of the capsule. During the inhalation, the high-speed air flow (normally around 60 L/min) from the inlet of the inhaler forces the capsule to rotate and collide frequently with the inhaler wall. This would, for a certain degree, help the de-agglomeration process of the particle clusters and enhance the performance of the DPI. The drug emptying process normally takes about several seconds. It is a rather complex process, involving the interaction of solid capsule, particles and air flow. At an even higher air flow, for example, over 100 L/ min, the shattering of the capsule may occur, which will further increase the complexity of the problem”.6 Although Capsule DPIs are relatively simple and low-cost device it is absolutely limited their use for patient with high inhaling force over 60L/min which is insufficient for babies, young children, elder people and many lung diseases patients. The result of shaking (rotating) the capsule inside the Capsule DPI still does not guarantee, according to studies, the solution to the agglomeration problem when inhaling into the lungs.
The Great Why Questions
• Why after decades of producing and using DPI's, still the fine particle lung deposition is so low?
• Why is the use of DPI's required complicated, especially for children, babies, the elderly, and those with disabilities?
• Why is the airflow resistance so high in capsule inhalers and is it not suitable for many populations that suffer from breathing problems?
• Why is the high price of inhalers that does not contribute to health in the third world where 3.2 million people die from lung diseases every year?
• Why is it forbidden to exhale into the inhaler and why is it impossible to breathe through the inhaler?
• Why do you need expensive digital inhalers just to cover up the inefficiency of delivering the drug to the lungs?
• Why practically the existing inhalers are not suitable for use for new drugs or biological drugs aiming as lifesaving drugs suffering from the combination of the DPI's inefficiency with the high price of the drug?
• Why is the use of hazardous medical cannabis cigarettes and VAPs still in use?
The answer to all questions is: Engineers, companies, and entrepreneurs are captive to old concepts and they only try to improve them and complicate them in more complex systems to cover the basic performances which do not contribute to users, medical insurance companies, and global health.
As Mr. Jacek Olczak, CEO, of Philip Morris International speech regarding stopping smoking, it's very true for DPI: "It's time to try something else, to try including an innovative approach"
Like years of using polluting vehicles, a new generation of hybrid and electric vehicles is starting!
Hence the revolutionary solution, the result of joint research and development by the author of the article, who has many inventions in the field of medical devices
with Prof. Dan Adler from the Jet Engine Department at the Faculty of Aeronautics in the Israeli Technion.
CanDapi revolutionary DPI is based on a VibraMeshTM technology activated only when the patient inhales; creating a highfrequency vibration of an internal leaf. The vibrations shake the drug chamber, which is covered by a sieve. The vibrations break powder agglomeration and release particles at a specific size that is streamed to the lungs until all powder is consumed. The device achieved the highest score of lung particle deposition scores vs. competitors during the development of the prototypes (~80% efficiency, Kiel University, Germany)7,8 and 91.8% with cannabinoids dry powder as tested by Copley lung simulator. The patient can breathe through the device with the lowest known airflow resistance without special warnings. The cost of the inhaler without the drug is less than 2€.
Key Message
The most important finding of The VibraMeshTM DPI:
• The most important finding of The VibraMeshTM DPI
• >90% efficiency
• Works only during inhaling (breath through DPI).
• Lowest air-flow resistance (0.01 KPa1/2/ l/min)
• Simple and easy-to-use DPI
• Suits everyone – Multi-age and disabled patients
• Customisable
• delivery of soluble cannabinoid powder
• Very low cost
• Very long shelf life
Regulatory & Marketplace Nasal & Pulmonary
De-agglomeration
According to “The Investigation of the “New Single-Use Dry Powder Inhaler” by Wagenseil, L., Menge, and Steckel, H. Department of Pharmaceutics and Biopharmaceutics, Christian Albrecht University, Grasweg 9a, 24118 Kiel, Germany,
the VibraMeshTM Dry powder inhalers are well-established in the treatment of various lung diseases and are also useful for systemic drug delivery. They can simply be adapted to the requirements of a single use device, which provides several advantages such as application in acute therapy, economic integration into treatment regimen in hospitals and onceonly use. Therefore, preferred features of a disposable device are, amongst others, a low resistance, an effective powder deagglomeration and a constant powder release at clinically relevant air flow-rates. Intuitive and simple use of the device is essential as well. The de-agglomeration efficiency of the device was calculated in percentage based on the obtained primary particle size distribution, which was assumed to be 100% de-agglomerated.
Grandma sifting flour
Clarinet single reed diffuser
Minimum airflow resistance and creating a "jet" air stream based on aeronautical principles
Low-cost materials and manufacturing
Summary of the scientific technology solution and testing results and achievements
Comparison of Fine particle others to CanDapi DPI
Analyzing VibraMeshTM air-flow resistance by Copley DFM4 – Flow Meter vs. Others9
Efficacy Test Results
Dose DPI: Air-flow Resistance Test
Nasal & Pulmonary
Cannabinoids VibraMeshTM Test Results
Note: The cannabinoids formulation is a water-soluble dry powder.
• > 90% of the cannabinoid-lactose formulations material was released from the ESD
• No increase in degradation, photodegradation or oxidation products in comparison to control (d8-THC, CBL or CBN).
• Cannabinoid profile remain with no change.
• Cannabinoids powder DPI test results shows excellent performance
Conclusion
The patented DPI based on VibraMeshTM, solved four major issues of the existing DPI:
1. Efficiency
2. Solving high airflow resistance issues
3. Solving DPI powder agglomeration issues
4. Simplicity of use for all ages and medical condition
5. Price for the end user, the healthcare provider and a solution for developing countries
6. Replacing harmful cannabis cigarettes and cannabis VAPEs with safer DPI solution
REFERENCES
1. Chronic obstructive pulmonary disease and the impact of poor inhalation technique with commonly prescribed dry powder inhalers in three European countries A. Lewis1, S. Torvinen2, P. N. R. Dekhuijzen3, H. Chrystyn4, A. T. Watson1, M. Blackney1* and A. Plich2, Lewis et al. BMC Health Services Research (2016) 16:251 DOI 10.1186/s12913-016-1482-7
2. The Confusing World of Dry Powder Inhalers: It Is All About Inspiratory Pressures, Not Inspiratory Flow Rates, Andrew R. Clark, PhD,1 Jeffry G. Weers, PhD,2 and Rajiv Dhand, MD3, JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY, Volume 33, Number 1, 2020, Mary Ann Liebert, Inc. 1–11, DOI: 10.1089/jamp.2019.1556
3. The Confusing World of Dry Powder Inhalers: It Is
All About Inspiratory Pressures, Not Inspiratory Flow Rates Andrew R. Clark, PhD, Jeffry G. Weers, PhD, and Rajiv Dhand, MD. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY Volume 33, Number 1, 2020 Mary Ann Liebert, Inc. Pp. 1–11, DOI: 10.1089/jamp.2019.1556
4. Dry powder inhalers and the right things to remember: a concept review, Roberto W Dal Negro, Dal Negro Multidisciplinary Respiratory Medicine (2015) 10:13, DOI 10.1186/s40248-0150012-5
5. Dry Powder for Pulmonary Delivery: A Comprehensive Review Birendra Chaurasiya and You-Yang Zhao Pharmaceutics 2021, 13, 31. https:// dx.doi.org/10.3390/pharmaceutics13010031
6. Flow and Particle Modelling of Dry Powder Inhalers: Methodologies, Recent Development and Emerging Applications by Zhanying Zheng, ORCID, Sharon Shui Yee Leung 2ORCID and Raghvendra Gupta, Pharmaceutics 2021, 13(2), 189; https://doi.org/10.3390/ pharmaceutics13020189
7. Wagenseil, L., Menge, A.‐K., Solomon, I., Steckel, H. "Optimisation and performance of the ResQhaler‐a single‐use disposable dry powder inhaler" DDL22, Edinburgh, Scotland (2011)
8. Investigation of Single-use disposable ResQhaler New Single-Use Dry Powder Inhaler, Wagenseil, L.1, Menge, A.-K., and Steckel, Department of Pharmaceutics and Biopharmaceutics, Christian Albrecht University, Grasweg 9a, 24118 Kiel, Germany, Kiel university publications
9. (EXPERT OPINION ON DRUG DELIVERY, 2017 VOL. 14, NO. 4, 499–512)
Amnon Kritzman Kadron, Founder and Inventor, Engineer (M.Sc.) From the Technion, Israel. Amnon is a serial entrepreneur and inventor with over 20 patents. CEO and CTO of several medical devices and biotechnology engineering companies based on his inventions. Amnon is currently the CEO and founder of CanDapi in Israel based on his invention of the innovative new Dry Powder Inhaler (DPI).
Amnon Kritzman Kadron
Emerging Trends in Nasal Spray Development: Tools for Future Success
A Brief History of Nasal Drug Delivery
Nasal drug delivery has significantly evolved since its inception several decades ago. Initially, it was primarily used for locally acting treatments related to decongestion and rhinitis, with some of the earliest multi-dose sprays appearing in the 1950s and 1960s. A major milestone was reached in 2003 with the approval of the first nasal vaccine, FluMist® Quadrivalent. Since then, pharmaceutical developers have pursued intranasal drug administration as a viable alternative to traditional routes. It has become an attractive option for various indications, including rescue therapies, infectious disease prevention, pain management, central nervous system conditions, and more.
Recent breakthroughs, such as Narcan® for opioid overdose reversal and the antidepressant Spravato® for treatment resistant depression, demonstrate a growing interest in novel nasal therapies. Additionally, the emergence of the COVID-19 pandemic in 2020 spurred vaccine development, renewing interest in intranasal and inhaled vaccine administration due to ease of administration and rapid action.
Advantages of Intranasal Drug Delivery
The nasal cavity is an ideal target for administering drugs locally and systemically, particularly to the central nervous system (CNS). The nasal mucosa is highly vascularised, providing a direct route for drug absorption into the bloodstream, which facilitates rapid onset of action. This efficient absorption can lead to reduced
dosages compared to oral administration, as it bypasses first-pass metabolism in the liver. Moreover, intranasal administration is non-invasive and can usually be selfadministered, enhancing patient compliance. These benefits underscore intranasal drug delivery as a valuable alternative for achieving rapid and effective therapeutic outcomes in various clinical settings.
Key Regulations and Consideration for Nasal Drug Development
The approval process for nasal drug products is governed by strict regulatory testing and development guidelines. Major regulatory bodies, such as the Federal Drug Administration (FDA) and the European Medical Association (EMEA), require comprehensive in vitro characterisation tests and in vivo clinical evaluations. While there are a variety of paths to approval depending on the drug product and region, there are several essential in vitro tests for nasal spray products that include Pump Delivery, Spray Pattern, Plume Geometry, Dose Content Uniformity, Aerodynamic Particle Distribution, and Droplet Size Distribution. These tests are essential for charactersing the emitted nasal spray and crucial for proper development and approval of nasal drugs products.1
When delivering drugs to the nasal cavity, it is critical to understand the relationship between the formulation, device, and the patient and to control their effects on both in vitro spray characteristics and potential implications for in vivo therapeutic outcomes. When developing a nasal drug product, it is essential to monitor the effects of these factors on spray characteristics to maintain equivalence to a reference product or internally defined product performance standards. These three factors dictate the
location, amount, and therapeutic outcome of nasal drug products, making their development uniquely challenging.
The Device
The device is more than primary packaging to contain and administer a formulation; it is a tool operated by a patient to deliver medication to the target site. A nasal spray device contains multiple components, such as the nasal pump, dip-tube, pump assembly, and internal swirl chamber, and has a specific nozzle length and diameter. Developers must consider all these mechanical factors during development, as they can influence the performance of nasal sprays both in vitro and in clinical studies. For example, changing the diameter of the orifice and swirl chamber design of a nasal spray, while all other factors are held constant, can lead to drastically different in vitro performance.2
The Formulation
Formulations play a significant role in nasal spray performance. In addition to the Active Pharmaceutical Ingredient (API), the physical characteristics of the emitted spray are critical to therapeutic outcomes and critical in vitro spray characteristics. Formulation factors, such as the viscosity and the presence of excipients, can lead to distinct spray characteristics and deposition and absorption behavior in the nasal cavity. We have shown that an increase in formulation viscosity decreases the Spray Pattern area and an increase in droplet size. The inverse of this relationship holds true, where a decrease in formulation viscosity leads to an increase in Spray Pattern area size and a decrease in particle size.3 From a therapeutic perspective, highly viscous formulations can remain in contact with nasal epithelium longer, allowing for increased time
Nasal & Pulmonary
for absorption before mucociliary clearance from the nasal cavity. While most nasal drugs are spray formulations, dry powders are also growing in use due to certain advantages, such as formulation stability, longer residence time in contact with nasal mucosa, and better control over particle characteristics.
The Patient Human Factors are another critical factor for nasal spray performance. Unlike other modalities, nasal drug products are highly influenced by human factors, making humanrealistic testing essential for product efficacy and approval. The way a patient administers the spray, known as actuation, can be studied and then replicated through specialised instruments known as Automated Actuators. Regulatory agencies recommend automated
actuation for in vitro characterisation tests to eliminate human variability in manual testing and create robust, repeatable methods for development and regulatory submission. It is important to define a demographically relevant actuation profile to be used throughout development and regulatory testing. For example, actuation profiles determined from adult and pediatric patients yield significantly different performance in critical in vitro tests like Spray Pattern and Droplet Size Distribution, when using the same device-formulation.4
Advancements and Opportunities in Targeted Nasal Drug Delivery
There is growing interest in targeted nasal delivery, which opens the nasal route to new therapies and innovative products. Specific
regions of interest include the olfactory and the Nasal Associated Lymphatic Tissue (NALT) regions.
• Olfactory Region: This region is thought to provide a non-invasive means to bypass the Blood Brain Barrier (BBB) for direct administration to the CNS.
• NALT Region: This region is an ideal target for effective non-invasive means for vaccine administration.
The rise of targeted nasal delivery products and the complex nature of the human nasal cavity make development testing under human-realistic conditions essential. The use of tools like human-realistic nasal casts and controlled administration are needed to advance nasal drug development. Using nasal casts early and throughout development with automated actuation reduces the risk and time to market by providing clinical indications before human testing begins. Advancements in manufacturing and imaging technology have improved the utility of nasal casts, enabling the creation of more complex and human-realistic geometries. The enhanced nasal casts provide deeper insights compared to traditional idealized models.
Pairing in vitro development testing with in vitro human-realistic deposition testing can enhance nasal product development, reducing the risk and time to market. Tools known as nasal casts have been used for decades to replicate human usage, considering factors such as insertion angle and depth, orientation, and the influence of breathing.
Anatomical Regions of the Human Nasal Cavity
Methodologies
Regulatory & Marketplace Nasal & Pulmonary
Navigating Development with Proveris Laboratories: The Proveris Advantage for Nasal Product Success
The successful development and launch of a nasal drug product requires strict adherence to regulatory guidelines and a thorough understanding of the three factors that influence nasal drug efficacy: the device, formulation, and patient. Proveris Laboratories, a division of Proveris Scientific Corporation, offers a comprehensive approach to in vitro nasal product development. Our Orally Inhaled and Nasal Drug Product (OINDP) expert scientists utilise the Proveris by Design™ approach, proven to reduce timelines and streamline submission. As targeted and novel nasal therapies rise, Proveris Laboratories not only adheres to industry best practices using gold-standard Proveris equipment, but also advances the science of aerosol and spray delivery with innovative tools.
Proveris Laboratories further enhances the nasal development process by integrating human-realistic nasal cast deposition testing early and throughout development. This method enriches nasal product development by providing deeper insights into product performance. Conducting nasal cast deposition studies during the formulation and device optimisation phases allows for an understanding of nasal deposition before progressing to in vivo testing, significantly reducing the risk of failure due to poor in vivo performance.
The Proveris Nasal Cast: Revolutionising Human-Realistic
Nasal Deposition Testing
The Proveris Nasal Cast is a human-realistic adult model of the nasal cavity, complete with five extractable regions and a nasopharynx filter, allowing for the quantification of API in each region. During nasal deposition testing,
Proveris Laboratories employs human-derived automated actuation and studies the effects of device insertion angle and depth, as well as human-realistic breathing, when applicable.
Proveris Nasal Cast studies provide rapid results and are particularly useful for those developing targeted nasal therapies, novel device designs, and innovative delivery techniques. The Proveris nasal cast is an accurate model of realistic complex geometries and barriers that are important to account for when targeting specific regions like the Olfactory region and the NALT region. Developers must ensure that the target region receives the required amount of drug products and nasal cast studies are the most efficient way to determine this without in vivo testing.
Next-Generation
Nasal Spray Products with Proveris Laboratories
The next generation of nasal spray products has advanced well beyond primarily localized treatments like decongestants and rhinitis therapies. Innovative products now target specialised regions within the nasal cavity with novel formulations and devices. By understanding and controlling the complex relationship between device, formulation, and patient, Proveris Laboratories offers a comprehensive development process that leverages innovative tests and technologies, reducing timelines, and enhancing insights.
Proveris Laboratories performs traditional testing with demographic-specific human actuation parameters, paired with HumanRealistic Nasal Cast testing, to elevate development. This approach allows for the assessment of therapeutic indications before clinical trials. Trust our experts to work for you and start a partnership with Proveris, an
industry leader in OINDP instrumentation and laboratory services, for over 25 years.
REFERENCES
1. FDA Guidance for Industry (2002) Nasal Spray and Inhalation Solution, Suspensions, and Spray Drug Product – Chemistry, Manufacturing, and Controls Documentation https://www.fda. gov/files/drugs/published/Nasal-Spray-andInhalation-Solution--Suspension--and-DrugProducts.pdf) Accessed June 27, 2024.
2. Impact of actuator design on multi-dose nasal spray characteristics. Poster presented at Respiratory Drug Delivery Conference 2018. Chauhan H, Liu-Cordero S, Liao L, Werbeck J; Respiratory Drug Delivery 2018. Volume 2, 2018: 497-502.
3. Synergies of inhalation product development and spray characterisation. Catalent Incorporated, Proveris Scientific Corporation. Workshop presented at Respiratory Drug Delivery Conference 2023 (RDD2023). RDD Europe 2023 –Workshops (rddonline.com)
4. Automated actuation of nasal spray products: effect of hand-related variability on the in vitro performance of Flonase nasal spray. Doughty, D. V., Hsu, W., & Dalby, R. N. (2013). https://doi.org/ 10.3109/03639045.2013.777735
Grant Thurston
Grant Thurston, Product Manager – Proveris Scientific is responsible for overseeing the development and enhancement of Proveris product lines, ensuring the organization continuously meets and exceeds customer expectations. Grant has a deep understanding of laboratory instrumentation and strong ability to communicate technical concepts effectively. Grant holds Bachelor and Master degrees in Biomedical Sciences.
Alyssa Rubino
Alyssa Rubino, Technical Data Scientist – Proveris Laboratories is responsible for ensuring appropriate study design, compiling and analysing analytical study data, deriving conclusions and key information, and generating reports for all contracted projects to ensure they meet internal, customer, and regulatory standards. Alyssa holds a Bachelor of Science in Mechanical Engineering.
Proveris Nasal Cast attached to Vereo® Automated Actuator
Nasal & Pulmonary
Advances in Intranasal Vaccine Delivery: A Promising Non-invasive Route of Immunisation
Vaccination is an effective weapon for disease prevention and has been proven to significantly reduce the transmission of infections and the number of deaths worldwide. Almost all the approved vaccines are injectables and consequently parenteral vaccination is the most used method for their administration. The continuous emergence of new pathogens and the resistance of microorganisms increase the trend towards the development of new strategies to produce longlasting immunity. Furthermore, the interest of the scientific community on the non-invasive methods of vaccine administration is risen constantly in the last decade. It is quite known that the nasal cavity is the first-contact area when the antigens are entered in the human body. Hence, the development of nasal vaccines may be a feasible alternative for a more effective immunisation.
The aim of this comprehensive review is to summarise and critically discuss the advances in the field of nasal vaccines to reveal their prospect as an alternative mode of immunisation. We assess the clinical applicability (protective and/or therapeutic) of IN vaccination in various diseases, such as influenza, pertussis, meningitis, hepatitis, based on in vitro, preclinical, and clinical studies.
Innovative Delivery Systems for Nasal Vaccines
Nanoparticulate Nasal vaccines
Several preclinical studies have focused on using nanoparticles as delivery systems and adjuvants either for intramuscular or nasal vaccination employing rodent or pig animal models. Particularly, the recent review of Nian et al. efficiently summarises the available types of mucosal adjuvants for nasal vaccines. Interestingly, through the nasal passage, nanoparticles can pass across mucus and interact directly with NALT cells. The stimulation of mucosal immune responses leads to the production of persistent immunological memory. The main reason for the use of
nanoparticulate vaccines is their ability to protect antigens from the proteolytic degradation and improve the cellular distribution. The sustained release of the antigen in the mucosa can also be achieved by its formulation into nanoparticles. Thus, the probability of antigen uptake by the mucosal and lymphatic cells is increased. Modifications in physicochemical properties increase the stability of nanoparticles in biological fluids, allowing the prolonged presentation of the antigen in the body. Furthermore, various types of antigens can be entrapped and higher loading capacities, not only for the antigens but also for the adjuvants, can be achieved.
It is also feasible to modify the physicochemical properties of the particles, such as their charge, shape and size, rendering them ideal carriers for protein delivery, as well as for mimicking the viruses’ properties. Specifically, it has been shown that the spherical shape, size around 100 nm, cationic charge and hydrophobic character favours the uptake of nasally administered antigens from APCs. Another useful feature of nanoparticles is the ability to integrate TLR ligands in their surface, leading to a prolonged TLR signalling and reducing the number of antigens and adjuvants needed for an efficient immune response. Τhis cascade of events can also reduce the incidences of toxicity in non immune cells.
Nanoparticle Types Used in Nanoparticulate Nasal Vaccines
Polymeric nanoparticles. Nanoparticles from natural and synthetic polymers have been extensively studied as antigen delivery systems. Chitosan and PLGA molecules are the main representatives. Particularly, their mucoadhesive properties have been shown to enhance the immune response at both mucosal and systemic level. This is also verified in preclinical studies in which nasal immunisations were performed involving surface antigens of the hepatitis B virus (HBsAg), ovalbumin molecules and strains of the influenza A virus. In the study of Zaman et al. the self-assembling of amphiphilic dendrimers from polyacrylate molecules is described. These polymers could form IN subunit vaccines, giving a self-adjuvanting effect against systemic group A streptococcus
infection. In the case of influenza A several chitosan-based mucosal vaccines have been tested in vivo, in poultry and pigs, having been delivered either orally or intranasally, triggering both mucosal and cellular immune responses.
Polysaccharide Nanoparticles
Polysaccharide nanoparticles are among the most well-established carriers of intranasally administered antigens. Due to their biocompatibility, polysaccharides such as starch and dextran, have been used to formulate nanoparticulate nasal vaccines. Cationic nanoparticles consist of maltodextrins (Supramolecular biovectors: SMBVTM) were evaluated as delivery systems of two recombinant proteins, either of a particulate (Hepatitis B surface antigen: HBsAg) or of a soluble (b-galactosidase) one, for their immunogenicity to mice. This type of vector comprises a polysaccharide core that can be further surrounded by a phospholipid layer, mimicking the size and structure of viruses. Thus, it can be used for protein and peptide delivery, especially through nasal sprays. The results showed high levels of serum IgG, mucosal IgA and Cytotoxic T lymphocytes (CTL)-mediated responses. Similar outcomes were observed after the IN immunisation of rabbits, with nanoparticles consisting of starch and Carbopol 974P, which served as influenza virus antigens’ carriers. Furthermore, dextran-based nanovaccines induced dendritic cells (DCs)mediated responses after IN administration of tetanus toxoid in rabbits. In another study, sweet corn-derived cationic alphaD-glucan nanoparticles were combined with a synthetic double RNA molecule and a tolllike receptor (TLR)-3 ligand to be delivered intranasally in pigs. This vaccine was found to induce the production of antibodies and the cytokine response more efficiently than the inactivated influenza A vaccine given either intranasally or intramuscularly.
Proteosomes
Proteasomes are protein-based multilayered structures, including hydrophobic outer membrane proteins. They are mainly used for the administration of subunit mucosal vaccines, such as influenza hemagglutinin glycoproteins. These glycoproteins include hydrophobic domains in their structure and
Regulatory & Marketplace Nasal & Pulmonary
thus, they can be non-covalently associated with the macromolecules of proteasomes, and successfully present these antigens to the mucosal immune system. In addition, the proteasomes have been successfully tested in phases 1 and 2 of clinical trials, as delivery and adjuvant systems.
Lipopeptides
Lipopeptides are a category of nanoparticles, consisting of one or more lipid moieties with antigenic and/or immunomodulatory peptides, forming amphiphilic liposomal or micellar structures. They can self-assemble in an aquatic environment, forming both nano- and micro-particle structures, due to the interactions between peptides and lipids. Lipopeptides were tested as vaccine delivery systems and evaluated for their immunogenicity against Group A Streptococcus. Their IN administration demonstrated that the appropriate size of vector and epitopes can induce epitopespecific antibody responses, as well as high IgG serum titers. Moreover, the entrapment of lipopeptides in cationic liposomes can induce both mucosal and systemic immunity, increasing IgA and IgG titers in mice. High levels of antibodies were also detected five months after the inoculation.
Nano-Emulsions For Nasal Vaccines
Emulsions are stable dispersions of two or more immiscible phases (oily and aqueous), containing small amounts of surfactants as stabilising agents. These formulations are considered ideal for the development of
nasal vaccines, as they can produce particles smaller than 200 nm. Nano-emulsified antigens are controlled release formulations, showing an increased resistance to enzymatic degradation. Moreover, the properties of the oils and surfactants used in their composition can increase the interaction with the nasal mucus. The total charge of the nanoemulsion, as well as the degree of their induction can vary, depending on the components of the formulation and the type of the immune response. Nanoemulsionbased nasal vaccine was developed and tested for its protective effects against hepatitis B. More precisely, surface antigens of the hepatitis B virus (HBsAg) were trapped into a soybean oil-based emulsion and administered intranasally to animal models. The nanoemulsion was able to cross the epithelial membrane intracellularly. Therefore, the opening of nasal mucosa tight junctions or the involvement of M cells were not required to achieve effective permeation. The results demonstrate that needle-free nasal immunisation with emulsion formulation can induce an effective Th1 associated cellular immunity, providing therapeutic benefit to patients with chronic hepatitis B infection, comparable to that derived by parenteral vaccination.
Dry Powders for Nasal Vaccines
Dry powders have also been tested as candidates for the formulation of nasal vaccines. They are considered as advantageous forms being less susceptible to physical, chemical, and thermal degradation,
than the liquid ones. Dry powder nasal vaccines have been widely tested in vivo for immunisation against various diseases, such as diphtheria, influenza, tetanus, anthrax, meningitis and gastroenteritis. Moreover, recently a nano-adjuvanted dry powder vaccine was assessed for its ability to induce mucosal immunisation against the Mycoplasma hyopneumoniae, producing higher IgA and IgG levels than the conventional intramuscular vaccine . To administer the Dry Powders Vaccines (DPVs) intranasally, appropriate devices are needed to ensure the stability and protection of the product by humidity. In addition, these devices should be aerosolized and improve the nasal deposition of DPVs, eliminating the lung deposition of vaccine particles. The most used devices are Dry Powder Inhalers (DPIs) and Monopowder single-dose disposable device Powder-Jet is their main representative.
The outbreak of severe coronavirus syndrome caused by the SARS-CoV-2 rendered the development of an effective vaccine the priority for health scientists globally. The SARS-CoV-2 is a single-stranded RNA virus comprising four main structural proteins as follows: S (spike), E (envelope), M (membrane) and N (nucleocapsid). The S glycoprotein forms trimeric spikes on the virion, enabling virus attachment on the membrane of the host cells after binding with the surface receptor of the angiotensin-converting enzyme 2 (ACE2). It has been found that most of ACE2 receptors
Nasal & Pulmonary
are located on the nasal epithelium and salivary gland ducts, while less are found in the alveoli. The virus replication takes place on the nasal mucosa before reaching the systemic circulation. Thus, the infection of the individual with the virus elicits the immune response of both upper and lower respiratory tract, mediated by the sIgA and serum IgG antibodies, located on the nasal mucosa and lungs, respectively.
Intramuscular vaccination results in the intense production of serum IgG, but not of the mucosal IgA. Although adequate amounts of IgG can be found on the mucous membranes of the upper respiratory tract, sIgA deficiency may leave a person vulnerable to covid-19 infection. IN vaccination can induce effective mucosal antibody response, providing sterile immunity in the upper respiratory tract.
The attempts of research groups to produce a nasal vaccine for SARS-CoV-2 were as follows:
1. King et al. (2020) developed AdCovid™, a nasally administered, single-dose vaccine using type 5 adenovirus to deliver the Receptor Binding Domain (RBD). In mice, it induced strong IgG serum neutralising activity, high mucosal IgA titers, and cell-mediated immunity. It's in phase 1 clinical trials.
2. Washington University School of Medicine created a nasal adenovirusbased vaccine encoding the stabilised S protein. In mice and non-human primates, a single dose provided complete protection of upper and lower airways, unlike intramuscular injections.
3. Codagenix Inc. developed COVI-VAC, a nasal vaccine containing a whole inactivated virus. It entered phase 1 clinical trials on January 12, 2021.
4. AstraZeneca/Oxford Jenner Inst. tested their approved intramuscular vaccine intranasally in rhesus macaques. It significantly increased neutralised antibodies and reduced viral RNA in oropharyngeal swabs compared to intramuscular vaccination.
5. Recombinant Vectored Newcastle Disease Virus (NDV): This vaccine encodes the SARS-CoV-2 S glycoprotein gene and can be administered intranasally. Preclinical studies showed amplified humoral and cell-mediated immune responses in lungs and serum, and total inhibition of viral presence and disease transmission four days post-vaccination.
6. University of Houston developed an IN-subunit vaccine with a trimeric or monomeric S protein and a stimulator of interferon genes (STING) as adjuvant, encapsulated in liposomes. In BALB/c mice, it produced high titers of secretory and S-specific IgA antibodies in NALT, lungs, and spleen.
7. Ku et al. (2021) explored a lentiviral vector for IN inoculation in mice with induced ACE2 receptors, which effectively decreased lung viral loads and local inflammation compared to intramuscular injection.
Most Recent Development: A lipid nanoparticle (NP)-based vaccine containing virus proteins or mRNAs combined with NP-monosodium urate adjuvant showed better respiratory tract disinfection and higher antibody levels in ferrets. NP-based vaccines with virus proteins upregulated proinflammatory cytokine expression and enhanced Th1 and Th2 immune responses.
Nasal vaccination is an advantageous mode of immunisation, as the active agents of vaccines are not exposed in extreme pH values and/or digestive enzymes of the gastrointestinal tract. However, the presence of specific physiological features of the nasal cavity constitutes an important limitation of vaccines’ effectiveness. Mucociliary clearance occurs continuously, impeding the attachment of the antigens on the epithelium. The soluble antigens when administered alone can be diffused through TLR receptors. Nevertheless, their uptake is often inefficient, revealing the need for particulate formulations.
In addition, the small capacity of the nasal cavity, the protein efflux systems, and the possible pathological conditions can reduce the extent of the antigen absorption. The anatomical position of the nasal cavity requires special care during the nasal
vaccine delivery, as the excipients used can often lead either to the irritation of the nasal cavity, or to respiratory infections and neurotoxicity. Specifically, in the case of the NasalFlu vaccine, which was approved in Switzerland in 2001, it was quickly withdrawn due to reports of several Bell’s palsy cases attributed to the adjuvant contained in the formulation. Τhe presence of the Escherichia coli heat-labile toxin as mucosal adjuvant and its direct transfer in the CNS, through the olfactory epithelium, led to this severe adverse effect. To avoid this event, accurate targeting of antigens is required, via the proper design of specialised nasal vaccine delivery devices.
Although several limitations accompany the existing products, the optimisation of the antigen form in the nasal vaccines using advanced delivery systems can increase its absorption and diffusion, as well as reduce the possible adverse effects and the events of toxicity. Nasal vaccination tends to gain ground against the conventional intramuscular administration, as a non-invasive method and thus, a promising alternative to increase patients’ compliance. Certainly, critical steps towards the improvement of delivery devices and added adjuvants are required to bend the last doubts against nasal vaccines.
Paraskevi Papakyriakopoulou
Georgia Valsami
Department of Pharmacy, School of Health Sciences, National and Kapodistrian University of Athens, 15784, Greece
*Corresponding author at: National and Kapodistrian University of Athens, Panepistimiopolis, 15784 Zografou, Greece.
Eleni Kehagia
UniSpray
Single-metered dose for systemic-acting drug administration
Nasal & Pulmonary
Accelerating The Go-To-Market of Nasal Combination Products with Integrated Solutions
Ear, Nose, Throat Market Trends Outlook
The overall Ear, Nose, and Throat (ENT) market was heavily impacted by Covid-19 pandemic due to masks-wearing, lockdown, and less social interactions, exposing people less to respiratory viruses as well as allergens. People also tend to avoid going to the drugstores for non-vital treatment during the pandemic, leading to a significant 9,5% drop in Over the Counter (OTC) volume demand in 2020. When we start returning to our normal life post-pandemic, the market need is clearly observed to rebound in 2021, reaching an increase of 5,7%, requiring an agile and fast turnaround from the pharmaceutical industry to secure supply for patients in need of medications. The ENT market is estimated at 1,9 billion devices, with the majority of drug products for nasal applications.1
The topical therapies still dominate the nasal market to treat allergic rhinitis, sinusitis, and nasal congestion. Allergic rhinitis is mostly seasonal and is usually relieved by nasal spray containing topicalacting medications. Corticosteroids are today the first line treatment for allergic rhinitis as they reduce swelling, inflammation, and mucus secretion in the nasal cavity. This long-standing historic market is still very dynamic following incoming pipelines from the generic players, representing opportunities for growth. In fact, today 65% of the topical treatments are generics.2
Most of these topical treatments are available Over the Counter (OTC). In many developed countries, OTC regulations have come a long way becoming more and more stringent, approaching Rx drugs dossier requirements, strengthening barriers to entry, ensuring patient’s safety. For instance, recent Medical Devices Regulations (MDR) for European Market opens an opportunity to upgrade the required standard on raw material compliance, control strategy and documentations on products as well as processes for the registration of OTC drug products, implying similar exigence level as per prescription bound solutions.
The nasal route is non-invasive and does not require healthcare professionals’ intervention. Unlike injectables, patients can self-administer their medication with a rapid onset. It offers better bioavailability as nasal administration avoids hepatic first-pass effect which could be encountered when taking medications orally. Nasal devices are needle-free hence increasing patient acceptance level, leading to positive therapy outcomes following improved patients’ adherence and compliance.
More recently, there has been a growing interest and further exploration in delivering drugs through the nose for systemic-acting drugs, by targeting nasal turbinates. Turbinate occupies a large surface area of the nasal mucosa and is highly vascularized, offering a convenient pathway for a systemic delivery.
A rising number of prescribed systemicacting drugs originally administered in injectable forms have been successfully repurposed and made available as unit-dose nasal sprays. Patients and/or their caregivers are now able to administer a one-shot spray easily and rapidly to manage emergency and crisis situations, such as overdoses, seizures, and migraines. This allows a broader patient population targeting, increasing its
accessibility to different end-user groups. To ensure patient’s safety, the regulatory bodies impose strict regulations on this alternative route to optimise its drug efficacy with equivalence as per injectables, especially for life-saving drugs. The reliability of the device plays a key role to ensure success in saving patients’ lives.
A Full Range of Pumps Solutions for Topical, Multidose Administration
Nemera has a long-standing experience in developing and manufacturing complex nasal devices. Our journey since several decades has proven our capabilities in bringing a wide range of successful market references across the globe, both in regulated and low-regulated countries, to ultimately improve patients’ quality of life. Every year, our state-of-the-art GMP manufacturing facility produces millions of multidose nasal spray pumps. Our market-proven devices have been commercialised for diverse formulations delivery, bringing our customers’ combination products to serve patients. Our expertise in spray characterisation and bioequivalence have made it possible to commercialise a number of combination products. Today, millions of patients rely on our devices to relieve symptoms from chronic conditions such as allergic rhinitis, sinusitis, or nasal congestion. (Figure 1)
Figure 1: UniSpray: safe and reliable single-metered spray for systemic-acting drug administration
Regulatory & Marketplace Nasal & Pulmonary
Excellent spray plume and characteristics
Single-Metered Dose for Systemic-Acting Drug Administration
Multi-dose nasal spray pumps find their place in chronic therapies, but not in acute applications. When the device use is punctual, repriming might be needed before use; also, a large volume of a drug content could be wasted as it’s a one-off usage. Consequently, novel therapies are starting to emerge with a precise, ready-to-use unit-dose nasal spray for emergency and crisis treatments.
Nemera’s UniSpray delivers a singlemetered 100µL dose spray and can be used for new, repurposed, or generic drugs. (Figure 2) UniSpray has gone through different human factor studies, design verification and processes to ensure device reliability and robustness, assuring patients’ safety and ease-of-use, complying to the regulatory requirement.
This single dose nasal spray is a ready-touse primeless device with 360° functionality, enabling one-handed activation. To ensure the correct use of the device, it offers an ergonomic and intuitive design. Once the device is activated, the plunger is locked in its position, preventing premature activation which is extremely critical in emergency use, visually telling users a clear message that the dose has been administered. The final locking also prevents device disassembly after activation.
Ergonomic finger rest for intuitive use
Compatible with existing primary packaging
Final locking position after activation
UniSpray is a customisable platform, offering flexibility for spray adjustment for new formulations and generics. To accelerate time to market, UniSpray is compatible with the existing marketed primary drug containers and is also adapted to fit conventional filling lines. In line with this objective, for generic drugs, the preliminary bioequivalence of selected molecules is
performed by Nemera which generate initial documentations, assuring spray characteristics equivalence and consistent performances; this foundation therefore must be completed by customers based on their formulation. (Figure 3) For new or repurposed drugs, spray performance adjustment can also be done to ensure drug administration efficacy.
Spray expertise and customization
Robust testing methodologies
Statistical analysis know-how
Regulatory expertise
Figure 2: Nemera's spray expertise assures successful bioequivalent projects for OTC and Rx products
Figure 3: Nemera provides a comprehensive range of multidose pumps and offers novel technologies to tap into new market needs
Accurate 100µl liquid dose delivery
Nasal & Pulmonary
Innovative Solutions to Explore Novel Therapies
The vaccine research and delivery are accelerated amid Covid-19 pandemic. The nasal route is seen as an alternative for vaccine delivery to conventional injectable forms, benefitting from Nasal Associated Lymphoid Tissue (NALT), a region associated to the lymphatic network that can induce a mucosal and systemic immune response. By obtaining mucosal immunity in the nasal cavity, our immunity response will be able to combat respiratory pathogens as soon as they enter the upper respiratory tract, preventing further infection. It is also more accessible for patients with needle-phobia, especially for younger age groups.
With our extensive experience in intranasal delivery and injectables, we are developing a reliable, easy to use and safe nasal vaccine solution. The concept device is compatible with existing prefilled luer lock syringes and is constituted with a nozzle and a dose divider. It contains two-doses, allowing vaccine delivery for each nostril.
Another promising route being explored nowadays is the nose-to-brain pathway, presenting opportunities to offer efficient drug delivery particularly for central nervous system (CNS) therapies. Direct brain delivery would mean lower doses therefore less toxicity and less off-target effects as the medications mitigate the first-pass metabolism. However, anatomically, our brain is protected by the blood brain barrier (BBB) which prevents >95% of molecules from entering the CNS from the bloodstream. Noseto-brain delivery poses a real challenge in targeting the olfactory region in the nose,
which is extremely difficult to reach as it represents a small surface area.
Electronic components are also being explored to foster patient’s safety, for instance to secure potent drug administrations. There is a wide range of drugs that are used for painkillers. As an example, opioid treatment such as fentanyl is used to relieve pain with its rapid onset of action through unit-dose or multidose nasal drug delivery.
Indeed, fentanyl is a potent drug, used to treat severe pain that has become the main driver of recent increases in synthetic opioid
deaths. The fentanyl painkiller is used on a regular basis by patients with cancer. For multi dose nasal spray presentations, this may lead to opioid overdose when it is not used according to the treatment posology. Given this, it is crucial to consider the mode and way of administration to ensure the patients’ safety.
On that account, Nemera explores how to manage drug overdosing using a smart nasal spray device. Nemera developed a smart electronic concept device with childresistant, dose counting and locking features, Safe'n Spray, offering solutions to prevent overdosing of potent drugs. It is an integrated device with a reusable electronic locking unit and fingerprint identification, to monitor drug dose delivery in a defined period to ultimately ensure patient’s safety.
Holistic End-To-End Partner for the Combination Product Journey
“We put patients first” is Nemera’s bottom line principle. Our development team works actively to understand patients’ needs through discussions and tests with patients individually or within groups and round tables with Key Opinion Leaders. Our early-stage development concepts are evaluated through the user test studies to ensure a profound understanding of patients’ unmet needs. This is to ultimately optimise the intuitiveness, ease-of-use, and usability of the device with the aims of reducing the occurrence of misuse and optimising the device performance when handled by patients.
Delivering drug products to the targeted site is challenging in the case of nasal delivery. Nemera continues to work on nasal
Figure 4: Full turnkey solutions to support nasal combination product development
Regulatory & Marketplace Nasal & Pulmonary
delivery system concepts aiming the delivery to specific areas of the nasal cavities for optimum drug efficacy. To illustrate, we use in vitro testing on nasal casts which replicate human anatomy to predict in vivo deposition. To support our internal projects, we have developed our own nasal cast, which can also support the joint development effort with customers towards a successful combination product.
The expertise on spray technology is also key to nasal drug delivery. Understanding the physics of the atomisation and achieving good control of the spray characteristics are two of the R&D pillars for nasal drug delivery. We actively work on design processes and tools to support our spray development and reduce the number of design iterations required.
In the case of establishing bioequivalence, following regulatory guidance is extremely crucial. Thanks to our in vitro test capabilities, we can support specific generic projects through a complete set of tests that meets the authority’s prerequisite. This data generation will be statistically analysed regarding USA and EU guidelines for eventual
Speaking of the fundamental idea of integrating patients within the combination product development, Nemera also provides a full understanding of the patient journey and recommends user-related activities to further optimise patients’ experience for a specific drug/device combination product. Thanks to our extensive human factor capabilities, the pharma company could ensure that their selected device, in combination with their drug, is appropriate, safe and effective for the target population. For instance, our support encompasses making a specific Instruction For Use (IFU) adapted to a certain target population, as well as supporting Human Factor activities in alignment with pharma company’s chosen regulatory path including ANDA versus NDA, or else.
Incorporating patients’ insights as early as possible to feed in our platform advancement is crucial. This is done by leveraging the patients’ journey throughout our drug delivery device research and development internally, to ensure our device answers the unmet needs of the end-users.
Through capabilities in human factors engineering, user experience design, engineering, lab services, statistical expertise, and regulatory support, Nemera is uniquely positioned to offer all the support that customers require through an integrated device platform and service program.
Conclusion
In line with the growing interest across localand systemic-acting treatments within the nasal route, our device platforms coupled with the integrated end-to-end approach enable us to support and bring drug/delivery combination solutions to the patients. Nemera is the utmost holistic partner and helps its customers succeed in the sprint to market. From early device strategy to state-of-the-art manufacturing, Nemera is committed to the highest quality standards. Agile and open-minded, Nemera works with our customers as partners. Together, they go the extra mile to fulfil their mission.
REFERENCES
1. IQVIA market data, total of devices sold, 2019–2021, worldwide
2. IQVIA market data, total of multidose devices sold, 2021, worldwide
Séverine Duband
Séverine Duband is Marketing Director for drug delivery devices at Nemera, steering overall category strategy, product portfolio, and innovation development for key delivery routes such as ophthalmics, inhalation, ear-nose-throat, dermal and parenteral. She joined Nemera Global Marketing team in 2018 as a Global Category Manager, focusing on the parenteral segment.
Email: severine.duband@nemera.net
Audrey Chandra
Audrey Chandra is the Category Project Manager at Nemera. Ms. Chandra graduated from the Faculty of Medicine Universitas Atma Jaya Indonesia and pursued her master’s degree in Strategy & Business Development at Toulouse School of Management, France. With her dual competence asset, she is in charge of Dermal Category at Nemera as well as providing strategic support for other various targeted marketing projects.
Email: audrey.chandra@nemera.net
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA’S DNA
Listen to industry experts on the latest in drug discovery, development, research, industry regulations and much more at Pharma,s DNA, the podcast channel by Senglobal Ltd., available on Sound Cloud, Spotify, iTunes and YouTube.
senglobalcoms.com
Advertisers Index
Page 27
Page 43
Page 70
IFC
Page 19
IBC
Page 55
Page 39
Page 41
Page 52
Page 63
Page 35
Page 5
Page 3
Page 15
OBC
Page 71
Page 13 & 17
Subscribe today at www.international-pharma.com or email info@senglobalcoms.com
COSMOGEN SAS
CPHI Worldwide 2024
DDL2024
EyeC GmbH
FUJIFILM Wako Chemicals U.S.A. Corporation
H&T Presspart
Intertek
Kahle Automation
Krautz Temax
MVIC (Medicon Valley Inhalation Consortium)
Nemera
Nipro
Novo Nordisk A/S
Owen Mumford
PCI Pharma Services
Proveris Scientific Corporation
Senglobal Ltd
Valsteam ADCA
I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry
IPI is also now active on social media. Follow us on: