Tecnologia Ceramica Piastrelle


Copyright 2025 - SACMI IMOLA S.C. Via Selice Provinciale 17/A - 40026 Imola (BO) Italy Tel. 0542/607111 - Fax 0542/642354 www.sacmi.com sacmi@sacmi.it
Volume non commerciabile 3a edizione - 10 2025
I diritti di traduzione, di memorizzazione elettronica, di riproduzione e di adattamento totale o parziale, con qualsiasi mezzo (compresi i microfilm e le copie fotostatiche), sono riservati per tutti i paesi.
I nomi e i marchi riportati in questo documento sono di proprietà delle rispettive società e sono citati solo a scopo illustrativo.
La tecnologia di fabbricazione delle piastrelle ceramiche rappresenta un patrimonio industriale e culturale che sta alla base del successo di un settore merceologico diffuso su scala mondiale e caratterizzato da continue evoluzioni, finalizzate da un lato a rendere sempre più efficiente il processo produttivo e dall’altro alla realizzazione di prodotti innovativi a più alto valore aggiunto tecnico ed estetico.
Con questi obiettivi, fin dalle origini dei moderni processi di produzione basati sulla pressatura di polveri e sulla cottura rapida in forni a rulli monostrato, SACMI ha riconosciuto l’importanza di promuovere una cultura tecnologica diffusa e condivisa, che potesse costituire un terreno fertile per lo sviluppo di produzioni ceramiche di qualità sempre crescente.

Forte di questa convinzione, già nel 1986 il Laboratorio Ceramico SACMI realizzò due volumi sulle tecnologie di fabbricazione delle piastrelle ceramiche. Il primo libro illustrava le tipologie di materie prime utilizzabili negli impasti (argille caolinitiche, illitiche, cloritiche, montmorillonitiche, sabbie, feldspati, carbonati di calcio e magnesio, talco, ecc.), i metodi di controllo e di analisi, le composizioni dei prodotti dell’epoca (maiolica, cottoforte, terraglia, monoporosa, monocottura greificata, grès rosso e grès porcellanato). Il secondo volume trattava invece le principali fasi di lavorazione: macinazione, atomizzazione, pressatura, essiccamento, smaltatura e scelta del prodotto finito. Interessante anche l’inserimento di un capitolo finale sui difetti riscontrabili in produzione, descrivendo in particolare quelli determinati da caratteristiche e parametri tecnologici non corretti. Una rivisitazione dei volumi di tecnologia è stata poi effettuata nel 2001, con ulteriori ristampe ed aggiornamenti negli anni immediatamente successivi, l’ultima delle quali nel 2006.
Da allora le tecnologie di produzione delle piastrelle ceramiche hanno subìto profonde trasformazioni, anzi vere e proprie rivoluzioni del processo produttivo che hanno reso non più procrastinabile la stesura di una terza edizione del volume sulle principali fasi di lavorazione.
Tra i macro-temi che hanno caratterizzato lo sviluppo tecnologico di questi ultimi vent’anni si possono ad esempio citare:
- l’affermazione del grès porcellanato smaltato in tutte le sue declinazioni
- la decorazione digitale mediante stampa a getto d’inchiostro
- la formatura di grandi e grandissimi formati
- il conseguente processo di taglio a crudo dei sottoformati modulari
- l’ampia gamma di spessori realizzabili in funzione della destinazione d’uso finale
- le post-lavorazioni di finitura delle superfici smaltate ad umido e/o a secco.
Parallelamente agli sviluppi di prodotto, l’introduzione in ceramica dei concetti dell’industria 4.0 ha determinato un forte aumento dell’automazione in tutte le fasi del processo, a partire dalla preparazione impasto, alla gestione ed al dosaggio delle polveri, al controllo dei semilavorati e del prodotto finito, agli stoccaggi intermedi fino alla logistica dei magazzini automatici.
Tutto questo ha comportato un sensibile miglioramento dell’efficienza degli impianti, una maggior costanza delle rese produttive, una maggior rapidità nei cambi di prodotto e quindi una più elevata flessibilità delle linee.
Sulla base di queste premesse, la nuova edizione del Volume I riporta, suddivisa in dodici capitoli, una trattazione anche storica delle materie prime utilizzate nella fabbricazione delle piastrelle e delle loro proprietà chimico-fisiche, oltre alla classificazione tecnica e merceologica dei prodotti per pavimenti e rivestimenti, con due ulteriori capitoli dedicati specificatamente al grès porcellanato ed alle lastre ceramiche.
Il Capitolo 1, relativo alla piastrella ceramica, descrive le proprietà di questo prodotto fondamentale per il mondo delle costruzioni in edilizia. Si passa poi da un excursus storico sulle origini della piastrella ceramica per giungere ai giorni nostri con i dati di produzione italiani e mondiali.
Il successivo Capitolo 2 sulle rocce è una trattazione geologica dei minerali che costituiscono la crosta terrestre con particolare riferimento a quelli di interesse per l’industria ceramica, per i quali vengono descritti i meccanismi di formazione e le strutture cristallografiche che poi ne determinano le proprietà.
A seguire nel Capitolo 3 vengono analizzate approfonditamente le materie prime per impasto ed in particolare il loro ruolo all’interno delle composizioni degli impasti ceramici. Infatti sono indubbiamente le tipologie e le caratteristiche delle materie a determinare la lavorabilità di un impasto in tutte le fasi del processo produttivo fino ad assicurare la qualità tecnica ed estetica del prodotto finito.
Parimenti, nel Capitolo 4 vengono illustrate le materie prime per fritte e smalti, certamente più complesse rispetto alle materie prime per impasti in quanto devono poter garantire la formazione di fasi vetrose con elevate proprietà tecniche ed estetiche. Basti pensare alla necessità di controllarne la fusibilità e l’accordo dilatometrico con il supporto, alla varietà di tipologie estetiche richieste, dai materiali trasparenti a quelli opacizzati, dai lucidi, ai satinati alle superfici matt.
Nel Capitolo 5 si riprende la trattazione dei materiali argillosi, con particolare riferimento al sistema acqua-argilla. Come noto, infatti, l’interazione dei minerali argillosi con l’acqua è determinante sia nei processi di lavorazione a umido degli impasti (macinazione, scioglitura, setacciatura e atomizzazione), sia nell’assicurare una adeguata plasticità in fase di formatura (pressatura o estrusione). Relativamente alle sospensioni acqua-argilla (barbottine) risultano di fondamentale importanza le grandezze reologiche come la viscosità, il limite di
scorrimento e la tissotropia. Vengono pertanto descritte in dettaglio le numerose tipologie di additivi deflocculanti utilizzati per ottimizzare la reologia delle barbottine, con un particolare focus sui loro meccanismi di azione.
Nel successivo Capitolo 6 si passa poi ad illustrare i metodi di caratterizzazione delle materie prime soprattutto attraverso tecniche strumentali, come la spettrometria a fluorescenza per l’analisi chimica, la diffrattometria a raggi X per la caratterizzazione mineralogica e le varie tipologie di analisi termiche imprescindibili per valutare il comportamento dei materiali durante la cottura, come ad esempio la loro decomposizione termica, i cambiamenti di fase e la fusibilità.
Tornando al prodotto finito, il Capitolo 7 descrive classificazione e caratterizzazione delle piastrelle ceramiche, riassumendo sinteticamente le norme ISO13006 ed i relativi metodi di prova ISO10545; si riporta poi una descrizione della recente norma ISO17889 che fornisce un sistema di valutazione della sostenibilità sulla base del rispetto di requisiti energetico-ambientali obbligatori e volontari.
Seguono i Capitoli 8 e 9 dedicati rispettivamente ai prodotti da rivestimento e da pavimento, nei quali sono descritte le specifiche materie prime utilizzate, le composizioni, i parametri di lavorabilità, le soluzioni impiantistiche ed alcuni dati statistici sulla loro distribuzione produttiva.
Il Capitolo 10 è invece dedicato al grès porcellanato, la tipologia di prodotto che, negli ultimi decenni, ha indubbiamente dominato il mercato delle piastrelle; anche in questo caso si riportano dati statici sulla produzione italiana e mondiale, le molteplici destinazioni d’uso, le caratteristiche, le composizioni e le soluzioni impiantistiche adottate in funzione delle tipologie produttive.
Infine il Capitolo 11 tratta in dettaglio le lastre di grande formato, la più recente classe di prodotti in grès porcellanato, realizzabili grazie a tecnologie d’avanguardia come la formatura continua su nastro e la decorazione digitale su pezzi di grandissime dimensioni. Oltre alle caratteristiche degli impasti, ai parametri di lavorazione ed all’impiantistica utilizzata, vengono evidenziate le prerogative estetiche e funzionali di questa tipologia di prodotti applicabili anche al settore dell’arredo (ad es. piani cucina) in sostituzione di altri materiali alternativi (engineered stone, solid surface e pietre naturali).
Da ultimo, il Capitolo 12 contiene tabelle e figure di possibile interesse per le attività quotidiane svolte dai tecnici ceramici.
Buona consultazione!
Il prodotto certamente più conosciuto e diffuso nell’ambito dei materiali ceramici [1] è senza alcun dubbio la “piastrella”, ovvero un manufatto di forma solitamente quadrata o rettangolare e di spessore contenuto, utilizzato nel settore delle costruzioni per realizzare pavimentazioni o per il rivestimento di pareti, sia in edifici residenziali che commerciali [2].
La piastrella ceramica, prodotta mediante un processo di cottura ad alta temperatura, si caratterizza rispetto ad altre tipologie di materiali alternativi (cementi, marmette, marmi e pietre naturali, ecc.) per numerosi vantaggi prestazionali, funzionali e produttivi, quali ad esempio:
- l’elevata resistenza meccanica intrinseca di un materiale ceramico che permette la realizzazione di piastrelle a spessore contenuto ed ottimizzato in funzione della tipologia di prodotto, ovvero in relazione alla sua destinazione d’uso
- l’ottima inerzia chimica della ceramica, costituita da ossidi inorganici consolidati ad alta temperatura, che garantisce igienicità, facile pulibilità e durabilità in tutti gli ambienti privati e pubblici
- le infinite possibilità di colorazione e finiture estetiche delle superfici: naturali, levigate o smaltate, lucide, satinate o matt, lisce o strutturate, decorate con grafiche digitali ad alta risoluzione
- la varietà dei formati e spessori realizzabili, da una formella 10 × 10 cm ad una lastra 180 × 360 cm, idonea a soddisfare qualsivoglia progetto abitativo ed architettonico
- l’affidabilità e la totale riproducibilità del processo produttivo, replicabile nel tempo per qualunque tipologia di prodotto a catalogo
- il costo di fabbricazione relativamente contenuto, in considerazione soprattutto delle tipologie di materie prime naturali utilizzate (argille, sabbie, feldspati, ecc.), presenti o comunque reperibili in tutte le aree geografiche industrializzate.
Bastano queste peculiarità esemplificative a render conto della diffusione su scala mondiale della piastrella ceramica, affermatasi da decenni come elemento costruttivo dell’edilizia abitativa dall’Europa alle Americhe, dal Medio Oriente al Far East.
Certamente la crescita economica mondiale, ed in particolare l’aumento del potere d’acquisto pro capite anche nei paesi emergenti ad alta densità demografica, costituisce un ulteriore fattore trainante per il mercato globale della piastrella ceramica.
Va infatti sottolineato come, da un lato, la piastrella possa rappresentare una soluzione a basso costo per la realizzazione di pavimenti e rivestimenti di qualità, una commodity idonea a soddisfare grandi volumi di domanda mentre, da un’altra prospettiva, la fascia alta di gamma sia un prodotto di assoluta eccellenza.
In tal caso la piastrella ceramica viene scelta non solo per le sue caratteristiche tecniche ma soprattutto per la sua valenza estetica, che la rende a tutti gli effetti un prodotto di design, soggetto alle tendenze evolutive del mondo della moda, alla stessa stregua di un capo di abbigliamento.
Le infinite combinazioni di formato, colore, decoro, lucentezza e finiture delle superfici consentono la realizzazione di ambientazioni di pregio in qualsivoglia stile architettonico, moderno o retrò, raffinato o rustico, eccentrico o minimalista.
Avremo ad esempio un pavimento di una grande sala in formato 80 × 80 cm, con uno smalto satinato ed un decoro sfumato tono su tono, il rivestimento di una cucina con piastrelle anticate di piccolo formato che richiamano una lavorazione artigianale, il rivestimento di un bagno con formati rettangolari squadrati e posati accostati per non avere soluzioni di continuità, il box doccia realizzato con un finto mosaico o, all’opposto, con grandi lastre che limitano quella zona specifica e la caratterizzano rispetto all’ambiente circostante.
Questa è la potenza espressiva, senza eguali, della piastrella ceramica.
L’etimologia della parola “ceramica” deriva dal greco keramikós che significa “fatto di argilla cotta” e, più anticamente, da una radice indoeuropea del verbo “bruciare”.
Risulta quindi evidente come la ceramica sia, fin dalle sue origini, il prodotto ottenuto dalla combinazione tra l’argilla ed il fuoco, ovvero una terracotta.
Da un lato, infatti, la peculiarità dell’argilla di essere facilmente modellata ha permesso già alle civiltà preistoriche di “creare” oggetti funzionali e ornamentali (vasi, statuette, ecc.) resi duri e durevoli grazie al loro consolidamento con il fuoco, una sorta di cottura ante litteram.
A riguardo alcuni tra i reperti più antichi risalgono infatti al neolitico come, ad esempio, un vaso in terra rossa del neolitico cinese (6.000 a.C.) esposto al Museo di Shanghai o una statuetta di donna in ceramica mesopotamica risalente al 5.000 a.C., oggi custodita al Metropolitan Museum di New York (vedi Fig. 1.1).

Le prime piastrelle ceramiche sono invece relativamente più recenti (Egitto, III millennio a.C.) in quanto collegate alla costruzione di edifici o di tombe funerarie, analogamente ai mattoni ma con una valenza decorativa già nell’antichità.

Fig. 1.2 Formelle dalla Tomba di re Djoser, Saqqara (2.600 a.C.) [5]
Ben presto, infatti, le piastrelle vennero decorate attraverso l’uso di smalti, colori e bassorilievi, come testimoniamo le formelle smaltate ritrovate nella tomba di re Djoser all’interno della piramide di Saqqara (2.600 a.C.) – vedi Fig. 1.2 – e, successivamente, le sontuose realizzazioni babilonesi e persiane della Porta di Ishtar e del palazzo di Dario a Persepoli (ricostruite al Pergamon Museum di Berlino) – vedi Fig. 1.3.

1.3
Si può pertanto affermare che la piastrella si è inizialmente sviluppata nei paesi islamici per diffondersi dapprima nella penisola iberica con l’invasione araba dal 711 d.C. al 1492, con le caratteristiche piastrelle smaltate e decorate con blu cobalto, denominate azulejos (Fig. 1.4).

Fig. 1.4 Azulejos nazaríes, Granada (XIII-XV sec.) [8]
Con l’avvento dell’era moderna, caratterizzata da una forte competizione anche commerciale tra le monarchie regnanti in Europa, l’utilizzo delle piastrelle decorative si diffuse ulteriormente.
In particolare, le piastrelle in terracotta calcarea biscottata, smaltata e decorata (le cosiddette “maioliche” o “faenze”) si svilupparono dal XIII secolo al Rinascimento e fino al XVIII secolo nella costruzione di palazzi nobiliari e chiese (vedi Fig. 1.5).

Fig. 1.5 Piastrelle maiolicate del Chiostro di Santa Chiara, Napoli (1739)
Seguirono le prime produzioni industriali in Olanda, Inghilterra, Germania e quindi anche in Italia con la Manifattura Dallari, poi Rubbiani (oggi Marca Corona), alla quale si aggiunsero altre aziende storiche come l’Appiani di Treviso (1873), la Cooperativa Ceramica di Imola (1874), la Società Ceramica Richard Ginori di Doccia (1896), e così via (Fig. 1.6).

Fig. 1.6 Piastrelle di maiolica in formato 15x15 cm, Sassuolo (fine ‘800) [9]
Fu però solamente dopo la seconda guerra mondiale che, sospinta dal boom economico e dallo sviluppo tecnologico (presse a ginocchiera poi a frizione per la formatura a secco delle polveri e forni a tunnel poi a rulli per una cottura più rapida), si concretizzò la forte crescita dell’industria ceramica italiana delle piastrelle, in particolare nel comprensorio sassolese.
Anche i prodotti ceramici subirono rapide trasformazioni: alle maioliche ed alle terraglie porose da rivestimento, si aggiunsero il grès rosso ed il cottoforte più adatti per pavimentazioni resistenti all’usura, poi soppiantati da composizioni a pasta bianca (la monocottura greificata) ottenute per macinazione ad umido ed atomizzazione, che permettevano la realizzazione di prodotti smaltati con una qualità superiore e soprattutto l’essiccamento e la cottura a cicli rapidi [10].
L’evoluzione ulteriore delle piastrelle da pavimento avvenne a metà degli anni ‘90 con l’introduzione del grès fine porcellanato, composizione in pasta bianca colorabile in massa, totalmente greificata a più alta temperatura (1200 °C), utilizzata dapprima come materiale tecnico non smaltato, poi smaltata e decorata, che per le sue elevate caratteristiche meccaniche si presta alla realizzazione di qualsivoglia formato (fino a 3600 mm) e spessore (da 3 a 30 mm).
Grazie a questa sua versatilità tecnico-estetica, ed al conseguente maggior valore aggiunto, il grès porcellanato si è affermato come tipologia di prodotto dominante, lasciando limitati spazi ai rivestimenti porosi smaltati (monoporosa e bicottura) ed alla monocottura greificata da pavimento prevalentemente in pasta rossa, per i segmenti più economici.
Il ciclo di fabbricazione delle piastrelle ceramiche [11] parte da una opportuna selezione e dall’approvvigionamento delle materie prime in funzione della tipologia di prodotto da realizzare, dal processo di lavorazione adottato, da considerazioni economiche che devono tener conto anche della logistica dei trasporti e della costanza delle forniture nel tempo. Comunque sia effettuata la scelta delle materie prime costituenti l’impasto, ovvero il corpo della piastrella, le successive fasi di lavorazione in fabbrica sono tipicamente le seguenti:
- eventuali lavorazioni ausiliarie delle materie prime (stagionatura, frantumazione, ecc.)
- dosaggio, miscelazione e macinazione della composizione di impasto
- preparazione dell’impasto al tipo di formatura (bagnatura, atomizzazione, ecc.)
- formatura dei semilavorati (piastrelle crude)
- essiccamento
- smaltatura e decorazione delle superfici mediante svariate tecniche applicative, digitali e analogiche
- cottura
- eventuali lavorazioni meccaniche (taglio, squadratura, lappatura, ecc.)
- scelta, confezionamento ed immagazzinamento.
Nella Fig. 1.7 sono riassunte le fasi dei più comuni processi produttivi utilizzati per la fabbricazione delle piastrelle, con particolare riferimento alle tecnologie di bicottura e monocottura.
Stoccaggio materie prime
Dosaggio
Macinazione a secco
Bagnatura Granulazione
Pressatura
Essiccamento
Cottura biscotto
Decorazione
Cottura vetrato
Scelta e confezionamento
Stoccaggio materie prime
Dosaggio
Macinazione a umido
Atomizzazione
Pressatura
Essiccamento
Decorazione
Cottura
Finitura
Scelta e confezionamento
Fig. 1.7 Principali schemi di processo per la produzione di piastrelle ceramiche
Così come le materie prime e la formulazione dell’impasto, anche tutte le fasi del processo produttivo devono essere ben progettate e controllate al fine di ottenere piastrelle finite della tipologia e qualità richieste.
A riguardo si considerano come riferimento le norme internazionali ISO 13006 (vedi Capitolo 7), anche se ogni produttore può stabilire criteri di scelta più stringenti o specifici in funzione della destinazione d’uso del prodotto.
Ne consegue che, in funzione della tipologia di piastrella, da rivestimento o pavimento, smaltata o no, da interno o da esterno, devono essere verificate e rispettate una serie di caratteristiche tecnologiche, tra le quali:
- assorbimento di acqua (porosità del supporto)
- calibro (dimensioni di fabbricazione)
- tono (colore/decoro)
- resistenza meccanica a flessione
- resistenza all’abrasione, alla macchia e ai prodotti chimici
- resistenza al gelo e allo shock termico.
È evidente come le proprietà finali del prodotto siano determinate da una corretta scelta e formulazione delle materie prime, come pure dalle condizioni di lavorazione adottate in tutte le fasi del processo produttivo.
Le lavorazioni semiartigianali che, fino a pochi decenni fa, caratterizzavano la fabbricazione delle piastrelle ceramiche, conferendo loro anche una connotazione artistica distintiva per ogni azienda produttrice, hanno oggi lasciato spazio ad una produzione industriale efficiente e oramai totalmente automatizzata.
Basti, ad esempio, evidenziare come negli anni ‘80 la produzione annua per addetto fosse di circa 10.000 m2/anno/addetto mentre oggi si raggiungono produttività superiori ai 35.000 m2/anno/addetto.
Le ragioni di questa vera e propria rivoluzione industriale sono molteplici e trasversali, riguardando da un lato le tipologie di materiali e di prodotti finiti e, dall’altro, le macchine di processo e soprattutto le tecnologie di automazione.
Sul fronte dei materiali, si può constatare come ci sia stata una standardizzazione degli impasti (in primis con il grès porcellanato smaltato) e, conseguentemente, nelle formulazioni delle materie prime e nelle tipologie di smalti utilizzati, semplificando notevolmente gli approvvigionamenti e le necessità di ottimizzazioni personalizzate.
Anche l’affermarsi della decorazione digitale inkjet con inchiostri ceramici “pronti all'uso” ha contribuito grandemente ad eliminare le numerose lavorazioni manuali e gli interventi correttivi richiesti dalle precedenti tecnologie di decorazione con paste serigrafiche autoprodotte.
Un ulteriore fattore di efficientamento delle linee è stato determinato dalla crescente produzione di formati medio-grandi, con minori dispersioni su formati piccoli che comunque possono essere ottenuti più convenientemente dopo cottura attraverso semplici operazioni di taglio e rettifica.
In questo contesto produttivo maggiormente standardizzato rispetto al passato, si è inserito lo sviluppo dell’automazione caratteristico dell’industria 4.0, ovvero una completa interconnessione delle macchine e l’introduzione di sistemi di controllo on-line che consentono una gestione delle linee totalmente automatizzata (la cosiddetta “fabbrica a luci spente”).
La moderna produzione industriale delle piastrelle ceramiche ha avuto inizio principalmente in Italia e in Europa con la ricostruzione successiva alla seconda guerra mondiale. Già negli anni ‘70 la produzione mondiale di piastrelle raggiunse i 500 milioni di m2/anno, il 64% dei quali fabbricati in Europa (Italia, Spagna, Germania, ecc.) e le restanti quote suddivise tra Asia (Giappone, ecc.) e Americhe (Brasile, ecc.).
La diffusione della piastrella nell’edilizia residenziale, il continuo sviluppo di tecnologie produttive sempre più efficienti, l’apertura dei mercati verso le esportazioni, il generale aumento del potere d’acquisto pro capite ed il forte incremento demografico dei paesi emergenti, hanno determinato una crescita esponenziale della produzione mondiale di piastrelle fino agli attuali 16,7 miliardi di m2/anno (2022) [12], come mostrato in Fig. 1.8.
Produzione (milioni m 2 )
Fig. 1.8 Crescita della produzione mondiale di piastrelle ceramiche (1970-2022)
La Cina per prima e a seguire l’India sono state artefici di questo straordinario sviluppo a partire dai primi anni 2000 quando hanno superato la produzione europea che fino ad allora era stata trainata da Italia e Spagna, grazie anche al continuo incremento delle esportazioni. In particolare, l’Italia ha raggiunto la sua massima capacità produttiva nel 2001 con 638 milioni di metri quadrati, affiancata poi superata dalla Spagna che per alcuni anni ha mantenuto volumi produttivi elevati, fino alla crisi finanziaria mondiale del 2008.
Fig. 1.9 Evoluzione delle aree di produzione di piastrelle ceramiche (2001 - 2022)
Già nel 2001 comunque, la produzione europea, che si attestava intorno a 1500 milioni di metri quadrati/anno, veniva superata dalla sola Cina. La Fig. 1.9 mostra una comparazione tra 2001 e 2022 in termini di distribuzione della produzione mondiale di piastrelle. Confrontando i due grafici a torta, si vede chiaramente lo spostamento della produzione mondiale verso il continente asiatico (solo la Cina produce oggi oltre 7 miliardi di m2!).
Lo scenario attuale (2022) vede una distribuzione produttiva delle piastrelle ceramiche dominata dall’Asia (Cina, India, Vietnam, Iran, ecc.) con il 73% sul totale, mentre l’Europa (Italia, Spagna, Turchia, ecc.) si attesta intorno a 11% e le Americhe (Brasile, Messico, Stati Uniti, ecc.) poco sotto a 10%. A completamento della produzione mondiale si registra un 5% del continente africano, grazie ad alcuni paesi a consolidata tradizione ceramica (Egitto, Tunisia, Algeria e Sud Africa), ma con auspicabili prospettive di crescita anche nelle aree subsahariane densamente popolate.
Le produzioni dei primi dieci Paesi produttori (2022) sono riportate nel grafico di Fig. 1.10.
Produzione (milioni m 2 )
Asia
Europa
Americhe
Africa
[1] G. Peco, I prodotti ceramici. Dalla tradizione all’alta tecnologia, Marzorati, 1991.
[2] P. G. Burzacchini, G. P. Emiliani e M. Morganti, Dizionario enciclopedico della ceramica, Ed. Polistampa, 2016.
[3] «The Shanghai Museum - A red pot with two ears, from the Peiligang culture» [Online]. Available: https://commons.wikimedia.org/wiki/File:PeiligangCulture-RedPotWithTwoEars-ShanghaiMuseum-May27-08.jpg.
[4] «Metropolitan Museum - The Collection» [Online]. Available: https://www.metmuseum. org/art/collection/search/327066.
[5] «Metropolitan Museum - The Collection» [Online]. Available: https://www.metmuseum. org/art/collection/search/543904.
[6] «Ishtar Gate - Pergamon Museum (Berlin)» [Online]. Available: https://commons. wikimedia.org/wiki/File:Ishtar_Gate_at_Berlin_Museum.jpg.
[7] «Persian warriors - Pergamon Museum (Berlin)» [Online]. Available: https:// commons.wikimedia.org/wiki/File:Berlin_-_Pergamon_Museum_-_Persian_warriors_-_20150523_6849.jpg.
[8] «Museo Arqueológico y Etnológico de Córdoba (España)» [Online]. Available: https:// es.m.wikipedia.org/wiki/Archivo:Azulejos_nazar%C3%ADes_%2816657400083%29. jpg.
[9] «Maioliche storiche Marca Corona» [Online]. Available: https://it.m.wikipedia.org/wiki/ File:Marca_Corona_maioliche_storiche_15x15.JPG.
[10] T. Emiliani e E. Emiliani, Tecnologia dei processi ceramici, Ceramurgica, 1982.
[11] G. P. Emiliani e F. Corbara, Tecnologia ceramica. La lavorazione (Vol. 2), Faenza Editrice, 2010.
[12] ACIMAC, «World production and consumption of ceramic tiles - 11th edition 2023».
CERAMICA
Materiale costituito da ossidi inorganici, solitamente caratterizzato dalla presenza di minerali argillosi che ne consentono la formatura con svariate tecniche (pressatura di polveri, estrusione, colaggio, modellazione a plastico) e da un processo di cottura ad alta temperatura che conferisce elevata resistenza meccanica e inerzia chimica.
PIASTRELLA
Manufatto di forma solitamente quadrata o rettangolare e di spessore contenuto, utilizzato nel settore delle costruzioni per realizzare pavimentazioni o per il rivestimento di pareti.
MAIOLICA
Ceramica a supporto poroso costituito da un impasto illitico-calcareo sottoposto ad una prima cottura a circa 1000 °C (biscotto), quindi smaltato con smalto bianco, decorato e ricotto a temperature analoghe.
Ceramica a supporto poroso costituito da un impasto caolinitico bianco, sottoposto ad una prima cottura ad alta temperatura (biscotto), quindi decorato e invetriato, poi ricotto a più basse temperature.
GRÈS ROSSO
Prodotto ceramico a bassa porosità costituito da argilla rossa, che veniva cotto in forni a tunnel per realizzare piastrelle da pavimento di piccolo formato.
COTTOFORTE
Prodotto ceramico poroso costituito da argille rosse carbonatiche che veniva sottoposto ad una prima cottura in forni a tunnel, quindi smaltato e ricotto, per realizzare piastrelle da pavimento per interni.
BICOTTURA
Prodotto ceramico a supporto poroso, costituito da un impasto carbonatico rosso o bianco, sottoposto ad una prima cottura a ciclo rapido (biscotto), quindi smaltato e decorato, poi ricotto in forno a rulli a ciclo rapido.
MONOPOROSA
Prodotto ceramico a supporto poroso, costituito da un impasto carbonatico rosso o bianco, smaltato e decorato in crudo, quindi cotto in forno a rulli a ciclo rapido.
Prodotto ceramico a supporto semi-greificato, costituito da un impasto non carbonatico rosso o bianco, smaltato e decorato in crudo, quindi cotto in forno a rulli a ciclo rapido.
Prodotto ceramico a supporto totalmente greificato, costituito da un impasto bianco non carbonatico, colorato tramite l’aggiunta di pigmenti inorganici, quindi cotto ad alta temperatura (1200 °C) in forni a rulli a ciclo rapido.
Prodotto ceramico a supporto totalmente greificato, costituito da un impasto bianco non carbonatico, smaltato e decorato in crudo, quindi cotto ad alta temperatura (1200 °C) in forni a rulli a ciclo rapido.
I minerali sono materiali naturali solidi ed omogenei con una composizione chimica ben definita ed una specifica struttura cristallina. Ad esempio, il quarzo è un minerale con formula chimica SiO2 ed una struttura composta da tetraedri [SiO4]4− legati tra loro in un reticolo ordinato e periodico secondo precise regole di simmetria.
Le rocce invece sono degli aggregati naturali e coerenti di uno o più minerali. Un esempio di roccia molto comune è il granito in Fig. 2.1 dove sono presenti minerali differenti, tra cui il quarzo (di colore grigio), il feldspato (rosa) e la biotite (nera).

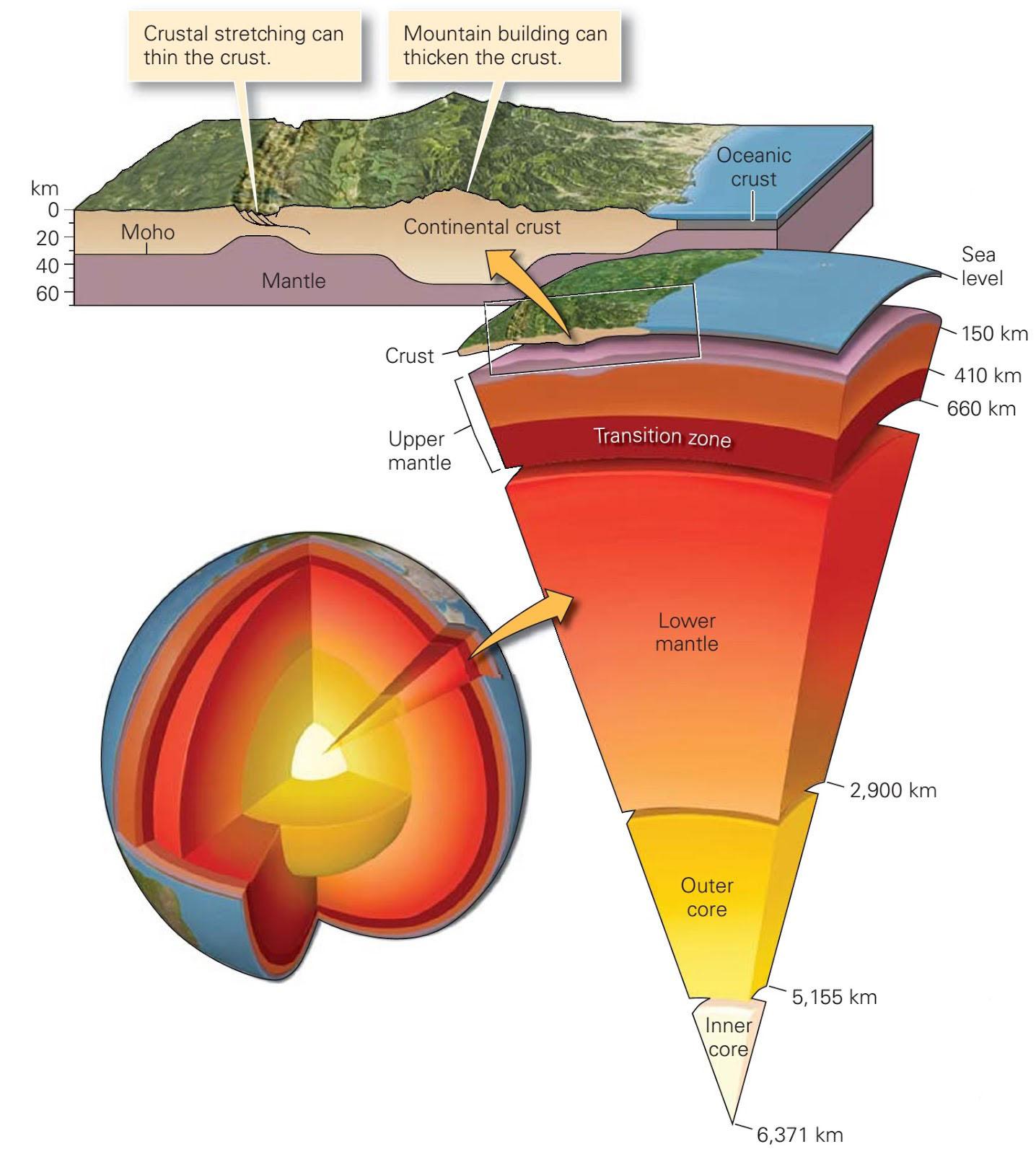
La struttura interna della Terra può essere descritta con una successione di strati concentrici, con caratteristiche chimiche e fisiche molto diverse. Le rocce costituiscono l’elemento base della parte solida della superficie terrestre chiamata crosta terrestre. La crosta terrestre ha uno spessore medio di 30 km ed è relativamente leggera e fragile con una densità compresa indicativamente tra 2,2 e 3,0 g/cm3. Sotto la crosta terrestre si trovano altri strati che compongono il mantello ed il nucleo a cui non possiamo accedere direttamente. La Fig. 2.2 illustra il modello attualmente accettato della stratificazione interna della Terra.
Gli elementi chimici principali nella crosta terrestre sono l’ossigeno ed il silicio e pertanto i minerali principali presenti nelle rocce appartengono alla famiglia dei silicati il cui elemento strutturale è il già menzionato tetraedro [SiO4]4−. I silicati più abbondanti nella crosta terrestre sono i feldspati (41%) e il quarzo (12%). In Tab. 2.1 viene riportata la composizione chimica media della crosta terrestre. Gli otto ossidi elencati nella prima colonna sono anche gli stessi che ritroviamo nella composizione chimica delle materie prime di utilizzo ceramico.
Gli
Composizione in elementi (% massa)
Composizione in ossidi (% massa) O 46,1 Si 28,2
0,2
Tab. 2.1 Composizione media della crosta terrestre
Le rocce possono avere composizioni e caratteristiche molto variabili in funzione dei minerali presenti e dei processi di formazione delle stesse.
Sulla base dei processi di formazione si distinguono tre famiglie principali di rocce: - rocce ignee (intrusive o plutoniche ed effusive) che si sono formate per solidificazione del magma. Costituiscono circa il 95% della crosta terrestre e contengono minerali importanti per il processo ceramico come feldspati e quarzo; - rocce sedimentarie di cui fanno parte le argille; - rocce metamorfiche che hanno origine dall’alterazione di rocce sedimentarie e ignee.
Considerato che le materie prime utilizzate per la produzione ceramica provengono da tutte e tre le tipologie di roccia, è importante per il ceramista avere alcune nozioni di base sulla loro formazione.
Il ciclo delle rocce (Fig. 2.3) è un modello semplificato che descrive le transizioni tra le diverse tipologie di rocce che sono avvenute nel corso delle ere geologiche in seguito ai cambiamenti di temperatura, pressione e condizioni ambientali.
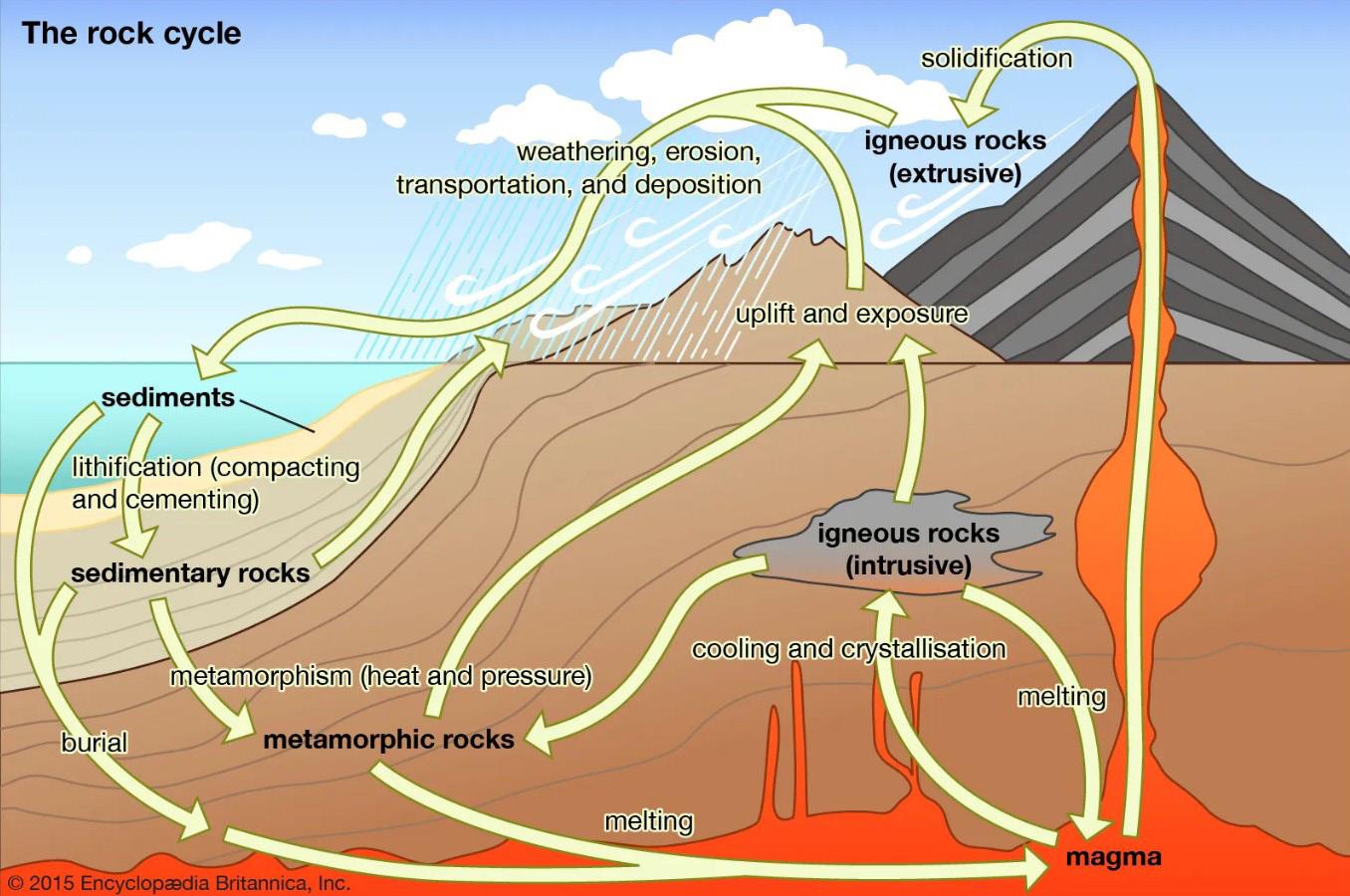
Fig. 2.3 Ciclo di alterazione dei magmi e delle rocce [3]
A causa dell’elevata temperatura e pressione, la parte esterna del mantello si trova allo stato fuso (magma). Quando il magma si muove verso le zone più fredde prossime alla superficie, può solidificare dando origine alle rocce ignee intrusive (es. i graniti). Le rocce intrusive possono in seguito affiorare in superficie a causa di spinte di origine tettonica.
Se il magma invece raggiunge direttamente la superficie, ad esempio mediante eruzione vulcanica, il raffreddamento avviene rapidamente in aria oppure in acqua, formando le rocce ignee effusive (es. i basalti).
Mentre per le rocce intrusive il raffreddamento lento porta alla formazione di minerali ben cristallizzati, nelle rocce effusive il raffreddamento rapido produce cristalli finissimi, invisibili ad occhio nudo, spesso accompagnati da fase vetrosa e sostanze volatili. Se la dimensione dei cristalli ha un impatto relativo per il processo ceramico, in quanto il materiale subisce sempre una macinazione, la presenza di fase vetrosa e di sostanze volatili è un fattore da considerare attentamente quando si selezionano le materie prime da utilizzare.
Il ciclo di trasformazione procede quando la parte superficiale delle rocce viene esposta a processi di alterazione (weathering) e di successiva erosione. A contatto con aria, acqua e anidride carbonica si verificano reazioni di ossidazione, dissoluzione ed idrolisi che, insieme alle variazioni di temperatura, gelo e disgelo, provocano alterazioni chimiche e fisiche del materiale, disgregando la roccia in una coltre detritica.
Un esempio classico di processo di alterazione è quello che porta alla caolinizzazione del feldspato potassico presente nelle rocce granitiche, secondo la reazione seguente (espressa in ossidi):
K2O·Al2O3·6SiO2 + 3H2O → Al2O3·2SiO2·2H2O + 2KOH + 4SiO2 feldspato potassico caolinite
Analoghe reazioni per feldspati di K, Na, Ca sono invece riportate di seguito:
KAlSi3O8 + H+ + 9/2 H2O → 1/2 Al2Si2O5(OH)4 + 2H4SiO4 + K+ ortoclasio caolinite
KAlSi3O8 + 2/3 H+ + 4H2O → 1/3 KAl3Si3O10(OH)2 + 2H4SiO4 + 2/3 K+ ortoclasio illite
NaAlSi3O8 + H+ + 9/2 H2O → 1/2 Al2Si2O5(OH)4 + 2H4SiO4 + Na+ albite caolinite
NaAlSi3O8 + 3/4 H+ + 7/2 H2O → 3/8 Na0.66Al2.66Si3.33O10(OH)2 + 7/4 H4SiO4 + 3/4 Na+ albite smectite
CaAl2Si2O8 + 2H+ + H2O → Al2Si2O5(OH)4 + Ca2+ anortite caolinite
I detriti prodotti (clasti) raramente rimangono stabili nel loro sito di origine; vento, piogge e ghiacciai esercitano sia un’azione di erosione che di trasporto, accumulandoli nei bacini di sedimentazione (alluvionali, lacustri o marini). Qui i detriti vengono interessati da processi di trasformazione (diagenesi) e di compattazione e di cementazione (litificazione) che portano alla formazione delle rocce sedimentarie
Le rocce sedimentarie possono formarsi anche per precipitazione chimica dei sali presenti nelle acque che si saturano in seguito all’evaporazione, oppure per sedimentazione di materiali di derivazione organica (fossili, gusci, ecc.) A prescindere che i sedimenti siano costituiti da detriti, da materiali di derivazione organica o da sali precipitati dalle acque, il loro accumulo avverrà in strati successivi per formare in certe condizioni tipologie di rocce ben litificate, come ad esempio le arenarie, o a volte sedimenti non litificati come le argille. La successiva sedimentazione di strati differenti spiega la difficoltà di trovare dei minerali argillosi puri; è più corretto, infatti, parlare di argille o di rocce argillose a prevalente composizione illitica, caolinitica o montmorillonitica in base al minerale argilloso principale. La genesi sedimentaria, l’effetto del trasporto e della selezione chimica e fisica sono alla base della tessitura estremamente fine dei minerali argillosi (di dimensioni colloidali, < 2 µm) e della loro morfologia tendenzialmente lamellare. L’estrema finezza e le caratteristiche strutturali conferiscono alle argille il tipico comportamento plastico, ovvero l’attitudine a lasciarsi modellare quando vengono impastate con una opportuna quantità di acqua.
Quando le rocce ignee o sedimentarie preesistenti vengono spinte a profondità maggiori, la variazione di temperatura e pressione porta alla formazione delle rocce metamorfiche (es. argille scistose, talcoscisti, ecc.). Queste a loro volta possono essere portate nuovamente in superficie e sottoposte ai fenomeni di degradazione descritti con formazione di nuove rocce sedimentarie.
Il ciclo delle rocce si chiude quando queste raggiungono profondità tali da arrivare a fusione (anatessi). Si originano così nuovi magmi, la cui composizione sarà diversa da quella dei magmi originari. Vediamo in breve come si classificano i magmi.
Il magma è un materiale parzialmente o totalmente fuso, quasi sempre ricco in silice, in cui sono presenti anche notevoli quantità di componenti volatili (vapore acqueo, ossidi di carbonio, di zolfo ecc.). Essendo alcune caratteristiche del magma (es. la viscosità) correlabili al contenuto di silice, il magma viene suddiviso in:
- magmi acidi ricchi in silice (SiO2 > 63%) e allumina, chiamati anche sialici (dall’unione delle parole silicio e alluminio). Vengono anche denominati felsici in quanto le rocce che ne derivano contengono principalmente feldspati e silice; - magmi neutri o intermedi, con un contenuto in silice compreso tra 52% e 63%. Danno origine a rocce generalmente più ricche in ferro e magnesio e pertanto più dense e tendenzialmente più scure; - magmi basici poveri in silice (SiO2 tra 52% e 45%), caratterizzati da un alto contenuto di ferro e magnesio da cui il nome di rocce mafiche o femiche. Tra i minerali che si formano dalla solidificazione di questi magmi possiamo citare ad esempio anfiboli, pirosseni, olivine; - magmi ultrabasici poverissimi in silice, danno origine a rocce definite appunto ultrabasiche o ultrafemiche, molto scure, essenzialmente costituite da silicati di ferro e magnesio, con densità molto elevate (es. peridotite 3,3 g/cm3).
La composizione del magma determina quindi la tipologia e la composizione delle rocce primarie da cui poi deriveranno anche i diversi tipi di minerali argillosi. La conoscenza delle caratteristiche delle rocce primarie diventa importante per comprendere meglio anche le caratteristiche dei minerali argillosi che derivano dalla loro trasformazione.
Di solito la composizione chimica delle rocce e dei minerali si esprime come percentuale in peso degli ossidi degli elementi, essendo l’ossigeno praticamente l’unico ione negativo presente. Questo consente di avere anche una rapida valutazione della bontà dell’analisi effettuata, in quanto la somma dei singoli ossidi deve essere molto prossima a 100%.
Il diagramma che normalmente si utilizza per la classificazione della maggioranza delle rocce ignee è quello di Streckeisen, denominato anche QAPF che è l’acronimo di Quartz, Alkali feldspar, Plagioclase, Feldspathoid. Si tratta di un diagramma basato sulla composizione mineralogica della roccia, piuttosto complesso, che richiede competenze specifiche e non sempre è possibile identificare con precisione le fasi cristalline specialmente quando nella roccia sono presenti anche fasi amorfe. Avendo a disposizione la sola analisi chimica, è invece possibile utilizzare il diagramma TAS (Total Alkali vs. Silica) che solitamente si utilizza esclusivamente per le rocce effusive o molto basiche.
In Fig. 2.4 viene illustrato il diagramma TAS (blu) ruotato e sovrapposto a quello QAPF (in rosso). Se consideriamo ad esempio una roccia la cui analisi chimica identifica sul diagramma TAS un rapporto tra SiO2 ed alcali totali (Na2O+K2O) che cade nella zona del basalto (roccia effusiva), sul diagramma QAPF avremo che la roccia intrusiva corrispondente sarà della famiglia del gabbro. Il riconoscimento di una roccia con la sola analisi chimica è certamente una grossa approssimazione ma può comunque dare qualche indicazione al tecnologo ceramico che non ha approfondite competenze geologiche.
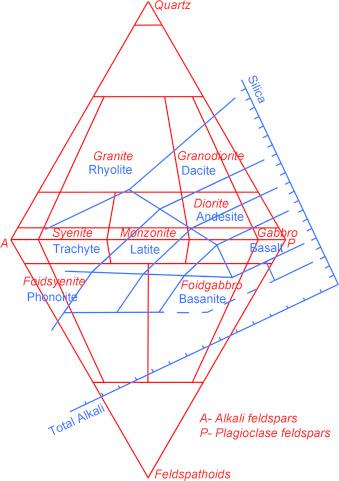
Fig. 2.4 Sovrapposizione dei diagrammi TAS e QAPF [1]
In Tab. 2.2 sono riportate le formule brute e le composizioni teoriche dei principali minerali presenti nelle rocce primarie. Quelli di maggiore interesse ceramico verranno descritti in modo approfondito nel Capitolo 3 dedicato alle materie prime.
Minerale
Albite (0÷10% anortite)
Anortite (0÷10% albite)
Ortoclasio Microclino
Sanidino Anortoclasio
Formula
FELDSPATI (abbondanza 41%)
Feldspati sodico-calcici (plagioclasi)
Na[AlSi3O8]
Ca[Al2Si2O8]
K[AlSi3O8]
(Na,K)AlSi3O8
68.7 % SiO2
19.5 % Al2O3 11.8 % Na2O
43.2 % SiO2
graniti, pegmatiti, dioriti alcaline, basalti
36.7 % Al2O3 20.1 % CaO rocce plutoniche mafiche
64.8 % SiO2 18.3 % Al2O3 16.9 % K2O
67.7 % SiO2 19.2 % Al2O3 4.4 % K2O
8.7 % Na2O
QUARZO (abbondanza 12%)
graniti, pegmatiti, sieniti
rioliti, trachiti, rocce vulcaniche felsiche
Quarzo SiO2 100 % SiO2 rocce felsiche
PIROSSENI (abbondanza 11%)
55.5 % SiO2
Diopside
Augite
Orneblenda
CaMgSi2O6
(Ca,Mg,Fe,Ti,Al)2 [(Si,Al)2O6]
18.6 % MgO
25.9 % CaO
48.3 % SiO2
6.1 % FeO
8.6 % Al2O3
3.38 % TiO2
15.35 % MgO
21.35 % CaO
1.31 % Na2O
ANFIBOLI (abbondanza 5%)
51.22 % SiO2
2.43 % FeO
(Ca,Na)2−3(Mg,Fe,Al)5 (Al,Si)8O22(OH,F)2
10.86 % Al2O3
19.63 % MgO
13.66 % CaO
2.19 % LOI
peridotite, basalto, andesite, rocce mafiche
gabbro, basalto, rocce mafiche e ultramafiche
granito, sienite, diorite, gabbro, basalto, andesite, gneiss, scisti
(abbondanza 3%)
39.19 % SiO2
Olivina (Mg,Fe)2SiO4
Muscovite
42.06 % MgO 18.75 % FeO
MICA (abbondanza = 5%)
45.3 % SiO2
KAl2[AlSi3O10] (OH,F)2
Leucite K[AlSi2O6]
Nefelina
38.4 % Al2O3 11.8 % K2O 4.5% LOI
(abbondanza < 1%)
55.06 % SiO2
23.36 % Al2O3 21.58 % K2O
41.1 % SiO2
KNa3[AlSiO4]4
34.9 % Al2O3
8.1 % K2O 15.9 % Na2O
gabbro, basalto, peridotite, rocce mafiche e ultramafiche
granito, sienite, gneiss, scisti
rocce vulcaniche mafiche e ultramafiche
sieniti, fonoliti
Tab. 2.2 Formula e composizione teorica dei minerali presenti nelle rocce primarie
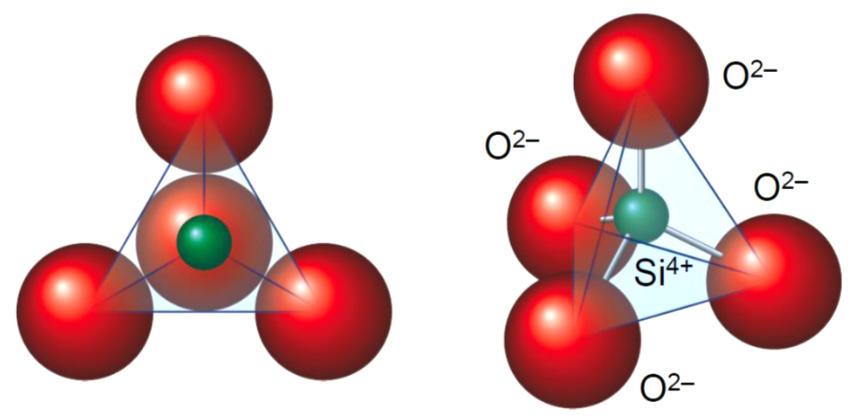
L’elemento base della struttura cristallina dei silicati è il tetraedro [SiO4]4− in cui lo ione silicio si dispone al centro della struttura tetraedrica circondato dai quattro atomi di ossigeno disposti ai vertici della stessa (Fig. 2.5). Avendo lo ione silicio una elevata densità di carica positiva, il suo legame con l’ossigeno risulta essere molto forte e l’energia necessaria per romperlo molto elevata. Il quarzo, ad esempio, ha una temperatura di fusione di circa 1700 °C.
2.5
In questa struttura, ogni atomo di ossigeno dispone di due elettroni liberi e può legarsi con un altro atomo di silicio. I silicati vengono classificati in funzione del numero di atomi di ossigeno in comune tra i tetraedri. In particolare:
- Nesosilicati (es. zircone, mullite e sillimanite) formati da tetraedri isolati [SiO4]4− collegati tra loro da cationi metallici interstiziali come Zr4+o Al3+ (verdi in figura). A causa dell’elevata forza di legame sono considerati in ceramica come materiali inerti.
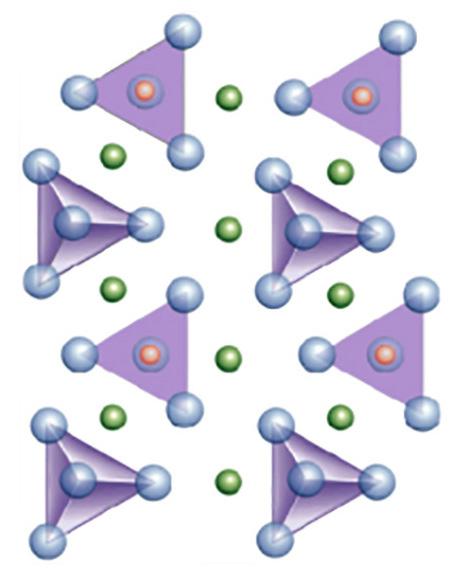
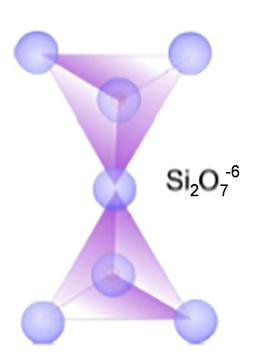
- Sorosilicati (es. gehlenite) in cui si ha unione di due tetraedri ad un vertice [Si2O7]6− Le coppie di tetraedri sono collegate tra loro da cationi metallici (es Ca2+).
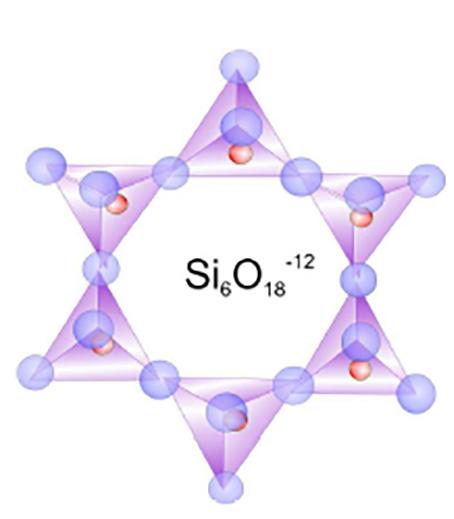
- Ciclosilicati (es. cordierite) in cui si ha unione ad anello (tri, tetra ed esa) tra tetraedri [SinO3n]2n−. Gli anelli sono collegati da ioni Mg2+ ed Fe2+.
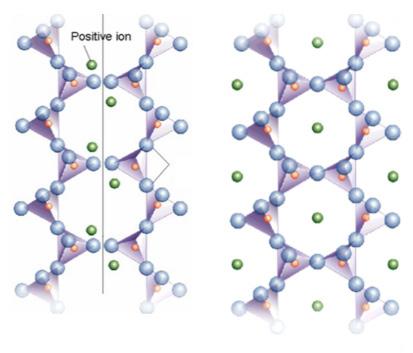
- Inosilicati (es. wollastonite, enstatite e diopside), fibrosi, aventi catene di tetraedri singole (pirosseni [SiO3]2−) o doppie (anfiboli [Si4O11]6−). Le catene si legano tra loro mediante l’inserimento di cationi (per es. Ca2+, Mg2+, Al3+, Fe2+). Dentro le catene doppie si ha spazio per l’interposizione di gruppi OH (acqua di idratazione) o di ioni F .
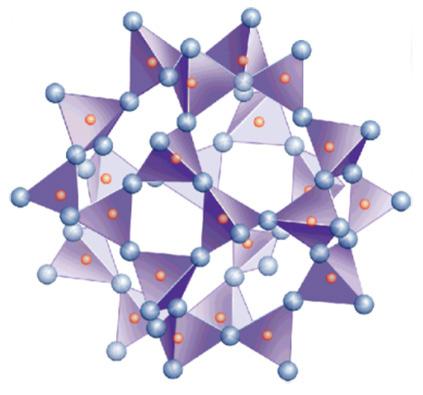
- Fillosilicati (argille, talco) in cui i foglietti di tetraedri si estendono nelle due dimensioni. Un tetraedro si collega ad altri tre formando esagoni regolari. Gli ossigeni non condivisi si dispongono dalla stessa parte del foglietto e si rendono disponibili per legare altri elementi. Si crea una struttura a strati successivi. L’unità di base è [Si2O5]2−. Nel prossimo paragrafo verrà approfondita la struttura dei minerali argillosi.
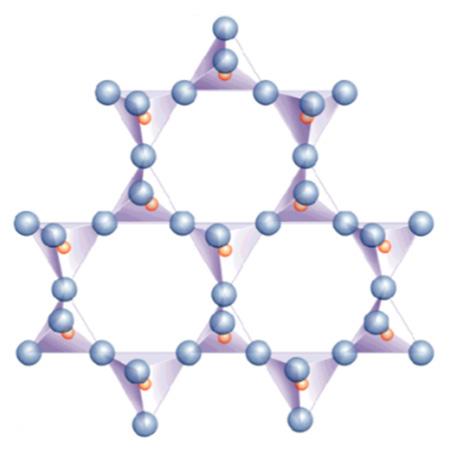
- Tectosilicati (quarzo, feldspati, nefelina, zeoliti), dove i tetraedri si uniscono a formare una struttura tridimensionale con reticolo continuo. Ogni tetraedro è collegato con altri quattro con un rapporto Si:O di 1:2. Nei feldspati si ha sostituzione di silicio con Al3+ e la neutralità elettrica è ottenuta con ioni alcalini (Na+ e K+) e alcalino-terrosi (Ca2+ e Ba2+) che si inseriscono negli spazi tra i tetraedri.
Come accennato, nel gruppo dei fillosilicati (dal greco phyllon, “foglia”), i tetraedri si uniscono in una struttura bidimensionale che viene denominata foglietto tetraedrico o foglietto T. Il foglietto tetraedrico raggiunge la neutralità elettrica legandosi ad un foglietto di ottaedri (O) il cui elemento base è appunto un ottaedro (Fig. 2.6) dove un atomo centrale di alluminio è coordinato a sei gruppi ossidrili OH . A volte l’alluminio può essere sostituito da un atomo di magnesio o ferro; in base al tipo di atomo centrale si avrà una struttura denominata gibbsitica o brucitica, riferendosi ai due idrossidi minerali puri, gibbsite Al2(OH)6 e brucite Mg3(OH)6. Nella struttura gibbsitica sono sufficienti due cationi Al3+ per neutralizzare i sei gruppi OH e quindi si parla di minerali di-ottaedrici. Nella struttura brucitica sono necessari tre cationi bivalenti e quindi avremo minerali tri-ottaedrici.
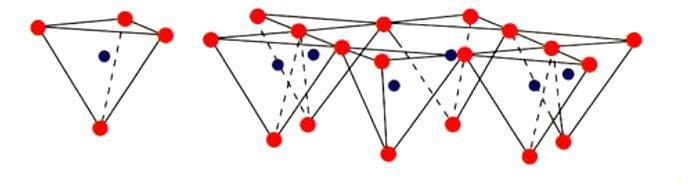
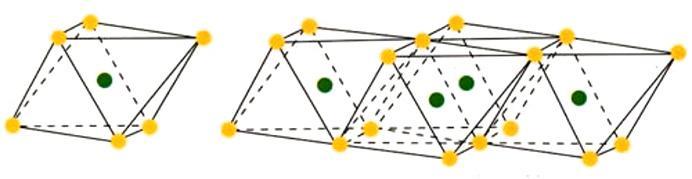
Fig. 2.6 Foglietto tetraedrico (T) ed ottaedrico (O)
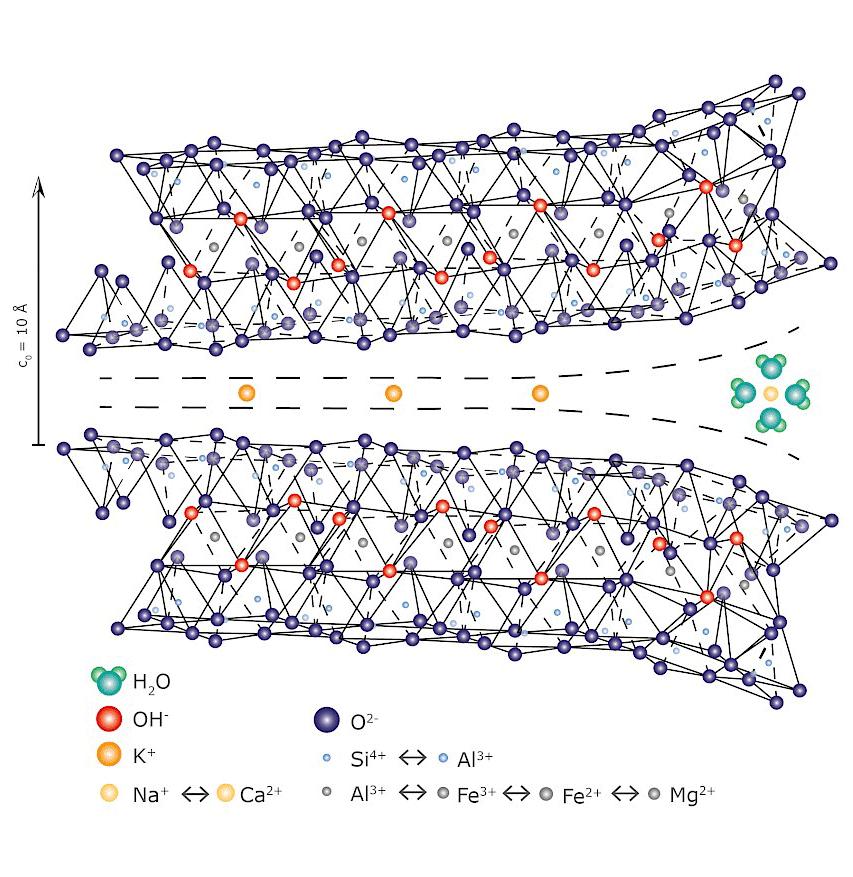
L’unione di foglietti T e O forma l’elemento base del reticolo cristallino che viene chiamato strato o pacchetto. La forza di legame interna ad un pacchetto è molto elevata mentre la coesione tra uno strato e quello adiacente è più debole (legami di Van der Waals). Questo conferisce ai minerali argillosi le particolari caratteristiche di plasticità (e di sfaldatura) dovute alla possibilità di scorrimento degli strati senza che si abbia la distruzione del reticolo cristallino. In seguito alle sostituzioni isomorfe degli atomi nel reticolo, ad esempio la già menzionata sostituzione di alluminio trivalente con magnesio bivalente, diventa necessario ristabilire la neutralità elettrica della struttura. Per questa ragione nello spazio interstrato vengono adsorbite molecole di acqua (chiamata acqua zeolitica o di interstrato) e ioni metallici che possano compensare la carica deficitaria. L’adsorbimento di acqua e ioni aumenta inoltre la distanza reticolare e conferisce alla struttura una maggiore possibilità di scorrimento tra i piani (si veda la Fig. 2.7). Questa particolare caratteristica è alla base del comportamento plastico delle argille e spiega la scarsa plasticità di una caolinite pura, che è priva di acqua zeolitica, rispetto a minerali argillosi come illite o smectite, altamente plastici, in cui vi è presenza di acqua nell’interstrato del reticolo [4].
interstrato
interstrato
(Al,Si)O4
(Al,Mg,Fe)O6
Fig. 2.7 Struttura con strati T−O−T e presenza di ioni idratati interstrato
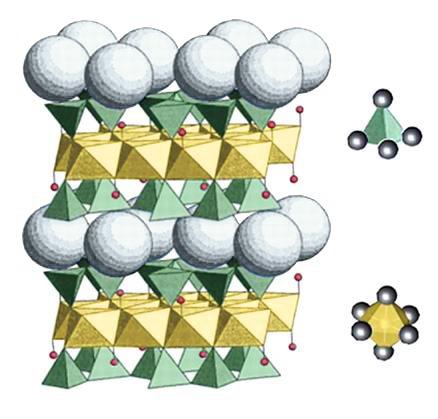
La combinazione più semplice tra foglietti è quella costituita dal pacchetto T−O. Se consideriamo anche lo spazio interstrato, lo spessore totale dell’unità strutturale è circa 7.2 Å (Fig. 2.8). Questa distanza è caratteristica dei minerali appartenenti al gruppo della caolinite e si misura mediante l’analisi diffrattometrica RX.
Fig. 2.8 Distanza reticolare della caolinite
La classificazione dei fillosilicati viene effettuata sulla base delle diverse combinazioni di foglietti T−O e delle relative distanze reticolari. Come evidenziato in Tab. 2.3 si possono distinguere tre gruppi principali di fillosilicati. Il gruppo della caolinite caratterizzato dal pacchetto T−O, quello della mica con pacchetto T−O−T e quello della clorite con una configurazione più complessa T−O−T + O (vedi Fig. 2.9). A questi tre gruppi fanno riferimento un numero elevato di minerali argillosi in base alle possibili sostituzioni dei cationi nel reticolo, all’interposizione di acqua e di ioni di dimensioni diverse, al modo e alla tipologia di disposizione degli strati, ecc. Si tratta in realtà di una classificazione molto semplificata ma ancora utile per inquadrare le diverse configurazioni delle strutture cristalline dei minerali argillosi. Per una classificazione rigorosa basata su layer e sottogruppi si rimanda ai testi specializzati.
Gruppo N° fogli per strato
distanza reticolare [Å]
Esempi triottaedrici (Mg2+)
Esempi diottaedrici (Al3+)
Caolinite 2 (T−O) 7.2 serpentino crisotilo caolinite halloysite
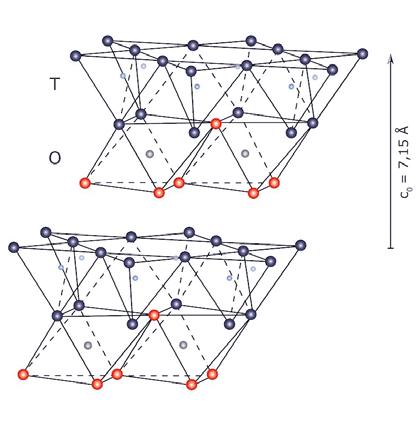
Mica 3 (T−O−T) 10 talco flogopite illite pirofillite muscovite montmorillonite
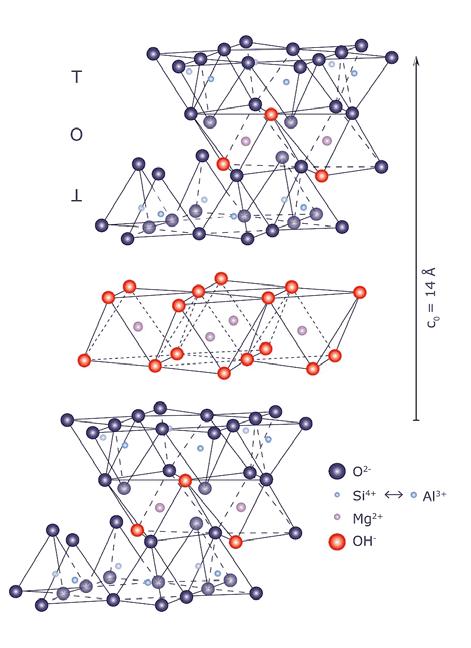
Clorite 4 (T−O−T + O) 14 clorite –
Tab. 2.3 Classificazione dei fillosilicati
mica
caolinite
clorite
Fig. 2.9 Struttura di caolinite, mica e clorite [5]
Nello spazio interstrato gli ioni possono essere adsorbiti in maniera preferenziale secondo il seguente ordine:
Na+ < K+ < Ca2+ < Mg2+
Lo ione calcio può quindi rimpiazzare lo ione sodio più facilmente di quanto quest’ultimo possa sostituire il calcio.
Fig. 2.10 Adsorbimento di ioni Na+ e Ca2+ nell’interstrato della montmorillonite
Questo effetto di scambio potrà variare in funzione della concentrazione ionica dell’acqua con cui l’argilla verrà in contatto. Una grandezza che può dare un’indicazione di quanto questi fenomeni avvengano in un determinato minerale argilloso è la capacità di scambio cationico CEC (Cation Exchange Capacity), che misura il grado di sostituzione dei cationi nell’interstrato del minerale.
In Tab. 2.4 si riportano valori indicativi di CEC per tre minerali argillosi molto differenti. Il valore di CEC può essere utilizzato come indicazione della plasticità di un minerale argilloso o della sua difficoltà ad essere deflocculato.
Minerale argilloso
CEC [meq/100 g]
Caolinite 5 ÷ 15
Illite 20 ÷ 50
Montmorillonite 50 ÷ 150
Tab. 2.4 Capacità di scambio cationico per tre diversi minerali argillosi
L’identificazione precisa dei minerali argillosi risulta però un’analisi piuttosto complessa e di fatto, in natura, l’unico minerale argilloso che si può trovare con un buon livello di purezza è la caolinite mentre le altre materie prime argillose sono solitamente costituite da diversi minerali e presentano pertanto caratteristiche tecnologiche molto variabili.
Da quanto esposto, è evidente come la descrizione di pochi minerali argillosi (cioè caolinite, illite, montmorillonite e cloriti), permetta una caratterizzazione pressoché completa di un generico minerale argilloso, tenendo conto della frequente possibilità di trovarsi di fronte a materiali polifillitici di inter-stratificazione (detti mixed layer), come ad esempio la bravaisite, consistente in una miscela di illite e montmorillonite.
[1] «Sand Atlas» [Online]. Available: https://www.sandatlas.org.
[2] «Learning Geology» [Online]. Available: https://geologylearn.blogspot.com/2016/01/ what-are-earth-layers-made-of.html.
[3] «Encyclopaedia Britannica» [Online]. Available: https://www.britannica.com/science/ magma-rock.
[4] F. Veniale e C. Palmonari, Clay mineralogy and ceramic processes and products, AIPEA, 1973.
[5] «Tonmineralogie - Dr. Krakow Rohstoffe» [Online]. Available: https://www.dr-krakow-labor.de/tonmineralogie.
ANATESSI
L’anatessi (dal verbo greco anatéko, “liquefarsi”) è il processo di fusione parziale di una roccia metamorfica che avviene a grande profondità nella crosta terrestre e dà origine ad un magma acido. Sinonimo di anatessi è ultrametamorfismo, che significa che la roccia è andata oltre il campo fisico di esistenza delle rocce metamorfiche entrando in quello delle rocce ignee.
LITIFICAZIONE
La litificazione è la trasformazione di un sedimento incoerente (disgregabile per immersione in acqua) in una roccia sedimentaria coerente (non disgregabile per immersione in acqua). Il termine indica l’insieme di tutti quei fenomeni che interessano il sedimento, a partire dalla sua deposizione e che conducono, per l’appunto, alla litificazione.
Si definisce metamorfismo l’insieme delle trasformazioni mineralogiche e/o strutturali che una roccia subisce quando viene a trovarsi in ambienti fisico-chimici diversi da quelli in cui si è originata. I fattori che determinano il metamorfismo sono i cambiamenti di temperatura e pressione, oltre che l’azione di fluidi come acqua e anidride carbonica.
La perdita al fuoco di un materiale ceramico (ad es. a 1060°C per 1 ora) costituisce il complemento di un’analisi chimica di un minerale espressa in ossidi inorganici, in quanto esprime la percentuale dei composti volatili persi nel trattamento termico. Questi consistono in “acqua combinata” (idrati e composti idrossilici labili) e anidride carbonica proveniente dalla decomposizione dei carbonati e dalla combustione delle sostanze organiche. È anche un semplice test che può essere utilizzato come indicatore della qualità/costanza della materia prima.
Lo stato cristallino di un materiale solido è caratterizzato da una struttura costituita da atomi disposti secondo una geometria periodica e ordinata (reticolo cristallino).
I punti del reticolo sono costituiti da una “base” (racchiusa all’interno di una cella unitaria), cioè da un insieme di uno o più entità molecolari (atomi, molecole o ioni), per cui la struttura atomica dei cristalli è definita dal reticolo e dalla base del reticolo.
Il suo opposto è lo stato amorfo, non ordinato, caratteristico ad esempio del vetro.
Il weathering (o degradazione meteorica) è il processo di disintegrazione e alterazione delle rocce e dei minerali affioranti sulla superficie terrestre, attraverso il contatto diretto o indiretto con gli agenti atmosferici. Rappresenta il primo stadio del processo sedimentario, al cui termine si ha la formazione di una nuova roccia sedimentaria. Il termine indica un fenomeno che avviene “senza movimento”, da non confondere con l’erosione, che invece è dovuta a movimenti e disintegrazione di rocce e minerali per effetto del movimento di acqua, vento, ghiaccio e della forza di gravità.
Le materie prime da utilizzare nella formulazione di un impasto ceramico vengono selezionate in base alla conoscenza di proprietà specifiche (composizione chimica, mineralogica, granulometria, ecc.) ed alla valutazione delle caratteristiche tecnologiche necessarie alle diverse fasi del processo. La formulazione di un impasto ceramico, anche quando particolarmente complessa, può essere schematizzata suddividendo i componenti in base a tre funzioni principali che questi conferiscono all’impasto finale: la funzione plastica, quella inerte e quella fondente.
Si può così effettuare una prima generica suddivisione dei materiali distinguendo tra: - materie prime argillose (plastiche) - materie prime non argillose (inerti o fondenti)
Questi componenti dovranno essere bilanciati correttamente per raggiungere le caratteristiche desiderate. Ad esempio, un impasto ceramico deve contenere una sufficiente quantità di componente plastico (argilla) tale da garantire una buona resistenza meccanica in crudo ed evitare rotture dei pezzi, senza però eccedere per non presentare problemi in fase di macinazione o essiccazione. In gergo ci si riferisce ad un impasto troppo “magro” quando il contenuto di argilla risulta in difetto o troppo “grasso” quando è in eccesso.
La componente plastica argillosa esplica a sua volta diverse funzioni: durante la fase di macinazione ad umido, ad esempio, esercita un’azione sospensivante che determina la stabilità e l’omogeneità della barbottina ceramica; in fase di formatura consente di mantenere la forma al manufatto e di conferire la necessaria resistenza meccanica al materiale crudo. Durante la cottura sono principalmente le argille a determinare il ritiro dell’impasto e la formazione di nuove fasi cristalline. Inoltre, è sempre l’argilla, insieme ai materiali fondenti, a produrre la fase vetrosa e a determinare la colorazione dell’impasto dopo cottura.
Nella categoria dei materiali non argillosi o “duri”, come spesso vengono chiamati, rientrano i materiali che costituiscono la componente inerte o quella fondente, come sabbie, feldspati, carbonati, ecc. Prima della cottura i materiali non argillosi esplicano una funzione “sgrassante” in quanto, a differenza delle argille, hanno una debole interazione con l’acqua e presentano una granulometria più grossolana. La loro introduzione controbilancia di fatto alcune proprietà delle componenti argillose nell’impasto, in quanto aumenta la permeabilità della massa ceramica e facilita le operazioni di essiccamento e di degasazione in cottura. I materiali duri inerti hanno inoltre la funzione di contenere il ritiro dell’impasto durante la cottura e di conferire struttura al manufatto. In funzione delle loro caratteristiche e delle condizioni di cottura, i fondenti in particolare contribuiscono alle trasformazioni chimiche con lo sviluppo di nuove fasi cristalline e della fase vetrosa.
Le argille possono presentare diversi gradi di plasticità. Le argille molto plastiche solitamente vengono chiamate ball clay in quanto in passato, durante l’estrazione a taglio, l’argilla si agglomerava su sé stessa appunto come una palla. Più propriamente, secondo la definizione di Wilson [1] una ball clay è “un’argilla sedimentaria, a granulometria fine, molto plastica, prevalentemente caolinitica, che dopo cottura assume un colore bianco o molto chiaro”. Pertanto, il termine ball clay si presta spesso a fraintendimenti ed è opportuno di definire con maggior chiarezza le terminologie comunemente utilizzate per indicare le diverse tipologie di argille.
China clay
Si tratta di argille che contengono una percentuale elevata di caolinite di origine primaria. Sono pertanto di tessitura grossolana e poco o per nulla plastiche. Vengono impiegate per ottenere un maggior grado di bianco dell’impasto e per aumentare il contenuto di allumina (Al2O3), conferendo una maggior refrattarietà.
Ball clay
Contengono una frazione di caolinite fino al 50%. Di origine secondaria, presentano una granulometria fine con un arricchimento in sostanze organiche che contribuisce ad aumentarne la plasticità. In crudo hanno una colorazione generalmente scura che, dopo cottura, diventa chiara. Sono utilizzate per conferire plasticità all’impasto ceramico.
Fire clay
Argille molto ricche in caolinite, non plastiche, principalmente utilizzate nella preparazione di prodotti refrattari per l’alto contenuto di allumina (Al2O3) e l’elevato punto di fusione (> 1400 °C).
Flint clay
Si tratta di argille compatte con frattura concoide (il flint è la selce). Sono argille quarzose dove il quarzo (SiO2) è presente in quantità variabili, con granulometria più o meno grossolana. Scarsamente o per nulla plastiche. Idonee per realizzare prodotti porosi a colorazione variabile. La loro refrattarietà è funzione del contenuto di quarzo e degli ossidi fondenti presenti. Generalmente si usano come argille correttive.
Argille alcaline
Sono le argille più utilizzate nella produzione del grès porcellanato. Sono argille prevalentemente di tipo illitico con elevata plasticità. Il contenuto di ossidi alcalini come K2O determina in cottura un’azione fondente con graduale rammollimento della massa fino a raggiungere la completa greificazione.
Common clay (o Heavy clay)
Sono argille rosse, ferruginose e possono essere caratterizzate dalla presenza di carbonati di calcio e/o magnesio (argille calcaree). Contengono composti di ferro che, oltre a colorare il biscotto, sono anche energici fondenti, specialmente in presenza di carbonati. Si tratta pertanto di argille molto fusibili. Hanno una plasticità variabile in funzione del tipo e della quantità di minerali argillosi presenti. Possono presentare problemi di reologia in fase di macinazione a umido.
Bentoniti
Si tratta di rocce caratterizzate da una prevalenza di minerali argillosi montmorillonitici. Utilizzate negli impasti in piccola percentuale ne aumentano la plasticità e la tenacità in crudo. Se introdotte in eccesso hanno effetto negativo sulla reologia della barbottina e in essiccamento possono produrre crepe o rotture. Fondono rapidamente in un intervallo termico molto ristretto.
Al fine di descrivere in dettaglio i singoli minerali argillosi, è opportuna una rapida panoramica delle loro caratteristiche principali e dei metodi analitici che ne consentono l’identificazione, rimandando l’approfondimento di tali metodi al Capitolo 6 dedicato alla caratterizzazione delle materie prime.
Le proprietà più significative per la caratterizzazione di un materiale argilloso sono le seguenti:
- plasticità
- granulometria
- reologia
- composizione chimica e mineralogica
- comportamento del pressato (in crudo e dopo cottura)
La plasticità si può semplicemente definire come l’attitudine di un materiale a lasciarsi deformare sotto l’azione di una forza e a mantenere la nuova forma una volta cessata la sollecitazione. Questa condizione avviene ad esempio durante la fase di foggiatura di un impasto ceramico a base argillosa. La plasticità di un’argilla è determinata principalmente dalla dimensione estremamente fine delle particelle primarie dei minerali argillosi (< 2 µm) e dalla loro struttura cristallina. La microstruttura a «foglietti» caratteristica dei fillosilicati permette infatti, in presenza di acqua, di garantire una buona adesione tra le particelle argillose consentendo anche un parziale slittamento tra i piani cristallini quando vengono sottoposti all’azione di una forza deformante. Granulometria e struttura cristallina spiegano sostanzialmente la differenza di plasticità che si osserva tra i principali minerali argillosi, oltre alla valutazione dell’eventuale presenza di sostanze organiche accumulate durante il processo di genesi sedimentaria.
Un impasto ceramico non può essere eccessivamente plastico e deve essere sempre corretto con materiali duri, non argillosi.
Nel settore ceramico la plasticità non viene misurata direttamente mediante, ad esempio, la registrazione di curve sforzo/deformazione, ma viene generalmente effettuata una sua stima in funzione di caratteristiche indirette, quali:
- resistenza meccanica in crudo
- espansione post-pressatura
- ritiro in essiccamento
- % di particelle < 2 µm
- contenuto di acqua caratteristico (limite plastico di Atterberg o di Pfefferkorn)
- penetrometro
- indice al blu di metilene
- capacità di scambio cationico.
Come già accennato, le argille sedimentarie presentano una granulometria inferiore a 2 µm e questo consente al materiale di avere una superficie specifica elevatissima da cui dipendono proprietà come il potere adsorbente, la plasticità, il ritiro in crudo e in cotto, la velocità delle reazioni e delle trasformazioni che avvengono durante la cottura. A differenza delle argille sedimentarie, i caolini primari hanno granulometrie più grossolane, con una mediana attorno a 7 ÷ 10 µm. I valori di superficie specifica per i caolini primari sono nell’ordine delle decine di m2/g mentre si raggiungono 100 ÷ 120 m2/g con le argille illitiche e si superano anche i 600 m2/g con le argille montmorillonitiche.
La misura della distribuzione granulometrica con valutazione della frazione < 2 µm si effettua con strumentazioni scientifiche di laboratorio come la diffrazione laser o la scansione RX della sedimentazione. Una tecnica più tradizionale e laboriosa, ma economica, è l’utilizzo della pipetta di Andreasen.
La dimensione micrometrica delle particelle argillose e la presenza di cariche elettriche residuali sulla superficie dei cristalli, fanno sì che in sospensione acquosa le argille abbiano alcuni comportamenti tipici dei materiali colloidali. In particolare, in presenza di acqua, l’argilla assume una struttura di tipo micellare che è alla base di tutti i fenomeni di flocculazione e deflocculazione, da gestire opportunamente in macinazione e nelle diverse fasi di lavorazione della barbottina. Questo argomento verrà approfondito nel Capitolo 5.
L’analisi chimica dei minerali argillosi si effettua solitamente mediante l’utilizzo di tecniche analitiche strumentali come la fluorescenza a RX (XRF) o la spettroscopia di emissione (ad es. ICP-AES), determinando gli elementi presenti sotto forma di ossidi (tipicamente: SiO2, Al2O3, Fe2O3, TiO2, CaO, MgO, Na2O e K2O).
L’analisi mineralogica viene effettuata per diffrazione a RX (XRD) ed anche attraverso l’analisi termica differenziale e gravimetrica (DTA-TG).
In aggiunta alle precedenti analisi, sui materiali argillosi è opportuno valutare anche il contenuto di carbonati mediante calcimetria Pizzarelli, la percentuale di zolfo, di carbonio totale, di carbonio organico e la quantità di sali solubili presenti (solfati, cloruri e nitrati).
Per selezionare una materia prima argillosa e stabilire la sua percentuale di utilizzo nell’impasto, non si può prescindere dalle caratteristiche tecnologiche che il materiale evidenzia dopo pressatura, sia in crudo che in cotto.
Sul provino pressato crudo solitamente vengono determinati i seguenti parametri:
- espansione di pressatura
- variazione dimensionale in essiccato
- carico di rottura in verde (MOR verde)
- carico di rottura in secco (MOR essiccato)
La caratterizzazione in cotto viene effettuata cuocendo alcuni campioni di materiale pressato in un determinato intervallo di temperature (cotture a gradiente). Sui campioni vengono determinati:
- assorbimento d’acqua
- variazione dimensionale
- carico di rottura (MOR cotto)
- colore
- sviluppo di cuore nero
In particolare, il valore di assorbimento d’acqua e di variazione dimensionale vengono espressi in funzione della temperatura di cottura, costruendo la cosiddetta curva di greificazione. Dall’esame della curva di greificazione è possibile distinguere le argille sulla base del loro comportamento alla cottura (refrattarie, fusibili o vetrificabili) al fine di valutare l’opportunità di un loro utilizzo e, nel caso, le relative proporzioni nella ricetta di uno specifico impasto.
Anche il colore del materiale dopo cottura è utile per effettuare una prima rapida selezione, distinguendo tra materiali cuocenti bianco e materiali cuocenti rosso.
Questi semplici criteri di classificazione sono importanti per stabilire in prima approssimazione la formulazione di un impasto, specialmente quando si deve effettuare una scelta tra un ampio numero di materie prime disponibili.
Di seguito si riporta una descrizione più approfondita dei singoli materiali argillosi.
Origine del termine
Il termine deriva dal cinese Gaoling, che significa “collina alta” ed indica, nella provincia di Jiangxi, il primo luogo ove questa materia prima fu estratta per utilizzi ceramici.
Struttura mineralogica
L’elemento base è costituito dall’associazione di un “foglietto tetraedrico”, formato da quattro atomi di ossigeno (ciascuno con due cariche negative) posti ai vertici dei tetraedri e da un atomo di silicio (avente quattro cariche positive) ubicato al centro, con uno ottaedrico, formato da sei gruppi ossidrili OH (ciascuno portante una carica negativa) che occupano tutti i vertici della struttura e da un catione solitamente trivalente (Al3+) in posizione centrale, sostituibile anche da un catione bivalente (vedi Fig. 3.1).
L’unità tetraedrica presenta quindi quattro cariche negative non saturate; anche l’ottaedrica è negativa con tre cariche non saturate (se il catione è trivalente) o quattro (se bivalente).
Nell’elemento base del reticolo, i due foglietti si “legano” procedendo alla sostituzione di un ossidrile OH– di ciascun ottaedro con la condivisione dell’atomo di ossigeno, posto ai vertici dei tetraedri, che dispone di una carica libera. Questo anione viene quindi condiviso tra un tetraedro e due ottaedri in quanto ciascun gruppo ossidrile OH– è bilanciato dai cationi di alluminio di due ottaedri contigui. Con questo accoppiamento la struttura della caolinite risulta elettricamente neutra; essa può ripetersi per un elevato numero di volte (in senso verticale) conservando la stessa distanza tra piani eguali contigui.
In questa “rigidità strutturale” risiede la spiegazione della non espandibilità del reticolo della caolinite, nonostante le forze di legame della struttura siano deboli (interazioni di Van der Waals e legami a idrogeno).
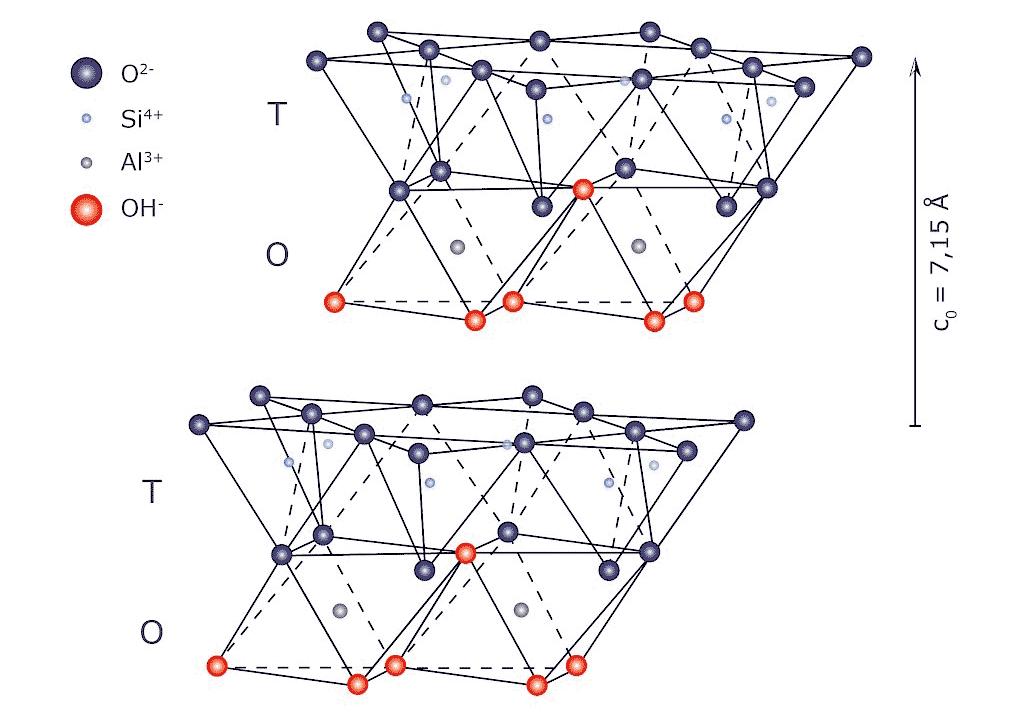
3.1 Struttura della caolinite [1]
Formula chimica
Al2(Si2O5)(OH)4
dove sia Al3+ in posizione ottaedrica che Si4+ in posizione tetraedrica possono essere sostituiti da cationi vicarianti a valenza più bassa che portano alla deficienza di una carica positiva della struttura.
Composizione teorica in ossidi
2SiO2∙Al2O3∙2H2O corrispondente alla composizione percentuale in peso:
46,54% SiO2 + 39,50% Al2O3 + 13,96% H2O
Genesi e caratteristiche dei depositi contenenti caolinite
L’origine dei giacimenti caolinitici è legata alla trasformazione delle rocce contenenti feldspati soggette al rilascio degli elementi alcalini (sodio e potassio).
Nel caso della formazione di un caolino particolarmente puro, originato da filoni feldspatici, questo presenta una colorazione bianco-latte, scarsa plasticità, superficie untuosa e lascia un velo biancastro persistente sulle dita. Più frequentemente però i giacimenti caolinitici sono miscele di caolinite, quarzo (inattaccabile dai fenomeni di alterazione e trasformazione), feldspati residuali, a cui talvolta si aggiungono ossidi di ferro (principali responsabili della colorazione del materiale) e miche.
I depositi di caolino sono generalmente di origine primaria, cioè permangono ove preesistevano le rocce madri e non hanno quindi subito trasporti; i giacimenti secondari sono stati invece oggetto di allontanamenti e deposizioni in ambiente acquoso (generalmente lacustre) che hanno concentrato e selezionato le particelle più fini inquinandole però con materiali di origine organica vegetale (foglie, rami, radici, elementi carboniosi).
È opportuno ricordare la differenza esistente tra i giacimenti di caolino puro (scarsamente plastico) e quelli di argille caolinitiche (ball clay). Queste ultime, sempre di origine secondaria, sono caratterizzate da grande plasticità, conseguenza della selezione granulometrica e dell’arricchimento in materiali plasticizzanti naturali (le parti vegetali e carboniose). Ad essi è da attribuirsi la colorazione nera dell’argilla tal quale, che presenta la caratteristica bianchezza solo dopo cottura, quando cioè tutti gli inquinanti organici sono stati ossidati.
Ubicazione dei principali depositi e tecniche di coltivazione
I più importanti giacimenti di caolino e di argille caolinitiche sono concentrati in Inghilterra (Devon e Cornovaglia), Germania (Westerwald e Baviera), Francia (Bretagna), Repubblica Ceca (Karlovy Vary) e Stati Uniti (Georgia e Carolina del Sud).
Il più antico metodo industriale impiegato nell’estrazione della caolinite dalla roccia madre si avvale di potenti getti di acqua ad alta pressione che vengono diretti contro le pareti del deposito con il risultato di allontanare, sospesa in acqua, la frazione caolinitica. La sospensione viene poi raccolta in grandi vasche di sedimentazione. Questa tecnica è stata inventata in Gran Bretagna ed è tuttora applicata ai giacimenti idonei a questa tipologia di estrazione. Per quanto riguarda i depositi di ball clay, la tecnica di estrazione avviene a cielo aperto. Nei siti estrattivi del Devon, dopo aver rimosso lo strato inerte superficiale mediante l’impiego di bulldozer e scraper, la coltivazione si sviluppa su banchi di spessore variabile tra 3 e 6 m mediante utilizzo di escavatori idraulici, dragline ed escavatori a tazze. Talvolta la compresenza di sedimenti di lignite fa aumentare lo scarto fino al 50% del materiale smosso. Le condizioni in cui si opera (in fossa) e la impermeabilità della materia prima sono tali da favorire gli allagamenti per cui è indispensabile ricorrere all’uso di pompe di grande portata che mantengono operativi i pozzi di coltivazione. Nel Westerwald, in Germania, si opera un’estrazione fortemente selettiva, preceduta da campionature sistematiche con trivellazioni ed esami di laboratorio che forniscono le indicazioni necessarie per procedere ad una corretta coltivazione. Le tecniche adottate prevedono uno sviluppo “a gradoni” con impiego di draghe a ruote idrauliche che operano in modo continuo alimentando una serie di nastri che trasportano l’argilla fino ai magazzini. In alternativa si utilizzano gli escavatori idraulici, che operano dal gradone in estrazione, caricando dei dumper parcheggiati sul gradone sottostante.
Analisi mineralogica e analisi termiche della caolinite
L’identificazione del minerale argilloso avviene tramite analisi diffrattometrica ai raggi X (Fig. 3.2) e comporta l’osservazione delle diffrazioni dei piani basali, riportate nella seguente tabella:
Caolinite
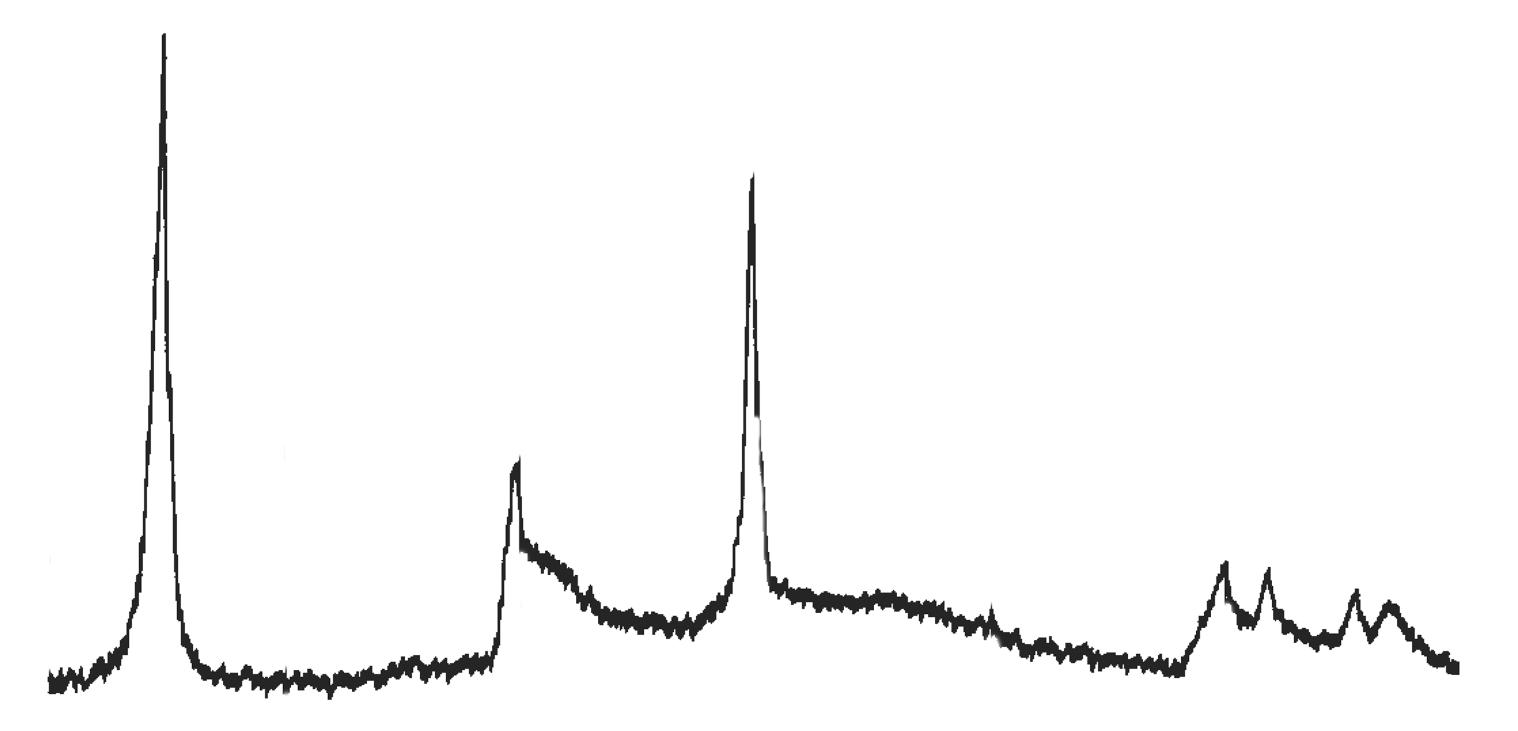
Fig. 3.2 Esempio di diffrattogramma di caolinite (K) altamente disordinata
Fig. 3.02
La presenza di cloriti può mascherare la presenza di caolinite a causa della coincidenza dei riflessi basali a 12,5 °(2θ) (corrispondenti a 7,18 Å). ln questo caso occorre verificare l’eventuale sdoppiamento che si verifica nella riflessione sul piano (002), dato che sussiste una piccola differenza tra la distanza reticolare della caolinite (3,47 Å) e della clorite (3,54 Å) a cui corrispondono posizionamenti dei picchi rispettivamente a 24,9 e 25,2 °(2θ).
Il diagramma dell’analisi termo-differenziale (DTA) di una caolinite (Fig. 3.3) presenta un netto picco endotermico a 580 ÷ 600 °C dovuto alla trasformazione della caolinite in metacaolinite con perdita dell’acqua di cristallizzazione; attorno a 980 °C si manifesta invece un marcato picco esotermico determinato dalla cristallizzazione di mullite 3Al2O3 ∙2SiO2 (vedi Fig. 3.4).
3.3 Curve DTA di alcune caoliniti
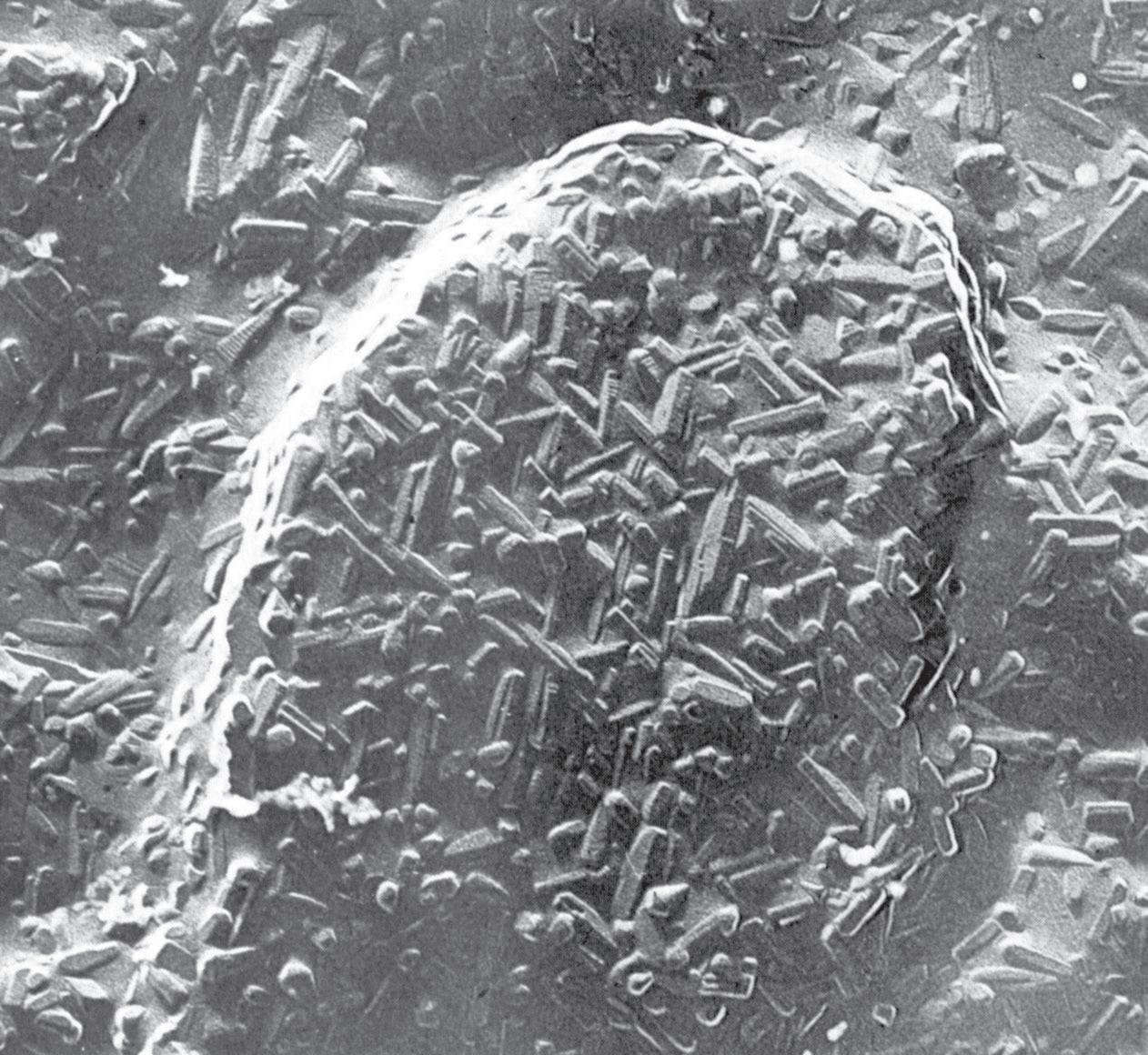
Fig. 3.4 Cristalli di mullite formati per riscaldamento di caolinite in matrice silicea (×37000) [2]
La caolinite, sottoposta ad analisi termo-gravimetrica (TG), presenta una forte perdita in peso attorno a 520 ÷ 580 °C, dove avviene la perdita dell’acqua di cristallizzazione; nessuna variazione si osserva invece verso i 980 °C, dato che la reazione esotermica di formazione di mullite è dovuta alla liberazione di energie di legame senza variazioni di massa.
Fig. 3.5 Curva dilatometrica di un campione di caolino puro, che mostra due brusche contrazioni nella fase di deossidrilazione e nella reazione di ricristallizzazione
Fig. 3.05 NEW
Fig. 3.6 Curve dilatometriche di caolini con contenuto crescente di quarzo (curve a → c), che gradualmente fa prevalere il suo effetto su quello della caolinite
La dilatometria (vedi Fig. 3.5, Fig. 3.6, Fig. 3.7) è caratterizzata fino 450 °C da una modesta dilatazione a cui fa seguito (oltre 550 °C) una prima fase di ritiro connessa alla eliminazione degli ossidrili OH (che si protrae fino a 880 °C) seguita da una seconda fase di contrazione, più marcata, dovuta all’inizio delle reazioni di sinterizzazione.
Fig. 3.06
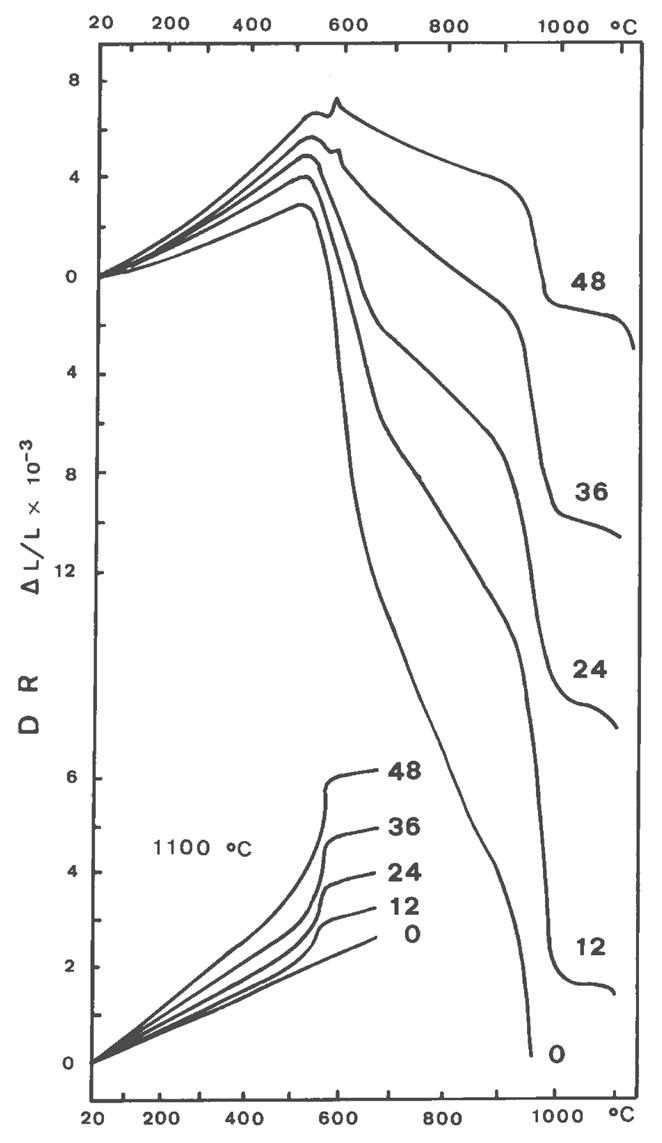
Fig. 3.7 Dilatazione e ritiro di miscele caolinite–quarzo (0 ÷ 48% di quarzo) [3]
In Tab. 3.1 sono riportate le analisi mineralogiche di alcune comuni materie prime caolinitiche.
Tab. 3.1 Esempi di analisi mineralogiche di argille caolinitiche
Caratterizzazione tecnologica di materiali caolinitici pressati
Come già accennato anche dal punto di vista tecnologico esiste una netta distinzione tra ball clay e china clay.
Più specificatamente le ball clay presentano:
- discreta espansione di post-pressatura (≈ 0,3%);
- ottima resistenza meccanica alla flessione in crudo (verdi > 1,2 N/mm 2; essiccati > 3,0 N/mm2);
- per cottura a 1100 °C la variazione dimensionale oscilla tra 2 ÷ 4% (ritiro) con porosità apparente del 10 ÷ 15%;
- i valori di resistenza meccanica in cotto a 1100 °C possono variare (da 10 a 20 N/mm2) in funzione delle “impurezze” presenti che possono favorire la sinterizzazione;
- il coefficiente di dilatazione termica lineare α del cotto a 1100 °C varia tra 50 e 70×10−7 °C−1.
Viceversa, i comportamenti tipici della china clay sono:
- espansione di post-pressatura maggiore (0,5 ÷ 0,7%);
- scarsa resistenza meccanica in verde e in secco;
- tendenza all’espansione in essiccamento;
- per cottura a 1100 °C si osservano modeste contrazioni dimensionali con valori di assorbimento d’acqua elevato (15 ÷ 20%), bassa resistenza meccanica (<15 N/mm2) e basso coefficiente di dilatazione termica lineare α (40 ÷ 60×10−7 °C−1) dipendentemente dal contenuto di quarzo. Nel complesso il materiale risulta altamente refrattario.
Utilizzi ceramici della materia prima
Le ball clay possono essere impiegate tal quali, oppure possono venire miscelate per ottenere blend con caratteristiche costanti, che meglio rispondano alle esigenze di qualità dei prodotti da realizzare.
Le ball clay trovano applicazioni in una vasta gamma di prodotti ceramici (sanitari, piastrelle, vasellame, oggettistica, ecc.) principalmente con funzioni leganti e plastificanti.
Le percentuali di impiego variano in un intervallo molto ampio da prodotto a prodotto ed anche all’interno della stessa tipologia di manufatto. A livello indicativo le quantità utilizzate sono le seguenti:
piastrelle (rivestimento)
piastrelle (pavimento)
grès porcellanato
vasellame (terraglia)
sanitari
20 ÷ 30%
20 ÷ 30%
25 ÷ 35%
20 ÷ 35%
20 ÷ 30%
Nel campo delle fritte e degli smalti, i materiali caolinitici vengono purificati per essere utilizzati come componenti delle miscele da fondere e come additivi sospensivanti aggiunti alle fritte in fase di macinazione.
I caolini meno puri, anche non trattati ed inquinati dalla presenza di ferro, vengono impiegati nella fabbricazione di refrattari alluminosi, dove agiscono da apportatori di allumina.
Solo una piccola percentuale di caolino trova sbocco nel campo ceramico mentre quasi il 90% è impiegato dall’industria della carta, dove viene utilizzato come carica inorganica e come ricoprente superficiale.
Numerosi sono poi gli altri impieghi minori come carica inorganica (pneumatici e gomma, vernici, ecc.), come supporto per uso farmaceutico (compresse), per cosmetici e dentifrici, per insetticidi in polvere e fertilizzanti.
Origine del termine
Il nome halloysite fu dato ad una roccia belga per la prima volta studiata dal geologo J.B. Omalius d’Halloy (1783 - 1875).
Struttura mineralogica
Si tratta di un minerale della famiglia della caolinite, costituito quindi dall’associazione di un foglietto tetraedrico con uno ottaedrico, avente la particolarità di essere idratato – vedi Fig. 3.8.
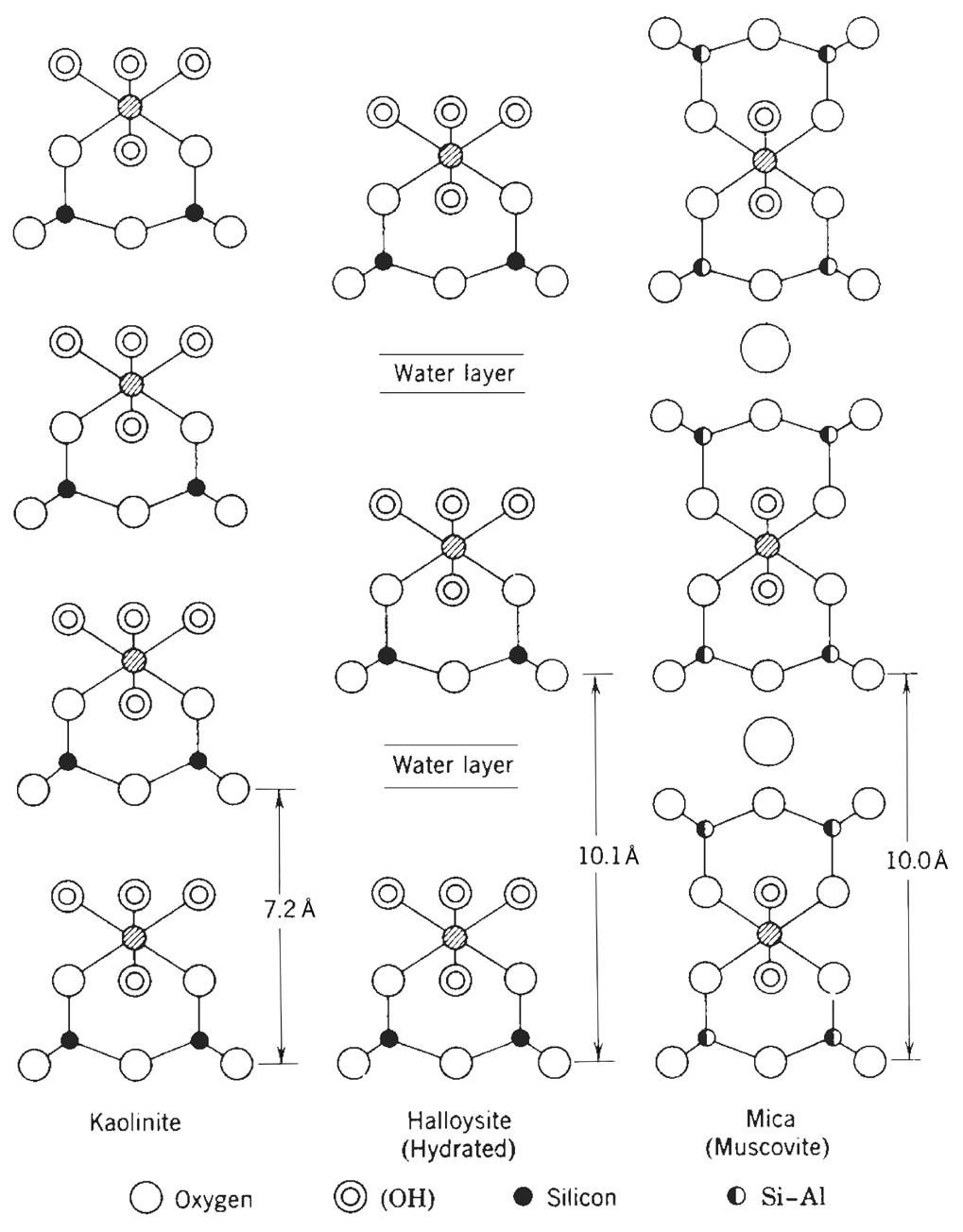
Fig. 3.8 Confronto tra le celle elementari di caolinite, halloysite e muscovite
La presenza di un interstrato di molecole d’acqua conferisce una certa mobilità ai foglietti che tendono così ad arrotolarsi in forme cilindriche come risulta chiaramente dall’esame al microscopio elettronico (Fig. 3.9).

Fig. 3.9 Struttura della caolinite e della halloysite
Per spiegare questo caratteristico comportamento si sono ipotizzate tensioni differenziali tra il foglietto tetraedrico e quello idrargillico (ottaedrico). L’attrazione tra i foglietti è evidentemente superiore alla tensione differenziale dato che in caso contrario ne deriverebbe la distruzione del reticolo stesso.
In seguito alla perdita dell’acqua di idratazione, la struttura arrotolata può appiattirsi formando metahalloysite (o halloysite-7A), caratterizzata da una distanza reticolare di 7,4 Å, contro i 10,1 Å dell’halloysite idrata (chiamata halloysite-10A). La metahalloysite perde tutta l’acqua d’interstrato attorno a 200 °C portando irreversibilmente ad una struttura simile a quella della caolinite.
Passando dalla caolinite alla metahalloysite e alla halloysite si assiste ad un progressivo aumento dell’idratazione a cui si accompagna anche un aumento del grado di disordine del reticolo.
È interessante osservare come le dimensioni dei cristalli dei vari minerali della famiglia della caolinite siano dipendenti dal grado di stabilità (e di ordine) della struttura. Si osserva così che le particelle più piccole appartengono appunto alla halloysite mentre quelle più grosse sono quelle della caolinite.
Composizione chimica
Al2(Si2O5)(OH)4·2H2O
dove non si osserva quasi mai la sostituzione del Si4+ con vicarianti tipo Al3+ o Fe3+ che, come spiegato, comportano la deficienza di una carica positiva nella struttura.
Genesi e caratterizzazione dei depositi
La halloysite è un costituente di numerosi giacimenti argillosi. Si può presentare in masse piuttosto compatte di colore biancastro (se priva d’inquinamenti cromofori) o con tonalità sul giallo o sul rosa ed è caratterizzata da aspetto lucido e da untuosità al tatto (come la caolinite). Tuttavia, è presente anche in materie prime con elevata plasticità.
La classica genesi della halloysite è riconducibile a rocce madri vulcaniche (da acide a neutre) e vulcanoclastiti (rocce piroclastiche e brecce vulcaniche).
In linea generale la trasformazione di queste rocce originali è simile a quella che porta alla formazione di caolinite. Come per questa ultima, infatti, avviene una separazione degli alcali (sodio e potassio) che solo in certi casi si realizza totalmente.
Frequentemente permangono anche residui importanti di quarzo; spesso si osservano associazioni con caolinite con possibile presenza di cristobalite ed alunite (solfoalluminato potassico idratato).
Anche nel caso dei depositi halloysitici si possono incontrare giacimenti primari (rimasti nel punto delle preesistenti rocce madri) o secondari (in questo caso le particelle sono state trasportate e rideposte in ambiente acquoso).
Inoltre, nella genesi dei depositi halloysitici è spesso presente alterazione idrotermale.
Ubicazione dei principali giacimenti e tecniche di coltivazione
Pur essendo piuttosto diffusa, raramente la halloysite costituisce la più importante specie mineralogica di un deposito.
In alcune zone degli Stati Uniti si coltivano giacimenti di grande capacità (Utah, Nevada, ldaho, North Carolina e Georgia).
I depositi europei più importanti sono segnalati in Francia (Dordogne) mentre sono conosciuti giacimenti minori in Cechia e nella penisola Balcanica.
Le cave operano a cielo aperto con l’impiego della classica attrezzatura per movimento terra (bulldozer, scraper, escavatori idraulici, ecc.).
Analisi mineralogiche di materie prime halloysitiche (tipici comportamenti di riconoscimento)
L’identificazione della specie tramite analisi diffrattometrica ai raggi X comporta esclusivamente l’osservazione delle diffrazioni dei piani basali (Fig. 3.10) caratterizzati in particolare da una intensa riflessione a circa 4,4 Å, spesso più forte di quelle basali a 7,3 Å e 3,6 Å, che risultano anche larghe e asimmetriche verso i bassi angoli
Nella seguente tabella si riporta la corrispondenza tra distanze reticolari e posizionamento dei picchi (angolo 2θ per radiazioni CuKα) per halloysite idrata e metahalloysite:
Halloysite idrata
Il diagramma dell’analisi termo-differenziale (DTA) di una halloysite (Fig. 3.10) presenta un caratteristico picco endotermico che si sviluppa prima di 200 °C connesso con la perdita dell’acqua di interstrato; successivamente l’andamento è simile a quello della caolinite; a partire da 450 °C si ha un nuovo picco endotermico, conseguenza dell’allontanamento degli ossidrili OH , mentre attorno a 980 °C avviene la cristallizzazione di mullite con un netto picco esotermico. Corrispondentemente, l’analisi termo-gravimetrica (TG) presenta una forte perdita di peso sia prima di 200 °C che verso 450 °C, in corrispondenza della eliminazione dell’acqua di interstrato e della perdita degli ossidrili OH .
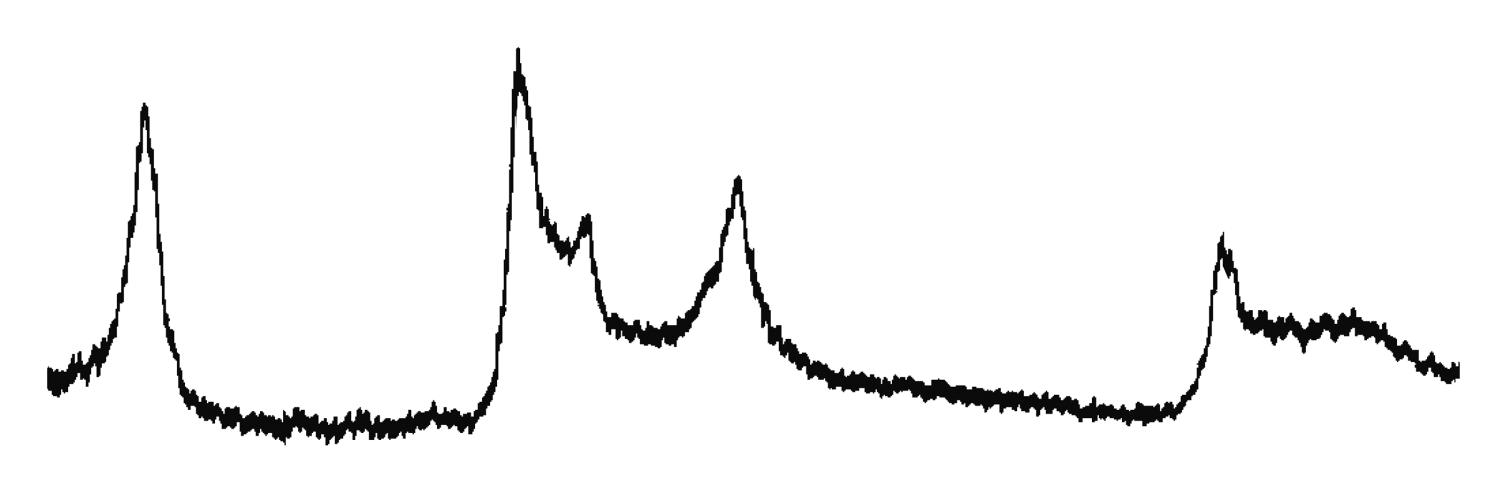
Fig. 3.10 Esempio di diffrattogramma della halloysite
Viceversa non si osserva nulla a 980 °C dato che questa reazione esotermica non contempla variazioni di materia ma solo liberazione di energia di legame.
Fig. 3.10
La dilatometria presenta una iniziale debole dilatazione contrastata da un marcato ritiro poco oltre 150 °C, causato dall’allontanamento dell’acqua dell’interstrato; superata questa fase, il ritiro continua moderatamente fino ai 450 °C dove si osserva la contrazione dovuta alla perdita degli ossidrili OH che si protrae fin verso 880 °C; a tale temperatura iniziano le reazioni di sinterizzazione e si riscontra un ritiro ancora più forte con conseguente rammollimento del provino.
Caratterizzazione tecnologica di materiali halloysitici pressati
Piastrelle eventualmente formate con una materia prima prevalentemente halloysitica mostrerebbero:
- normale espansione di post-pressatura;
- buona resistenza meccanica alla flessione in crudo (verdi ed essiccati);
- sensibile ritiro di essiccazione;
- in cottura a 1100 °C, la contrazione dimensionale è già rilevante (3 ÷ 8%) con porosità apparente del 20 ÷ 25%;
- a 1100 °C i valori di resistenza meccanica sono generalmente compresi tra 10 e 15 N/mm2;
- il coefficiente di dilatazione termica lineare α (per campioni cotti a 1100 °C) è molto basso (anche inferiore a 35×10−7 °C−1).
Si evidenzia che il forte ritiro di essiccazione rende assolutamente problematico l’allontanamento dell’acqua comportando molto spesso la formazione di crepe nei manufatti.
Utilizzi ceramici
L’halloysite viene raramente impiegata per produrre piastrelle in associazione con altre materie prime per le notevoli complicazioni in essiccazione che questa comporta.
Talvolta trova applicazione nella produzione di mattoni (specie negli Stati Uniti), fire-bricks e refrattari, essendo una materia prima ad alto contenuto di allumina.
In talune produzioni l’impiego di halloysite può trovare giustificazione qualora si intenda aumentare la plasticità della miscela. Si evidenzia però come questo minerale abbia un pessimo comportamento reologico e, nell’eventuale impiego in processi produttivi che prevedono la macinazione ad umido delle materie prime, peggiora sensibilmente la deflocculazione dell’impasto.
In sintesi, l’utilizzo di una materia prima halloysitica deve sempre essere molto contenuto (5 ÷ 20%); in queste basse percentuali può comunque trovare applicazione in produzioni di vasellame, piastrelle porose e vetrificate.
Utilizzi in settori differenti
Taluni impieghi risultano alternativi a quelli del caolino; in particolare l’halloysite può venire usata come carica inorganica nella produzione di carta, pneumatici e gomma.
Anche le Fuller’s earth (letteralmente “terra di Fuller”), polveri minerali con proprietà assorbenti, sono argille sedimentarie costituite da significative percentuali di halloysite e, in più larga misura, da montmorillonite (vedi Paragrafo 3.1.5).
Origine del termine
Il termine fu proposto da Grim nel 1937 quando questa tipologia di minerali fu descritta per la prima volta a Fithian, un villaggio dell’Illinois. Inizialmente il termine illite si riferiva ad una famiglia di minerali argillosi cosiddetti mica-like, con una distanza reticolare (001) di circa 10 Å. Negli anni la definizione dell’illite è stata modificata e perfezionata fino a consentire di definire con il termine illite un minerale specifico. Essendo l’illite pura un minerale molto raro, la definizione di Grim resta tuttora quella più utilizzata anche in ambito ceramico.
Struttura mineralogica
L’illite presenta la caratteristica struttura dei fillosilicati, simile a quella delle miche; l’unità base strutturale è formata da due “foglietti” tetraedrici che ne racchiudono uno ottaedrico (Fig. 3.11), e risulta sostanzialmente identica a quella della montmorillonite salvo che alcuni atomi di silicio, in coordinazione tetraedrica, sono sempre sostituiti da alluminio a cui si aggiunge uno ione potassio per bilanciare l’equilibrio elettrico della struttura.
Pertanto il potassio è fortemente legato e non risulta facilmente sostituibile come accade con la montmorillonite.
La differenza strutturale tra l’illite e una mica (ad esempio la muscovite) risiede in una minor sostituzione di silicio con alluminio e quindi in una inferiore concentrazione di potassio nell’illite. Inoltre, l’illite può contenere molecole d’acqua interstratificate.
Per alterazione chimico-fisica, le illiti si trasformano parzialmente in montmorillonite e/o vermiculite, fillosilicati caratterizzati da una più accentuata struttura a interstrato, che conferisce ancora maggior plasticità alle argille illitiche. Il rapporto SiO2 / Al2O3 varia da 2 a 4; frequentemente risulta pari a 3. Un’illite particolare è la glauconite (illite ferrifera) che si genera per alterazione della biotite.
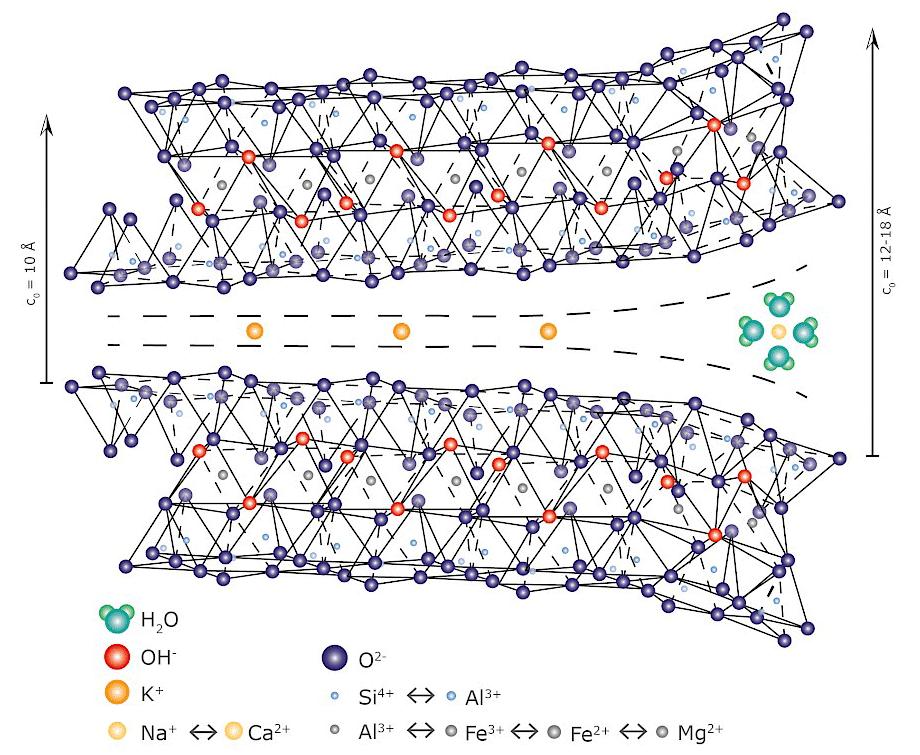
Fig. 3.11 Struttura cristallografica dell’illite [1]
Composizione chimica
La formula della illite non è definibile ma si può semplificare come segue:
K xAl2(Si4-xAlxO10)(OH)2
La formula evidenzia che la saturazione massima di potassio caratteristica della mica si riduce di una frazione x nelle diverse tipologie di illite, in base alla quantità di silicio che viene sostituito dall’alluminio.
Un esempio di composizione può essere il seguente:
K0,65Al2(Si3,35Al0,65O10)(OH)2
anche se in natura le argille illitiche inglobano sovente nella loro struttura significative percentuali di magnesio, calcio e ferro.
Genesi e caratteristiche dei depositi
L’origine dei giacimenti è legata a deposizioni sedimentarie. Nella pratica però, non si incontrano mai depositi costituiti esclusivamente da illite, bensì associazioni con altri minerali, come: clorite, montmorillonite, caolinite, quarzo, calcite, dolomite, ecc.
Ubicazione dei principali giacimenti e tecniche di coltivazione
I giacimenti argillosi a prevalente contenuto illitico, storicamente più utilizzati dall’industria ceramica, sono quelli dell’Appennino emiliano (Italia).
Qui troviamo le argille “grigio-azzurre” del Pliocene ed il cosiddetto Complesso Emiliano costituito dalle marne di Monte Piano (argille rosse e verdi note con il nome di red-beds) e le serie di Antognola (marne ed argille grigio-azzurre); le composizioni chimiche di rocce argillose di questo tipo sono rappresentate nei diagrammi ternari di Fig. 3.12 e Fig. 3.13, a confronto con le argille illitico-caolinitiche chiare, presenti in Europa.
Argille illitico-cloritiche
Argille illitico-caolinitiche: tedesche inglesi francesi
Fig. 3.12 Diagramma ternario SiO2 / Al2O3 / altri ossidi con gli intervalli composizionali delle argille rosse e chiare [4]
Fig. 3.12 IT NEW
Argille illitico-cloritiche
Argille illitico-caolinitiche: tedesche inglesi francesi
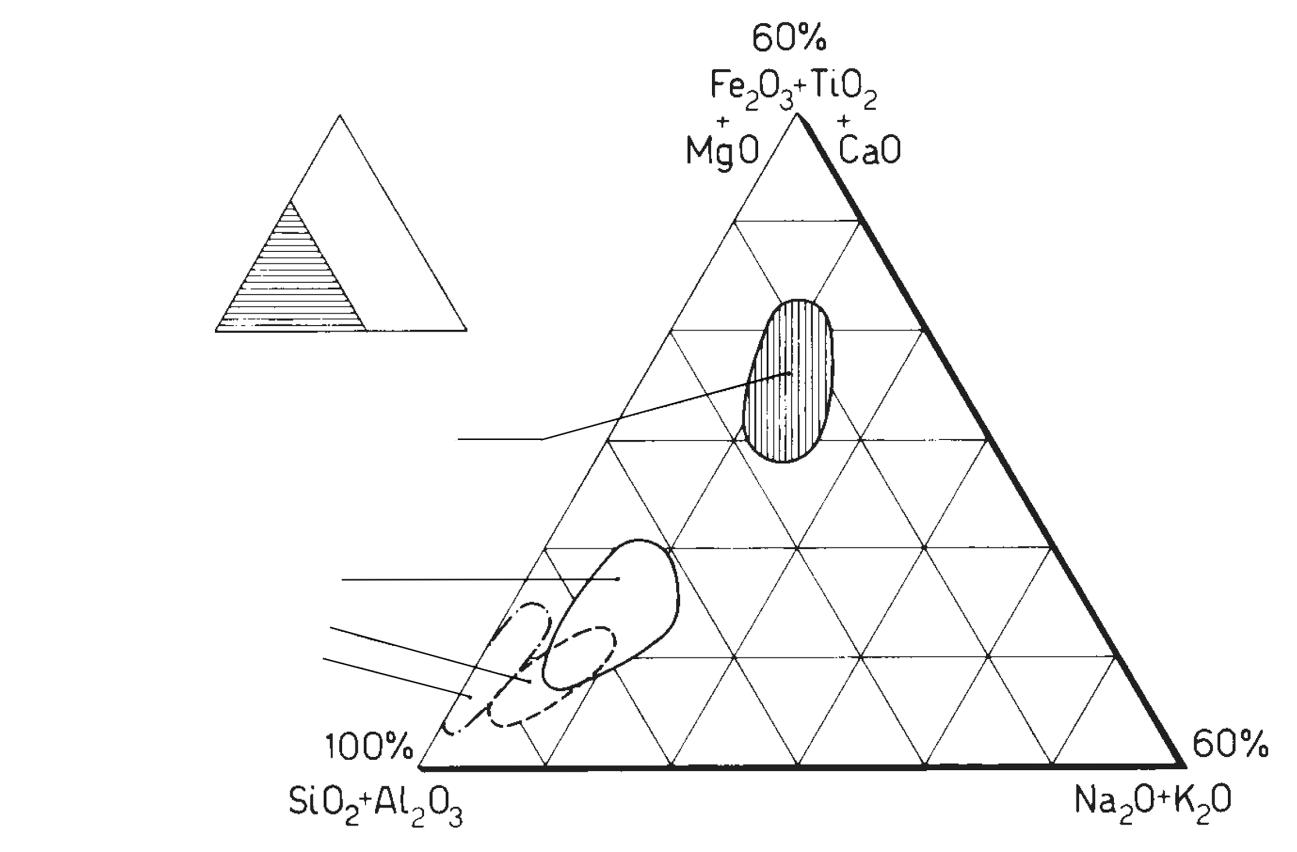
Fig. 3.13 Diagramma ternario SiO2+Al2O3 / Na2O+K2O / altri ossidi con gli intervalli composizionali delle argille rosse e chiare [4]
Nella Fig. 3.14 è invece riportato un diagramma ternario relativo ai minerali complementari.
Fig. 3.13 IT NEW
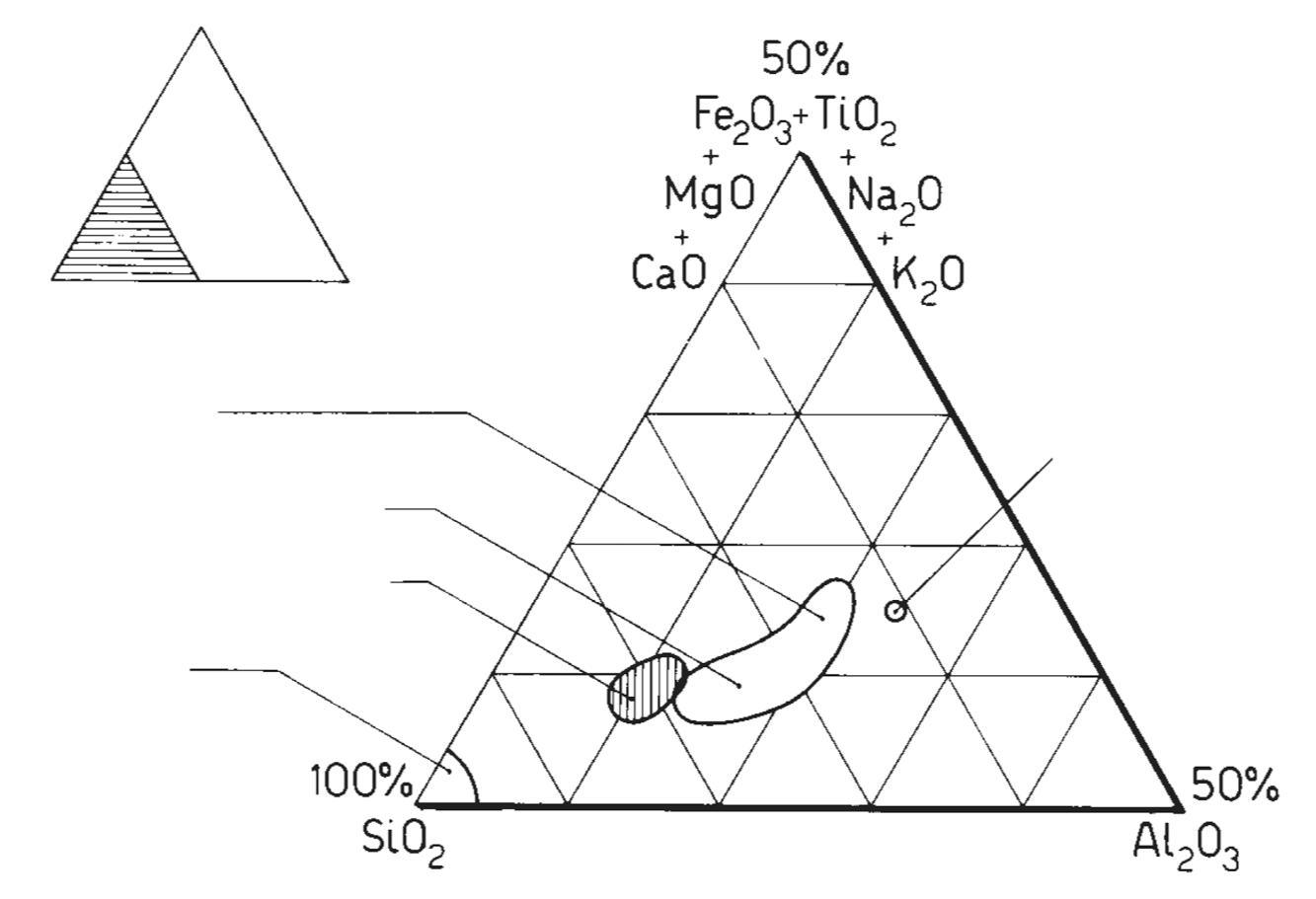
Feldspati
Rocce feldspatiche
Sabbie quarzo-feldspatiche
Sabbie quarzifere
Sienite nefelinica
Fig. 3.14 Diagramma ternario SiO2 / Al2O3 / altri ossidi con gli intervalli composizionali delle materie prime complementari [4]
Fig. 3.14 IT NEW
Inoltre, sempre in Europa, sono ben noti i giacimenti ungheresi di illite bianca associata a montmorillonite e di argilla plastica miocenica ceca, oltre ai vasti giacimenti in Ucraina di argille illitico-caolinitiche utilizzate dai produttori di grès porcellanato e monocottura chiara. Negli Stati Uniti invece, nello stato dell’Illinois, è da lungo tempo utilizzato un giacimento di illite per la produzione di argilla espansa.
Per quanto concerne la tecnica di coltivazione, nel caso di aree collinari, si utilizzano rippaggio e ruspaggio successivi di “gradoni” aventi solitamente larghezza superiori a 20 m e lunghezza di varie decine di metri oppure si opera su piani inclinati impiegando per lo più bulldozer di grande potenza.
Nelle zone semi pianeggianti, nel caso in cui l’area di scavo sia di notevoli dimensioni, all’utilizzo del bulldozer si preferisce quello dello scraper. Questo veicolo compie lunghe “raschiate” sul terreno raccogliendo e caricando la parte centrale di materiale proveniente da una fascia piuttosto vasta. Con questo sistema si ottiene un’ottima omogeneizzazione della materia prima, tra l’altro favorita dallo stesso sistema di caricamento del veicolo.
Anche il metodo del rippaggio e ruspaggio è apprezzato in quanto consente di operare su vaste superfici realizzando grandi movimenti di materiale e di impiegare le medesime macchine operatrici per lo spostamento ed il trattamento dell’argilla estratta (sminuzzamento, omogenizzazione ed essiccazione).
Quest’ultima serie di interventi ha luogo sul piazzale di cava dove il materiale viene distribuito con uno spessore di 15 ÷ 20 cm, lasciandolo tutto il giorno esposto al sole ed avendo cura di smuoverlo per esporre ai raggi anche la parte sottostante che verrebbe a trovarsi coperta.
Analisi mineralogiche di materie prime illitiche; tipici comportamenti di riconoscimento
L’identificazione della specie tramite analisi diffrattometrica ai raggi X comporta essenzialmente l’osservazione delle diffrazioni dei piani basali come da tabella seguente:
Illite
Posizione angolare dei picchi °(2θ) 8,8
Le Fig. 3.15 e Fig. 3.16 riportano il diagramma dell’analisi termo-differenziale (DTA) di una illite e di una mica muscovite, rispettivamente. L’illite mostra un primo picco endotermico attorno a 140 °C (allontanamento dell’acqua zeolitica) a cui ne fa seguito un secondo verso 600 °C (eliminazione degli ossidrili), con picchi di minor intensità rispetto ad altri minerali argillosi. Verso 900 °C si ha la formazione di silico-alluminati alcalini e, in presenza di calcio, di alluminati di calcio, silicati di calcio (wollastonite) e ferriti di calcio; tali neoformazioni vengono evidenziate da un debole picco esotermico.
Coerentemente l’analisi termo-gravimetrica (TG) evidenzia moderate perdite di peso in corrispondenza dell’allontanamento dell’acqua zeolitica e degli ossidrili.
Fig. 3.15 Tipica curva DTA per una illite
Fig. 3.16 Tipica curva DTA per una muscovite
La dilatometria di una miscela illite/quarzo riportata in Fig. 3.17, presenta una espansione fino a 600 °C; da 600 °C a oltre 800 °C si osserva il tipico “pianerottolo” (condizione di assenza di variazioni dimensionali) a cui fa seguito una contrazione quasi verticale dovuta alla deformazione plastica del provino in fusione.
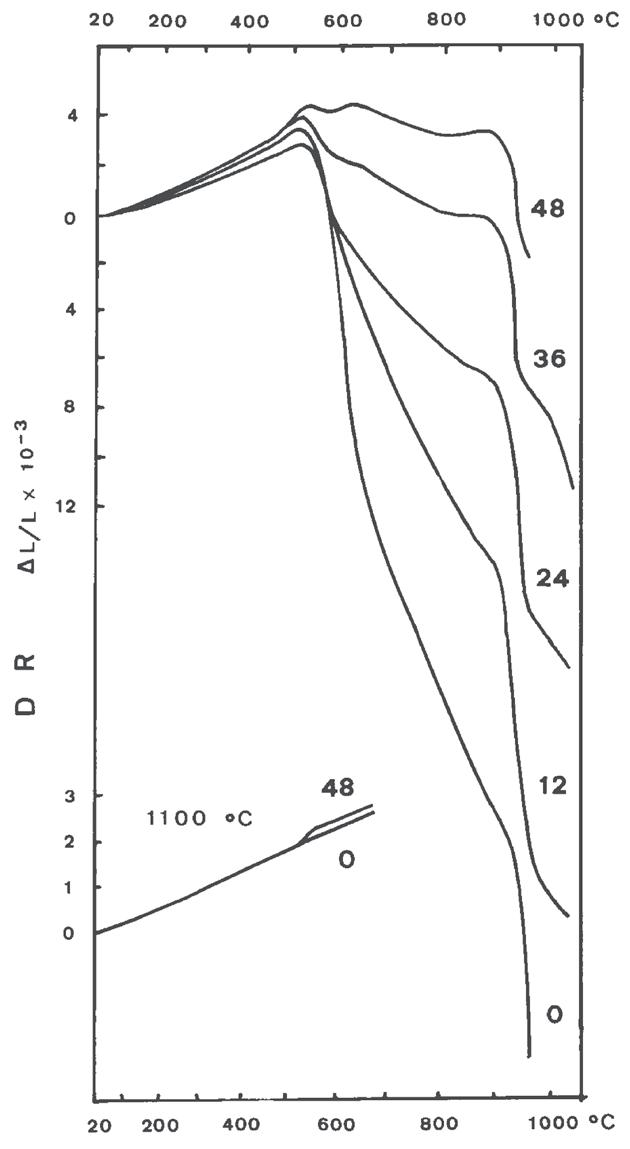
Fig. 3.17 Dilatazione e ritiro di miscele illite-quarzo (0 ÷ 48% di quarzo) [3]
Di seguito si forniscono le analisi mineralogiche semiquantitative di tre tipi di argille dell’Appennino emiliano:
Illite Clorite Montmor. Caolinite Quarzo Feldspati Carbonati Mica
A. argille grigio-azzurre del Pliocene-Pleistocene
B. argille della serie Antognola
C. red-beds
Come si può osservare il quadro mineralogico è piuttosto costante ed a grandi linee si può affermare che la sola grossa differenza riguarda il contenuto in carbonati che passa da un massimo del 20 ÷ 23% delle argille grigio-azzurre Plioceniche e Pleistoceniche al 13 ÷ 16% di quelle della serie Antognola allo 0 ÷ 6% delle red-beds. Per quanto riguarda le red-beds, si segnalano però anche eccezioni in cui si raggiungono percentuali di carbonati molto rilevanti (oltre il 16%).
Un’ultima osservazione riguarda la plasticità naturale (cioè della materia prima tal quale e non macinata); le argille della serie Antognola e le red-beds hanno subito processi di diagenesi che hanno inibito, in parte o completamente, questa proprietà tipica dei materiali argillosi.
Caratterizzazione tecnologica di materiali illitici pressati
Piastrelle formate partendo da materie prime prevalentemente illitiche presentano i seguenti comportamenti:
- normale espansione di post-pressatura;
- buona resistenza meccanica alla flessione dei crudi (verdi ed essiccati);
- per cottura a 1100 °C la variazione dimensionale (ritiro) è nell’intorno del 5 ÷ 8% con valori di assorbimento d’acqua da 0 a 5%; tuttavia, nel caso frequente della compresenza di un rilevante tenore di carbonati (15 ÷ 20%) la variazione dimensionale presenta una leggera espansione o un leggero ritiro (da +1 a −1%) mentre la porosità si attesta a 15 ÷ 22%;
- anche la resistenza meccanica dei cotti risente marcatamente della presenza o dell’assenza dei carbonati: nel primo caso si registrano valori decisamente più bassi (da 10 a 20 N/mm2), nel secondo si superano facilmente 20 N/mm2;
- il coefficiente di dilatazione termica lineare α (a 1100°C) vale generalmente 60 ÷ 75×10−7°C−1
Nel complesso le piastrelle ottenute da queste materie prime risultano piuttosto greificabili. Quelle prive di carbonati raggiungono la completa vetrificazione a 1050 ÷ 1080 °C, con palier molto ristretti; quelle carbonatiche risultano leggermente più refrattarie e passano allo stato vetroso oltre 1100 °C con crollo improvviso e completa assenza di palier (caratteristica dei carbonati).
Nel caso delle argille ricche in carbonati, i colori dei provini cotti variano dal rosa salmone, al giallastro, al verde-giallastro (a vetrificazione); invece le argille illitiche non carbonatiche presentano tonalità del rosso molto carico alle basse temperature, passando ad un colore bruno testa di moro quando avvengono le reazioni di vetrificazione.
Utilizzi in settori differenti
Le argille illitiche cuocenti bianco possono essere impiegate in alternativa alle argille caolinitiche, ad esempio come cariche inorganiche in polvere.
Alcune tipologie “cuocenti rosso” trovano impiego nella produzione di materiali espansi leggeri utilizzati dall’edilizia; in questo caso devono contenere sostanze che in cottura facciano espandere il materiale (preventivamente pellettizzato) o, in alternativa, devono essere additivate con materiali espandenti a basso prezzo.
Origine del termine
Il nome montmorillonite deriva dalla località Montmorillon (Francia) dove questo minerale argilloso fu localizzato e successivamente identificato. La montmorillonite appartiene al gruppo dei fillosilicati identificati come smectiti.
Con il termine bentonite s’intende invece una roccia a prevalente natura montmorillonitica caratterizzata da un marcato rigonfiamento quando posta a contatto con acqua. L’origine della parola è connessa con la località di Fort Benton (Wyoming, Stati Uniti) dove si trova un importante giacimento di questa materia prima.
Struttura mineralogica
La struttura base è formata da due piani tetraedrici che ne racchiudono uno ottaedrico (Fig. 3.18). Alcune specie della famiglia della montmorillonite derivano dalla sostituzione del silicio con alluminio nei tetraedri e da quella di magnesio e ferro bivalente al posto dell’alluminio negli ottaedri.
Le forze che legano i cristalli dei minerali di questo gruppo sono particolarmente deboli. Infatti, non vi sono legami idrogeno (come nella caolinite) in quanto non vi è possibilità di contatto tra strati ottaedrici e tetraedrici appartenenti a strutture base differenti, essendo i primi “prigionieri” all’interno dei secondi. Il solo legame esistente è costituito dalle deboli forze di Van der Waals e pertanto risulta estremamente facile l’inserimento di molecole d’acqua negli interstrati con il risultato di far “espandere” la struttura cristallina fino a quasi sei volte il volume originale. La maggior reattività della famiglia della montmorillonite (rispetto, ad esempio, a quella della caolinite) deriva dalla disponibilità delle facce interne che delimitano ciascuna “struttura base” in quanto tali elementi sono facilmente separabili tra loro. Risulta poi molto accentuata la cosiddetta “sostituzione isomorfa”, cioè la capacità di rimpiazzare un catione con un altro di carica differente. Se, per esempio, uno ione trivalente (alluminio) sostituisce il silicio dei tetraedri (tetravalente), l’equilibrio elettrico viene raggiunto solo con l’assorbimento di un catione esterno mono o bivalente. In questo secondo caso il numero di cationi bivalenti sarà pari alla metà delle cariche negative liberatesi con la sostituzione isomorfa.
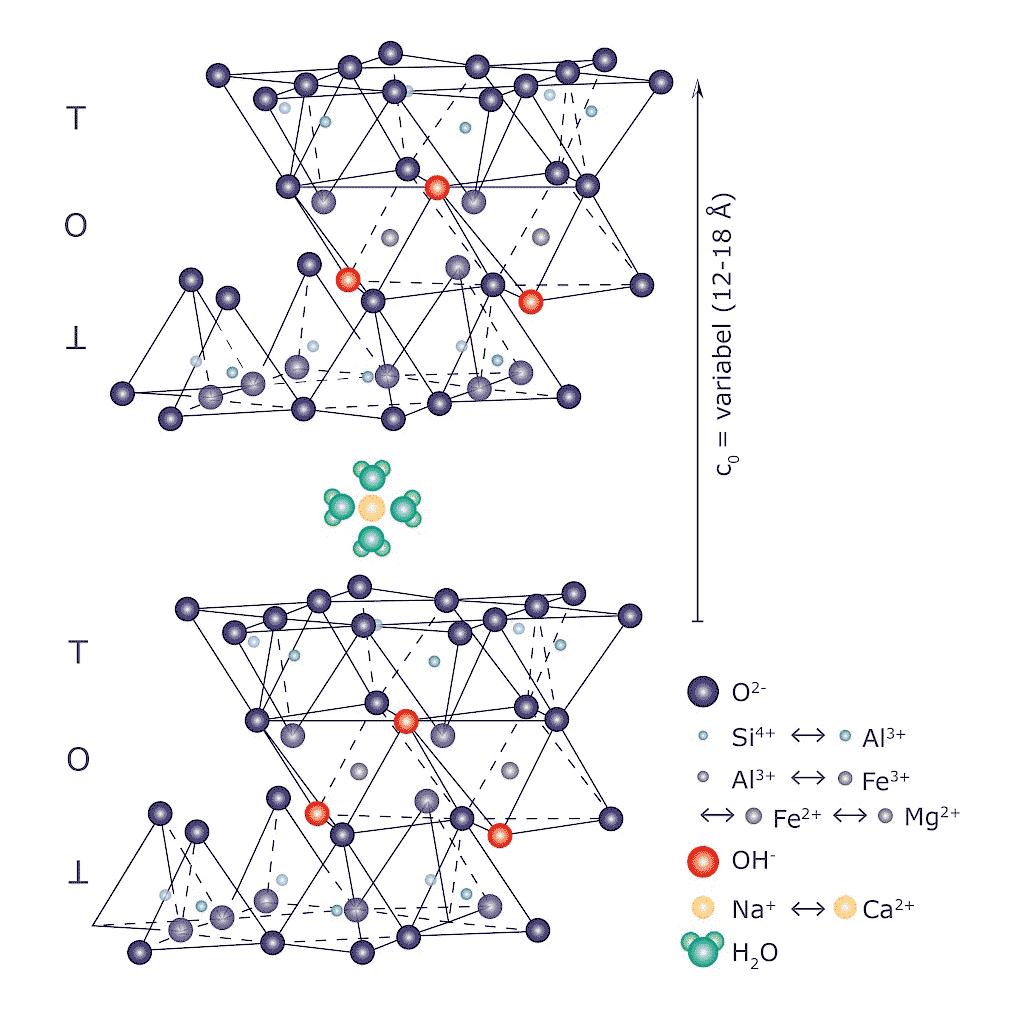
Fig. 3.18 Struttura base della montmorillonite [1]
Una caratteristica della montmorillonite che rende conto di una parte dei suoi comportamenti peculiari sono le dimensioni delle particelle estremamente ridotte. Vi è poi una forte diversità tra le montmorilloniti sodiche e calciche, in quanto le prime fanno registrare valori di plasticità e di capacità di scambio ionico molto più elevati.
È poi piuttosto frequente incontrare strutture miste dovute alla associazione di montmorillonite ed illite (interstrati). Questa struttura può realizzarsi in quanto i “foglietti” dei due minerali sono simili.
Composizione chimica
X2Y4O10(OH)2
X = catione alluminio trivalente, parzialmente sostituto da magnesio bivalente (nel foglietto ottaedrico) con conseguente assorbimento di cationi mono o bivalenti per bilanciare le cariche elettriche non equilibrate.
Y = catione silicio tetravalente, parzialmente sostituto da alluminio trivalente (nel foglietto tetraedrico) con conseguente assorbimento di cationi mono o bivalenti per bilanciare le cariche elettriche non equilibrate.
Composizione teorica
66,7% SiO2 + 28,3% Al2O3 + 5,0% H2O
Genesi e caratteristica dei depositi
Una parte rilevante delle argille montmorilloniche si è formata per alterazione di rocce vulcaniche (rocce piroclastiche e brecce vulcaniche) come: rioliti, trachiti, daciti, andesiti, basalti e lipariti. La bentonite del Wyoming deriva da alterazione in situ di ceneri vulcaniche accumulatesi in una formazione cretacea. Diffusa è pure la genesi sedimentaria. I giacimenti sedimentari di montmorilloniti si presentano con strati spessi anche vari metri; quelli di origine idrotermale possono avere forme molto irregolari che ne complicano l’attività estrattiva.
Analisi mineralogiche di materie prime montmorillonitiche; tipici comportamenti di riconoscimento
L’identificazione della specie tramite analisi diffrattometrica ai raggi X comporta essenzialmente l’osservazione delle diffrazioni dei piani basali (Fig. 3.19). L’analisi risulta piuttosto complessa in quanto presenta un debole riflesso a ca. 6,9 °(2θ) (corrispondente ad una distanza reticolare di 12,5 Å) che, dopo glicolazione del campione, si sposta verso un angolo più basso (4,8 ÷ 5 °(2θ)) fino a collassare a circa 8,8 °(2θ) dopo riscaldamento a 550 °C. Spesso però la smectite è presente come interlaminato associato all’illite. In questo caso la banda di riflessione che ne deriva è molto allargata attorno a 7,2 °(2θ), espande ancora a 4,8 ÷ 5,2 °(2θ) e collassa a circa 8,8 °(2θ) dopo riscaldamento a 550 °C.
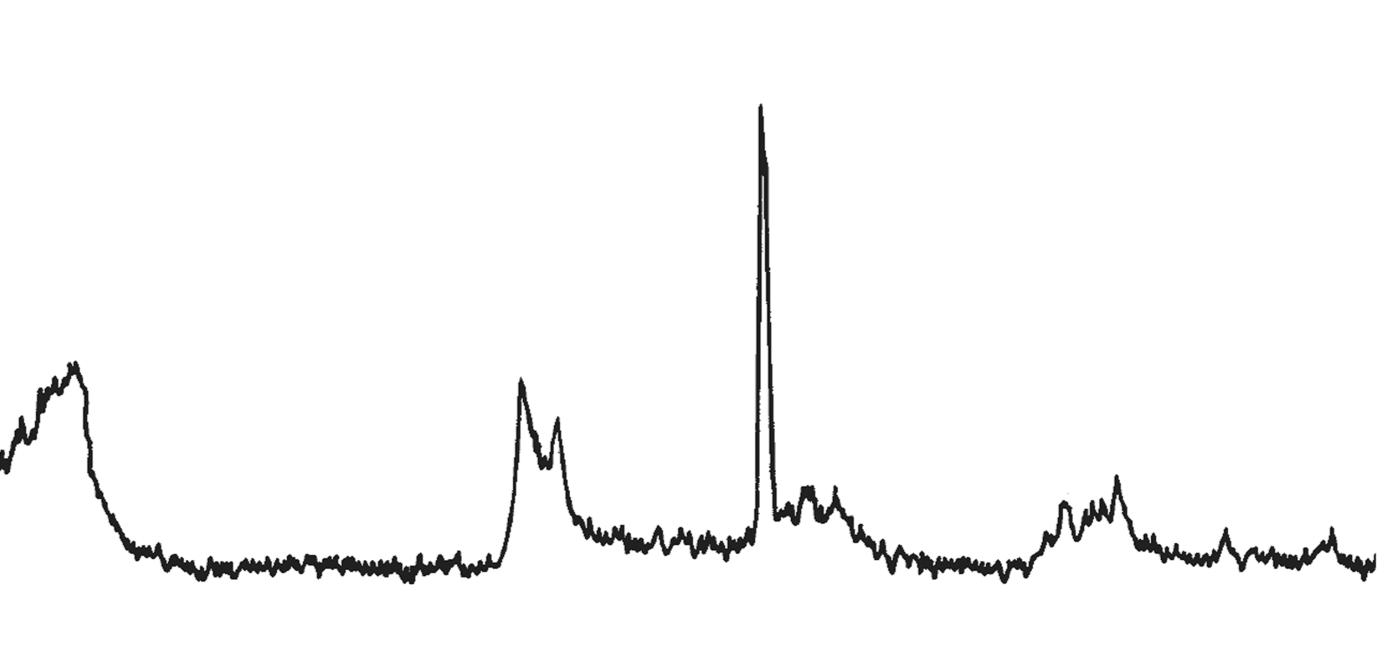
Fig. 3.19 Analisi diffrattometrica di una montmorillonite (M) con presenza di quarzo (Q)
Il diagramma dell’analisi termo-differenziale (DTA) della montmorillonite presenta due picchi endotermici rispettivamente a 180 °C e tra 450 °C e 650 °C dovuti, il primo all’allontanamento dell’acqua di interstrato ed il secondo all’allontanamento degli ossidrili OH (Fig. 3.20 e Fig. 3.21). Il picco relativo alla disidratazione a bassa temperatura (100 ÷ 250 °C) è il più intenso e caratteristico, e la sua forma varia molto in funzione del catione di interstrato. Le altre smectiti diottaedriche (beidellite e nontronite) hanno curve simili a quelle della montmorillonite.
Fig. 3.19
L’analisi termo-gravimetrica (TG) mostra, in corrispondenza dei picchi endotermici delle DTA, due perdite di peso a 180 °C e nell’intervallo 450 ÷ 650 °C.
La dilatometria, dopo un breve tratto iniziale in leggera espansione, presenta una prima contrazione verso 180 °C a cui fa seguito una nuova espansione (che si accentua in prossimità dei 573 °C qualora la materia prima contenga anche quarzo libero); verso i 900 °C ha inizio la contrazione legata allo sviluppo delle reazioni di sinterizzazione ed al rammollimento del provino.
Fig. 3.20 Curve DTA per la montmorillonite: a) Ca-montmorillonite; b) Na-montmorillonite
Montmorillonite
3.20
Hectorite
Saponite
Nontronite
Nontronite
Vermiculite
Vermiculite
Vermiculite
Fig. 3.21 Curve DTA di alcuni minerali smectitici di varia provenienza
Fig. 3.22
Ubicazione dei principali giacimenti e tecniche di coltivazione
I giacimenti più importanti coltivati in Europa sono localizzati in Gran Bretagna (Surrey, Somerset e Bedfordshire), Francia (Limousin), Germania (Baviera), Ungheria (sulle colline del Tokaj), Italia (Sardegna ed isola di Ponza), Repubblica Ceca, Caucaso, Kazakistan, Grecia (Milos e Mikonos) e Penisola balcanica. Negli Stati Uniti sono assai noti i depositi cretacei del Wyoming (bentonite); altri sono ubicati in Arizona, Oklahoma, Texas e Nevada. Ancora in Nord-America si possono ricordare i giacimenti del Canada (Alberta e Manitoba) e del Messico (regioni di Puebla e Monterrey). Altri importanti giacimenti bentonitici sono segnalati in Sud-America (Argentina), Asia (India e Giappone) e Sud-Africa.
Le cave di giacimenti argillosi prevalentemente montmorillonitici operano “a cielo aperto”. Si tratta quindi di un’attività di movimento terra condotta con metodi tradizionali.
Tuttavia, la coltivazione di questi depositi non può prescindere dall’altissima plasticità della materia prima e della grande facilità d’imbibizione. Non è infrequente che si debba operare con materiale contenente il 15 ÷ 25% d’acqua. In queste condizioni l’enorme plasticità rende praticamente impossibile l’uso di bulldozer con apripista, dato che questi mezzi tendono a formare grossi “materassi” arrotolati che si accrescono enormemente sino a bloccare il veicolo. Di fronte a questi problemi talvolta si preferisce operare con escavatori idraulici o con draghe a ruota idraulica.
Tale equipaggiamento inoltre consente un’estrazione più selettiva e permette di coltivare con accuratezza ampi banchi con spessori non particolarmente rilevanti.
Caratterizzazione tecnologica di materiali montmorillonitici pressati
Generalmente i giacimenti montmorillonitici sono caratterizzati da un’importante presenza di particelle colloidali e da un’elevata plasticità. Piastrelle formate da materie prime costituite in percentuale rilevante da minerali della famiglia della montmorillonite hanno i seguenti comportamenti:
- espansione di post-pressatura generalmente bassa (< 0,5%);
- elevatissima resistenza meccanica dei crudi (verdi ed essiccati);
- fortissimo ritiro di essiccazione (> 1%);
- sui cotti ottenuti già alla temperatura di 1020 °C, la variazione dimensionale oscilla dal 5 al 10% (ritiro) mentre la corrispondente porosità può variare da valori bassissimi a più del 5%, dipendendo dal grado di espansione (bloating) che avviene col procedere della sinterizzazione. Questi valori variano notevolmente anche in funzione dell’eventuale contenuto di ioni alcalini negli spazi inter-reticolari;
- sempre a 1020 °C i valori della resistenza meccanica possono variare in modo molto ampio (da 15 a 25 N/mm2), in funzione del grado di espansione raggiunto.
Utilizzi ceramici
Mentre l’impiego di materie prime argillose contenenti montmorillonite in strati misti con illite è consolidato nella produzione di piastrelle, laterizi e vasellame, i materiali quasi esclusivamente montmorillonitici presentano forti limiti a causa del loro tipico comportamento. In particolare la montmorillonite interferisce negativamente ed in modo molto marcato sulle proprietà reologiche delle barbottine. La bentonite, quando idratata, rigonfia enormemente fino a formare un gel colloidale.
Pertanto l’utilizzo ceramico delle montmorilloniti più pure è solo come additivo plasticizzante; in questo caso le percentuali di impiego superano molto raramente il 5%.
Utilizzi in settori diversi
L’impiego prevalente della bentonite è come costituente principale dei fanghi che vengono utilizzati per le trivellazioni petrolifere. Come noto il compito di tali fanghi è quello di portare alla superficie il materiale litoide che viene rimosso nel corso della trivellazione. L’utilizzo di un fluido con viscosità più elevata di quella dell’acqua favorisce l’operazione a condizione che risulti pompabile. Per tale impiego si preferisce la montmorillonite sodica; l’argilla deve essere particolarmente pura e risultare priva di particelle abrasive (quarzo). Un altro impiego delle materie prime montmorillonitiche è quello della decolorazione degli oli alimentari e lubrificanti; in questo caso il materiale impiegato è noto con il nome di Fuller’s earth ed è prevalentemente costituito da montmorillonite con presenza di attapulgite e, in minor misura, di halloysite.
Un altro interessante settore d’impiego della montmorillonite è quello della chiarificazione di bevande alcoliche (vino, birra, liquori, ecc.). L’operazione consiste nella coagulazione e rimozione delle impurità colloidali presenti in sospensione. La montmorillonite, preferibilmente sodica, è aggiunta direttamente alle bevande e, dopo agitazione, separata con l’uso di filtropresse.
Origine del termine
Deriva dal greco clorós (“verde”) ed è stato introdotto da Werner per definire questo minerale in conseguenza del colore prevalente verdastro, dovuto ad una elevata presenza di ferro ridotto.
Struttura mineralogica
Si tratta di minerali lamellari aventi un aspetto che ricorda quello della mica. La struttura classica della clorite è formata dalla combinazione alternata di un foglietto di tipo micaceo (dove un piano ottaedrico con Al3+ o Mg2+ si trova tra due piani esagonali di tetraedri di silice) con un ulteriore piano ottaedrico di tipo brucitico – vedi Fig. 3.22.
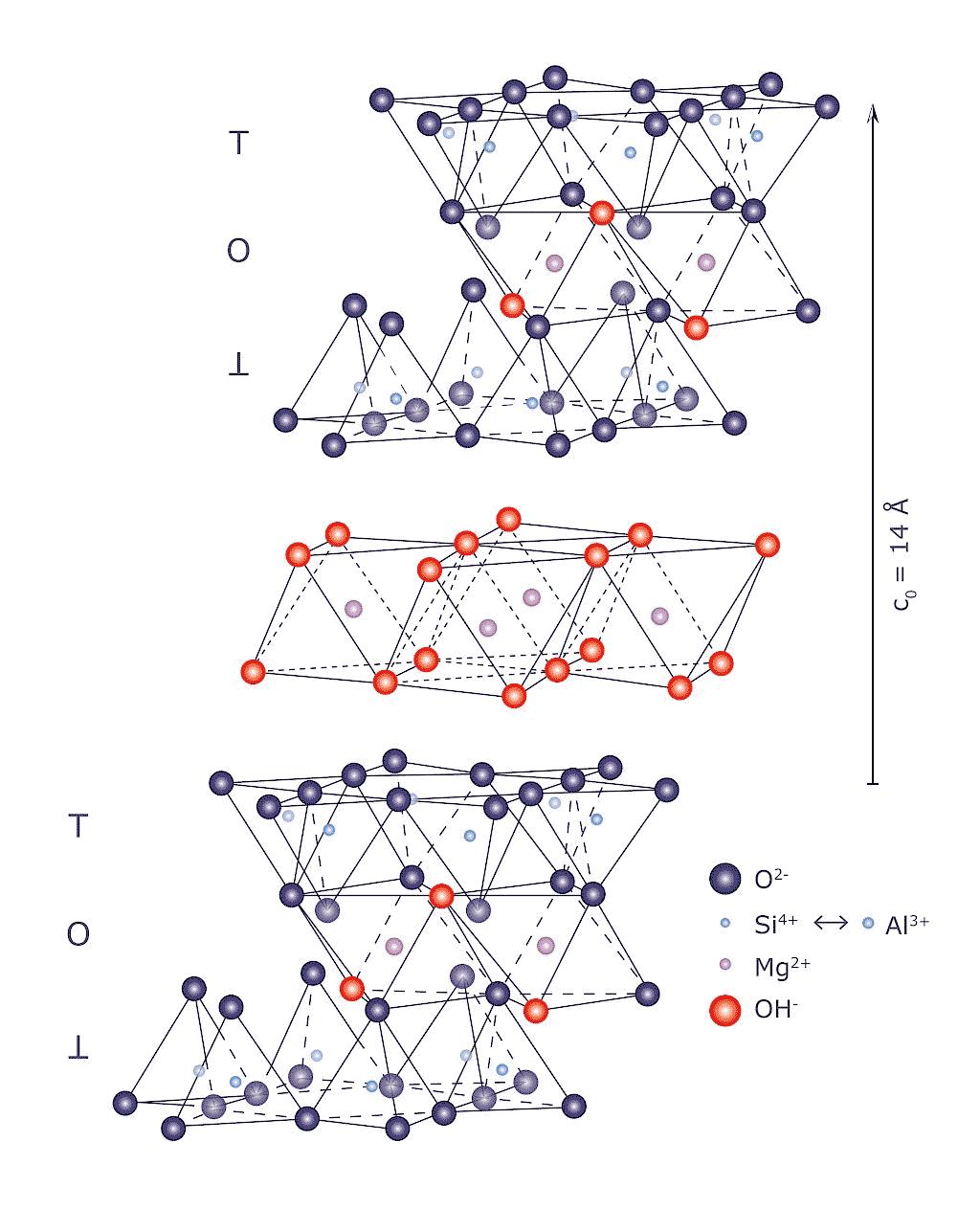
Fig. 3.22 Struttura elementare della clorite [1]
La maggior parte dei minerali cloritici ha struttura triottaedrica, tuttavia sono state identificate anche forme diottaedriche. Vi sono inoltre casi di cloriti degradate, caratterizzate da una espansione del reticolo conseguente all’assorbimento di acqua zeolitica durante fenomeni di weathering che hanno agito sul foglietto brucitico.
Si segnala inoltre l’esistenza di interlaminati clorite-vermiculite ed illite-clorite.
Composizione chimica
La struttura mineralogica non è originale, ma una combinazione dei due foglietti micaceo e brucitico.
La composizione dei foglietti micacei è:
X8Y6O10(OH)4
dove: X è costituito da Si4+ o Al3+
Y è costituito da Mg2+ o Fe2+
Quella dei foglietti brucitici è invece:
X6(OH)12
dove: X è costituito da Mg2+, Al3+ o Fe2+
Quando nella struttura micacea un Si4+ sostituisce un Al3+, la differenza di carica elettrica che si viene a creare è equilibrata da un eccesso di carica negativa del foglietto brucitico che si genera in conseguenza della sostituzione di Al3+ con Mg2+
La formula teorica della vera clorite (o leptoclorite) è la seguente: Mg3(Mg3−x Alx)(Si4−x Alx)O10(OH)8
L’oscillazione del valore di x (che può variare da 1 a 2) e la natura dei sostituenti portano alla formazione di differenti varietà, quali ad esempio: bavalite (clorite ferrosa), clinocore, pennina e proclorite.
Genesi e caratteristiche dei giacimenti
L’origine dei depositi contenenti cloriti è legata a sedimentazioni soprattutto in ambiente marino.
I minerali della famiglia delle cloriti possono derivare direttamente dalla roccia madre (minerali detritici) o formarsi da silicati ricchi di Fe e Mg, quali ad esempio biotite ed anfiboli in presenza di un dilavamento poco spinto.
Non si hanno comunque riscontri di giacimenti costituiti prevalentemente da cloriti.
Ubicazione dei principali giacimenti e tecniche di coltivazione
Probabilmente alcuni dei giacimenti più ricchi di questi minerali sono quelli dell’Appennino emiliano (Italia). Infatti, la frazione argillosa di tali depositi è formata soprattutto da illite e, in sott’ordine, dalle cloriti. Si tratta del così detto Complesso Emiliano, costituito da:
- marne del Monte Piano (argille rosse e verdi note con il nome di red-beds) accumulate su formazioni precedenti e successivamente scomposte per fenomeni orogenetici locali; - serie di Antognola (marne ed argille grigio-azzurre).
Per quanto concerne la tecnica di coltivazione, nel caso di aree collinari si basa sul rippaggio e ruspaggio successivi su “gradoni” ad esaurimento (aventi solitamente larghezza superiore a 20 m e lunghezza di varie decine di metri) o su piani inclinati impiegando per lo più bulldozer di grande potenza.
Nelle zone semi pianeggianti, nel caso in cui l’area operativa sia di notevoli dimensioni, all’utilizzo dei bulldozer si preferisce quello dello scraper. Questo veicolo compie lunghe “raschiate” sul terreno raccogliendo e caricando la parte centrale di materiale proveniente da una fascia piuttosto vasta. Con questo sistema si ottiene un’ottima omogeneizzazione della materia prima, tra l’altro favorita dallo stesso sistema di caricamento del veicolo.
Anche i metodi di “rippaggio” e “ruspaggio” presentano caratteristiche apprezzabili; essi infatti consentono di:
- operare su vaste superfici realizzando grandi movimenti di materiale con riduzione dei costi unitari;
- condurre i lavori (ove necessario) con notevole elasticità isolando eventuali aree non idonee;
- impiegare le medesime macchine operatrici per lo spostamento ed il trattamento della argilla estratta (sminuzzamento, omogeneizzazione ed essiccazione).
Quest’ultima serie di interventi ha luogo sul piazzale di cava ove il materiale viene distribuito con uno spessore di 15 ÷ 20 cm lasciandolo tutto il giorno esposto al sole ed avendo cura di smuoverlo onde esporre ai raggi anche la parte sottostante che verrebbe a trovarsi coperta.
Composizione mineralogica di depositi contenenti cloriti
L’identificazione delle specie mineralogiche tramite analisi diffrattometrica ai raggi X comporta essenzialmente l’osservazione delle diffrazioni dei piani basali (Fig. 3.23). Le riflessioni angolari caratteristiche (per radiazioni CuKα) sono rilevabili come in tabella; si notano inoltre altri picchi differenziati rispetto al piano basale 001, contrassegnati con i numeri 1 ÷ 5.
Posizione angolare dei picchi °(2θ) 6,2
Distanze reticolari Å
La diffrazione di 12,5° corrisponde anche a quella del piano 001 della caolinite, per cui non risulta facile una corretta interpretazione. Per evitare questo inconveniente si ripete l’analisi trattando il campione a 550 ÷ 600 °C; in questo modo si rinforza notevolmente il picco a 6,2° mentre scompaiono gli altri riflessi basali.
L’analisi termo-differenziale (DTA), Fig. 3.24, mostra forti variazioni per i vari tipi di clorite; generalmente si nota un primo picco endotermico verso 400° quando avviene l’allontanamento degli ossidrili dal foglietto brucitico; segue poi tra 600 e 800 °C un secondo picco endotermico che corrisponde all’allontanamento degli ossidrili del foglietto micaceo.
Corrispondentemente a questi intervalli di temperatura, l’analisi termo-gravimetrica (TG) presenta una marcata perdita di peso. Qualora vi sia una forte alterazione della struttura reticolare, anche la dilatometria del crudo può evidenziare un flesso in corrispondenza dell’allontanamento dell’acqua zeolitica.
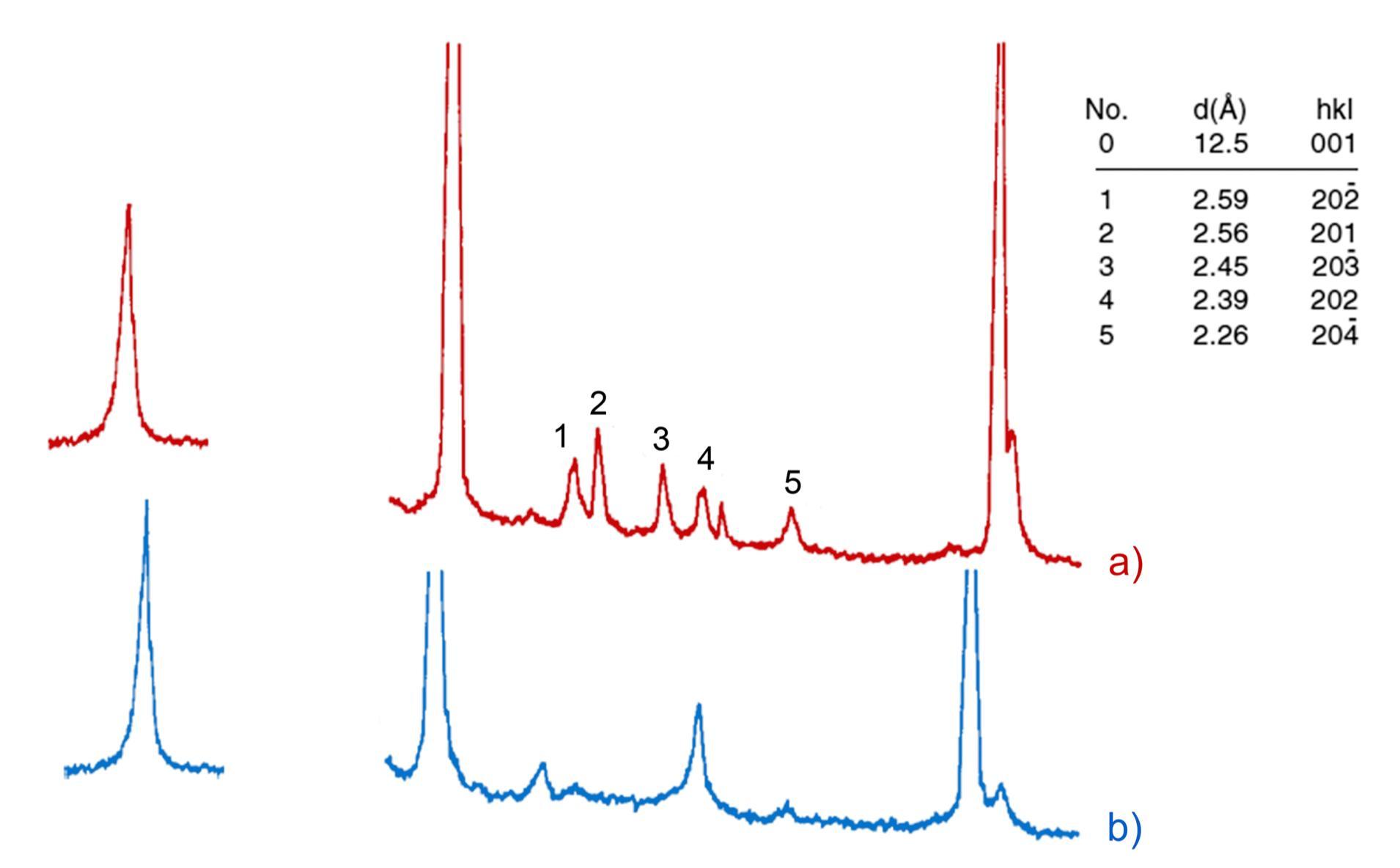
Fig. 3.23 Diffrattogrammi ai raggi X di due diversi tipologie di clorite a) e b)
Fig. 3.24
Fig. 3.24 Esempi di DTA di due tipologie di cloriti, con evidenza del picco esotermico anticipato a causa di un maggior contenuto di ferro (curva b)
Caratterizzazione tecnologica di provini pressati preparati con materie prime cloritiche Per la valutazione del comportamento di argille costituite da una frazione importante di minerali cloritici è opportuno fare riferimento alle materie prime dell’Appennino emiliano, in quanto tali minerali sono costituiti da clorite e illite. I comportamenti che si rilevano sono pertanto analoghi a quelli evidenziati dalle argille illitiche:
Fig. 3.25
- normale espansione di post-pressatura;
- buona resistenza meccanica alla flessione dei crudi (verdi ed essiccati);
- per cottura a 1100 °C la variazione dimensionale (ritiro) è nell’intorno del 5 ÷ 8% con valori di assorbimento d’acqua da 0 a 5%; tuttavia, nel caso frequente della compresenza di un rilevante tenore di carbonati (15 ÷ 20%) la variazione dimensionale presenta una leggera espansione o un leggero ritiro (da +1 a −1%) mentre la porosità si attesta su valori compresi rispettivamente tra il 22 ed il 15%;
- anche la resistenza meccanica dei cotti risente marcatamente della presenza o dell’assenza dei carbonati: nel primo caso si registrano valori decisamente più bassi (da 10 a 20 N/mm2), nel secondo si superano facilmente 20 N/mm2; - il coefficiente di dilatazione termica lineare α (a 1100 °C) si attesta generalmente su valori compresi tra 60 ÷ 75×10−7 °C−1
Nel complesso le piastrelle ottenute da queste materie prime risultano piuttosto fusibili. Quelle prive di carbonati raggiungono la completa vetrificazione a 1050 ÷ 1080 °C con palier di sinterizzazione molto ristretti.
Quelle carbonatiche risultano leggermente più refrattarie e passano allo stato vetroso oltre 1100 °C con una caduta verticale della curva dilatometrica (caratteristica dei carbonati). Nel caso delle argille ricche di carbonati, i colori dei cotti variano dal rosa salmone al giallastro ed al verde giallastro (a vetrificazione); le non carbonatiche presentano invece intense tonalità di rosso (a temperature più basse), passando al bruno testa di moro quando avvengono le reazioni di vetrificazione.
Utilizzi ceramici
Con riferimento alle materie prime argillose italiane a natura prevalentemente illitico-cloritica, i loro impieghi nelle produzioni di piastrelle ceramiche sono stati i seguenti:
- argille grigio-azzurre del Pliocene-Pleistocene utilizzate per supporti rossi ad alta porosità (maiolica) per rivestimento smaltato;
- argille grigie della serie di Antognola utilizzate per supporti colorati a media porosità (cottoforte), da pavimento e rivestimento smaltati, ed anche per supporti non smaltati tipo “cotto toscano”;
- red-beds utilizzate per supporti rossi antigelivi e comunque a bassa porosità (monocottura greificata), smaltati o non smaltati, impiegati per pavimenti di costruzioni abitative o industriali anche da esterni.
Le argille grigio-azzurre del Pliocene, caratterizzate da una plasticità naturale più elevata, sono tuttora utilizzate nella produzione di vasellame rustico (maiolica), di oggettistica e nella fabbricazione di laterizi trafilati di grandi dimensioni (pignatte e tavelloni).
3.2.1
Origine del termine
Dal latino silex che significa “pietra dura”.
Formula chimica
Struttura mineralogica
La struttura base della silice è formata da un tetraedro avente ai vertici gli anioni ossigeno ed, al centro, l’atomo di silicio; nell’insieme quindi un singolo tetraedro possiede quattro cariche negative (Fig. 3.25).
La silice anidra si presenta sotto tre forme cristalline (quarzo, tridimite e cristobalite) ed una amorfa (vetrosa), come illustrato in Fig. 3.26 e Fig. 3.27. Ciascuna specie fondamentale presenta caratteristiche stabili entro determinati intervalli di temperatura.
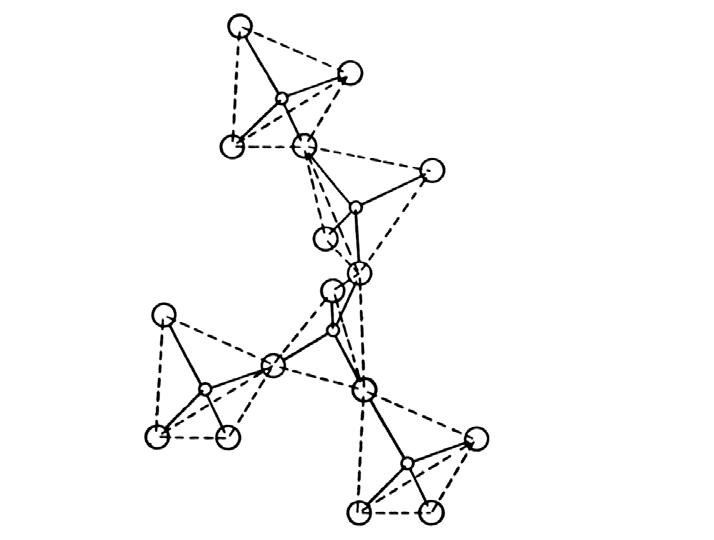
Fig. 3.25 Disposizione strutturale dei tetraedri di silice nel quarzo
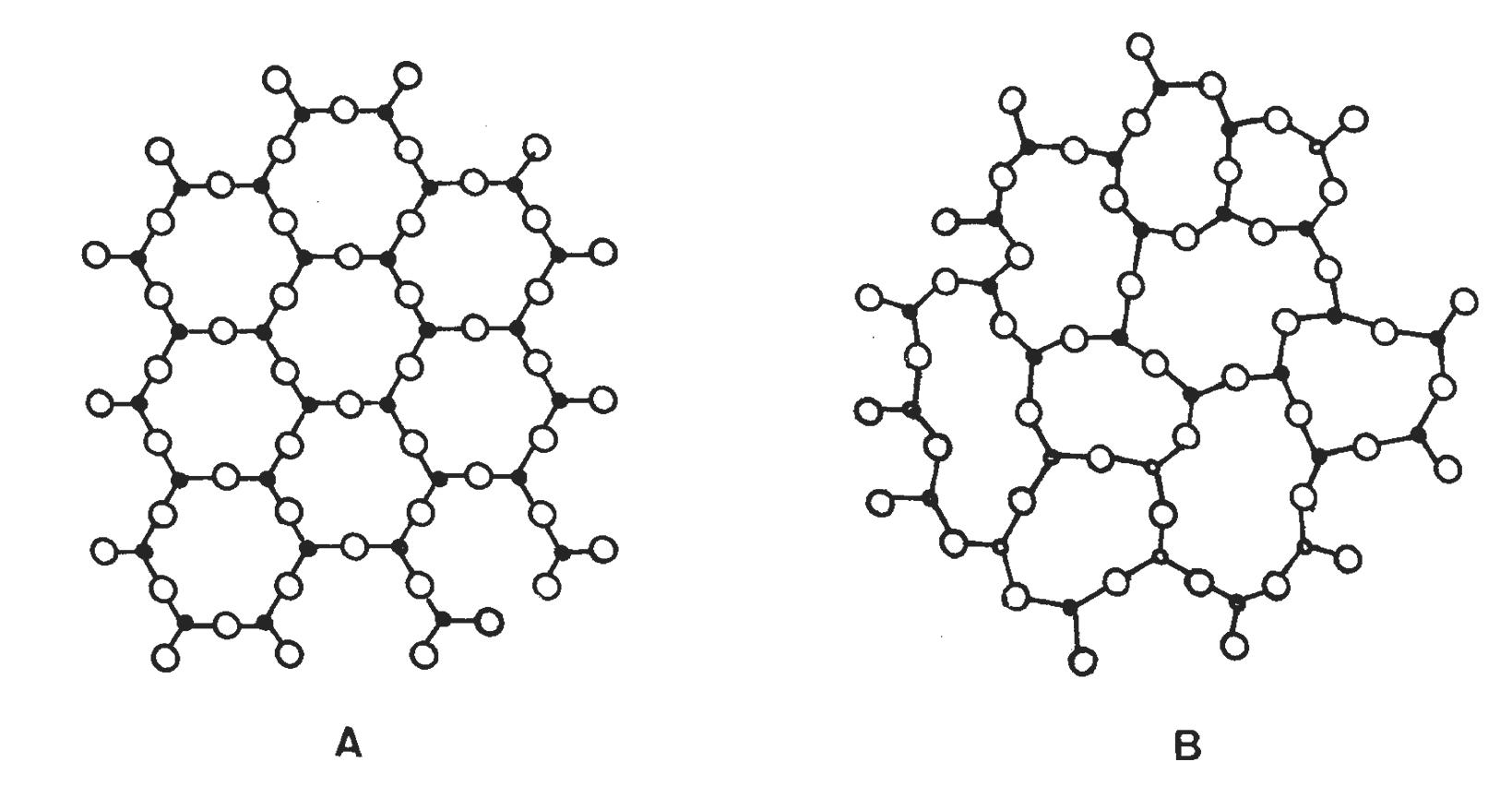
Fig. 3.26 Struttura cristallina (A) e struttura vetrosa (B) [5] [6]
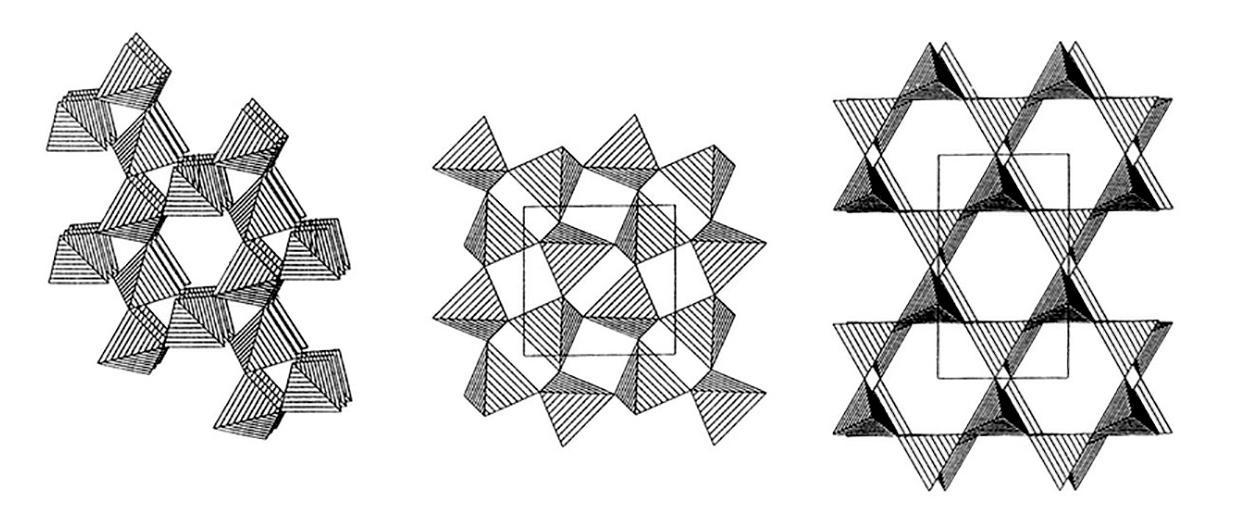
Tridimite Cristobalite
Fig. 3.27 Le tre forme cristalline della silice
Le temperature alle quali avvengono le trasformazioni più importanti (Fig. 3.28) sono le seguenti:
Fig. 3.28 IT NEW
Quarzo → Tridimite 870 °C
Quarzo α → Quarzo β 573 °C
Tridimite → Cristobalite 1470 °C
Tridimite α → Tridimite β 117 ÷ 163 °C
Cristobalite α → Cristobalite β 220 ÷ 270 °C
Fig. 3.28 Diagramma di stato della silice, incluse le fasi metastabili [2]
Genesi e caratteristiche dei depositi
I depositi di silice si distinguono in giacimenti primari (quarziti), secondari (sabbie e arenarie) e di origine vulcanica (perliti).
I depositi primari si presentano con forme filoniane in pegmatiti o scisti oppure in zone di contatto con corpi magmatici (aureole). Questi filoni possono avere dimensioni molto variabili (da pochi cm a varie decine di metri). Naturalmente solo quelli più importanti rivestono interesse e possono rendere economica la coltivazione. I depositi più puri mostrano colorazione biancastra e tipico aspetto translucido. La presenza di diverse impurezze porta ad ammassi di colore molto vario (da rosato a grigio, da giallastro a bruno). Con il termine “sabbie” ci si riferisce invece a sedimenti clastici, sciolti, con granuli di dimensioni comprese tra 2 e 0,25 mm. Si tratta quindi di depositi sedimentari relativamente grossolani, non cementati, omologhi delle arenarie, rocce che hanno identica struttura granulometrica ma si distinguono per una forte cementazione che fa loro assumere aspetto e consistenza litoide. I depositi sabbiosi possono essere di origine alluvionale, lacustre, marina ed anche di accumulo eolitico, con riferimento all’ambiente di deposizione delle particelle, mentre per i primi tre casi il fattore comune è il trasporto fluviale in condizioni di piena.
Infatti, solo un fiume od un torrente ingrossato da cospicue precipitazioni raggiunge velocità tali da riuscire a “sostenere” e, quindi, trasportare particelle di tipo grossolano quali sono appunto quelle sabbiose. Le precipitazioni nei laghi e nei mari hanno sempre luogo nei pressi della riva o, meglio, nelle vicinanze degli estuari dei fiumi quando cioè il corso d’acqua perde velocità disperdendosi nelle acque del bacino. Nel caso dei mari sono poi frequenti le azioni di rimaneggiamento, fenomeni che comportano una ridistribuzione dei sedimenti a seguito di forti correnti o di mareggiate. Le deposizioni fluviali hanno invece luogo nei punti ove si registrano rallentamenti di velocità; è il caso del delta, ove il corso d’acqua risente dell’azione frenante delle maree o del punto in cui il fiume raggiunge la pianura ed inizia a meandrizzare.
Piccoli accumuli si osservano poi sulle parti interne delle curve fluviali laddove la velocità dell’acqua è di molto inferiore a quella delle parti esterne (forza centrifuga) dove, al contrario, si sviluppano con continuità fenomeni erosivi.
Ovviamente le differenti condizioni di sedimentazione si riflettono sullo spettro granulometrico del materiale e sulla presenza o meno di elementi inquinanti più fini (argille) o più grossolani (ghiaie).
Durante le fasi di trasporto, sedimentazione e compattazione, i depositi sabbiosi subiscono un processo di “maturazione” in conseguenza di attacchi di natura per lo più chimico-fisica. Tali azioni portano all’alterazione, trasformazione e dissoluzione degli elementi mineralogici meno stabili e modificano la tessitura delle particelle (dimensione, forma, ecc.).
Solitamente i depositi silicei secondari contengono maggiori quantità di impurezze rispetto ai primari; tra queste sono spesso presenti: miche, feldspati, ossidi ed idrossidi di ferro, materiali argillosi e sostanze vegetali.
Si osservi però che con il termine quarziti ci si riferisce anche a depositi sedimentari di quarzo molto puri ad altissimo grado di cementazione ed a giacimenti metamorfici caratterizzati da fenomeni di ricristallizzazione dei granuli originali.
Un ultimo tipo di deposito siliceo assai specifico è costituito dalle diatomiti. Si tratta di rocce sedimentarie formate prevalentemente da gusci silicei delle diatomee (minuscole alghe marine). Esse si caratterizzano per grande porosità e friabilità. Quelle deposte in acque dolci sono dette “farine fossili”, quelle sedimentate in ambiente marino “tripoli”.
Ubicazione dei principali depositi e tecniche di coltivazione
Interessanti giacimenti filoniani noti per la loro purezza sono coltivati in Germania (Monti Taunus), Brasile (formazioni di età paleozoica), Macedonia (Strumica) e Stati Uniti (Connecticut).
I sedimenti silicei europei scarsamente cementati di più ampia utilizzazione sono ubicati in Francia (Fontainebleau), Belgio (regione di Namur) e Germania.
Noti ed importanti depositi silicei fortemente cementati sono ubicati in molti stati degli Stati Uniti (principalmente lungo la catena degli Appalachiani e negli stati del South East); in Europa particolarmente conosciuti sono quelli inglesi (Sheffield), tedeschi (ancora sui monti Taunus, in Turingia ed in Slesia), francesi (Bretagna, massiccio centrale e Normandia).
Le tecniche di coltivazione variano in funzione delle caratteristiche genetiche dei giacimenti. I depositi filoniani possono essere coltivati esclusivamente con l’uso di esplosivi. Talvolta si è anche costretti a procedere in galleria. Nei casi più fortunati l’estrazione avviene all’aperto eseguendo “volate” (abbattimento con esplosivo) su fronti piuttosto ampi. Il tout-venant è poi trasportato presso impianti ove avviene la separazione tra i frammenti silicei e lo sterile. In passato tale selezione veniva effettuata manualmente lungo i nastri trasportatori; più recentemente si stanno utilizzando metodi di riconoscimento ottico.
L’esplosivo è impiegato anche per depositi metamorfici e per quelli sedimentari ad alto grado di cementazione. A volte ci si limita all’esecuzione di un “preminaggio” (serie di piccole cariche che scuotono la roccia fratturandola intimamente) a cui è possibile far seguire rippaggio e ruspaggio.
Sui terreni sedimentari poco cementati si opera esclusivamente con i mezzi per movimento terra (bulldozer, scraper ed escavatori idraulici).
Tipici comportamenti di riconoscimento
L’identificazione mineralogica della specie tramite analisi diffrattometrica ai raggi X comporta essenzialmente l’osservazione delle diffrazioni dei piani basali.
Sul diffrattogramma specifico e per radiazione CuKα si formano picchi nelle seguenti posizioni angolari: 26,6° (corrispondente ad una distanza reticolare di 3,34 Å) e 20,8° (che corrisponde a 4,26 Å) (Fig. 3.29).
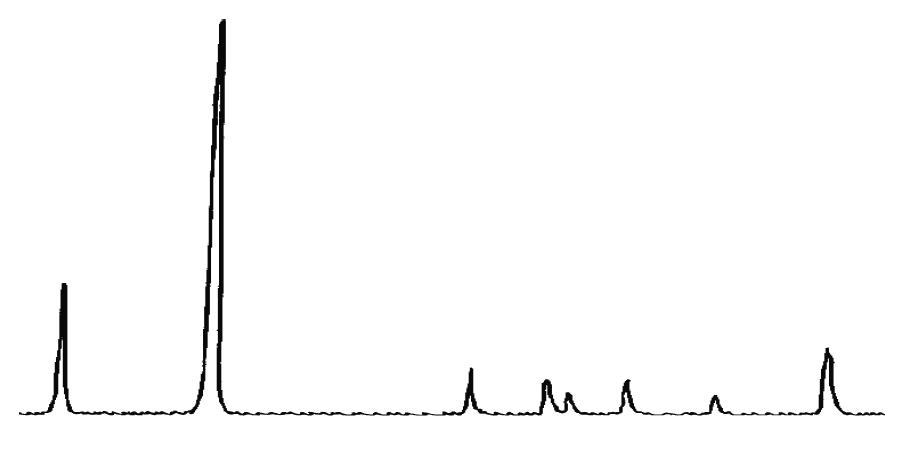
Fig. 3.29 Analisi diffrattometrica (DRX) del quarzo (Q)
Fig. 3.30
Le analisi termo-differenziali e termo-gravimetriche di una silice anidra presentano un andamento influenzato soltanto dagli effetti termici connessi alle modificazioni strutturali corrispondenti alle transizioni di fase (Fig. 3.30). Il quarzo mostra un picco endotermico (in riscaldamento) ed esotermico (in raffreddamento), con un ritardo o un anticipo rispetto alla temperatura teorica di 573°C a causa dell’inerzia termica del sistema di misura.
Fig. 3.30 Analisi termo-differenziale (in riscaldamento ed in raffreddamento) di un quarzo
La dilatometria (Fig. 3.31) mostra, in fase di riscaldamento, un forte incremento a 573 °C in corrispondenza del passaggio da quarzo α a quarzo β che, come noto, avviene con un significativo aumento di volume (circa +1%).
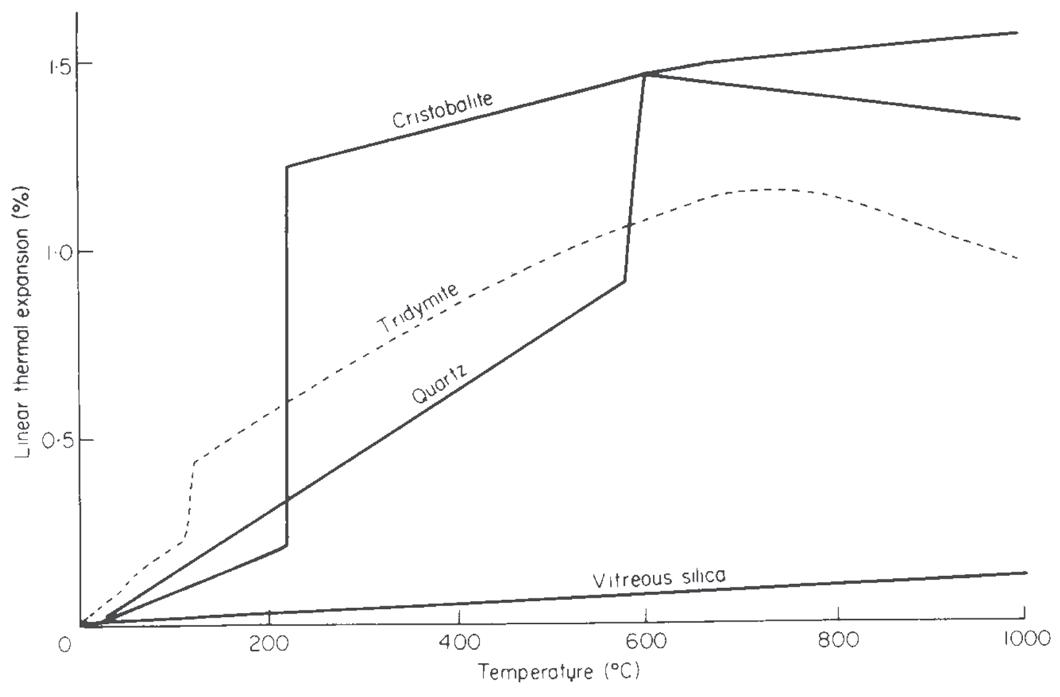
Fig. 3.31 Andamento dilatometrico delle tre forme cristalline della silice [7]
Il fenomeno, in modo ancora più marcato avviene anche poco oltre i 220 °C, quando ci si trova in presenza di cristobalite (piuttosto rara) che passa dalla forma α a quella β Durante il raffreddamento, qualora in cottura non tutta la silice cristallina si sia combinata per formare nuovi composti minerali (quali ad esempio wollastonite o mullite), si ha il passaggio da quarzo β a quarzo α con una forte contrazione dimensionale. Si tratta di un momento particolarmente critico per le produzioni ceramiche, dato che un raffreddamento troppo rapido può portare alle tipiche rotture dei pezzi (sfilo).
Trattamenti industriali dei tout-venant
Solitamente le sabbie vengono sottoposte a processi di purificazione e classificazione allo scopo di ottenere prodotti con caratteristiche rispondenti all’uso previsto. Si tratta di impianti che differiscono tra loro in relazione con il tipo di deposito e con le richieste del mercato. I principali trattamenti a cui sono sottoposte le sabbie sono: slimamento (lavaggio con eliminazione della frazione argillosa), attrizione (sfregamento delle particelle con distacco delle patine superficiali inquinanti), separazione magnetica, macinazione, flottazione (trattamento con reattivi anionici o cationici ed allontanamento delle impurezze), lisciviazione (attacco degli elementi inquinanti mediante impiego di acidi).
Nel caso delle quarziti l’operazione principale (e più costosa) consiste nella macinazione dei frammenti rocciosi, a cui fa seguito una classificazione granulometrica.
Sistemi a separazione magnetica possono contribuire alla rimozione delle impurità costituite da minerali ferromagnetici. Spesso però l’efficacia di questi metodi viene compromessa dalla carica elettrostatica delle polveri. In questi casi, se si opera con elevati campi magnetici, si rischia di scartare una parte rilevante della produzione. Spesso le società estrattive coltivano giacimenti cristallini che contengono contemporaneamente quarzo e feldspati. In questi casi l’impianto di trattamento si complica notevolmente ed è concepito in modo da separare le due specie mineralogiche nella maniera più efficace.
Si consideri che certi utilizzi, tipici ad esempio nell’industria vetraria, necessitano di requisiti granulometrici particolarmente stringenti in quanto le dimensioni delle particelle devono essere comprese solamente tra 0,8 e 0,1 mm.
Utilizzi nel settore ceramico
Le piastrelle per monocottura, sia di pasta bianca che rossa, impiegano piccoli quantitativi di sabbie (10 ÷ 15% massimo), mentre le piastrelle in pasta bianca per rivestimento smaltato prevedono percentuali di circa 10 ÷ 20%.
I supporti colorati possono utilizzare anche sabbie poco pure (ad esempio con sensibile presenza di ferro) senza particolari problemi. Spesso le composizioni per monocottura chiara non prevedono la diretta introduzione di sabbie silicee, ma queste sono già presenti nelle miscele naturali di feldspato, argilla e quarzo tipiche dei “feldspati parzialmente caolinizzati”. Anche nelle formulazioni per grès porcellanato le percentuali di sabbie quarzose sono minime. Nel settore delle piastrelle l’aggiunta di sabbie quarzose permette di “smagrire” le miscele favorendo la degasazione di eventuali impurità presenti nelle materie prime (sostanze organiche) e l’allontanamento dell’acqua durante il processo di essiccazione. Inoltre, si ottiene una riduzione della contrazione dimensionale e delle deformazioni in cottura.
Un’osservazione importante riguarda la granulometria e lo stato cristallino o amorfo dei frammenti quarzosi utilizzati. I produttori di piastrelle tendono ad impiegare sabbie fini per ridurre i tempi di macinazione dell’impasto e pertanto il materiale viene premacinato. Tuttavia, anche in natura esistono giacimenti silicei costituiti da particelle molto fini: si tratta di alcuni depositi piroclastici e delle diatomiti. Purtroppo, tali materiali interferiscono in maniera molto negativa sulle proprietà reologiche delle barbottine ceramiche e non possono essere impiegati. I settori che producono sanitari in vitreous-china e porcellana richiedono materiali silicei particolarmente puri. Le quantità utilizzate nelle composizioni di impasto sono di circa 20 ÷ 25%. L’aggiunta della silice negli impasti per vitreous-china e porcellana ha lo scopo di equilibrare la compresenza di silice e allumina e, quindi, di raggiungere i corretti rapporti stechiometrici che, in cottura, portano alla formazione di mullite.
La silice viene inoltre utilizzata nella produzione di refrattari acidi. I mattoni silicei vengono fabbricati partendo da quarziti, macinate secondo uno spettro granulometrico opportunamente assortito e con la sola aggiunta di piccole percentuali di leganti.
I semi-silicei prevedono l’introduzione di un 10% di allumina; i fire-clay sono formati da un 30 ÷ 40% di allumina con la parte rimanente costituita da quarzo.
Le diatomiti sono utilizzate per produrre mattoni isolanti.
Infine, la silice trova un ulteriore impiego nella preparazione delle fritte ove viene introdotta in percentuali comprese tra 20 e 40%; per lo più si utilizza quarzo ventilato, tuttavia certe formulazioni prevedono l’impiego, anche in misura rilevante, di sabbie fini.
Utilizzi in altri campi
Il principale utilizzatore della silice è l’industria vetraria.
Le specifiche richieste dalle caratteristiche del materiale variano in funzione del prodotto che s’intende ottenere. Per le bottiglie comuni di colore verde è possibile impiegare una sabbia contenente anche 1% di Fe2O3, mentre per le semi-bianche la percentuale scende allo 0,2%.
I vetri per lastre richiedono un contenuto di ossido di ferro inferiore a 0,3%, mentre per i bicchieri bianchi la quantità scende sotto 0,013%.
Infine, i più pregiati vetri per ottica richiedono sabbie di elevata purezza (SiO2 > 99,5%) con un bassissimo tenore di Fe2O3 (< 0,008%). È molto importante anche garantire un ridotto contenuto di TiO2 (meno dello 0,05%) e che il cromo e il cobalto siano nell’ordine di pochi ppm (parti per milione).
Un altro campo di grande impiego della silice è quello metallurgico (leghe speciali).
Con le diatomiti (rocce silicee sedimentarie formatesi dalle diatomee) si realizzano “letti filtranti” molto efficaci, utilizzati nel settore alimentare.
Origine del termine
La parola è di origine tedesca e viene da Feldspat cioè “spato di campo”, dove per “spato” s’intendono grossi cristalli che sfaldano secondo le forme elementari.
Per quanto riguarda l’etimologia delle rocce feldspatiche più diffuse possiamo citare:
- nefeline: dal greco neféle (“nube”), perché formano una massa gelatinosa quando decomposte con acidi;
- pegmatiti: dal greco pégma (“concrezione”);
- apliti: dal greco aplós (“semplice”) in riferimento alla loro composizione mineralogica; - felsiti: dal tedesco Felsenbildner che significa “formatore di roccia”.
Struttura mineralogica
La struttura base del feldspato è un anello formato da quattro gruppi tetraedrici (Fig. 3.32); nel caso dei feldspati potassici e sodici vi sono tre tetraedri di silicio ed uno di alluminio mentre nel feldspato calcico i quattro gruppi tetraedrici sono metà con il silicio e metà con l’alluminio. Il feldspato potassico può presentarsi in due forme cristalline: ortoclasio (monoclino) e microclino (triclino); il feldspato sodico si identifica come albite (triclino).
Fig. 3.32 Cella elementare del feldspato
La nefelina (un importante feldspatoide sodico-potassico) cristallizza nel sistema esagonale e possiede una struttura del tutto simile a quella della tridimite (forma allotropica della silice stabile tra 870 e 1470 °C); da questa si distingue solo per la sostituzione di un Si4+ con un Al3+ a cui si abbina l’introduzione di Na+ e, in misura molto minore, di K+ per mantenere la neutralità elettrica della struttura.
I cristalli si presentano come prismi esagonali molto appiattiti e sono stabili a basse temperature.
La carnegieite, una forma allotropica della nefelina stabile ad alte temperature, deriva per arricchimento in sodio dalla cristobalite (altra forma allotropica della silice). La nefelina è contenuta insieme ai feldspati alcalini in rocce chiamate sieniti, caratterizzate da una carenza di silice.
Composizione chimica
La formula generale dei feldspati è la seguente:
XY4O8
dove: X è generalmente costituito da Na+, K+, Ca2+
Y rappresenta quasi sempre Al3+ e Si4+, ma talvolta viene parzialmente sostituito da Fe3+
I tre principali feldspati (potassico, sodico e calcico) hanno le seguenti formulazioni:
- ortoclasio, microclino, sanidino K (AlSi3O8)
- albite
- anortite
Na (AlSi3O8)
Ca (Al2Si2O8)
Si osservi comunque che sono frequenti le soluzioni solide tra questi tre feldspati; in particolare albite ed anortite formano, ad alta temperatura, una serie di soluzioni cristalline che si mantengono stabili anche dopo raffreddamento (plagioclasi), note con le seguenti denominazioni (tra parentesi il rapporto albite/anortite):
- oligoclasio (7/1)
- andesina (2/1)
- labradorite (1/2)
- bytownite (1/7)
Anche l’ortoclasio contiene spesso, in soluzione solida, rilevanti percentuali di albite.
I feldspati di Na e K hanno la seguente composizione teorica (in ossidi):
SiO2 Al2O3 Na2O K2O
Na feldspato
La formula generale della nefelina è invece la seguente:
X4(Al4Si4O16)
dove X è costituito prevalentemente da Na con presenza di K che non supera il rapporto di 1/3.
La composizione teorica della nefelina risulta pertanto la seguente:
41,5% SiO2 + 35,2% Al2O3 + 17,5% Na2O + 5,8% K2O
Genesi e caratteristiche dei giacimenti
I giacimenti di feldspati, apliti, nefeline, pegmatiti, felsiti, ecc. sono di origine ignea, cioè il risultato del consolidamento di fluidi magmatici; esistono comunque anche depositi parzialmente feldspatici di origine sedimentaria (sabbie ed arenarie).
Nei giacimenti plutonici (cioè di consolidamento profondo) le rocce feldspatiche si presentano sotto forma filoniana e sono generalmente associate con quarzo e muscovite. La genesi di questi filoni sarebbe connessa a segregazioni magmatiche con uno specifico chimismo (arricchimento di alcali) oppure dovute a fusioni e ricristallizzazioni che si sono sviluppate in conseguenza di fenomeni metamorfici. Ad esempio, nel caso delle pegmatiti, di solito associate ad intrusioni granitoidi, lo sviluppo di cristalli di grandi dimensioni (feldspati
e mica) è verosimilmente dovuto alla presenza di liquidi fusi a bassa viscosità che hanno saturato le fratture di rocce preesistenti.
Le apliti, caratterizzate da cristallizzazione molto più minuta, si sarebbero invece formate da fasi liquide a viscosità più elevata probabilmente a causa dell’allontanamento dei composti basso fondenti che, diversamente, avrebbero mantenuto più fluida la soluzione favorendo lo sviluppo di fenocristalli (cristalli di grandi dimensioni). Ad esempio, il feldspato inglese proviene principalmente dalla regione di St. Austell (Cornovaglia) dove affiora una roccia granitica più o meno caolinizzata nota con il nome di Cornish stone. Il colore della roccia varia notevolmente passando dal bianco al rosso-purpureo, al cuoio. La parziale caolinizzazione sarebbe il risultato di azioni di weathering sviluppatesi durante il Cretaceo ed il Terziario. La genesi del feldspato di Pinzolo (Trento) sembra invece connessa ad una segregazione filoniana dal magma. L’aplite di Campiglia Marittima (Grosseto) costituisce un esempio di differenziazione magmatica acida (quarzo-feldspatica) cristallizzata in assenza di sostanze volatili. Le nefeline (o meglio le sieniti nefeliniche) sono rocce plutoniche che si sono formate per raffreddamento di fluidi sottosaturi in silice.
Le felsiti sono rocce eruttive caratterizzate da una struttura micro o cripto- cristallina con o senza fenocristalli.
Il sanidino (con una struttura cristallina deformata rispetto all’ortoclasio) si trova invece in rocce vulcaniche recenti (cioè magmi consolidati all’aria) ma difficilmente raggiunge concentrazioni tali da renderlo economicamente interessante per una sua estrazione.
Al contrario, depositi sedimentari coltivati per il loro elevato contenuto in alcali sono le arenarie feldspatiche e le sabbie feldspatiche. Entrambi sono il prodotto della disgregazione e del successivo accumulo in ambiente acquoso (delta fluviali, laghi o zone marine prossime alla costa) di rocce madri acide (graniti, pegmatiti e metamorfiti feldspatiche). Spesso in questi giacimenti si è già sviluppata naturalmente la separazione dei feldspati dagli altri minerali (solitamente quarzo e mica); il trasporto però può comportare una contaminazione di elementi inquinanti, tipicamente minerali argillosi e idrossidi di ferro, oltre a possibili concentrazioni di minerali con metalli pesanti.
Ubicazione dei principali giacimenti; tecniche di coltivazione e di arricchimento
Filoni feldspatici e pegmatitici sono coltivati in molte parti del mondo; alcuni tra i più noti sono ubicati in Turchia, in Scandinavia, Russia (Carelia e penisola di Kola), Ucraina, Gran Bretagna (Cornovaglia ed isola di Man), Italia (Pinzolo, Dervio e Vibo Valentia), Germania (Oberfranken, Oberpfalz e Hagendorf), Francia (Perpignan), Macedonia, Stati Uniti (North Carolina, Georgia, Connecticut e California), Grecia, Canada (Ontario e Quebec), Messico, Giappone, India, Sud-Africa ed Australia. Depositi di aplite sono poi segnalati negli Stati Uniti (Virginia e North Carolina), Giappone, Italia (Campiglia Marittima) e Germania (Hagendorf).
I depositi più importanti di sienite nefelinica sono ubicati in Russia (Carelia, Siberia e penisola di Kola) , Canada (Quebec, Ontario e Columbia britannica), Norvegia, Finlandia, Brasile (Minas Gerais e San Paolo) e Stati Uniti (Arkansas).
Per quanto concerne la coltivazione delle rocce feldspatiche, la loro compattezza rende solitamente indispensabile l’utilizzo di esplosivo. Nei casi di parziale caolinizzazione ci si può limitare al solo preminaggio (azione di “fracassamento” della struttura del deposito) e procedere quindi all’asportazione con mezzi meccanici; solo alcuni giacimenti possono essere direttamente coltivati con mezzi meccanici. Il tout-venant viene scelto manualmente
disponendo il materiale su nastri trasportatori o, più recentemente, mediante sistemi di selezione automatizzati con riconoscimento ottico.
Si procede quindi con frantumazioni successive fino ad ottenere un materiale parzialmente polverizzato che viene separato in diverse frazioni granulometriche. Talvolta s’introducono anche separatori magnetici che riescono a trattenere con facilità particelle metalliche (derivanti dall’usura dell’impianto) ma che risultano poco efficaci sui materiali scarsamente magnetici come ad esempio la biotite, spesso presente tra i principali inquinanti. In questo caso sarebbe necessario utilizzare un campo magnetico ad elevata intensità, incorrendo nel rischio (reale) di attrarre anche particelle feldspatiche caricatesi elettricamente durante l’operazione di macinazione. Vi sono poi tipologie di inquinanti che rendono incoltivabili certi giacimenti; il caso più clamoroso è quello del Cornish stone inglese per il quale la crescente profondità della coltivazione ha portato ad operare su aree meno caolinizzate e decisamente più ricche di minerali del fluoro. Come noto il fluoro causa grossi problemi di inquinamento ambientale e la roccia non può essere utilizzata se non previo un costoso trattamento di flottazione che la pone così fuori mercato rispetto ad altri materiali feldspatici. Questo è il motivo che ha portato alla sospensione della coltivazione in questo importante giacimento.
L’arricchimento del tenore in alcali con l’impiego della flottazione è ampiamente diffuso negli Stati Uniti. La tecnica permette di procedere alla separazione del quarzo dal feldspato impiegando opportuni reagenti chimici schiumanti che allontanano, in sospensione, uno dei due minerali.
Per quanto riguarda poi i depositi sedimentari (sabbia ed arenarie feldspatiche) il trattamento più diffuso consiste in un lavaggio (che allontana le particelle fini prevalentemente argillose) a cui fa seguito l’essiccazione in forno rotativo. Qualora il giacimento sia scarsamente cementato si procede all’estrazione con impiego di macchine per movimento terra (bulldozer e scraper) e non si rende necessaria l’operazione di frantumazione.
In caso contrario si ricorre al preminaggio e si utilizzano frantoi per ridurre le dimensioni dei frammenti.
Riconoscimento mineralogico di feldspati
Il posizionamento angolare dei picchi caratteristici che si rilevano nelle analisi diffrattometriche (radiazione CuKα) di alcuni minerali feldspatici sono riportati nella seguente tabella:
Albite
Ortoclasio
Distanze
Nefelina
Leucite
Per quanto concerne le analisi DTA, da letteratura l’albite presenta piccoli e bruschi picchi endotermici tra 820 e 900 °C in conseguenza di una sua trasformazione allotropica. Picchi analoghi tra 780 e 820 °C sono presenti nell’ortoclasio e nella labradorite (una varietà dell’anortite). Nel caso della nefelina si nota una reazione termica a circa 1250 °C connessa con una trasformazione allotropica del minerale. Viceversa, l’analisi TG non fornisce alcuna informazione in quanto i feldspati non manifestano reazioni che comportino una perdita di materia.
La dilatometria non presenta variazioni fino a 1050 ÷ 1100 °C, poi si osserva l’inizio della contrazione dovuta alla progressiva formazione di fasi liquide fino alla completa fusione.
Utilizzi ceramici
I materiali feldspatici trovano ampio impiego in tutti i prodotti ceramici che richiedono un elevato grado di vetrificazione dopo cottura; nel caso delle piastrelle, l’effetto fondente dei feldspati (da 25 a 55%) è necessario nella produzione sia di grès porcellanato che di monocotture chiare a bassa porosità. Anche nei sanitari in vitreous-china i feldspati vengono introdotti in percentuali da 20 a 30% e nelle porcellane da 17 a 37%.
Naturalmente le percentuali di utilizzo, oltre che variare con le diverse composizioni, dipendono dal tenore in alcali del materiale feldspatico introdotto. La scelta tra feldspato potassico e sodico viene fatta sulla base delle specifiche esigenze dei differenti manufatti, tenendo presente che il feldspato potassico è un fondente meno energico del feldspato sodico ma consente di ottenere un intervallo di vetrificazione più ampio (vedi Fig. 3.33 e Fig. 3.34).
Fig. 3.33 Determinazione della fusibilità al microscopio riscaldante per un feldspato sodicopotassico (Alavus): inizio del rammollimento a 1240 °C, punto di fusione a 1400 °C
Talvolta si aggiungono materiali potassici anche in composizioni studiate per la produzione di piastrelle a porosità medio alta; classico è il caso della terraglia forte (oggi in disuso). Inoltre, l’addizione di feldspato produce un abbassamento del coefficiente di dilatazione termica contrastando la tipica deformazione convessa caratteristica delle piastrelle in cui lo smalto è posto troppo in compressione dal supporto.
Un altro settore ceramico dove si impiega il feldspato è quello della produzione di fritte. In questo caso le percentuali di utilizzo variano dal 20% al 40%.
La nefelina trova gli stessi campi d’impiego del feldspato, tuttavia, avendo un costo nettamente superiore, specie quando le produzioni ceramiche sono ubicate lontano dal punto d’origine, il suo utilizzo è limitato ai casi in cui risulta indispensabile disporre di un materiale con un più elevato potere fondente.
Alavus 3,4 11,6
Forshammar 3,7 4,9
Dervio 6,8 2,2
Vibo Valentia 11,0 0,1
Lakefield 10,3 4,5
Fig. 3.34 Intervallo di rammollimento di fondenti feldspatici al microscopio riscaldante (A inizio del rammollimento, B punto di fusione)
Utilizzi in settori diversi
Fig. 3.35
Più del 50% della produzione mondiale di materiali feldspatici è assorbita dall’industria vetraria che, pertanto, risulta essere il principale cliente. Solitamente però questo settore impiega solo i materiali a più elevato tenore in alcali ed a contenuto di ferro molto basso (in relazione al tipo di vetro che si intende produrre). Pertanto, i feldspati flottati e soprattutto la nefelina vi trovano un utilizzo molto più ampio. In particolare l’introduzione di quest’ultima, grazie all’elevato tenore di sodio, porta all’ottenimento di vetri meno viscosi e più facilmente lavorabili.
Origine del termine
Deriva dall’associazione di due parole greche pyrós (fuoco) e phyllo (foglia), probabilmente per la tendenza del minerale a “sfogliare” in conseguenza del riscaldamento.
Struttura mineralogica
Si tratta di un minerale argilloso a struttura base tipo mica, costituita da due piani esagonali di tetraedri di silice contenenti un piano ottaedrico di Al3+ (Fig. 3.35); essa è simile a quella del talco ed è caratterizzata dalla presenza degli ossigeni del foglietto [SiO4]4- nella superficie esterna delle particelle, mentre gli ossidrili si trovano sempre all’interno, protetti dai due piani tetraedrici. Questa condizione comporta la neutralità strutturale ed una notevole stabilità ed inerzia chimica del minerale.
Composizione chimica
La composizione della pirofillite è la seguente: Al2Si4O10(OH)2
Non hanno luogo sostituzioni vicarianti e quindi neppure l’introduzione nel reticolo di cationi alcalini o alcalino-terrosi.
Genesi dei giacimenti e loro caratteristiche
La formazione di pirofillite è connessa a processi metamorfici e idrotermali. Spesso si trova associata a sericite (minerale che a sua volta deriva dall’alterazione della biotite).
Si tratta di una roccia tenera (durezza 1 Mohs), di peso specifico piuttosto elevato (2,8 kg/dm3), che presenta superfici lisce e patinate analoghe a quelle del talco.
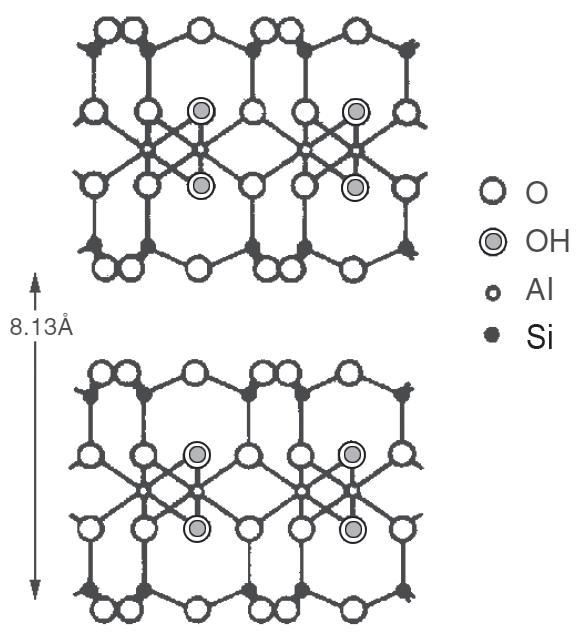
Fig. 3.35 Cella elementare della pirofillite
Ubicazione dei principali giacimenti e tecniche di coltivazione
I depositi di pirofillite relativamente pura non sono molto frequenti. Importanti giacimenti sono comunque coltivati in Giappone, Russia (gli scisti pirofillitici degli Urali), Stati Uniti (tra i tufi e le brecce della Carolina del Nord), Brasile e Sud-Africa, dove è presente una varietà nota con il nome di stumatite che ha la caratteristica di poter essere lavorata ottenendo oggetti con forme ben precise.
La friabilità del minerale consente di procedere all’estrazione con l’impiego di macchine movimento terra di notevole portata (bulldozer, escavatori idraulici, ecc.). Nel caso del giacimento sud-africano la coltivazione è manuale ed avviene con grande cautela dato che si vogliono ottenere frammenti integri di dimensioni rilevanti.
Tipici comportamenti di riconoscimento
La diffrattometria RX (Fig. 3.36) evidenzia le riflessioni dei piani basali alle seguenti distanze reticolari:
Pirofillite
Posizione angolare dei picchi °(2θ)
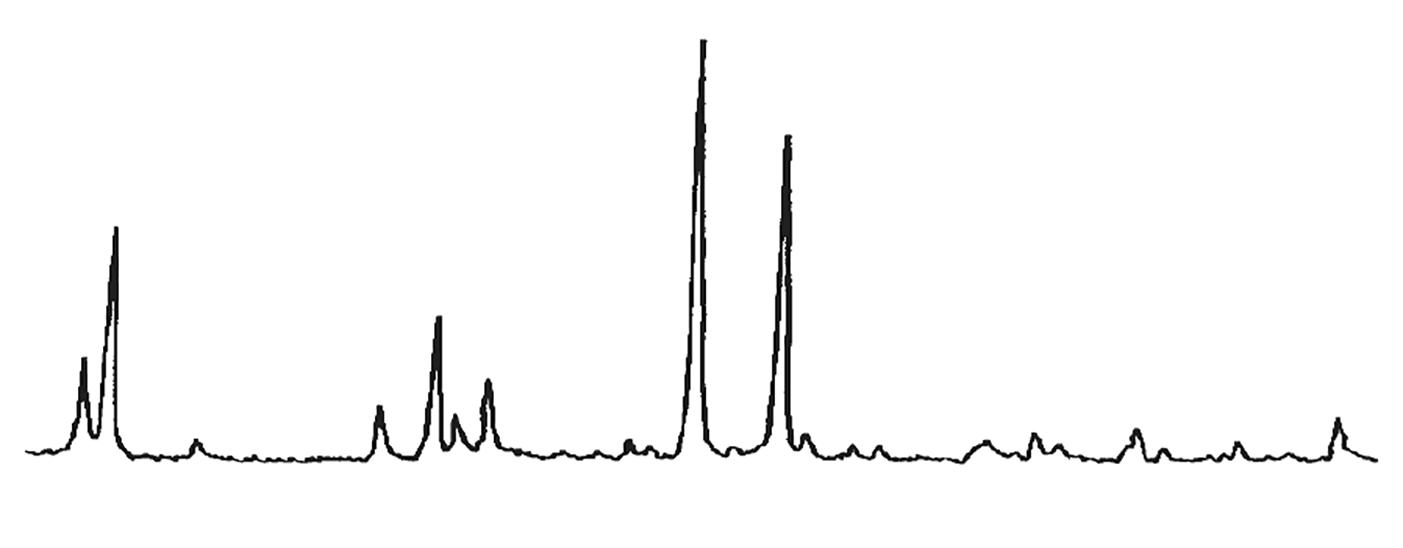
Fig. 3.36 Analisi diffrattometrica RX di una pirofillite (P=pirofillite, Q=quarzo, M=muscovite, K=caolinite)
L’analisi termo-differenziale (DTA), Fig. 3.37, mostra un picco endotermico tra 640 e 850 °C (con un massimo a 690 ÷ 780 °C) dovuto alla perdita degli ossidrili OH . Alle stesse temperature, sul diagramma della curva termo-gravimetrica (TG) si osserva una marcata perdita in peso (5% per una pirofillite pura).
Fig. 3.37
Fig. 3.37 Analisi termo-differenziale (DTA) della pirofillite
Fig. 3.38
Sottoposto ad analisi dilatometrica (Fig. 3.38), il minerale è caratterizzato da una fortissima fase espansiva che ha inizio alla temperatura di trasformazione allotropica del quarzo, prolungandosi con lo stesso gradiente di espansione fino a 750°, poi con minore intensità fino a 850 °C. L’inizio della fase di contrazione si osserva oltre 950 °C.
Fig. 3.38 Analisi dilatometrica della pirofillite
Caratterizzazione tecnologica di provini pressati preparati con materie prime pirofillitiche
Fig. 3.39 NEW
Si tratta di un minerale di notevole refrattarietà che ricristallizza in mullite (formula bruta Al6Si2O13 corrispondente alla formula in ossidi 3Al2O3∙2SiO2) + silice intorno ai 1200 °C e fonde a 1630 °C.
Le piastrelle formate partendo dal materiale pirofillitico presentano i seguenti comportamenti:
- normale espansione di post-pressatura;
- scarsa resistenza meccanica dei crudi (specie dell’essiccato);
- forte espansione in essiccazione;
- sul cotto ottenuto a 1020 °C si registra una forte espansione (3,3%) con assorbimento di acqua prossimo al 27%; a 1200 °C non si registrano variazioni dimensionali con assorbimento d’acqua di circa il 19%;
- a 1020 °C la resistenza meccanica è praticamente inconsistente; solo a 1150 °C si superano 12 N/mm2.
Utilizzi ceramici
La notevole refrattarietà della pirofillite ne condiziona i campi d’impiego. Si hanno rari esempi di un suo utilizzo in composizioni per piastrelle in pasta bianca da rivestimento e pavimento smaltati in produzioni brasiliane. Anche in questo caso però le percentuali introdotte sono minime.
In generale la presenza di pirofillite porta ad un abbassamento del coefficiente di dilatazione e riduce il ritiro in cottura: in questo senso è ipotizzabile un suo uso nella produzione di impasti greificabili ad alta temperatura (per pavimentazione) con un ritiro controllato se cotti a temperatura inferiore (quindi adatti per rivestimento).
La pirofillite viene impiegata anche nella produzione di vasellame da tavola, in quanto conferisce una caratteristica lucidità al supporto cotto. Il principale suo utilizzo è comunque nella produzione di refrattari, isolatori ceramici e crogiuoli refrattari.
Utilizzi in settori diversi
Trova impieghi alternativi al caolino per cui viene utilizzata come carica inorganica della gomma e dei materiali plastici, oltre che come supporto per insetticidi e vernici.
Del tutto originale è poi l’impiego che si fa della stumatite sudafricana che, data la sua lavorabilità, viene impiegata per piccole produzioni di oggettistica. I pezzi opportunamente sagomati sono poi cotti a 1300 °C senza alcun apprezzabile ritiro, ottenendo manufatti caratterizzati da notevole igroscopicità.
Origine del termine
Deriva dalla parola araba talq
Struttura mineralogica
Si tratta di un minerale avente una struttura del tipo mica, con foglietto elementare costituito da due piani esagonali di tetraedri di silice contenente un piano ottaedrico di Mg2+ (Fig. 3.39).
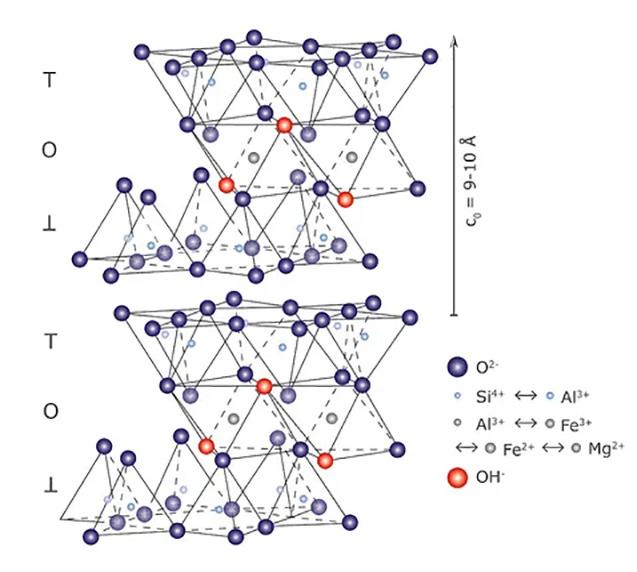
Fig. 3.39 Struttura elementare del talco [1]
L’elemento strutturale di base risulta neutro e gode di una grande stabilità. Il talco, in conseguenza della propria struttura, può essere considerato come appartenente alla famiglia dei minerali argillosi. Ha una caratteristica struttura scistosa lamellare (talcoscisti) e presenta una marcata untuosità al tatto. Una roccia molto diffusa ad elevato contenuto di talco è nota con il nome di steatite
Composizione chimica
Il talco è un metasilicato acido di magnesio avente formula:
Mg3 Si4O10(OH)2
Non essendo possibile alcuna sostituzione vicariante con elementi canonici a carica deficitaria non si hanno quindi introduzioni di cationi alcalini.
La sua composizione teorica in ossidi è la seguente:
63,5% SiO2 + 31,7% MgO + 4,8% H2O
Tuttavia, le rocce talcose contengono generalmente altri minerali (clorite, serpentino, magnesite, dolomite, ecc.) e varie impurezze che ne influenzano marcatamente le proprietà, risultando mediamente formate da circa il 50% di silice, fino al 30% di magnesio e poco meno del 10% di allumina.
Genesi e caratteristiche dei giacimenti
Il talco può avere un’origine metamorfica o idrotermale. In questo secondo caso si trova spesso associato a dolomite e magnesite; da questa associazione è stata ottenuta la chiave interpretativa di una delle genesi del minerale che risulta conseguente a reazioni tra soluzioni idrotermali acide (ricche di silice) ed i carbonati magnesiaci.
Talvolta invece è associato a rocce ultrabasiche (quali olivina, anfiboli ed augite) avendo preso origine (come clorite e serpentino) dalla loro trasformazione per azioni metamorfiche.
Spesso i giacimenti hanno natura filoniana.
Nel caso del più importante giacimento italiano (quello di Fontane nella Val Germanasca, in Piemonte) si osserva un solo grande banco di lunghezza superiore a 2 km e di profondità variabile da meno di 1 m a 15 m, compreso tra gneiss, micascisti e calcari (alla base) e micascisti, cloritoscisti (al tetto).
Lo stesso banco contiene però inclusioni inquinanti costituite da dolomia, anfiboliti e lembi delle rocce incassanti.
Ubicazione dei principali giacimenti e tecniche di coltivazione
Numerosi sono i depositi coltivati in molte aree geografiche, tra le quali si possono menzionare: Germania (Göpfersgrün e Fichtelgebirge), Italia (Piemonte, Lombardia e Sardegna), Austria, Spagna (Regione di Barcellona), Francia (massiccio di Saint Barthélemy), Grecia, Finlandia, Russia (monti Urali), Stati Uniti (New York, California, Georgia, Montana, Texas, Virginia, ecc.) Canada (Ontario), India, Corea, Cina (Zhejiang e Shandong) e Sud-Africa. In conseguenza alla natura filoniana richiamata in precedenza, è spesso necessario procedere a coltivazioni in galleria al fine di evitare la rimozione di enormi quantitativi di sterile. Il tout-venant subisce processi di selezione e di macinazione per ottenere le granulometrie richieste dagli utilizzatori.
Tipici comportamenti di riconoscimento
L’identificazione della specie mediante diffrazione a raggi X si basa sull’osservazione dei picchi relativi ai piani basali (Fig. 3.40):
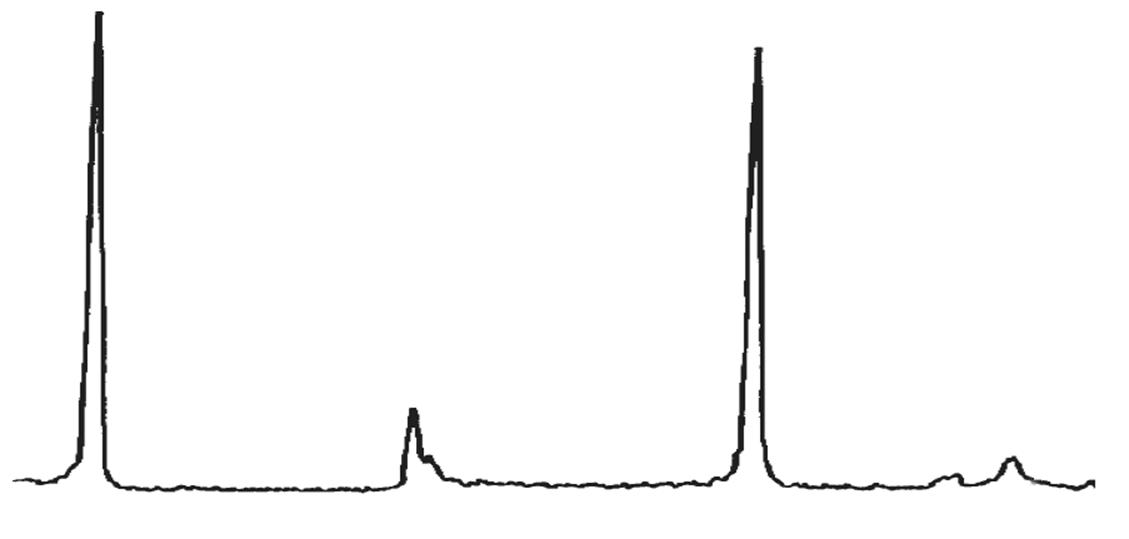
Fig. 3.40 Analisi diffrattometrica RX del talco (T=talco)
L’analisi termo-differenziale (DTA) di un talco puro mostra un solo picco endotermico tra 900 e 1000 °C, dovuto all’allontanamento degli ossidrili OH . Corrispondentemente la TG indica una significativa perdita di peso (Fig. 3.41). In alcuni casi è invece ben rilevabile la presenza di clorite e la perdita d’acqua zeolitica.
Fig. 3.41
Anche la dilatometria è spesso influenzata dalla presenza di altri minerali, in particolare dalla clorite, che provoca un netto incremento della dilatazione tra 600 e 800 °C (Fig. 3.42).
3.41 Analisi TG e DTA del talco
Fig. 3.42
3.42 Analisi dilatometrica del talco
Utilizzo e caratterizzazione tecnologica
3.43 NEW
I talchi possono essere convenientemente impiegati nella produzione di piastrelle. Normalmente questi minerali presentano: espansione di post-pressatura, scarsa resistenza meccanica in verde ed essiccato, sensibile espansione dopo essiccazione, assorbimento molto elevato (> 25%) per cottura a 1020°C con leggera espansione dimensionale; i provini cotti sono praticamente inconsistenti. Il coefficiente di dilatazione termica lineare α per materiali cotti a 1020 °C è molto elevato (90×10−7 °C−1).
In cottura il talco, soprattutto se abbastanza puro ed a basso tenore di ferro, si comporta praticamente da inerte fino a 1050 °C mentre a temperatura più elevata inizia a partecipare alle reazioni svolgendo un’azione fondente. In questo senso anche piccole percentuali in impasto (2 ÷ 5%) possono svolgere un’utile azione fondente, tramite la formazione, con gli alcali presenti nei feldspati, di miscele eutettiche a temperatura di fusione particolarmente bassa. Tali effetti sono spesso ricercati per la produzione di materiali compatti a bassissimo assorbimento di acqua, come le piastrelle di grès porcellanato.
Utilizzi ceramici
L’introduzione del talco negli impasti ceramici consente anche di controllare l’espansione termica. L’aspetto dilatometrico assume particolare importanza nella produzione di piastrelle. Alle temperature più basse (prima cioè dello sviluppo delle reazioni allo stato liquido) il talco comporta un elevato coefficiente di dilatazione che favorisce l’accordo dei supporti porosi con gli smalti e, pertanto, agisce come sostitutivo dei carbonati.
A temperature più elevate determina invece un effetto opposto, tanto che viene introdotto per migliorare la resistenza agli shock termici dei prodotti ceramici che sinterizzano a tali temperature.
Pertanto il talco viene utilizzato con diverse funzioni specifiche nelle composizioni di numerosi materiali ceramici:
- supporti porosi da rivestimento e pavimento: in certi casi l’utilizzo può superare anche il 40%; le formulazioni a base di talco sono molto diffuse negli Stati Uniti per il costo mediamente basso che presenta questo materiale in quelle aree. Oltre a favorire l’accordo
dilatometrico tra smalto e supporto, il talco riduce sensibilmente la post-espansione del prodotto cotto conseguente a reazioni di idratazione e, quindi, il rischio del cavillo tardivo. Gli impasti ricchi in talco sono particolarmente adatti a cotture rapide in quanto, pur mantenendo ottime caratteristiche generali, presentano una minima emissione di gas. L’uso generalizzato è però limitato dal suo maggior costo rispetto ai materiali carbonatici;
- vasellame di Faenza: sono composizioni calcaree o calcaree-magnesiache con sostituzione di parte dei carbonati con talco (dal 10 al 15%). Ne deriva una migliore lavorabilità degli impasti a crudo e la diminuzione delle rotture in raffreddamento specie quando si estraggono i pezzi dal forno ancora caldi;
- grès e vetrificati monocotti: il talco, in piccole percentuali (inferiori, di norma, al 5%) consente di ridurre la temperatura ed il ciclo di cottura. Le composizioni in pasta bianca subiscono un’azione sbiancante (forse per la formazione di ferrite di magnesio che “sequestra” il ferro presente). Naturalmente in questo caso si deve impiegare un talco di notevole purezza;
- sanitari in vitreous-china: piccole aggiunte (2 ÷ 3%) migliorano la resistenza agli shock termici;
- refrattari: nel caso di silico-alluminosi l’impiego di talco in quantità variabile dal 2 al 10% porta ad un miglioramento della resistenza agli shock termici (ma causa una diminuzione della refrattarietà) ed aumenta la resistenza meccanica dei crudi. L’utilizzo del talco è praticamente indispensabile nella produzione di piastre e supporteria in quanto spesso tali elementi sono sottoposti a forti gradienti termici tra le facce opposte. Questo è ancora più vero nel caso vengano impiegati in forni che operano con cicli rapidi o in forni intermittenti. Il limite di impiego è rappresentato dalla formazione di eutettici che possono abbassare anche marcatamente le caratteristiche refrattarie del materiale;
- isolatori elettrici: in questo caso il talco è la principale materia prima in quanto questi manufatti sono costituiti (dopo cottura) da cordierite (5SiO2∙2Al2O3·2MgO) e clinoenstatite (SiO2∙MgO). Per questa produzione spesso viene preferita la steatite;
- vasellame da tavola: porta ad una caratteristica traslucidità e rende i pezzi più resistenti;
- laterizi: piccole aggiunte di talco migliorano l’estrusione alla filiera agendo come lubrificante. Si rilevano aumenti della resistenza meccanica di crudi e cotti e diminuzione delle rotture in cottura.
Ancora, il talco viene impiegato nella produzione di certi smalti magnesiaci, mentre nei sanitari viene spolverato sugli stampi di gesso in quanto favorisce il distacco dei pezzi (specie nel caso di manufatti di grandi dimensioni). Per molti di questi utilizzi si richiedono talchi di grande purezza, mentre ciò non è necessario nell’impiego in composizioni per piastrelle in pasta colorata.
Utilizzi in settori diversi
Il talco trova impiego come carica inorganica della carta e dei materiali plastici ed inoltre nell’industria delle vernici. Quando raggiunge una elevata purezza trova spesso utilizzi alternativi a quelli del caolino. Un uso caratteristico è come supporto per le polveri deodoranti dell’industria cosmetica.
Origine del termine
È stata così chiamata in onore del chimico e fisico inglese W.H. Wollaston (1766 - 1828).
Struttura mineralogica
È quella dei sorosilicati in cui due tetraedri di silicio si collegano formando una coppia con uno ione ossigeno in comune (Fig. 3.43).
Solitamente il minerale ha aspetto fibroso (i singoli cristalli hanno forma aghiforme e talvolta anche tabulare), peso specifico 2,9 g/cm3, durezza Mohs 4,5 ÷ 5 e colore bianco.
A temperatura ambiente è stabile la forma naturale, per poi passare alla forma allotropica (pseudo-wollastonite) a 1125 °C.
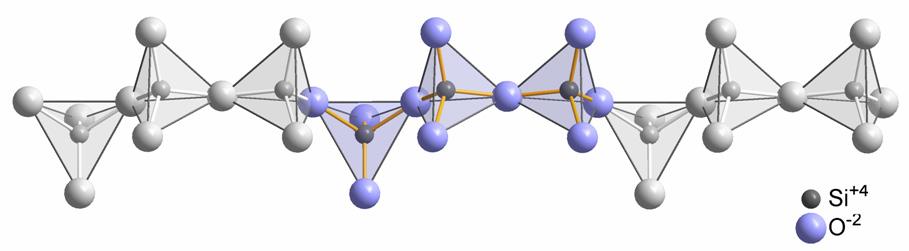
Composizione chimica
È un metasilicato di calcio, con formula:
La wollastonite teorica è pertanto costituita da:
48,3% CaO + 51,7% SiO2
Genesi e caratteristiche dei giacimenti
Si tratta di un minerale abbastanza diffuso originatosi da calcari per contatto con rocce ignee (metamorfismo di contatto), raramente coltivabili. Di solito si presenta assieme a calcite, granati (alluminosilicati alcalino-terrosi), quarzo, feldspati, diopside, tremolite, ecc.
Spesso si osserva una compresenza di wollastonite ed altri minerali che porta a fenomeni di mutua inter-crescita, rendendo nella pratica non economicamente vantaggioso separare il minerale.
Nel caso del giacimento più noto (Willsboro, nello stato di New York, Stati Uniti) la situazione litologica è caratterizzata da fasce di skarn (roccia metamorfica di contatto a mineralogia ibrida) costituite prevalentemente da calcari, dolomie e silice, addossate a rocce feldspatiche (anortositi).
Dallo skarn si estrae la wollastonite attraverso un processo di frantumazione, classificazione, flottazione (che porta alla concentrazione del minerale utile) e separazione magnetica ad alta intensità (che consente di separare il granato).
Nel giacimento di Lappeenranta (Finlandia) le fasce mineralizzate si trovano nella zona di contatto tra granito e rocce carbonatiche (calcare e dolomite) che risultano attraversate anche da filoni di anfibolite e pegmatite. I granati sono completamente assenti. Il processo di trattamento prevede macinazioni successive, la separazione della wollastonite da calcite e quarzo (per flottazione) e la deferrizzazione magnetica per eliminare i minerali di ferro. In natura è invece piuttosto rara la pseudowollastonite che si forma per devetrificazione di vetri.
Ubicazione dei principali giacimenti
I principali depositi in coltivazione si trovano negli Stati Uniti (New York e California), Finlandia (Lappeenranta), Messico, Kenya, Russia, India (Rajasthan), Sud-Africa, Sudan, Spagna, Giappone, Nuova Zelanda e Serbia.
Wollastonite sintetica
Viene prodotta artificialmente partendo da calcite e fireclay silicea (argilla caolinitica a struttura disordinata).
Composizione mineralogica di rocce wollastonitiche e metodi di riconoscimento
La diffrattometria ai raggi X (Fig. 3.44) permette di riconoscere le specie mediante osservazione delle diffrazioni dei piani basali. Quando si utilizzano radiazioni CuKα si ottengono i valori della seguente tabella:
Wollastonite
Posizione angolare dei picchi °(2θ) 23,2 25,3
reticolari
Si tratta di un minerale molto stabile che non dà luogo a perdite di materiale o trasformazioni allotropiche fino a 1000 °C per cui non si possono ottenere informazioni dalla DTA o dalla TG. Naturalmente qualora siano presenti anche minerali carbonatici, si osserveranno le loro specifiche reazioni termiche. La dilatometria del crudo presenta un andamento progressivo molto lineare e con modesto gradiente.

Caratterizzazione tecnologica di impasti contenenti wollastonite Piastrelle formate con wollastonite grezza (fino a un massimo del 40%) e umidificazione con acqua mostrano:
Fig. 3.45
- normale espansione di post-pressatura;
- minima resistenza meccanica di crudi ed essiccati;
- nessun ritiro di essiccazione;
- minimo ritiro dimensionale dei cotti a 1020 °C (0,3%);
- resistenza meccanica dei cotti molto bassa fino a 1060 °C, poi > 10 N/mm2.
Utilizzi ceramici
Il principale utilizzo è nella fabbricazione di piastrelle da rivestimento e pavimento. Il suo impiego è rilevante, specialmente negli Stati Uniti e nei paesi nordici, visto il costo limitato del minerale in queste aree geografiche.
In monocottura l’aggiunta di wollastonite contribuisce al miglioramento della resistenza meccanica dei supporti, diminuisce il ritiro in cottura e consente di ridurre i cicli dei forni; nel grès porcellanato è utilizzata anche con funzione sbiancante.
La wollastonite si comporta in crudo da smagrante (come le sabbie) mentre in cottura agisce (oltre 1050 °C) da fondente e, durante la fase di raffreddamento, non presenta la variazione di volume a 573°C caratteristica del quarzo e causa di possibili rotture (sfilo).
Si può inoltre rimarcare come l’introduzione di questo minerale in composizioni ceramiche migliori l’uniformità dimensionale dei pezzi e faciliti l’accordo con gli smalti.
Ulteriori caratteristiche dei supporti ceramici ottenuti da impasti a base wollastonitica sono: bassa dilatazione termica, lucentezza, superficie liscia e minima tendenza al rigonfiamento.
La wollastonite è inoltre impiegata nella formulazione di smalti (dal 5 al 20%), in particolare per sanitari e ceramica fine, in quanto migliora l’intervallo di fusione ed aumenta la lucentezza.
In piccole percentuali viene introdotta nelle composizioni di alcuni prodotti refrattari (ad esempio caselle e supporti) per migliorarne la resistenza agli shock termici e agli urti.
Utilizzi in settori diversi
Circa il 50 ÷ 60% della produzione mondiale di wollastonite trova utilizzo nel campo ceramico (supporti e smalti), il rimanente viene impiegato come filler per vernici, plastica e nell’industria vetraria.
Origine dei termini
Calcite: il nome deriva dal latino calx, termine usato dagli antichi Romani per indicare la calce, cioè l’ossido di calcio (CaO) ottenuto per trattamento termico del calcare (CaCO3).
Dolomite: dal cognome del geologo francese D. Dolomieu (1750-1801) che per primo la distinse dal calcare.
Aragonite: dalla regione spagnola di Aragona dove venne identificato il minerale.
Natura e composizione chimica
I principali minerali carbonatici sono:
Calcare
Aragonite
Dolomite
CaCO3
CaCO3
CaMg(CO3)2
Magnesite MgCO3
Struttura cristallina
I differenti minerali carbonatici cristallizzano nei seguenti sistemi:
Calcite romboedrico
Aragonite ortorombico
Dolomite romboedrico
Magnesite romboedrico
L’aragonite si trasforma irreversibilmente in calcite intorno a 500 °C.
Genesi e caratteristiche dei giacimenti
Calcare
Si origina in ambiente marino per deposizione di tipo chimico (precipitazione) da soluzioni saline soprassature e per accumulo di resti minerali carbonatici di organismi marini morti. Le condizioni che favoriscono la sua formazione sono: acque calde, temperatura ambientale elevata, ventilazione e scarso ricambio delle acque (mari chiusi).
A questa fase sedimentaria fa seguito un processo diagenetico durante il quale i costituenti originali scompaiono e vengono sostituiti da calcite ricristallizzata.
Spesso i giacimenti calcarei presentano impurità dovute a vari materiali (dall’argilla alla sabbia) sedimentati contemporaneamente alla precipitazione chimica e all’accumulo organogeno (resti di organismi animali e vegetali).
Talvolta nello stesso giacimento si osservano zone laterali o verticali nelle quali si concentra la presenza di impurezze. Taluni inquinanti (fosfati e solfuri) possono anche rendere inutilizzabile il giacimento.
Dolomite
La genesi delle dolomiti è simile a quella dei calcari; tuttavia, durante la diagenesi, circa la metà dei cationi Ca2+ vengono sostituiti da Mg2+ (presenti nelle soluzioni marine saturanti), dando luogo a regolari stratificazioni tra i due carbonati. L’aumento della pressione che si accompagna all’accumulo del materiale favorisce la sostituzione in quanto il magnesio è di dimensioni minori del calcio e trova una più facile sistemazione nel reticolo cristallino.
Magnesite
La genesi è identica a quella della dolomite con la quale si trova spesso associata. Si può dire che la magnesite rappresenta lo stadio finale della sostituzione del calcio da parte del magnesio. Si distingue un tipo cristallino da un altro criptocristallino. Per quest’ultimo la genesi ipotizzata è attribuita a reazioni tra acque ricche in carbonati e rocce silicatiche ricche in magnesio.
Ubicazione dei principali giacimenti e tecniche di coltivazione
Calcare
La diffusione di queste rocce è ubiquitaria; in Europa la loro genesi è concentrata in alcune ere geologiche (Devoniano, Triassico, Giurassico, Cretaceo e Terziario) ed ha portato alla formazione di parti rilevanti di importanti catene montuose (Alpi, Appennini, Carpazi, ecc.).
Alcuni depositi recenti, concentrati ad esempio nel Nord-Africa (tufo) e nel mar dei Caraibi (caliza arrecifal) non hanno subito fenomeni diagenetici e si presentano in uno stato semicoerente. Per l’estrazione delle rocce calcaree, pur non particolarmente dure, è comunque necessario ricorrere all’uso di esplosivi, mentre i depositi semicoerenti possono essere coltivati con l’impiego di mezzi per movimento terra.
Dolomite
Anche i giacimenti dolomitici (emblematici della omonima catena montuosa italiana) sono ampiamente diffusi. La loro genesi si è concentrata nelle ere Siluriana, Devoniana, Triassica e Giurassica. Anche per la coltivazione delle dolomiti è necessario ricorrere all’uso di esplosivi.
Magnesite
I principali giacimenti con mega-cristallizzazione sono ubicati in Austria, Russia, Cechia, Spagna, Brasile, Cina, Stati Uniti, Canada e Australia. I depositi criptocristallini hanno forma filoniana e sono contenuti in serpentini e rocce ultrabasiche. I principali depositi di questo tipo sono ubicati in Grecia (Macedonia), regione Balcanica, Austria (Stiria), Turchia (Eskişehir), India, Stati Uniti, Russia e Canada.
Tipici comportamenti di riconoscimento
Calcare
Dall’analisi diffrattometrica (vedi Fig. 3.45) si evidenziano i seguenti picchi:
Calcare Posizione
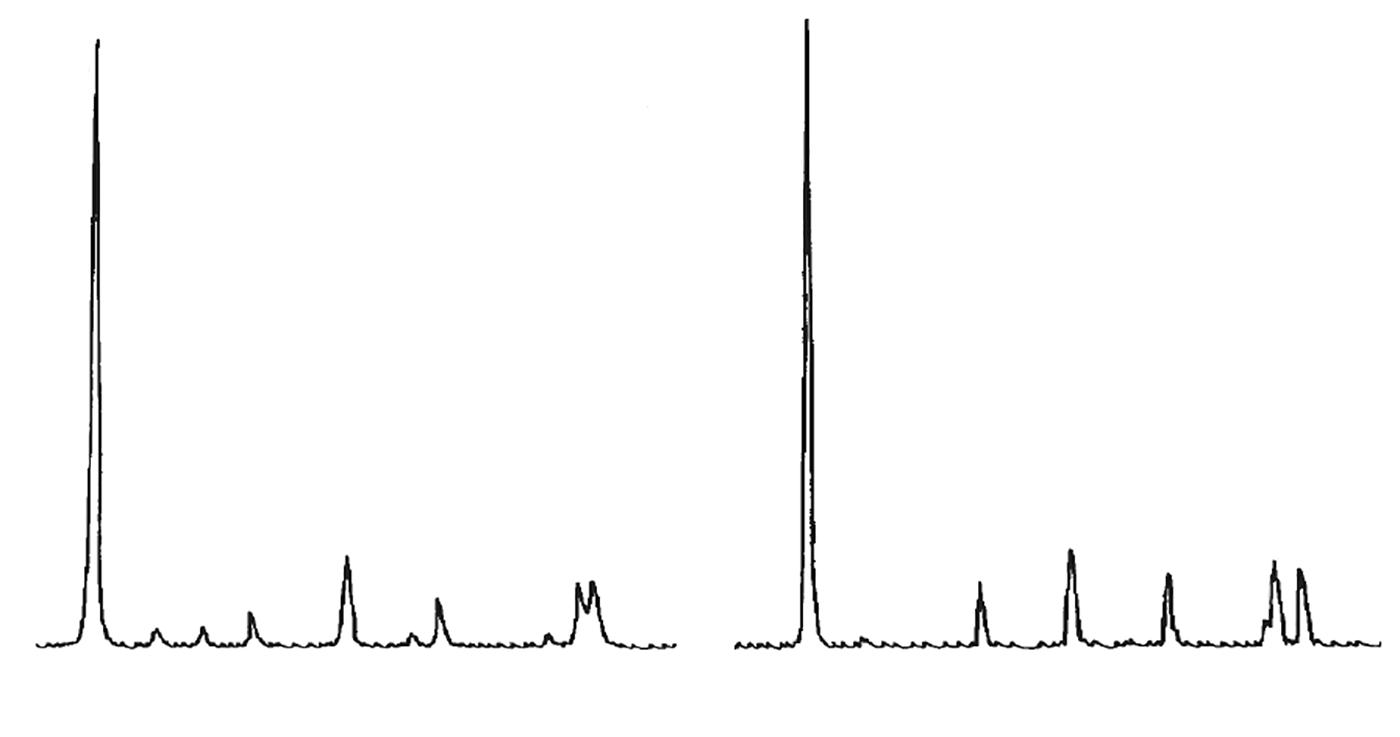
Fig. 3.45 Analisi diffrattometrica della dolomite e della calcite (D=dolomite, C=calcite)
Fig. 3.46
L’analisi termo-differenziale (DTA) di Fig. 3.46 mostra un marcato picco endotermico attorno a 920 °C in corrispondenza della dissociazione del minerale e dell’allontanamento dell’anidride carbonica (CaCO3 → CaO + CO2). Alla stessa temperatura l’analisi termo-gravimetrica mostra una marcata perdita di peso dovuta all'emissione della CO2
Dolomite
L’analisi diffrattometrica (vedi Fig. 3.45) evidenzia i seguenti picchi caratteristici: Dolomite
L’analisi termo-differenziale (DTA) mostra chiaramente come nel caso della dolomite la dissociazione abbia luogo in due tempi; rispettivamente a circa 780 e 920 °C.
Anche la TG (termo-gravimetria) evidenzia l’allontanamento dell’anidride carbonica alle medesime temperature. Il comportamento del minerale è quello di una miscela tra carbonati di calcio e di magnesio.
Fig. 3.46 Curve termiche differenziali di calcite (a) e dolomite (b)
Magnesite
L’analisi termo-differenziale (DTA) presenta il picco endotermico connesso con la decomposizione del minerale e l’allontanamento della CO2 ad una temperatura variabile tra i tra 600 e 730 °C. Corrispondentemente la termo-gravimetria (TG) mostra un forte gradiente in tale intervallo.
3.47
Influenza sui materiali pressati
Le variazioni di comportamento tecnologico che sussistono tra una argilla priva di carbonati ed una miscela di questa con carbonati (calcite e dolomite) sono particolarmente marcate durante la cottura. Infatti, la dissociazione e la liberazione di anidride carbonica comportano l’aumento della porosità e l’espansione dimensionale del supporto. Ad esempio, in Tab. 3.2 si forniscono i dati di cottura di due argille dell’Appennino emiliano, una quasi priva di carbonati (B) ed una che ne contiene il 15% (A), entrambe macinate a secco.
Tab. 3.2 Influenza dei carbonati in due argille dell’Appennino emiliano
L’argilla A è una classica composizione per cottoforte ottenuta mescolando in cava un’argilla molto carbonatica (circa il 20%) con una argilla quasi priva, come quella del tipo B. È importante osservare che la presenza di carbonati tende a restringere il palier di vetrificazione. È pertanto fondamentale che le composizioni per supporti a basso assorbimento (< 2%) non contengano più del 3 ÷ 4% di carbonati. Una presenza ridotta entro questi termini può risultare utile ai fini dell’allontanamento delle sostanze organiche contenute nelle argille. In questo modo si possono ridurre i fenomeni di rigonfiamento e di cuore nero.
Utilizzi ceramici
I calcari e la dolomite sono materie prime fondamentali per la produzione di materiali ceramici a basso ritiro, come biscotti porosi da rivestimento e piastrelle smaltate prodotte in monocottura per pavimento e rivestimento (monoporosa). Nel biscotto, la loro presenza (fino a 20 ÷ 22%) consente di produrre supporti con valori di assorbimento ideali (particolarmente ai fini della smaltatura) e praticamente senza ritiro (o con un ritiro massimo di 0,5 ÷ 0,6%). Questa ultima caratteristica ha consentito di adottare, nel passato, una tecnologia di cottura su alte pile che, in caso di ritiro di cottura più elevato non avrebbe potuto essere praticabile. In monocottura da rivestimento i carbonati sono utilizzati (quale correttivo di argille calcaree naturali) fino ad una percentuale di CaCO3 del 12 ÷ 15% e conferiscono, grazie alla formazione dei silicati di calcio e magnesio, un bassissimo ritiro in cotto delle piastrelle (< 1%); la loro presenza, però, complica la progettazione e gestione della cottura (soprattutto in preriscaldo), poiché la reazione di dissociazione termica dei carbonati provoca emissione di CO2, che deve poter attraversare lo strato di smalto senza formare bolle e crateri (pinhole). È possibile anche effettuare l’aggiunta di carbonati nella barbottina sotto forma di polvere a granulometria già fine, convertendo ad esempio un impasto per grès porcellanato, in un nuovo impasto idoneo per la produzione di piastrelle porose da rivestimento.
La presenza di carbonati porta poi, solitamente, ad un aumento del coefficiente di dilatazione che meglio si adatta alla maggior parte degli smalti in commercio.
I carbonati sono largamente utilizzati per la produzione di maiolica e terraglia.
La dolomite e la magnesite vengono anche utilizzate per produrre refrattari basici.
Argille ricche in carbonati vengono solitamente impiegate nella fabbricazione di materiali trafilati. In queste produzioni si può presentare il fenomeno cosiddetto dei “calcinelli”, termine con cui si definiscono frammenti calcarei di dimensioni che possono anche superare il centimetro e che derivano da deposizioni di origine chimica. Talvolta sono dovuti anche a frammenti di gusci di antichi molluschi marini (bivalvi, gasteropodi, brachiopodi, ecc.). In entrambi i casi, qualora le materie prime che contengono questi composti calcarei vengano trafilate o macinate in maniera grossolana (per via secca), si ha l’insorgenza di difetti superficiali. Nel caso delle piastrelle si osserva la formazione sul supporto di punti bianco-giallastri affondati che deteriorano la superficie del biscotto e portano a puntinature dello smalto.
Nel caso dei laterizi i calcinelli, parzialmente decomposti dopo cottura, possono espandere (anche quando il prodotto è già posato) per idratazione con acqua o per riassorbimento di CO2 dell’aria, provocando scagliature e rotture dei pezzi particolarmente critiche nel caso di muri “faccia a vista”.
Utilizzi in settori diversi
Il principale impiego del calcare è la produzione di cemento e di calce; per questi prodotti è basilare la minima presenza di magnesio. Ultimamente, calcari molto puri stanno sostituendo il caolino come carica inorganica della carta, in quanto più economici. La magnesite trova invece impiego come carica minerale inorganica nella fabbricazione di materiali isolanti e come supporto per fertilizzanti e prodotti chimici.
3.3.1
Questo termine comprende varie tipologie di fillosilicati tra le quali, oltre ai minerali illitico-cloritici già descritti, le più diffuse sono:
biotite
muscovite
6SiO2·Al2O3·6(Mg,Fe)O·K2O·2H2O
6SiO2·3Al2O3·K2O·2H2O
Dalla decomposizione di queste, per un processo di idratazione, deriva poi la sericite
6SiO2·3Al2O3·K2O·4H2O
La struttura di questi minerali è simile a quella dell’illite; si hanno infatti due piani di tetraedri silicei, tra i quali è compreso un piano di ottaedri di alluminio. Le parziali sostituzioni del silicio con alluminio che creano squilibri di cariche elettriche vengono compensati dall’introduzione di nuovi cationi.
La biotite (o mica ferrifera) ha colore bruno-nerastro, è lucida ed è uno dei principali costituenti dei graniti insieme a quarzo e feldspati, per cui è spesso presente, come inquinante, nei giacimenti di feldspato coltivati. Durante il processo di alterazione delle rocce madri (weathering, metamorfismo di contatto, ecc.) la biotite tende ad alterarsi con una certa facilità trasformandosi in sericite, per cui difficilmente si ritrova nelle rocce sedimentarie.
La muscovite (o mica potassica) si presenta sotto forma di lamelle di aspetto metallico, molto brillanti, flessibili, facilmente sfaldabili e di forma esagonale. Tali lamelle possono raggiungere dimensioni enormi (anche oltre il metro) come nel caso di giacimenti indiani. Anche questo minerale è presente in certi tipi di graniti e, per alterazione, si trasforma in illite; quindi è piuttosto facile ritrovarla in rocce sedimentarie (argille e sabbie) dove talvolta raggiunge percentuali rilevanti (anche oltre il 10%).
La sericite si presenta sotto forma di frammenti piatti allungati o di aghi con colore verdastro. In talune rocce sedimentarie di natura litoide e non carbonatiche (come certe arenarie) costituisce, unitamente agli idrossidi di ferro, il cemento che lega i vari componenti.
L’analisi diffrattometrica (radiazioni CuKα) per biotite e muscovite evidenzia i seguenti picchi:
Biotite
Muscovite
La DTA delle miche (biotite e muscovite) è sostanzialmente piatta fino al picco endotermico di deidrossilazione (rimozione dei gruppi OH–) tra 850 e 920 °C. Nel caso della biotite, qualora questa contenga ferro bivalente, si può osservare un marcato picco esotermico tra 400 e 500 °C dovuto alla sua ossidazione (Fig. 3.47).
Muscovite
Biotite
Zinnwaldite
Glauconitic clay
Illite (Fithian) Illite (OECD)
Fig. 3.47 Curve termiche differenziali di alcune tipologie di miche
La biotite costituisce un notevole problema per i giacimenti di feldspato; si cerca di allontanarla operando con separatori magnetici ad intensità molto elevata. Non è però infrequente che i separatori attraggano anche particelle di feldspato e di quarzo caricatesi elettricamente durante la macinazione. In questi casi ci si trova di fronte all’alternativa di una separazione molto parziale della biotite (con il risultato di commercializzare un prodotto che dopo cottura sarà di colore grigio-nerastro e presenterà puntinature nere), oppure di una perdita consistente della produzione a causa delle quantità di materiale scartato dai separatori magnetici.
Fig. 3.48
Anche la muscovite, quando raggiunge percentuali rilevanti, può costituire un problema notevole. È ben noto il caso del Cornish stone inglese la cui estrazione è cessata proprio perché il giacimento è andato via via arricchendosi di questo materiale. La muscovite può essere allontanata dagli altri componenti mediante “separazione a vento” e flottazione. Tuttavia, spesso tali processi risultano troppo costosi e pongono il prodotto così trattato fuori mercato rispetto ad altri materiali concorrenti che non richiedono impianti di separazione.
Come accennato in precedenza, la muscovite è spesso presente anche nelle rocce sedimentarie, ad esempio in varie argille dell’Appennino emiliano (Italia). Si tratta comunque sempre di percentuali molto contenute che non costituiscono alcun problema pratico produttivo.
Sui supporti porosi cotti a temperature di 1000 ÷ 1020 °C la presenza della mica è facilmente rilevabile ad occhio nudo. Nel caso dei supporti greificati la componente potassica agisce da fondente e contribuisce alla vetrificazione del materiale. Inoltre, è opportuno evi-
denziare come la struttura lamellare della muscovite la renda difficilmente macinabile; questo comportamento è più marcato nel caso della tecnologia di macinazione “ad umido”, dove possono verificarsi intasamenti delle reti dei setacci durante la vagliatura della barbottina. È pertanto consigliabile avvalersi di vagli che eliminano con particolare efficienza la frazione grossolana di scarto.
Infine, si evidenzia come nelle miche avvenga una parziale sostituzione degli ossidrili OH– con F–; il fluoro, rilasciato nei fumi del forno nella fase di cottura sotto forma di acido fluoridrico HF, verrà poi abbattuto dai filtri a calce.
Si tratta principalmente degli idrossidi di:
- alluminio: gibbsite (o idrargillite) Al(OH)3 diasporo AlO(OH) boehmite Al2O3·H2O
- ferro: goethite FeO·OH
Gli idrossidi di alluminio sono i principali costituenti delle bauxiti.
Le curve relative alle analisi termo-differenziali e termo-gravimetriche indicano che, per le varie specie mineralogiche, la disidratazione ha luogo alle seguenti temperature:
Intervallo di disidratazione (°C) gibbsite
240 ÷ 380 diasporo
410 ÷ 570 bohemite
450 ÷ 580 goethite > 250
Gli idrossidi sono caratterizzati da importanti frazioni colloidali e rappresentano un problema durante la fase di essiccazione e preriscaldo in quanto l’eliminazione degli ossidrili comporta variazioni dimensionali dei pezzi.
La Tab. 3.3 riporta gli indici diffrattometrici (distanze reticolari e intensità relative) per vari ossidi e idrossidi. In Fig. 3.48 si riporta invece la DTA di alcuni idrossidi minerali.
Tab. 3.3 Principali riflessioni degli ossidi e idrossidi di ferro, alluminio, magnesio e titanio
Con tale termine si identifica una serie di composti inorganici (carbonati, solfati, cloruri alcalini e alcalino-terrosi, ecc.) che hanno in comune la proprietà di solubilizzarsi in acqua. La loro solubilità varia sensibilmente da caso a caso, passando ad esempio da valori molto elevati per il cloruro di sodio (357 g/L a 20 °C) a valori minimi per il carbonato di calcio (13 mg/L a 20 °C), tanto da ritenerlo praticamente insolubile. Nella Tab. 3.4 si riportano i valori della solubilità in acqua dei principali sali di interesse ceramico.
Solubilità in acqua [g/L] NaCl
MgCl2
Mg(OH)2 0,009
MgSO4 260
CaCl2 595
CaCO3 0,013
CaMg(CO3)2 0,32
Ca(OH)2 1,85
CaSO4 2,09
CaSO4·2H2O 2,41
BaCl2
Tab. 3.4 Solubilità in acqua di alcuni sali minerali
I più importanti sali solubili sono: - solfati di Na, K, Ca, Mg, Al e Fe - solfati doppi di Na+Al e K+Al - carbonati di Na, K e Ca
- cloruri di Na e K.
In particolare il carbonato di calcio, poco solubile in acqua, è largamente presente in tutte le argille impiegate nella produzione di supporti porosi. Un altro sale solubile comunemente presente nelle argille rosse è il solfato di ferro che si genera per ossidazione dei solfuri (pirite e marcasite) secondo la reazione:
FeS + H2O + 7/2 O2 → FeSO4 + H2SO4
La formazione dei giacimenti di materie prime ceramiche sedimentarie è strettamente legata a quella dei composti minerali solubili. La minor o maggior presenza di questi minerali è dovuta al raggiungimento di condizioni di precipitazione nelle acque marine contemporaneamente alla deposizione di fini particelle da sospensioni di provenienza continentale. Tali meccanismi si riferiscono alla presenza di sali minerali nei depositi argillosi di origine sedimentaria. Tuttavia, anche i giacimenti di genesi continentale (per lo più lacustri) che derivano dal disfacimento di rocce preesistenti, possono essere a loro volta di origine sedimentaria e quindi contenere sali solubili. Spesso si tratta di distribuzioni diffuse, microscopiche, non visibili se non nei periodi di siccità quando l’evaporazione dell’acqua lascia un deposito salino sulla superficie dei banchi argillosi che si presentano con caratteristici veli biancastri. Talvolta si osservano concentrazioni rilevanti in corrispondenza di depressioni del terreno alla base delle formazioni argillose, dove si accumulano le acque piovane che costituiscono il veicolo dei sali. In altri casi le dimensioni diventano macroscopiche, come nel caso di cristalli di gesso di qualche millimetro, dai caratteristici abiti lenticolari, a ferro di lancia o prismatici. In questo raggruppamento si possono poi includere anche i calcinelli, agglomerati carbonatici di forma arrotondata e colore chiaro, ben noti ai fabbricanti di prodotti trafilati.
In generale la distribuzione dei minerali solubili è casualmente dispersa su uno o più banchi di una formazione argillosa. A volte invece si osservano deposizioni concentrate in spessori di pochi millimetri che seguono esattamente i piani di sedimentazione, separando due banchi contigui. In altri casi ancora i sali si concentrano lungo fratture presenti nel deposito a causa dei fenomeni di dissoluzione e ricristallizzazione esercitate dalle acque d’impregnazione (diagenesi), senza raggiungere le condizioni metamorfiche che trasformerebbero questi giacimenti in scisti o shale
Un aspetto molto importante da considerare nel processo ceramico riguarda le interferenze dei sali solubili sulla reologia delle barbottine; ad esempio, la presenza dei sali di sodio e potassio può favorire la deflocculazione, mentre quelli di calcio, magnesio e ferro provocano flocculazione.
Anche la tissotropia delle sospensioni ceramiche, che dipende dalle proprietà superficiali delle particelle colloidali di argilla, è influenzata in maniera sensibile dagli elettroliti che modificano le cariche elettriche delle particelle. È noto ad esempio che la quantità di cloruri alcalini presenti è direttamente proporzionale alla tendenza alla gelificazione di sospensioni di argille caolinitiche, mentre è inversamente proporzionale nel caso di barbottine costituite da montmorilloniti. Risulta quindi evidente che la presenza di sali solubili può agire in maniera contrapposta dipendentemente dai minerali argillosi presenti.
I comportamenti in cottura dei principali sali solubili sono caratterizzati da pirolisi con conseguente eliminazione della fase gassosa e da fenomeni di disidratazione. Ad esempio, la perdita delle molecole d’acqua del gesso avviene a bassa temperatura e si sovrappone a quelle dei minerali argillosi a reticolo espandibile, contribuendo così al ritiro totale del pezzo nella fase di essiccazione e preriscaldo.
A riguardo, l’analisi termo-differenziale (DTA) ci offre utili indicazioni circa le reazioni eso- ed endo-termiche che hanno luogo durante il riscaldamento. Il gesso (solfato di calcio bi-idrato) mostra un primo picco endotermico a 140 °C (trasformazione in semi-idratato), un secondo, più piccolo, a 150 °C (trasformazione in anidrite) ed uno, minuscolo, esotermico, a 350 °C in corrispondenza del passaggio dell’anidrite solubile in insolubile; a 1125 °C l’anidrite passa dal tipo α a quello β (piccolo picco endotermico) ed hanno inizio i fenomeni di dissociazione in CaO e SO3.
La pirolisi dei cloruri invece si completa entro 700 °C; il solfato di magnesio comincia a dissociarsi verso 900 °C, mentre nel caso del solfato di sodio tali reazioni proseguono anche oltre 1000 °C. Pertanto, ne deriva che la neoformazione dei solfati alcalini ed alcalino-terrosi vengono annullate quasi completamente nei supporti cercamici che sinterizzano a temperature superiori a 1000 °C.
Infine, nel caso del solfato di ferro l’inizio dei fenomeni di pirolisi ha luogo verso 170 °C, mentre per il solfato di magnesio si osserva solo dopo 400 °C.
Le conseguenze dei fenomeni citati trovano rilevanza crescente con l’aumentare della rapidità dei cicli di cottura. Infatti, certe reazioni quasi trascurabili nei cicli dei tradizionali forni a tunnel, nei cicli rapidi ed ultrarapidi delle attuali monocotture possono determinare problematiche tali da richiedere l’esclusione di certe materie prime. Ad esempio, nel caso di piastrelle prodotte in monocottura, lo smalto crudo ricopre un supporto non stabilizzato da una prima cottura. Qualora le reazioni di pirolisi con emissione di gas avvengano in una fase della cottura in cui lo smalto non abbia una adeguata fluidità, si possono formare piccoli “crateri” che permangono poi nel prodotto finito. In pratica gli smalti da monocottura si lasciano attraversare dalle bolle di gas senza difficoltà solo nei casi estremi di alta e bassa fusibilità. Nel primo caso l’elevata fluidità a bassa temperatura permette la richiusura dei crateri formatisi con la rottura delle bolle in superficie. Nel secondo, lo smalto è fusibile solo ad alta temperatura e pertanto, durante i processi di pirolisi dei sali solubili, ha ancora una struttura granulare porosa che viene facilmente attraversata dai gas.
È stato anche ipotizzato un concorso dei sali solubili al problema del “cuore nero” la cui origine risiede comunque nella presenza di carbonio organico (non minerale) e di ferro.
Una ulteriore conseguenza della presenza dei sali solubili sui pezzi cotti, non smaltati, è la comparsa di efflorescenze. Con tale termine ci si riferisce ad una deposizione salina che ha luogo sulle superfici del biscotto (ma talvolta anche dell’essiccato) per precipitazione dei sali minerali conseguente al raggiungimento delle condizioni di saturazione.
Tale fenomeno, che è caratteristico dei laterizi, ma può comparire anche sulle piastrelle trafilate e pressate, si manifesta solo quando il corpo ceramico abbia porosità sufficiente per consentire, al suo interno, migrazioni di soluzioni saline che, una volta raggiunta la superficie, sono soggette ad una rapida evaporazione dell’acqua. Naturalmente è necessario che la materia prima contenga un contenuto relativamente elevato di tali sali (oltre a ossidi alcalini e calcare) o che questi si formino in cottura per pirolisi della pirite o in presenza di zolfo nel combustibile. La problematica della presenza di sali ha una elevata probabilità di manifestarsi quando il loro contenuto nell’impasto supera lo 0,5%.
In ordine di frequenza (sui materiali essiccati e cotti) le efflorescenze sono costituite da: solfati di sodio, calcio, potassio e magnesio, carbonati degli stessi metalli, ed ancora cloruri e nitrati alcalini. Nei supporti cotti possono anche essere presenti solfoalluminati e carbonati alcalini.
La formazione delle efflorescenze è condizionata dai fattori che determinano la diffusione delle soluzioni saline all’interno dei supporti cotti (dimensioni e numero dei pori) e dalle modalità di evaporazione dell’acqua che in certi casi può aver luogo, anziché sulla superficie, internamente al corpo ceramico.
Nel caso della migrazione di sali sulla superficie di piastrelle pressate, tipicamente sui biscotti destinati alla bicottura per rivestimento, si può così riscontrare un accumulo di sali, contenenti alcali, nelle zone soggette a maggiore riscaldamento in essiccazione, cioè ai bordi della piastrella; questo fenomeno, che avviene anche a causa della imbibizione del biscotto
durante la smaltatura, ha come conseguenza estrema l’alterazione della composizione chimica degli smalti in prossimità del bordo della piastrella, a causa di un maggior apporto di sali fusibili, che rendono tale zona ancora più sensibile alle variazioni di temperatura. Ciò può portare ad una “cornice” nelle piastrelle cotte, per la comparsa di una zona troppo fusa al bordo, il cui aspetto risulta in genere più opaco a causa delle bollicine di aria contenute nel vetro.
Un analogo fenomeno dovuto alla presenza di migrazione di sali è riscontrabile nella “rigatura” delle piastrelle di grès porcellanato, a causa dei sali che si accumulano in corrispondenza degli appoggi delle piastrelle durante l’essiccazione, generando strisce con maggior contenuto di fasi vetrose dopo cottura, di aspetto più lucido.
In entrambi i casi la possibile soluzione, oltre che, ovviamente, cercare di eliminare le materie prime apportatrici di sali, consiste nel variare drasticamente il ciclo di essiccamento, accelerandolo al limite della tenuta meccanica dei pezzi, per favorire una rapidissima evaporazione dell’acqua, che non consenta la migrazione e l’accumulo di sali in zone preferenziali delle piastrelle.
Le sostanze vegetali presenti nelle materie prime argillose (si vedano alcuni esempi in Fig. 3.49) possono essere distinte in due gruppi principali: - materiali non carbonizzati - materiali carbonizzati.
Tra i primi possiamo annoverare radici, frammenti legnosi, foglie ed acidi umici; si tratta di elementi presenti per lo più in depositi recenti o connessi con lo strato pedologico e con una imperfetta pulizia del terreno vegetale prima dell’inizio dell’attività estrattiva.
Queste sostanze vegetali vengono normalmente eliminate durante il processo di cottura, eventualmente anche con l’aiuto di additivi ossidanti appositamente aggiunti nell’impasto; la loro combustione ha inizio attorno a 250 °C e termina a 450 °C circa.
Fig. 3.49 Struttura di cellulosa e lignina
3.50
Il secondo gruppo è costituito da vari tipi di carbone e generalmente si trova in formazioni geologiche più antiche, risultando notevolmente più stabile. Questi materiali restano normalmente passivi al trattamento con ossidanti e, in cottura, bruciano a temperature comprese tra 300 e 600 °C.
La presenza delle sostanze vegetali causa vari problemi sia nella produzione di supporti per bicottura che per monocottura.
Nel caso di monocottura rapida, specie con cicli inferiori ad un’ora nella produzione di supporti a basso assorbimento e quando si è in presenza di minerali di ferro (pasta rossa), si osserva la formazione di macchie ellissoidali nere centrali (“cuore nero”) e, nei casi più gravi, di forti rigonfiamenti. L’origine di questo fenomeno deriva dall’incompleta combustione delle sostanze organiche con formazione di carbonio grafitico che annerisce il corpo ceramico mentre la CO2 della parte combusta, che non riesce ad attraversare il pezzo, provoca il rigonfiamento.
In presenza di sali inorganici poi, si ha la contemporanea formazione di composti come Na2SO4, che contribuiscono alla formazione di fasi vetrose, rese nere o scure dalla presenza di cationi cromofori, quali: Fe, Cu, Cr, ecc.
Anche in presenza di solfuri minerali, ad es. di ferro o rame, si verificano complesse reazioni che portano a combinazioni tra la silice ed i prodotti di decomposizione dei solfuri (Fig. 3.50) ed all’ottenimento di un vetro nero molto fusibile.
FeS2 + O2 → FeS + SO2
4FeS + 9O2 → 2Fe2O3 + 4SO3
2FeSO4 + 1/2O2 → Fe2O3 + 2SO3
[350 ÷ 450 °C]
[500 ÷ 800 °C]
[560 ÷ 775 °C]
Naturalmente, nel verificarsi o meno del fenomeno, concorrono altre cause quali: umidità e dimensioni delle polveri, pressione di formatura, curva di cottura ed atmosfera del forno.
Gli interventi correttivi per ridurre la problematica del “cuore nero” possono essere i seguenti:
- introduzione di smagranti nella composizione (sabbie, lapilli, chamotte);
- addizione di ossidanti nella composizione (MnO2, nitrati, ecc.);
- macinazione meno spinta dei componenti l’impasto;
- umidificazione più contenuta delle polveri (compatibilmente con la resistenza meccanica dei crudi);
- pressione di formatura inferiore;
- curva di cottura più appropriata, ovvero prolungando al massimo l’intervallo tra 250 e 600 °C;
- atmosfera dei forni quanto più possibile ossidante.
3.3.5
Talvolta certe materie prime feldspatiche o argillose contengono zolfo nativo di colore variabile tra il biancastro ed il giallo tipico. Solitamente si tratta di materie prime aventi origine connessa ad aree vulcaniche. Classico è l’esempio dei materiali di questo tipo esistenti nel Lazio (Italia centrale).
Lo zolfo porta notevoli inconvenienti in quanto in cottura si trasforma in anidride solforosa ed attacca la struttura interna dei forni; la presenza di anidride solforosa comporta anche problemi di tipo ambientale sia all’interno che all’esterno dello stabilimento.
La presenza di solfuri di ferro (pirite e marcasite) non è rara in molte argille utilizzate nella fabbricazione di piastrelle come, ad esempio, in quelle dell’Appennino emiliano (Italia).
La pirite causa la formazione delle classiche “pulci”, cioè di crateri con un nucleo centrale di colore scuro che interessano il biscotto e generalmente provocano una antiestetica puntinatura sugli smalti. Il meccanismo che determina questo difetto è probabilmente dovuto alla presenza di cristalli di pirite in prossimità della superficie dei supporti; attorno a 400 °C avviene l’azione ossidante che trasforma il solfuro in ossido con allontanamento di SO2. Tale processo è accompagnato da aumento di volume e provoca la “scagliatura” della porzione di supporto che ricopre il cristallo, causando la formazione di un mini-cratere.
L’alunite è un solfoalluminato di potassio idrato avente la seguente formula:
K2O∙3Al2O3∙4SO3∙6H2O
Si tratta di un minerale che si genera nelle condizioni idrotermali che spesso portano a caolinizzazioni dei filoni feldspatici. Per tale motivo l’alunite è spesso presente congiuntamente al caolino, come nel caso del famoso giacimento caolinitico di Djebel Debar (Algeria). Osservando la DTA di questo minerale si riscontra un primo picco endotermico a 550 °C (perdita dell’acqua) ed un secondo a 850 °C (allontanamento di parte dell’anidride solforica), con formazione di allumina e di solfato di potassio. Inoltre, è presente un picco esotermico poco oltre i 700 °C.
La liberazione in fase di cottura dell’anidride solforica comporta gli inconvenienti già richiamati in precedenza.
[1] «Tonmineralogie - Dr. Krakow Rohstoffe» [Online]. Available: https://www.dr-krakow-labor.de/tonmineralogie.
[2] W. D. Kingery, Introduction to Ceramics, Wiley, 1976.
[3] G. Peco, I prodotti ceramici. Dalla tradizione all’alta tecnologia, Marzorati, 1991.
[4] C. Fiori e B. Fabbri, «Mineralogical composition of the clay bodies used in the Italian tile industry», Applied Clay Science, vol. 4, n. 5-6, pp. 461-473, 1989.
[5] W. Zachariasen, Journal American Ceramic Society, n. 17, p. 3841, 1932.
[6] B. Warren, Journal American Ceramic Society, n. 17, p. 249, 1934.
[7] D. Worrall, Clays and ceramic raw materials, Elsevier, 1986.
[8] A. Ravaglioli e G. Pollifrone, Materie prime ceramiche - Vol.1 (materiali argillosi), Faenza Ed., 1977.
[9] A. Ravaglioli e G. Pollifrone, Materie prime ceramiche - Vol.2 (materiali non argillosi), Faenza Ed., 1978.
[10] C. Fiori, B. Fabbri e A. Ravaglioli, Materie prime ceramiche, Faenza Ed., 1989.
[11] G. P. Emiliani e F. Corbara, Tecnologia ceramica - Vol.1 (Le materie prime), Faenza Ed., 1989.
[12] Società Ceramica Italiana, Materie prime ceramiche, Litostampa, 2016.
[13] R. C. MacKenzie, Differential Thermal Analysis - Vol.1 (Fundamental Aspects), Academic Press, 1970.
[14] W. Smykatz-Kloss, Differential Thermal Analysis. Application and Results in Mineralogy, Springer-Verlag, 1974.
ACQUA ZEOLITICA
Acqua presente negli interstrati argillosi, debolmente legata, che viene eliminata con un blando riscaldamento (100 ÷ 200 °C) a differenza della acqua di costituzione proveniente dagli ossidrili legati alle unità strutturali del reticolo cristallino la cui distruzione avviene a più alte temperature (500 ÷ 650 °C).
BALL CLAY
Argille caolinitiche ad alto contenuto di allumina, caratterizzate da una elevata plasticità dovuta alla loro granulometria fine ed alla presenza di sostanze organiche carboniose; idonee per impasti bianchi greificabili.
Formazione di bolle e rigonfiamenti visibili sulla superficie di un materiale argilloso o di un impasto ceramico dovuti ad una temperatura di cottura troppo elevata o ad un gradiente termico eccessivamente rapido.
Grumi di ossido di calcio talvolta riscontrabili nei prodotti ceramici porosi a composizione calcarea che, per la loro granulometria grossolana, in cottura non hanno reagito con la silice e l’allumina. Costituiscono un difetto non solo estetico ma anche strutturale, in quanto i calcinelli si idratano con l’umidità e reagiscono con la CO2 (carbonatazione), aumentando di volume e provocando nel tempo la rottura del manufatto già in esercizio.
Materiale argilloso costituito prevalentemente da caolinite, formatosi per degradazione di rocce madri feldspatiche e pertanto spesso caratterizzato da elevata purezza. Per la sua struttura e la granulometria grossolana presenta una minor plasticità rispetto agli altri minerali argillosi, alto contenuto di allumina (fino al valore teorico del 39,5%) ed elevata bianchezza.
Sinonimo di caolino, indica un’argilla caolinitica molto pura, simile a quella utilizzata per le porcellane cinesi.
COEFFICIENTE DI DILATAZIONE
È una misura della variazione di lunghezza o del volume di un materiale in funzione della temperatura. Il coefficiente di dilatazione termica lineare (α) si calcola dalla formula:
α = (L L0 )
L0 (T T0 )
e si esprime in °C−1 (ad es. 60×10−7 °C−1). Il coefficiente di dilatazione termica volumica (o cubica) si approssima con 3α (ad es. se α=60×10-7 °C-1, allora 3α=180×10-7 °C-1).
FOGLIETTO
Struttura a foglio tipica dei fillosilicati, costituita ad esempio da uno strato di tetraedri con un atomo di silicio al centro e atomi di ossigeno ai vertici o da uno strato di ottaedri con un atomo di alluminio al centro e atomi di ossigeno ai vertici.
Roccia sedimentaria composta da una frazione argillosa e da una frazione carbonatica, generalmente carbonato di calcio, presente dal 35 al 65%.
MINERALE
Composto inorganico naturale solido caratterizzato da una composizione chimica ben definita e solitamente da una struttura cristallina.
Permanenza a temperatura circa costante in un processo termico; per esempio, il palier di massima temperatura di cottura oppure il palier per favorire la degasazione in modo da evitare la formazione di cuore nero.
RIPPAGGIO
Metodo di coltivazione di cave che prevede lo sgretolamento e la successiva rimozione della roccia dal suo giacimento mediante speciali aratri a forma di coltelli (dall’inglese ripper).
RUSPAGGIO
Asportazione del materiale di superficie e livellamento del terreno tramite escavatore o pala meccanica.
TOUT-VENANT
Con questo termine si definisce il misto naturale di cava, costituito da ghiaie grosse alluvionali di natura mineralogica prevalentemente calcarea, che non ha subito processi di selezione.
La finitura estetica della piastrella ceramica è normalmente realizzata mediante l’applicazione di uno strato superficiale di materiali fusibili (smalti) che, dopo cottura, formano una copertura vetrosa intimamente unita con il substrato ceramico di base. Questo strato vetroso presenta ottime caratteristiche igienico-sanitarie (pulibilità, resistenza all’attacco chimico, impermeabilità, ecc.) ed estetiche (decoro, colore, lucentezza, ecc.), conferendo alla piastrella il valore e le funzionalità che la rendono idonea per molteplici applicazioni e destinazioni d’uso.
Con smalto ceramico si intende una miscela di materie prime e fritte vetrose che viene applicata sulla superficie dei supporti ceramici, biscottati o solo essiccati, sotto forma di sospensioni acquose ottenute per macinazione ad umido. Questa miscela durante la cottura vetrifica, passando allo stato liquido ed eliminando le porosità superficiali; nella fase di raffreddamento solidifica, lasciando uno strato compatto e duro. In particolare, il termine smalto si riferisce a tutti i rivestimenti vetrosi dotati di opacità. Caratteristico fra questi è lo smalto denominato “maiolica”, che viene applicato sul supporto ceramico, in spessori medio-alti, al fine di conferire al prodotto finito un aspetto bianco e brillante. L’opacizzazione è ottenuta con un agente opacizzante, normalmente silicato di zirconio introdotto direttamente nella fritta.
La fritta ceramica è un semilavorato ottenuto dalla fusione di specifiche miscele di materie prime in forni fusori (Fig. 4.1) che possono essere di tipo continuo (forni a bacino) o discontinuo (forni rotativi). In generale le fritte sono costituite da silice e vari altri ossidi utili ad ottenere le caratteristiche desiderate.
Le fritte trasparenti o opache più comuni vengono fuse portando la miscela a temperature dell’ordine dei 1400 ÷ 1530 °C; il materiale fuso viene quindi fatto colare in acqua, provocando la solidificazione istantanea del vetro e al tempo stesso la sua frammentazione in piccoli grani. La differenza sostanziale tra una fritta ed un vetro consiste nel grado di omogeneità. I vetri, infatti, vengono prodotti in speciali forni a bacino che consentono di ottenere un materiale perfettamente amorfo ed omogeneo, privo di residui di materie prime non dissolte e di bolle.
Per le fritte questi aspetti non sono così fondamentali, in quanto esse costituiscono, contrariamente ai vetri, un semilavorato alla cui disomogeneità si sopperisce attraverso la successiva fase di macinazione ad umido per la preparazione dello smalto finale.

4.1
L’obiettivo del frittaggio è quello di:
- rendere insolubili alcuni componenti (derivati del boro, sali alcalini, derivati del piombo) che, se introdotti allo stato crudo, andrebbero in soluzione in fase di macinazione in acqua; - eliminare completamente tutte le impurezze di natura organica, allontanando quindi tutti i componenti volatili mediante reazioni termo-ossidative che altrimenti avverrebbero in fase di cottura del prodotto ceramico; - disperdere completamente nella massa alcune impurezze di natura ferrosa e/o metallica che, se pur contenute in percentuali molto basse, possono originare piccoli difetti locali. La fusione fa sì che queste particelle inquinanti vengano ripartite nella massa fusa ed inglobate nella composizione, eliminando difetti quali puntinature da ferro, rame, ecc.; - utilizzare temperature molto elevate (1400 °C ed oltre) per favorire le reazioni di formazione di composti vetrosi che, altrimenti, si svilupperebbero solo parzialmente alle normali temperature di cottura dei supporti ceramici.
Un particolare tipo di vetro, utilizzato soprattutto nei prodotti da rivestimento, è la cristallina, che viene applicata sulle decorazioni sopra lo smalto per conferire maggiore brillantezza e profondità alla superficie.
Nel caso specifico della monocottura porosa, oltre alle sopracitate caratteristiche di opacità e brillantezza, le fritte utilizzate devono presentare elevate temperature di rammollimento in modo da permettere l’emissione dei gas (CO2) provenienti dal supporto durante la cottura, evitando possibili danni o difetti negli smalti.
Per quanto invece riguarda gli smalti da pavimento per monocottura greificata e grès porcellanato, oltre a opacità e finitura (matt, satinata, semilucida, lucida), si devono considerare altri fattori quali le caratteristiche di durezza, di resistenza all’abrasione, all’attacco chimico, ecc.
In questo capitolo verranno trattati dapprima gli aspetti teorici relativi alla natura del vetro, per poi passare in rassegna le diverse tipologie di fritte e gli smalti in composto.
La scelta delle materie prime da impiegare per la produzione delle fritte ceramiche deve tener conto dei seguenti criteri:
- costanza nel tempo della composizione chimica
- costanza nel tempo della granulometria
- basso contenuto di Fe e Cr
- assenza di minerali di difficoltosa fusione (come cianite e sillimanite, che restano nella fritta sotto forma di infusi)
- economicità e facile reperibilità.
Le materie prime più importanti e quindi più usate risultano pertanto:
- quarzi e sabbie
- feldspati sodici
- feldspati potassici
- acido borico
- borace pentaidrato
- colemanite (borato di calcio)
- borace anidro (borato di sodio)
- ulexite (borato di sodio e calcio)
- carbonato di calcio
- silicato di zirconio (farina di zirconio)
- ossido di zinco
- carbonato di bario
- dolomite
- caolino (ventilato).
Oltre a queste, sono normalmente usati, in quantità inferiore, altri prodotti quali:
- nitrato di potassio
- carbonato di sodio
- carbonato di potassio
- carbonato di magnesio
- biossido di titanio.
Una varietà di materie prime, come quelle sopra riportate, implica una scelta sul tipo di stoccaggio e manipolazione da adottare. Infatti, talune delle materie prime citate non sono facilmente gestibili nei sili di un impianto produttivo. Inoltre, materiali come carbonato di bario, biossido di titanio e nitrato di potassio non sono reperibili allo stato sfuso.
L’ossido di zinco, nel tipo leggero, non risulta insilabile o estraibile dai silos, per cui è norma ormai diffusa l’uso del cosiddetto ossido di zinco “pesante” che non presenta particolari difficoltà di trattamento.
Per le difficoltà menzionate o per il fatto di essere impiegate in piccola quantità, queste materie prime vengono utilizzate in sacchi, mentre le altre possono essere ricevute dal fornitore allo stato sfuso su autotreno.
Nel Paragrafo 4.3 sono riportate maggiori informazioni su tipologia dei minerali da cui vengono estratte le materie prime, processi di raffinazione, parametri composizionali e granulometrici che rendono tali composti idonei alla fusione.
I materiali vetrosi utilizzati in ceramica (fritte) sono il risultato della fusione di composti inorganici precursori di specifici ossidi, aventi le seguenti funzioni (vedi Tab. 4.1):
- vetrificanti
- fondenti
- stabilizzanti
- opacizzanti
- devetrificanti.
Principali ossidi costituenti le fritte ceramiche
Vetrificanti SiO2 B2O3
Fondenti Na2O K2O PbO B2O3 Li2O
Stabilizzanti CaO BaO MgO PbO Al2O3 ZnO
Opacizzanti ZrO2 SnO2 TiO2
Devetrificanti ZnO CaO BaO MgO TiO2
Tab. 4.1 Costituenti di un vetro per uso ceramico
La caratteristica fisica fondamentale dei vetri è l’isotropia, a differenza dei corpi solidi a struttura cristallina che sono anisotropi. Per tale motivo, in un primo tempo si ipotizzò che i vetri fossero composti totalmente amorfi; successivamente, gli studi di Zachariasen [1] e di Warren [2] stabilirono che la coordinazione tetraedrica caratteristica del silicio è mantenuta anche nel vetro. Mentre nei cristalli la disposizione di questi tetraedri corrisponde rigorosamente ad una regolare costruzione geometrica, nei vetri la loro disposizione è completamente caotica e quindi priva di periodicità e di simmetria. Possiamo quindi parlare di reticolo anche per il vetro ma di un reticolo disordinato e contorto, costituito essenzialmente da atomi di silicio e ossigeno.
Le rappresentazioni schematiche bidimensionali della disposizione tetraedrica della silice cristallina e della silice fusa sono rispettivamente illustrate nella Fig. 4.2.
Fig. 4.2 Struttura cristallina (A) e struttura vetrosa (B) della silice [1] [2]
Analogamente al vetro di silice, il vetro comune ha la stessa disposizione irregolare di tetraedri, oltre alla presenza di ioni degli altri elementi costitutivi nei vuoti lasciati dal silicio e dall’ossigeno.
I legami che formano il reticolo del vetro non sono tutti equivalenti, come nel caso dei reticoli cristallini; di conseguenza l’energia richiesta per spezzarli è differenziata. Ne risulta che con innalzamento della temperatura aumenta l’energia di agitazione termica, fino a raggiungere i valori corrispondenti alla rottura dei legami più deboli. Progredendo nell’incremento di temperatura si ottiene il graduale disgregamento del reticolo, con una riduzione della viscosità ed una progressiva liquefazione del vetro.
Ad ogni temperatura corrisponde quindi una struttura del vetro caratteristica. Nel processo inverso di solidificazione del fuso, il vetro aumenta rapidamente la sua viscosità senza però raggiungere un equilibrio stabile del reticolo, con la conseguente possibile formazione dei composti cristallini elencati in Tab. 4.2.
Anortite
Gehlenite
Sfene
CaO·Al2O3·2SiO2
2CaO·Al2O3·SiO2
CaO·TiO2·SiO2
Gahnite ZnO·Al2O3
Willemite 2ZnO·SiO2
Cristobalite SiO2
Tridimite SiO2
Spodumene
Li2O·Al2O3·4SiO2
Magnesio titanato MgO·TiO2
Wollastonite CaO·SiO2
Rutilo TiO2
Cordierite
2MgO·2Al2O3·5SiO2
Forsterite 2MgO·SiO2
Enstatite MgO·SiO2
Diopside
Zirconio silicato
CaO·MgO·2SiO2
ZrO2·SiO2
Zirconio ossido ZrO2
Celsiana
Leucite
BaO·Al2O3·2SiO2
K2O·Al2O3·4SiO2
Tab. 4.2 Specie cristalline riscontrabili in fritte devetrificate
I cationi formatori di reticolo, che dai loro ossidi possono essere portati allo stato vetroso per semplice riscaldamento, sono Si4+ e B3+. Mentre il silicio (coordinazione 4) forma dei tetraedri collegati con i vertici, il boro (coordinazione 3) forma dei triangoli equilateri al cui centro è sistemato lo ione B3+. Poiché lo ione silicio dispone di quattro legami, mentre quello di boro soltanto di tre, ne risulta che il vetro borico è molto meno viscoso e quindi più fusibile.
I cationi fondenti, chiamati anche modificatori di reticolo, hanno l’effetto di spezzare i ponti di legame fra i tetraedri, una volta che essi siano aggiunti sotto forma di ossidi. Ad esempio:



introduzione



Tali ioni si sistemano in generale negli interstizi che separano i poliedri silicei. Quanto più grande è il numero di ioni sodio introdotti, tanto maggiore è il numero di rotture apportate, cosicché si ottiene una diminuzione progressiva della viscosità del vetro. Inoltre, l’elevato numero di rotture fra i tetraedri finisce col compromettere l’esistenza dello stesso stato vetroso perché quanta maggiore libertà acquistano i tetraedri, tanto più spiccata è la loro tendenza ad assumere la struttura regolare dei cristalli e di conseguenza il vetro devetrifica nelle specie cristalline di Tab. 4.2.
I cationi stabilizzatori sono essi pure dei modificatori di reticolo come nel caso di un catione bivalente:
A differenza dei cationi alcalini, che per il loro debole potenziale ionico sono meno legati al reticolo e quindi facilmente asportabili con conseguente alterabilità del vetro, quelli alcalino-terrosi, con un potenziale ionico quasi doppio, sono assai più legati e quindi rinforzano la struttura reticolare del vetro, agendo da stabilizzanti.
La sostituzione di uno ione modificatore monovalente (Na+) con uno di dimensioni equivalenti ma con carica elettrica superiore (Ca2+) provoca:
- un aumento di densità, perché la maggior forza attrattiva, esercitata sugli ioni ossigeno limitrofi, determina una più elevata compattezza;
- un aumento dell’indice di rifrazione, come conseguenza della maggiore densità; - una diminuzione della conduttività elettrica, come risultato della diminuita mobilità dei cationi per un’incrementata energia del legame;
- un aumento della viscosità per la stessa ragione precedente.
Il boro invece è un formatore di reticolo con numero di coordinazione 3; tuttavia non è mai impiegato come vetrificante unico, mentre è molto più utilizzato nei vetri silicei.
Nei vetri esclusivamente borici, il reticolo è formato da triangoli equilateri collegati nei loro vertici con ossigeni che fanno da ponte; nei vetri silico-borici invece, per quantitativi crescenti di B2O3, si formano dapprima tetraedri di BO4 (passaggio della coordinazione del boro da 3 a 4) che generano una struttura analoga a quella di un vetro silicioso. Quando il B2O3 supera però un certo valore, non si formano più i tetraedri BO4 ma i triangoli caratteristici della struttura del vetro borico puro.
L’alluminio di per sé non è uno ione che si possa considerare formatore del reticolo; tuttavia, come nei feldspati, i suoi ioni elettropositivi possono sostituirsi allo ione silicio nella struttura dei tetraedri. Analogo comportamento può avere l’alluminio nel vetro e tale consolidamento dei tetraedri ha l’effetto di apportare al vetro una maggiore viscosità, una più alta resistenza chimica, una più accentuata stabilità dello stato vetroso.
Risulta evidente dal comportamento dell’alluminio che non si può stabilire una netta distinzione fra ioni formatori di reticolo e ioni modificatori del reticolo. In determinate condizioni anche questi ultimi possono assumere il ruolo di ioni formatori.
A riguardo, sulla base delle teorie di Dietzel [3], i cationi che presentano nei confronti dell’anione ossigeno le più elevate forze di legame (si veda la Tab. 4.3) si comportano da formatori di reticolo (Si4+, B3+); quelli che manifestano invece i più bassi valori funzionano da modificatori del reticolo (Pb2+, Ca2+, Ba2+, Li+, Na+, K+) ed infine quelli che hanno valori intermedi possono assolvere ambedue le funzioni (Fe3+, Al3+, Mg2+, Zn2+).
Tab. 4.3 Forze di legame calcolate per alcuni ossidi
1 - Silice (SiO2)
La silice viene introdotta sotto forma di quarzo, sabbie quarzifere, sabbie feldspatiche o feldspati.
Rappresenta l’elemento principale delle composizioni vetrose, grazie alla sua proprietà di vetrificare sotto l’azione dei fondenti entro un larghissimo intervallo di temperatura. I fondenti o modificatori sono: PbO, B2O3, K2O, Na2O, Li2O. I rivestimenti ceramici molto ricchi in silice sono dotati di grande resistenza agli agenti chimici e di elevata durezza. Quanto maggiore è il contenuto di silice in uno smalto tanto superiore sarà la sua temperatura di cottura.
2 - Anidride borica (B2O3)
L’anidride borica viene introdotta sotto forma di acido borico, borace sodico, colemanite (borato di calcio) e ulexite (borato di calcio e sodio).
Dopo la silice, il boro è l’elemento più importante per la sua proprietà vetrificante. Da solo non potrebbe però essere utilizzato in quanto formerebbe dei vetri fortemente solubili. Sui vetri silicei, il boro agisce da fondente, risulta indispensabile per vetri senza piombo e a basso punto di fusione, dissolve molti coloranti, conferisce lucentezza, diminuisce la viscosità ed abbassa il coefficiente di dilatazione nei vetri in cui è introdotto.
3 - Ossido di piombo (PbO)
L’ossido di piombo impartisce al vetro:
- alta fusibilità;
- aumento dell’indice di rifrazione;
- aumento della densità;
- aumento della brillantezza.
Le vetrine al piombo, per contro, presentano:
- bassa viscosità;
- elevata tossicità, proporzionale al contenuto del piombo ed alla forma con cui è legato al vetro;
- elevata sensibilità all’attacco degli acidi se il contenuto in ossido supera certe proporzioni.
Per ragioni di tossicità per l’uomo oggi il piombo non è più utilizzato nelle composizioni di smalti per piastrelle ceramiche.
4 - Alcali (K2O, Na2O, Li2O)
Sono introdotti sotto forma di nitrati, carbonati o feldspati. Gli alcali sono dei modificatori del reticolo, la loro introduzione indebolisce la struttura reticolare del vetro abbassandone il punto di fusione.
Gli ioni Na+ e K+ si piazzano negli interstizi che separano i tetraedri. Gli ioni K+ che hanno dimensioni più grandi degli ioni Na+ si legano con più ossigeni e quindi conferiscono maggior stabilità al vetro. Da questo deriva la facile alterabilità dei vetri sodici tanto che vetri molto sodici sono facilmente solubili in H2O. Gli alcali in genere aumentano il coefficiente di dilatazione dei vetri ad eccezione del litio che, essendo molto fusibile, si utilizza in percentuali molto basse (molto inferiori al sodio ed al potassio).
Gli alcali, ed in particolare il litio, conferiscono brillantezza ai vetri; non possono però da soli costituire l’intera parte basica della composizione di un vetro a causa della loro attitudine alla devetrificazione e per la possibile solubilità in acqua dei silicati che si formano.
5 - Ossido di calcio (CaO)
Si introduce sotto forma di calcio carbonato, dolomite e wollastonite.
L’ossido di calcio è uno stabilizzante in quanto, aggiunto ad un silicato alcalino, riduce l’alterabilità del vetro. Infatti, mentre da solo forma silicati ad alta temperatura di fusione (> 1400 °C), con altri silicati alcalini dà luogo alla formazione di masse vetrificate. Elevate percentuali di questo ossido portano alla devetrificazione (matt al calcio). Introdotto nelle giuste proporzioni (5 ÷ 10% in ossidi) il calcio, oltre a dare stabilità, migliora la resistenza meccanica e l’aderenza dei vetri al supporto.
Nei vetri cotti ad alta temperatura provoca una diminuzione della viscosità.
6 - Allumina (Al2O3)
Si introduce generalmente sotto forma di allumina calcinata o idrata, feldspati, caolino, corindoni. L’allumina introdotta nei vetri ceramici nelle dovute proporzioni (4 ÷ 8%), nel caso di smalti a basse temperature conferisce le seguenti caratteristiche:
- aumenta la viscosità;
- diminuisce la tendenza a devetrificare;
- aumenta la resistenza meccanica;
- abbassa il coefficiente di dilatazione;
- aumenta la resistenza agli acidi;
- migliora l’opacità (se introdotta in alte percentuali compatibilmente con la temperatura di cottura dello smalto).
Questo ossido possiede l’attitudine, in quanto sostanza anfotera, a combinarsi tanto con la silice quanto con gli ossidi basici. È quindi il più efficace degli stabilizzanti.
La percentuale di Al2O3 introdotta in uno smalto può essere tanto maggiore quanto più alta è la sua temperatura di cottura e viceversa. Sarà inoltre superiore negli smalti matt o satinati, minore negli smalti brillanti.
Nel valutare la quantità di Al2O3 addizionabile ad uno smalto bisogna anche tener conto della sua granulometria: quanto più fine è l’allumina, tanto minore è la percentuale utilizzabile e viceversa.
Viene normalmente introdotto nelle composizioni da fondere sotto forma di carbonato di bario (BaCO3). Quest’ossido aumenta la densità e l’indice di rifrazione e di conseguenza conferisce brillantezza al vetro. Risulta inoltre un ottimo fondente nella fusione dei vetri silicatici e per questa sua proprietà può in parte sostituire efficacemente l’ossido di piombo, ma presenta anch’esso un’elevata tossicità.
In alte percentuali, l’ossido di bario indurisce le fritte e induce la devetrificazione. Un vetro al bario fonde molto più rapidamente ed è meno viscoso di uno al calcio.
8 - Ossido di magnesio (MgO)
Si introduce mediante dolomite, carbonato di magnesio e talco. Il comportamento dell’ossido di magnesio nei vetri è molto simile a quello dell’ossido di calcio. Si differenzia solo perché dà luogo a vetri più viscosi. Non lo si può usare in percentuali troppo alte in quanto innalzerebbe la temperatura di cottura del vetro. Il magnesio riduce il coefficiente di dilatazione ma aumenta la tensione superficiale del vetro.
9 - Ossido di zinco (ZnO)
L’ossido di zinco esplica azioni anche molto diverse tra loro in funzione della sua percentuale, ad esempio:
- a basse percentuali: aumenta la brillantezza dei vetri e dei colori, ad eccezione dei verdi e dei rosa; insieme con l’allumina migliora l’opacità e la bianchezza degli smalti, purché il tenore di CaO sia basso ed il B2O3 assente; riduce inoltre il coefficiente di dilatazione; - ad alte percentuali: devetrifica dalla massa vetrosa impartendo alla superficie dello smalto il caratteristico aspetto matt, favorito se lo smalto ha carattere basico (elevato contenuto di ossidi modificatori);
- a percentuali molto elevate: cristallizza e si separano corpuscoli cristallini costituiti da silicato di zinco. Vetri molto ricchi di questo ossido sono fortemente attaccati dagli acidi. In vetrine acide e ad alto contenuto di allumina, l’ossido di zinco esplica azione fondente.
L’ossido di titanio migliora la resistenza chimica e quella al cavillo. Quest’ultimo requisito si manifesta già con piccole percentuali di ossido e permane anche quando le percentuali aumentano. Le aggiunte di TiO2 colorano il vetro: già il 2% dà una tonalità che vira al giallo. Contemporaneamente la superficie della vetrina assume un aspetto matt, fino a divenire dura e rugosa per percentuali crescenti di ossido.
Ha proprietà opacizzanti che migliorano in assenza di B2O3, specialmente se la composizione del vetro è ricca in Al2O3 e se aggiunto al mulino; in tali condizioni anche il colore schiarisce.
Queste caratteristiche sono particolarmente evidenti se il TiO2 è introdotto nel vetro sotto forma di anatasio, mentre se è introdotto sotto forma di rutilo perde le caratteristiche devetrificanti fino ad arrivare, con alte percentuali e alte temperature di cottura, a dare origine a formazioni cristalline aghiformi.
La cristallizzazione avviene prevalentemente in vetri ad elevata fusibilità.
11 - Ossido di stagno (SnO2)
Opacizzante per eccellenza anche in basse percentuali (6 ÷ 10%) è però poco utilizzato come tale per il suo elevato costo.
L’opacità apportata da questo ossido è dovuta alla sua sospensione nella massa vetrosa sotto forma di particelle finemente disperse. Quindi il suo potere opacizzante dipende dalla purezza dell’ossido, dalla finezza delle sue particelle e dalla natura della massa vetrosa alla quale esso è aggiunto. Dannosi per una buona opacizzazione con questo ossido sono gli elementi alcalini ed il boro. È consigliabile aggiungere lo stagno nello smalto al mulino e non in fusione.
12 - Ossido di zirconio (ZrO2)
Si utilizza sotto forma di silicati di zirconio a varie granulometrie. Ottimo opacizzante, anche se non al livello dello stagno, rispetto al quale ha però il vantaggio di essere molto più economico. È sicuramente l’opacizzante più usato industrialmente.
Alte percentuali di questo ossido comportano un innalzamento della temperatura di cottura del vetro in cui è introdotto. I silicati di zirconio si prestano come opacizzanti per tutti i tipi di smalto cotti nell’intervallo di temperatura fra 940 e 1300 °C. Solo una frazione dei silicati di zirconio introdotti si solubilizza e si combina con gli altri componenti, mentre la parte preponderante rimane tal quale.
La frazione che si combina contribuisce ad aumentare la resistenza al cavillo dello smalto. Lo zirconio ha la proprietà di stabilizzare i colori. Gli ossidi di calcio e magnesio aiutano lo zirconio a migliorare l’opacizzazione, come pure lo zinco e l’allumina. Esistono in commercio diversi tipi di silicati di zirconio che si differenziano soprattutto per la granulometria:
- silicati di zirconio micronizzati (molto fini);
- silicati di zirconio sotto forma di farina (più grossolani);
- silicati di zirconio sotto forma di sabbia (molto grossolani).
Come opacizzanti per smalti si utilizzano prevalentemente quelli micronizzati, mentre le farine si usano nella fusione delle fritte e le sabbie vengono utilizzate più come indurenti o come zavorre. Smalti per basse temperature di cottura opacizzati con zircone presentano spesso superfici puntinate a causa del carattere fortemente viscoso di questi smalti.
Nella Tab. 4.4 viene riportato un quadro sintetico rappresentativo delle tipologie di minerali che permettono l’introduzione dei diversi ossidi.
Ossidi
Materie prime usate
SiO2 Quarzo, feldspato, China-clay, zirconio silicato
B2O3 Acido borico*, borace*, colemanite
PbO Minio, litargirio
Na2O Feldspati, borace*, Na2CO3*
K2O Feldspati, KNO3*, K2CO3*
Li2O Feldspati, Li2CO3*
CaO Wollastonite, CaCO3, dolomite, colemanite
BaO BaCO3
MgO MgCO3, talco, dolomite
Al2O3 Al2O3, Al(OH)3, China-clay, feldspato
ZnO Ossido di zinco
SnO2 Ossido di stagno
TiO2 Ossido di titanio, sabbia di rutilo
ZrO2 Zirconio silicato, ossido di zirconio
* composti solubili in H2O, pertanto utilizzabili solo in fusione
Tab. 4.4 Composti più utilizzati per la formazione di vetri e smalti
Con il termine fritta si usa comunemente indicare, nella pratica industriale, una miscela vetrosa fusa raffreddata bruscamente in acqua. Le fritte vengono utilizzate come base nelle vetrine e negli smalti a bassa temperatura per rendere insolubili i componenti e per ottenere un vetro sufficientemente omogeneo.
In commercio si possono trovare molte varietà di fritte con diverse caratteristiche di fusibilità, brillantezza, opacizzazione e mattizzazione.
Per tipologia e caratteristiche, si possono raggruppare come indicato di seguito.
1) Fritte brillanti, trasparenti, viscose
Sono comunemente note con il termine “cristalline” e variano in base al prodotto ceramico da realizzare:
a) bicottura rapida
Si utilizzano fritte che fondono a bassa temperatura. Sono composte da un’alta percentuale di SiO2 (50 ÷ 60%) e da una bassa percentuale di elementi fondenti (10 ÷ 20% fra Na2O, K2O, PbO, B2O3). Il restante della composizione è costituito dagli elementi stabilizzanti (Al2O3, ZnO, CaO, BaO, MgO), quasi sempre tutti presenti in percentuali singolarmente basse (in totale 10 ÷ 25%).
Queste fritte vengono impiegate prevalentemente nella preparazione delle cristalline e saltuariamente sono introdotte in piccole percentuali in alcuni smalti per basse temperature di cottura. Possono essere utilizzate anche per smalti cuocenti alle temperature superiori ai 1100 °C, anche se solo in basse percentuali, nella preparazione di quasi tutti gli smalti, con la funzione di vetrificare e dare fusibilità.
b) monocottura porosa
Le fritte per monocottura porosa devono presentare una composizione diversa dalle precedenti, per far fronte alle specifiche esigenze di questa tipologia di impasti che generalmente contengono una percentuale di materiali carbonatici compresa fra 8 e 15%.
Ciò ha comportato lo studio e la messa a punto di fritte “alto fondenti” con temperature di rammollimento superiori ai 1020 °C, in modo da permettere la completa emissione dell’anidride carbonica formatasi durante la decomposizione della calcite e/o della dolomite. Queste fritte, osservate al microscopio riscaldante, presentano un punto di rammollimento decisamente elevato congiuntamente ad un valore di punto di sfera (fusione) molto vicino al precedente.
Per ottenere un tale comportamento rispetto alle formulazioni delle fritte per bicottura, è stata aumentata la percentuale di ossidi quali CaO, ZnO, MgO e BaO, in modo da favorire una fusione “eutettica”. Congiuntamente è stato necessario diminuire la percentuale di ossidi fondenti come gli alcali e l’anidride borica. Le fritte di questo tipo vengono definite “rapide”, cioè in grado di fondere ad alta temperatura ma repentinamente.
c) monogreificata e grès porcellanato smaltato
Molte fritte possono trovare impiego anche per queste tipologie produttive, tuttavia, considerate le alte temperature di cottura, le loro percentuali negli smalti sono ridotte a favore di materie prime di minore costo. Qualora però si renda necessaria una elevata trasparenza e brillantezza, si utilizzano fritte opportunamente formulate per temperature di cottura di 1180 ÷ 1220 °C.
2) Fritte brillanti, opacizzate, viscose
Sono note con il nome di “bianchi allo zirconio” o “maioliche”:
a) bicottura rapida
Queste fritte differiscono da quelle del gruppo precedente unicamente in quanto opacizzate. Hanno caratteristiche e composizioni identiche; l’opacizzazione è dovuta al silicato di zirconio, introdotto nella composizione in percentuali variabili da 8 a 14%.
Queste fritte vengono utilizzate prevalentemente nella preparazione degli smalti bianchi brillanti, sia per le basse temperature di cottura che per le alte. Ovviamente negli smalti cuocenti ad alte temperature, la percentuale di fritta utilizzata diminuisce notevolmente a favore delle altre materie prime. Queste fritte trovano uno scarso utilizzo nella preparazione di smalti diversi dai bianchi brillanti.
Analogamente alle fritte brillanti trasparenti, anche in questo caso la cottura rapida ha richiesto una modifica sostanziale della formulazione per rendere compatibile la fusibilità del prodotto con la cottura rapida.
I coefficienti di dilatazione delle fritte bianche opacizzate ed anche delle cristalline trasparenti adatte per la bicottura rapida sono nell’intervallo 60 ÷ 70 × 10−7 (coefficiente di dilatazione termica lineare α).
b) monocottura porosa
Valgono gli stessi concetti espressi precedentemente per le fritte brillanti, a prescindere dall’utilizzo nella formulazione del silicato di zirconio.
c) monogreificata e grès porcellanato smaltato
Le fritte opacizzate per temperature di cottura di 1180 ÷ 1220 °C avrebbero viscosità molto alte, rendendo difficile e poco produttiva la loro fusione. In conseguenza di ciò si preferisce ottenere l’opacizzazione dello smalto aggiungendo silicato di zirconio micronizzato nella fase di macinazione delle fritte trasparenti brillanti idonee per queste temperature di cottura.
3) Fritte matt (CaO, BaO, ZnO)
Si distinguono principalmente in funzione dell'impiego per rivestimento o per pavimento:
a) bicottura rapida
Appartengono a questo gruppo tutte quelle fritte dove viene introdotto in quantità, nell’appropriata base vetrosa, un elemento che favorisca la devetrificazione. Gli elementi devetrificanti più utilizzati sono il calcio, il bario e lo zinco. I primi due normalmente devetrificano in basi vetrose alcalino-boriche mentre lo zinco devetrifica in basi piombiche.
I matt di calcio e bario sono normalmente apiombici; queste fritte sono inoltre viscose e nel caso del calcio possono presentare opalescenza.
b) monocottura porosa
Le formulazioni di fritte matt per monoporosa non si differenziano sostanzialmente da quelle idonee per la bicottura rapida, in particolare per le tipologie con elevate percentuali di calcio e bario.
La devetrificazione può portare ad effetti diversi dipendendo dall’accrescimento dei cristalli nello smalto, passando da superfici satinate con pochi cristalli di dimensioni medio-grandi, a superfici matt ottenute da molti cristalli di piccole dimensioni.
Anche per queste fritte, vi è la necessità di garantire un’elevata temperatura di rammollimento per lasciare uscire i gas dal supporto, mantenendo allo stesso tempo la bassa viscosità necessaria ad un veloce accrescimento dei cristalli.
A seconda della tipologia di cristallo che si separa dalle fasi vetrose dello smalto si ottengono smalti matt opalescenti o anche smalti matt trasparenti, condizione che avviene quando l’indice di rifrazione dei cristalli è molto vicino a quello della matrice vetrosa che li contiene.
c) monogreificata e grès porcellanato smaltato
Le fritte matt utilizzate per queste temperature elevate sono tipicamente a base calcio -bario con elevate percentuali di Al2O3. I cristalli che si formano devono resistere e non fondere durante la permanenza alle massime temperature di cottura. La quantità di alcali in queste fritte deve essere limitata per evitare spillature. Anche in questa tipologia di prodotti le fritte trovano impiego soprattutto quando viene richiesta una elevata trasparenza e compattezza degli smalti dopo cottura a 1180 ÷ 1220 °C.
Nella Tab. 4.5 sono riportate le composizioni indicative e le caratteristiche principali di alcune fritte usate nelle produzioni di bicottura rapida, monocottura porosa, monogreificata e grès porcellanato smaltato, suddivise nelle tipologie: trasparenti, brillanti opache (bianco lucido) e matt.
a) impiego per bicottura rapida
b) impiego per monoporosa
c) impiego per monogreificata e grès porcellanato smaltato
Tab. 4.5 Composizione e caratteristiche delle fritte più comuni
Il mercato delle piastrelle ceramiche offre innumerevoli tipologie di prodotti fabbricati con varie tecnologie. In funzione del tipo di tecnologia considerato, gli smalti devono presentare caratteristiche specifiche, ottenute variando la loro composizione chimica e di conseguenza le loro proprietà fisiche e prestazionali. Inoltre l’ampia differenziazione delle proposte commerciali richiede lo sviluppo di smalti con molteplici effetti estetici. Solitamente ciascuna tipologia produttiva richiede specifiche tipologie di smalti, anche se in alcuni casi vi sono smalti che possono essere utilizzati in più tecnologie produttive.
A riguardo, la Fig. 4.3 riassume in forma schematica la correlazione tra le tipologie di smalti utilizzati nei processi produttivi in funzione del tipo di prodotto (bicottura/monoporosa e monocottura/grès porcellanato).
BICOTTURA
Ingobbio
Smaltobbio
Bianco allo zirconio Maiolica
Cristallina alcalina
Matt
Sbobba trasparente
Graniglia trasparente
Fig. 4.3 Tipologie di smalti correlate alle esigenze dei diversi prodotti Ciascuna classe di prodotto, per rivestimento o pavimento, è ulteriormente suddivisibile in numerose tipologie che richiedono smalti con caratteristiche chimico-fisiche specifiche. Va inoltre evidenziato che oggi tutti gli smalti (bianchi allo zirconio, cristalline, matt, ecc.) devono essere idonei alla decorazione digitale e massimizzare il risultato cromatico degli inchiostri a base di pigmenti ceramici con le finezze granulometriche richieste per la stampa inkjet.
Fig. 4.03
Questa tecnologia produttiva prevede la pressatura del supporto e la sua cottura in forno a rulli a temperature dell’ordine di 1100 °C. Il supporto ceramico così ottenuto (biscotto) è dotato di un’elevata porosità e capacità di assorbimento, che lo rendono idoneo alla smaltatura ed alla successiva ricottura (cottura vetrato) a temperature dell’ordine di 1000 ÷ 1100 °C.
L’utilizzo di nuovi materiali a bassa inerzia termica nella costruzione dei forni e l’adozione della movimentazione a rulli delle piastrelle (senza supporti), ha permesso di velocizzare i cicli di cottura (fino a soli 25 ÷ 30 min) e ciò ha ovviamente richiesto la riformulazione degli smalti rispetto alle composizioni storicamente utilizzate nelle cotture tradizionali.
Bianco lucido allo zirconio
Smalto bianco composto quasi esclusivamente dalla caratteristica fritta allo zirconio, con elevata viscosità ed in grado di generare superfici coprenti, brillanti, speculari. Alla fritta vengono aggiunte quantità variabili da 3 a 10% di materie prime plastiche quali caolini di elevata qualità o argille bianche selezionate, in modo da ottenere in macinazione barbottine stabili e non soggette a sedimentazione durante il processo produttivo.
Le sospensioni acquose di questo tipo, costituite in misura preponderante da particelle vetrose, spesso richiedono anche piccole percentuali (da 0,01 a 0,3%) di composti ionici che, variando le proprietà elettrolitiche del mezzo disperdente (acqua), siano in grado di modificare la reologia della barbottina; la loro azione sospensivante e fluidificante permette infatti di regolare la viscosità in funzione delle specifiche esigenze applicative.
La scelta dello smalto bianco allo zirconio deve essere effettuata principalmente in base alle condizioni di cottura adottate. Buona norma è quella di utilizzare smalti con la temperatura di sfera simile a quella massima di cottura del forno. Non sono necessari particolari accorgimenti per quanto concerne la fase di preriscaldo, in quanto il supporto ha un comportamento inerte. Il coefficiente di dilatazione α ha generalmente valori compresi nell’intervallo 63 ÷ 70 ×10−7 °C−1 per mantenere lo smalto in giusta compressione ed evitare il fenomeno del cavillo, anche in condizioni di utilizzo particolarmente critiche.
Cristallina trasparente
Questa tipologia di fritta viene normalmente utilizzata per la produzione di piastrelle smaltate da rivestimento (chiamate in modo improprio "porcellanato"), con effetto estetico di un vetro trasparente e profondo. La sequenza delle applicazioni prevede di solito un primo strato di ingobbio composto da fritte (bianchi allo zirconio e/o cristalline alcaline addizionate con materie prime feldspatiche), silicati di zirconio (per migliorare la coprenza), argille e caolini (per adeguare la reologia). Successivamente all’ingobbio può essere applicata direttamente la cristallina.
Le decorazioni digitali con inchiostri pigmentati vengono normalmente realizzate sullo strato di cristallina per ottenere un effetto di profondità.
I parametri chimico-fisici delle cristalline non si differenziano da quelli dei bianchi se non per l’assenza del silicato di zirconio e di conseguenza per l’aspetto trasparente della cristallina. Anche l’additivazione chimica si effettua con le stesse modalità indicate per il bianco allo zirconio.
Smalti matt
La loro formulazione è studiata in funzione di una devetrificazione che sviluppi una superficie matt. La devetrificazione è normalmente causata da un eccesso di calcio. Il modo migliore per ottenere superfici devetrificate è l’introduzione di elevate percentuali di calcio in fritte specifiche. È invece da evitare la sua introduzione sotto forma di carbonato o dolomite, in quanto la decomposizione termica di questi minerali può causare l’inglobamento nello smalto di bolle di CO2.
Gli ossidi di zinco, titanio, calcio, bario e magnesio mattizzano per cristallizzazione mentre l’ossido di alluminio e in qualche caso il silicato di zirconio mattizzano per indurimento.
Gli smalti mattizzati con gli ossidi di zinco e titanio sono composti normalmente da una base vetrosa fusibile e piombica.
Gli smalti mattizzati con gli ossidi alcalino-terrosi si presentano sempre con colorazioni bianche e sono normalmente molto viscosi. Gli smalti mattizzati per indurimento sono sempre composti da basi vetrose fusibili e fortemente indurite con allumina, corindone e silicato di zirconio.
Quando l’elemento mattizzante è allumina o corindone si ottengono smalti satinati, mentre quando l’elemento mattizzante è silicato di zirconio si ottengono smalti tipo pietra.
Gli smalti matt (ZnO e CaO) a bassa temperatura sono composti direttamente dalle relative fritte in alte percentuali mentre, per temperature di cottura più elevate, si ricorre all’aggiunta di materie prime a crudo.
Ingobbi
Sono prodotti che precedono l’applicazione degli smalti e che esplicano diverse funzioni. Sono ad esempio in grado di uniformare l’assorbimento dell’acqua delle successive applicazioni, ovvero di ridurre i danni causati da un assorbimento non costante su tutta la superficie del supporto (sfondini).
Hanno anche la funzione di rallentare l’assorbimento dell’acqua, impedendo la formazione di bolle d’aria nelle applicazioni successive. Dal punto di vista ceramico, gli ingobbi hanno la capacità di rallentare e soprattutto frazionare le bolle di gas che salgono dal supporto durante la cottura per effetto di contaminanti presenti nei supporti non purificati (piriti, ematiti, calcopiriti, calcinelli, ecc.).
Un’altra loro peculiarità è quella di neutralizzare il colore del supporto, grazie all’elevato grado di coprenza, rendendo possibile l’uso successivo di cristalline colorate o smalti senza interferenze cromatiche dovute al supporto (in particolare se rosso).
Un ultimo effetto è quello di favorire l’aggancio dello smalto con il supporto, di limitarne la reattività e di modificare il valore medio del coefficiente di dilatazione dell’interfaccia per ridurre eventuali problemi di accordo dilatometrico tra smalti e supporti. La messa a punto delle proprietà degli ingobbi viene effettuata attraverso un corretto utilizzo delle fritte e delle materie prime aggiunte.
La formulazione degli ingobbi richiede quindi approfondite conoscenze tecnologiche, come per gli stessi smalti. Le fritte impiegate per favorire l’aggancio dello smalto al supporto appartengono alla categoria dei bianchi e delle cristalline, mentre le fritte speciali ad alta dilatazione vengono utilizzate per armonizzare il comportamento dilatometrico; la loro percentuale globale varia da 0% a 80%.
Le materie prime che vengono aggiunte per completare la formulazione sono: silicato di zirconio per la coprenza, feldspati e quarzi di buona qualità per diminuire il costo e regolare il coefficiente di dilatazione, materie prime plastiche (argille e caolini di buona qualità) per la reologia. Anche in questo caso è richiesta una fluidificazione spinta per garantire una buona stesura in fase di applicazione.
Questa tecnologia si è sviluppata negli anni ‘80 e ’90 a seguito dei grandi benefici tecnologici conseguiti con la monocottura greificata da pavimento. È stato così raggiunto l’obiettivo di ottenere anche piastrelle a supporto poroso attraverso una cottura unica di supporto e smalto, conservando le caratteristiche necessarie nel rivestimento, ovvero: basso peso al metro quadrato, basso spessore, calibro e planarità ben controllati, tipologie estetiche simili alla bicottura.
Gli impasti da rivestimento sono generalmente costituiti da una miscela di argille carbonatiche opportunamente selezionate o da argille cuocenti bianco addizionate con calcite, oltre che da quarzo e feldspatoidi.
Le piastrelle crude, dopo essere state essiccate e smaltate, vengono cotte in un’unica fase. Le temperature di cottura generalmente oscillano tra 1090 e 1130 °C, i cicli di cottura da 30 a 60 min, in relazione ai formati e agli spessori richiesti.
Bianco lucido allo zirconio
Rispetto alle formulazioni da bicottura rapida, per favorire ed innescare una rapida fusione “eutettica”, nella monoporosa è stato necessario aumentare la percentuale di ossidi devetrificanti, quali CaO, ZnO e MgO. Parallelamente si è diminuita la percentuale di ossidi fondenti come alcali e anidride borica.
L’obiettivo è quello di poter disporre di fritte definite “rapide”, cioè in grado di fondere ad alta temperatura ma molto repentinamente. Quanto detto è valido anche per le cristalline. Per quanto concerne le altre caratteristiche restano validi i concetti espressi per la bicottura rapida.
Riguardo alla reologia, oltre ai fluidificanti, è necessario aggiungere anche uno 0,3% di CMC (carbossimetilcellulosa) con lo scopo di favorire l’ancoraggio dello smalto crudo con il supporto, che altrimenti non sarebbe in grado di mantenere la sua adesione nella fase di preriscaldo. Attualmente si utilizzano anche CMC a bassa viscosità che consentono di ridurre o rendere superfluo l’uso dei fluidificanti.
Cristallina
Vale quanto detto per lo smalto bianco lucido. Anche in questo caso alla sospensione di smalto viene aggiunta una quantità di CMC tra 0,2 e 0,4%.
Smalti matt
Le formulazioni non si discostano sostanzialmente dagli smalti per la bicottura rapida se non per l’uso di sole fritte ad alta temperatura di rammollimento e per la possibilità di introdurre carbonati per favorire la mattizzazione. Gli smalti matt a base di fritte allo zinco-piombo non sono idonei per queste temperature. L’additivazione è la medesima dei bianchi e delle cristalline, anche se frequentemente sono necessari maggiori quantitativi di fluidificanti.
Ingobbi
Anche in questo caso resta valido quanto specificato per la bicottura rapida. Date le temperature di cottura più elevate, si riduce l’impiego di fritte a favore di sabbie e feldspati di buona qualità. Occorre di solito una percentuale di argilla bianca e plastica maggiore rispetto alla bicottura ed è normalmente sconsigliato l’uso di alte percentuali di caolino. Gli additivi sono costituiti dai fluidificanti utilizzati in quantità maggiori rispetto alla bicottura e, al bisogno, anche da CMC.
Sono le tipologie attualmente più diffuse per la produzione di piastrelle da pavimento. La tecnologia si è affinata negli anni, fornendo prodotti sempre più sofisticati dal punto di vista estetico e sempre più avanzati sotto il profilo qualitativo. La smaltatura su piastrella calda è indispensabile per far evaporare l’acqua della sospensione di smalto e quindi favorire il suo essiccamento.
L’aggiunta di carbossimetilcellulosa nella sospensione di smalto è indispensabile per favorire l’aggancio nell’interstrato smalto-supporto fino alla cottura.
Generalmente gli impasti cuocenti chiaro vengono cotti a temperature nell’ordine di 1160 ÷ 1220 °C con cicli termici di 35 ÷ 55 min.
Gli impasti cuocenti rosso vengono generalmente cotti alla temperatura di 1120 ÷ 1160 °C con cicli termici analoghi ai precedenti. Infine, gli impasti rossi ad alta porosità sono cotti a 1080 ÷ 1120 °C con cicli termici di 30 ÷ 45 min.
Di seguito si riporta una descrizione degli smalti più frequentemente utilizzati.
Cristallina trasparente
Con questo termine si intende una categoria di smalti, utilizzata di frequente in monocottura, che presenta una superficie brillante. Il loro grado di coprenza è intermedio tra quello di un bianco e quello di una cristallina; solitamente vengono applicati a vela prima della decorazione digitale. Vengono invece definiti come “protettivo lucido” prodotti completamente trasparenti applicati ad airless, successivamente alla decorazione con inchiostri digitali (inkjet), che dopo cottura presentano una superficie sufficientemente lucida e brillante.
Questi smalti vengono additivati di CMC per l’ancoraggio con il supporto; la quantità di deflocculanti dipende dal grado di macinazione, dalla densità e dal dispositivo applicativo utilizzato.
Bianchi lucidi opacizzati
Questi smalti differiscono in modo netto da quelli usati in bicottura. Sono generalmente formulati utilizzando fritte ad alto contenuto di calcio o ad alto tenore di zirconio. Le materie prime in aggiunta di solito sono silicati di zirconio micronizzati, argille e caolini. Possono venire applicati subito dopo l’ingobbio e prima delle eventuali applicazioni di altri smalti. La decorazione è sempre effettuata sopra-smalto. L’applicazione viene effettuata a vela o ad airless.
Smalti matt
Questa categoria di smalti si è sviluppata negli anni in modo estremamente articolato. Si tratta di smalti con superficie non brillante, morbida al tatto e matt-satinata alla riflessione della luce oppure gessosa ruvida e marcatamente matt.
Gli smalti matt vengono formulati con fritte ad elevato contenuto di calcio, bario e zinco accompagnate da una grande variabilità di altre fritte; le materie prime in aggiunta impiegate anche in alta percentuale sono: feldspati, quarzo, argille, caolino, wollastonite e allumina calcinata o corindone. I feldspati e la nefelina si usano per conferire vetrosità agli smalti, mentre le argille ed i caolini per regolare la reologia. Il livello di coprenza è molto variabile e dipende dalla frequente necessità di utilizzare questi smalti dopo la decorazione digitale, nel qual caso prendono il nome di “smalto protettivo matt” e vengono applicati con airless. I livelli estetici raggiunti sono molto raffinati in quanto, il possibile impiego di supporti strutturati ed il decoro digitale non a contatto consentono di sviluppare un infinito numero di soluzioni estetiche.
Ingobbi
Le loro funzioni sono le medesime descritte per le altre tecnologie. Sono formulati con un’elevata percentuale di materie prime argillose, caolini, sabbie e feldspati.
Smaltobbio
Si tratta di un materiale che presenta sia le caratteristiche dell’ingobbio che dello smalto e, conseguentemente, permette di ottenere con una sola applicazione un fondo vetroso idoneo allo sviluppo del colore degli inchiostri applicati con la decorazione digitale inkjet. Trova impiego prevalentemente nel grès porcellanato smaltato e nella monocottura greificata bianca, mentre non è suggerito il suo utilizzo nel caso di monocottura greificata con impasto rosso vista la ridotta opacità e la scarsa coprenza del colore del supporto.
Sbobba trasparente
Questa tipologia di smalto si è sviluppata per ridurre il consumo di graniglie, pur mantenendo un’ottima trasparenza e permettendo di realizzare prodotti “full lappato”. La sbobba è costituita da micro-graniglie di fritte trasparenti, mantenute in sospensione mediante colle e veicoli a base acquosa. La trasparenza è necessaria visto il suo impiego dopo la decorazione digitale inkjet. Inizialmente l’applicazione veniva eseguita esclusivamente con la vela a causa della difficoltà nel mantenere stabili le sospensioni ed evitare sedimentazioni della micro-graniglia. Il solo impiego di colle e sospensivanti organici comporta un elevato tempo di asciugatura dell’applicazione e difficoltà nell’evaporazione dell’acqua residua di smaltatura. La ricerca per l’ottimizzazione delle formulazioni e lo sviluppo della tecnica applicativa ha portato ad una riduzione delle problematiche applicative e dei difetti conseguenti, permettendo oggi l’applicazione con airless di una formulazione mista. In questo caso la sbobba prevede l’utilizzo delle micro-graniglie aggiunte ad uno smalto trasparente macinato ad umido. La possibilità di apportare modifiche composizionali della parte macinata ad umido permette una più facile messa a punto dell’accordo dilatometrico col supporto, aspetto indispensabile per garantire una planarità ottimale ai fini della successiva lavorazione di lappatura (dopo cottura). Queste nuove formulazioni hanno permesso una diffusione di questa tecnica applicativa su larga scala per realizzare prodotti “full lappato”.
Con lastre di grande formato si intendono i prodotti ceramici con dimensioni dopo cottura a partire dal 120 × 120 cm fino a 180 × 360 cm ed oltre, in un ampio intervallo di spessori (generalmente 6 ÷ 20 mm). Meno comuni sono le produzioni di lastre con spessori inferiori a 6 mm o superiori a 20 mm.
All’impiego classico delle lastre nelle pavimentazioni interne ed esterne, grazie alle possibilità estetiche offerte dalla decorazione digitale, si aggiungono anche il rivestimento di pareti di grandi ambienti interni e altri utilizzi nel settore dell’arredo, come le superfici di lavoro (tavoli e piani cucina).
La quasi totalità delle lastre di grande formato si realizza con impasti di grès porcellanato smaltato macinato ad umido ed atomizzato, che garantiscono le necessarie caratteristiche meccaniche di resistenza e rigidità del prodotto cotto, indispensabile per mantenere sotto controllo la planarità del materiale e consentire le successive lavorazioni di rettifica, taglio in sotto-formati e l’eventuale lappatura superficiale.
Le dimensioni massime sono tuttora in evoluzione in relazione agli sviluppi tecnici di macchine e impianti (in particolare per le fasi di formatura, smaltatura, decorazione, cottura, rettifica, lappatura e confezionamento), oltre che degli impasti e degli smalti stessi.
Le difficoltà tecnologiche degli smalti per la produzione delle grandi lastre sono principalmente dovute alla necessità di un perfetto accordo tra i coefficienti di dilatazione termica di tutti i materiali costituenti (impasti, smaltobbi e smalto finale che include ovviamente sbobbe e graniglie). Va ricordato che una minima variazione del coefficiente di dilatazione porta a modifiche permanenti della planarità delle lastre, pregiudicandone il trasporto e la posa in opera.
I principali smalti impiegati nella produzione delle lastre sono i seguenti:
- smaltobbio
- protettivo matt o lucido
- sbobba
- graniglia a secco.
Le quantità di smalti applicate sono mantenute le più basse possibili, compatibilmente con il risultato tecnico-estetico richiesto per ciascun prodotto, in modo da limitare le variazioni di planarità. In merito alle loro formulazioni si cerca di evitare per quanto possibile l’impiego di fritte con bassa temperatura di rammollimento e di materie prime con scarsa purezza o che possano causare problemi di planarità del prodotto (come calcite, dolomite o elevate percentuali di caolino).
Oggi giorno l’industria ceramica ha adottato universalmente la decorazione digitale per l’applicazione dei motivi grafici superficiali mediante stampanti a getto di inchiostro (si veda in dettaglio il Paragrafo 5.3 del Volume II). Le testine piezoelettriche utilizzate in queste stampanti funzionano con inchiostri liquidi aventi caratteristiche chimico-fisiche molto stringenti, certamente molto più critiche rispetto a quelle delle paste serigrafiche usate in passato per la decorazione. Nella Tab. 4.6 sono riportati alcuni valori di riferimento per le principali caratteristiche degli inchiostri inkjet, a confronto con i valori tipici delle paste serigrafiche.
Proprietà
Densità
Viscosità
Solvente
Apolare (idrocarburi, eteri, ecc.)
Polare (acqua + glicole)
Tab. 4.6 Caratteristiche degli inchiostri per inkjet e serigrafia a confronto
Come si può osservare, gli inchiostri inkjet sono molto meno viscosi dei corrispondenti serigrafici, in quanto devono potere attraversare i piccoli canali all’interno delle testine di stampa (sezioni di passaggio dell’ordine di 20 ÷ 50 µm). Inoltre, viscosità e tensione superficiale sono parametri fondamentali per il buon funzionamento di una testina inkjet, con intervalli di lavoro molto stretti.
Densità e contenuto di solido possono variare in un intervallo maggiore; ovviamente si cerca di ottenere una concentrazione di particelle solide (pigmenti) più elevata possibile, per raggiungere uno sviluppo colore (dopo cottura) particolarmente intenso. A questo proposito, vista l’elevata finezza dei pigmenti per inkjet (vedasi la dimensione mediana delle particelle D50), la capacità di sviluppo colore è particolarmente critica: maggiore il grado di macinazione, maggiore la superficie specifica delle particelle, minore l’intensità di colore in quanto aumenta l’aggressione chimica durante la cottura.
Gli inchiostri per stampa digitale, a differenza delle paste serigrafiche, devono essere colori primari il più possibile puri ed intensi. A causa della natura chimico-fisica dei sistemi ceramici e della limitata disponibilità di ossidi e complessi cromofori, i colori primari ceramici risultano cromaticamente inferiori rispetto agli standard della stampa su carta (che si realizza con coloranti organici a temperatura ambiente).
Nell’esempio di Fig. 4.4, i colori ceramici per stampa digitale (a destra) evidenziano purezza ed intensità inferiori ai colori organici per stampa in quadricromia tradizionale CMYK (a sinistra). In particolare, giallo e magenta in ceramica sono decisamente lontani dall’equivalente organico per carta o plastica.

Per questa ragione, al fine di ampliare la resa cromatica finale della decorazione digitale ceramica, è indispensabile aumentare il numero di colori primari (e quindi i canali di stampa) passando dalla classica quadricromia (Ciano, Magenta, Giallo, Nero) a sei-otto colori, con possibilità di utilizzare fino a dodici barre di stampa. Così facendo, è possibile aggiungere inchiostri con alcune specifiche tonalità, presenti con maggior frequenza nei soggetti da stampare (ad es.: rosa, arancio, verde, turchese, nocciola o marrone scuro).
La necessità di ridurre la dimensione media delle particelle solide di oltre un ordine di grandezza ha richiesto la ricerca e la sintesi di nuovi pigmenti, in grado di resistere alle alte temperature di cottura del grès porcellanato (fino a 1220 °C). Recentemente sono anche state sviluppate formulazioni specifiche di dimensione nanometrica (nano-pigmenti).
Nella Fig. 4.5 è riportato un esempio dei possibili pigmenti coloranti disponibili per decorazione digitale ad alta definizione.
Colore
Blu intenso
Azzurro
Turchese
Marrone rossastro
Marrone scuro
Giallo oro
Giallo pallido
Nero
Beige
Co-Si
Zn-Co-Al
Zr-Si-V
Zn-Fe-Cr-Al / Zr-Si-Fe
Zn-Fe-Cr-Ni
Zn-Fe-Cr-Al-Si
Zr-Si-Pr
Co-Cr-Fe-Ni
Zn-Fe-Cr-Al-Si
Rosa Ca-Sn-Cr-Si
Verde
Cr-Al-Si
Fig. 4.5 Pigmenti coloranti usati nella decorazione digitale ceramica
La preparazione dei pigmenti e la loro fine macinazione (con dimensione mediana D50 inferiore a 1 µm) richiedono impianti dedicati e tecnologie molto avanzate, in generale non disponibili presso le industrie ceramiche. Si tratta infatti di preparazioni altamente specializzate, che vengono svolte dai principali Colorifici e Aziende chimiche che operano sul mercato mondiale.
Fig. 4.05
Anche il sistema liquido dove i pigmenti devono essere dispersi è oggetto di continui perfezionamenti e ricerche. La fase liquida deve favorire il processo di stampa inkjet, mantenendo il più possibile costanti i parametri di viscosità e di tensione superficiale, così importanti ai fini delle buone prestazioni delle testine inkjet. Il mezzo disperdente deve inoltre mantenere in sospensione le particelle (aventi densità molto superiore al liquido) in modo stabile e per lungo tempo, evitando pericolose precipitazioni e sedimentazioni nei circuiti o nelle testine.
Negli anni si è assistito ad un progressivo sviluppo della chimica di questi sistemi liquidi, passando da composti a base di idrocarburi alifatici, lineari e ciclici, fino alle attuali composizioni con derivati di oli e grassi vegetali (eteri ed esteri). Attualmente la tendenza è quella di sostituire almeno in parte i solventi organici con acqua, anche per una migliore sostenibilità e riduzione dell’impatto ambientale; questa operazione però non è semplice, in quanto l’elevata polarità dell’acqua non è compatibile con le testine inkjet di tipo piezoelettrico.
Una composizione tipica di un inchiostro per stampa digitale ceramica a base solvente (apolare) è riportata come esempio in Tab. 4.7. In questo caso, la parte solida (pigmento e fritta) complessivamente vale il 45% in massa; oltre ai solventi alifatici, sono presenti additivi in piccola percentuale, utili a correggere le proprietà del sistema liquido/solido, evitando coalescenze e formazione di schiuma nel circuito idraulico.
Tipologia
Componente
Colorante Pigmento ceramico 35
Fondente Fritta ceramica 10
Solvente 1 Idrocarburi alifatici ciclici 35
Solvente 2 Idrocarburi alifatici lineari 18
Stabilizzante Acido policarbossilico 0,8
Disperdente Acido poliestere 0,8
Antischiuma Poliacrilato 0,4
Tab. 4.7 Composizione indicativa di un inchiostro ceramico per inkjet
Gli inchiostri per stampa digitale, in virtù delle loro particolari caratteristiche e complessità, hanno un costo decisamente superiore agli inchiostri ed alle paste per serigrafia tradizionale. Grazie però alla modalità di applicazione, appunto digitale, è possibile dosarne l’utilizzo in modo estremamente preciso, controllando per via elettronica ogni singola goccia applicata. Lo spreco di inchiostro è quindi praticamente nullo, in quanto la stampa digitale permette un perfetto controllo della sincronizzazione con il supporto ceramico da decorare.
Come risultato, i consumi di inchiostro per metro quadrato di prodotto ceramico decorato sono decisamente bassi, nell’ordine di 10 ÷ 25 g/m2 sommando complessivamente tutti i colori delle varie barre di stampa, valore paragonabile a quanto scaricato da un solo canale con le macchine di decorazione tradizionali. In definitiva, la stampa digitale inkjet, pur permettendo una elevatissima qualità di stampa e flessibilità di produzione, ha determinato anche una riduzione dei costi operativi ed un aumento di efficienza del reparto di decorazione.
Parallelamente alla diffusione della tecnica inkjet per la decorazione, in tempi più recenti si è assistito all’utilizzo della stessa tecnica anche per applicazioni di smalti e materie, sempre per via digitale. In questo caso il problema non è la presenza del pigmento (l’effetto cromatico è assente o molto basso) quanto la necessità di apportare grandi quantità di materia solida (fino a 100 ÷ 200 g/m2 con una o più barre dedicate).
Ai fini della stampabilità con inkjet piezoelettrico restano valide tutte le considerazioni sopra riportate per gli inchiostri. Le caratteristiche chimico-fisiche sono quindi le stesse di Tab. 4.6, mentre una composizione tipica è quella riportata come esempio in Tab. 4.8. Il contenuto in massa di solido è circa 45%, la parte liquida (solvente apolare) è una miscela di idrocarburi minerali e derivati di grassi vegetali. Infine, sono presenti additivi in piccola percentuale per correggere le proprietà della sospensione.
Tipologia
Materia Fritta ceramica
45
Solvente 1 (apolare) Idrocarburi aromatici 40
Solvente 2 (apolare) Esteri di acidi grassi vegetali 10
Stabilizzante
Disperdente
Acido policarbossilico 2
Acido poliestere 2
Antischiuma Poliacrilato 1
Tab. 4.8 Composizione indicativa di uno smalto ceramico per inkjet (effetto brillante)
Sono disponibili sul mercato varie formulazioni di smalti lucidi, matt, matt trasparenti, lustri e ingobbi, tutti applicabili con le medesime macchine digitali (vedi prossimo paragrafo), nelle quali vengono allestite barre di stampa con versioni di testine inkjet ad alto scarico. Tali testine hanno particolari configurazioni microfluidiche che massimizzano il volume della goccia, arrivando a scaricare gocce fino a 100 ÷ 200 picolitri, ideali per coperture a campo pieno come richiesto per smalti ed effetti.
Le grandi quantità applicate per ogni strato di smalto e/o effetto (100 ÷ 200 g/m2) comportano un elevato consumo di materia (anche se ovviamente sempre inferiore al caso di smaltatura tradizionale) e quindi un costo superiore. Per contro, la smaltatura digitale consente una completa gestione automatica di questa fase, con ricette e applicazioni perfettamente ripetibili.
Altro tema collegato agli elevati volumi scaricati è l’impatto ambientale. I solventi utilizzati evaporano durante le prime fasi della cottura in forno e possono essere trascinati verso l’ingresso dai fumi in controcorrente, ancora parzialmente incombusti. Questa situazione può portare alla formazione di composti odorigeni molesti per gli operatori della linea e, in uscita dal camino fumi, anche per l’ambiente circostante. Per questa ragione, ultimamente, i produttori di inchiostri stanno modificando la parte solvente sostituendola con acqua e altri composti polari, in modo da limitare la produzione di sostanze indesiderate ed anche l’emissione di CO2.
La tecnica dell’applicazione a secco si è sviluppata per far fronte a nuove esigenze estetiche e tecniche, oltre alla auspicata prospettiva di ridurre l’impatto ambientale dei processi di smaltatura e di preparazione degli smalti nell’industria ceramica.
Le operazioni di smaltatura avvengono senza l’utilizzo di liquidi disperdenti e quindi senza la produzione di fanghi. La smaltatura a secco di graniglie non prevede operazioni di macinazione, in quanto già preparate a secco dal colorificio. Tale processo comprende una selezione granulometrica e lo scarto delle frazioni a monte e a valle dell’intervallo prefissato.
Le graniglie di fritta sono utilizzate per varie tipologie commerciali che vanno dal lucido al semilucido, al matt. L’applicazione richiede un primo smalto di fondo applicato ad umido, seguito dall’applicazione per caduta della graniglia a secco. Se si ha l’avvertenza di effettuare tale applicazione quando lo smalto sottostante è ancora umido, cioè non ancora essiccato, si ottiene già una buona stesura e adesione della graniglia alla superficie della piastrella. Tuttavia, si preferisce spesso applicare una sospensione di colla sulla graniglia per evitare che una sua frazione possa staccarsi per effetto delle correnti d’aria del forno. In alternativa si sostituisce tale applicazione di colla con quella di un ulteriore smalto per migliorare l’adesione. Le graniglie di fritta possono anche essere usate su un fondo in cui è stata preventivamente applicata una colla speciale con tecnica serigrafica o digitale (inkjet). In questo modo, si realizza il successivo fissaggio della graniglia solamente dove era stata preventivamente applicata la colla ed è sufficiente una aspirazione per asportare la graniglia non fissata ottenendo effetti estetici di decoro tridimensionali.
Uno dei principali problemi delle graniglie di fritta è quello degli “infusi”. Si tratta di corpi estranei alla composizione che, a volte, sono presenti in piccolissime percentuali nelle fritte. La natura di questi “inquinamenti” non è sempre la medesima. Si tratta a volte di impurezze colorate, altre volte di pezzetti di refrattario provenienti dal rivestimento dei forni fusori. La presenza di infusi miscelati nella graniglia genera molto frequentemente difetti puntuali sulla superficie della piastrella finita.
È possibile miscelare diversi tipi di graniglia anche di differenti famiglie e caratteristiche tecniche, moltiplicando i possibili effetti cromatici e ceramici; inoltre, modificando i rapporti tra le graniglie si può regolare il grado di fusibilità ed il coefficiente di dilatazione termica. Vale la pena ricordare che gli intervalli granulometrici delle diverse graniglie non possono essere molto diversi per evitare separazioni e stratificazioni nelle fasi di stoccaggio ed applicazione. Le fasce granulometriche tipiche per queste graniglie sono comprese fra 50 e 300 µm. Di seguito si riporta una rassegna delle diverse tipologie di graniglie per applicazioni a secco.
Questa tipologia di graniglie è la più sviluppata e diffusa nel mercato per l’alto grado di trasparenza ottenibile. Si presta per superfici che prevedono una fusione della graniglia fino alla sua stesura completa. In altri casi invece si producono piastrelle dove la fusione delle graniglie è parziale.
Bianco lucido
Si tratta di una graniglia ottenuta da una fritta meno fusibile solitamente opacizzata con silicato di zirconio.
Bianco matt
Questo prodotto presenta inconvenienti sotto forma di graniglia da fritta, a causa di frequenti inquinamenti da infusi difficilmente controllabili. Un altro problema generale in questi casi è l’ottenimento di superfici ben stese.
Graniglie speciali
In questa gamma si annoverano le graniglie con effetto iridescente (risultano molto buone quelle ottenute dalla fritta), con effetto cristallizzato e con effetti di trasparenze colorate molto profonde. È sempre possibile miscelare diversi tipi di graniglia moltiplicando gli effetti cromatici e ceramici ottenibili. Gli intervalli granulometrici non possono essere molto diversi per evitare possibili separazioni e stratificazioni nelle fasi di stoccaggio ed applicazione.
Graniglie per prodotti tecnici
Sono solitamente graniglie da fritta impiegate in granulometrie fini. L’applicazione avviene su smalti di fondo specifici ed in spessori molto bassi. Gli smalti di provenienza appartengono alla famiglia dei “prodotti tecnici”, quindi con elevata resistenza all’abrasione e con buona compattezza anche all’interno della massa vetrosa. Sono in grado di superare il test ISO 10545-7 Classe V.
[1] W. Zachariasen, Journal American Ceramic Society, n. 17, p. 3841, 1932.
[2] B. Warren, Journal American Ceramic Society, n. 17, p. 249, 1934.
[3] A. Dietzel, «Glass Structure and Properties», Glass science and technology, vol. 22, p. 1187, 1968.
[4] A. Bresciani, C. Ricci, B. Spinelli e W. Medici, «Digital decoration of ceramic slabs with high colorfulness» in Qualicer 2016, Castellón (ES), 2016.
[5] S. Englise e W. Turner, «Relationship between chemical composition and the thermal expansion of glasses», Journal of American Ceramic Society, vol. 10, n. 8, pp. 551-560, 1927.
[6] A. Winkelmann e O. Schott, «Dependence of the thermal resistance of various glasses from the chemical composition», Annalen der Physik, vol. 287, n. 4, pp. 730-746, 1894.
[7] A. Knecht, «Control of crazing in sanitary ware glazes by Mayer And Havas coefficient of expansion factors», Journal American Ceramic Society, vol. 23, pp. 61-63, 1940.
[8] N. Tozzi, Smalti Ceramici, Faenza Editrice, 1992.
[9] G. Merendi e C. Gardenghi, Gli smalti ceramici - influenza delle materie prime, C&C, 2022.
La carbossimetilcellulosa è un polimero derivato della cellulosa per funzionalizzazione con gruppi carbossimetilici (–CH2–COOH) e successiva trasformazione in sale sodico. È comunemente usata come modificatore di viscosità o come addensante per stabilizzare le sospensioni ceramiche (barbottine di smalti).
Prende il nome dalla caratteristica trasparenza e brillantezza superficiale tipica del “vetro cristallo” che questa tipologia di fritta presenta dopo cottura ad alta temperatura. Può essere applicata sia prima che dopo i decori, per conferire un aspetto di vetro cristallino, omogeneo e brillante. Esiste anche una particolare tipologia di “cristallina matt” che, pur rimanendo estremamente trasparente, ha una finitura superficiale non brillante (matt).
Si definisce eutettico una specifica miscela composta da due o più componenti che presenta una temperatura di fusione inferiore alle temperature di fusione dei singoli componenti puri; quindi la miscela è più “facile da fondere” rispetto ai singoli materiali. Solitamente si identificano come eutettici le composizioni con le minime temperature di fusione.
Si tratta di una miscela di ossidi ceramici portata a fusione in appositi forni e raffreddata bruscamente tramite colata in acqua (dal cui tipico rumore deriva il nome "fritta"). A seguito del raffreddamento istantaneo il vetro solidificato si frantuma in piccoli pezzi, che vengono poi impiegati nella preparazione, per macinazione, delle composizioni di smalti ed ingobbi.
In ambito ceramico con graniglia si intende una miscela di fritte macinate ad una granulometria compresa tra 50 e 300 µm. In particolare, la graniglia di fritte trasparenti è utilizzata per elevati spessori di copertura sopra i decori digitali, ottenendo uno strato protettivo idoneo alla lappatura a specchio.
Dal francese engober, ovvero ricoprire con uno strato di terra il corpo ceramico. Infatti, la funzione primaria dell’ingobbio è coprire il supporto con uno strato opaco (normalmente di colore bianco) per limitare i difetti dovuti a eventuali contaminazioni e favorire una buona adesione tra impasto e smalto.
Una tipologia totalmente differente è invece un “ingobbio marca”, costituito da una barbottina a composizione refrattaria che viene applicata nella faccia inferiore delle piastrelle (che riporta la “marca” del produttore), con lo scopo di limitare la sporcatura dei rulli ceramici durante la cottura.
Il pigmento colorante in campo ceramico è un composto inorganico cristallino solitamente costituito da uno o più ossidi di metalli cromofori, insolubile nella composizione, tipicamente vetrosa, nel quale viene incorporato. La stabilità del pigmento nel vetro o negli smalti durante la cottura è condizione necessaria per mantenere l’integrità del reticolo cristallino e permettere le transizioni elettroniche che determinano lo sviluppo del colore caratteristico.
SBOBBA
La sbobba ceramica è una sospensione acquosa con un alto contenuto di materia solida. Il termine viene utilizzato per smalti liquidi, applicabili su piastrelle o lastre già decorate in linea di smaltatura, costituiti prevalentemente da una o più miscele di micro-graniglie trasparenti di fritte ceramiche in una base acquosa addizionata con leganti e sospensivanti sia organici che ceramici. Queste miscele risultano idonee all’applicazione a spruzzo airless oppure con cortina (vela).
Rivestimento vetroso che ricopre il corpo ceramico, lo rende impermeabile e gli conferisce una maggiore valenza estetica. Dopo cottura, lo smalto può risultare bianco, trasparente o colorato, con una finitura superficiale lucida, satinata o matt. È costituito da una miscela di fritte e materie prime minerali e viene solitamente preparato ed applicato sotto forma di sospensione acquosa prima della cottura.
Termine che deriva dall’unione di smalto e ingobbio, introdotto a seguito dello sviluppo della decorazione digitale su grès porcellanato. La sua funzione principale è quella di coprire il colore dell’impasto con uno strato solitamente bianco, idoneo anche allo sviluppo cromatico dei colori degli inchiostri digitali.
Il vetro è un solido amorfo formatosi dal rapido raffreddamento di un liquido fuso ad alta temperatura, processo che non ha permesso una completa cristallizzazione ma ha generato un reticolo distorto e disordinato, con caratteristiche tipiche di un solido trasparente. In campo ceramico il vetro è costituito da silicati di vari ossidi (Ca, Na, Zn, ecc.) e risulta insolubile, resistente e chimicamente stabile.
Nel processo ceramico l’acqua svolge un ruolo di grandissima importanza, dovuto in particolare alla sua influenza sulle proprietà dei minerali argillosi. Già dal punto di vista geologico, la genesi sedimentaria delle argille avviene in ambiente acquoso nei depositi alluvionali o, nel caso del caolino, attraverso l’alterazione di rocce primarie per azione dell’acqua.
L’interdipendenza tra acqua e argilla prosegue poi nelle principali fasi di lavorazione (si veda il Volume II), a partire dai processi di macinazione ad umido con la preparazione di una barbottina le cui caratteristiche di stabilità e di viscosità, quindi di lavorabilità, sono totalmente determinate dalla interazione argilla-acqua.
Un’altra proprietà fondamentale del sistema acqua-argilla è la plasticità dei minerali argillosi, ovvero la loro capacità di essere deformati con l’applicazione di sforzi di taglio e di compressione, come avviene nelle fasi di formatura per estrusione (impasti con 15 ÷ 20% di acqua) e per pressatura a secco (polveri con 5 ÷ 8% di acqua).
Come descritto nel Capitolo 2, la struttura dei minerali argillosi, costituita da strati di foglietti tetraedrici e ottaedrici, consente l’interposizione di molecole d’acqua e di ioni, determinando una maggior o minor coesione delle particelle stesse, ovvero le loro proprietà reologiche in sospensione e la loro lavorabilità allo stato plastico.
Le proprietà dei materiali ceramici, crudi o cotti che siano, sono in gran parte correlabili alla loro composizione chimica e mineralogica, ma altrettanta importanza rivestono altri parametri, come la dimensione e la forma delle particelle, il loro impaccamento e la loro interazione nel formare la struttura del corpo ceramico. Tutto ciò, infatti, influenza la possibilità di cambiamenti chimici e fisici a seguito dell’energia fornita nel processo di cottura.
Per questo motivo i componenti degli impasti ceramici devono essere sottoposti ad una ben definita riduzione delle dimensioni delle particelle, in modo da ottimizzarne i processi di trasformazione; ciò avviene, normalmente, attraverso una serie di metodi fisici di lavorazione, detti di comminuzione.
Un processo di comminuzione consiste nell’applicare sufficiente energia ad una particella, sino a causare la sua rottura o la separazione in particelle di dimensioni inferiori; il comportamento di ciascuna particella sarà, ovviamente, differente in funzione della sua resistenza alla compressione ed all’abrasione, della sua durezza, della sua elasticità, della sua forma, della sua microstruttura, parametri determinati anche dalla storia “geologica” del minerale stesso. Senza entrare nel merito dei principi teorici che governano la macinazione (descritti nel Volume II a cui si rimanda), i principali meccanismi di comminuzione sono i seguenti:
- Schiacciamento o compressione delle particelle, per azione di sistemi macinanti realizzati in materiali particolarmente duri. Teoricamente tale processo di comminuzione è più o meno efficace in funzione della resistenza a compressione del materiale processato, anche se la forma irregolare delle particelle risulta maggiormente soggetta alle forze di taglio e di tensionamento. Il prodotto di questa azione consiste normalmente nell’ottenimento di particelle di dimensioni simili, con scarsa presenza di polveri.
- Macinazione delle particelle, che implica differenti meccanismi, dove la comminuzione avviene attraverso lo sfregamento delle particelle fra loro e con i corpi macinanti, determinandone la frantumazione con riduzione graduale delle dimensioni medie e contemporanea produzione di abbondante particolato fine; per tale motivo la distribuzione granulometrica finale risulta piuttosto ampia.
- Frattura per impatto, che coinvolge ulteriori meccanismi di comminuzione, tramite l’azione di forze applicate su spigoli e facce delle particelle, particolarmente efficaci sui piani di frattura delle particelle stesse. Normalmente la rottura avviene lungo le linee di minor resistenza della struttura, dovute alla natura mineralogica del materiale o a piani di contatto fra specie minerali differenti.
In generale i macchinari utilizzati per la comminuzione di materiali ceramici, come illustrato nel Volume II, utilizzano una combinazione di tutti i principi fisici sopra descritti, con la generazione di particelle in un intervallo dimensionale molto ampio; pertanto si prediligono sistemi con vaglio a ricircolo, in modo da eliminare il più possibile le parti già comminute dal sistema di macinazione e reintrodurre le frazioni ancora grossolane.
Una materia prima per ceramica composta da particelle di varie dimensioni e varie forme può essere considerata un sistema particolato, la cui la distribuzione granulometrica, determinata da processi di macinazione a secco e soprattutto ad umido, è di grande importanza tecnologica. Tuttavia, a causa della forma irregolare delle particelle (non perfettamente sferiche), della loro eterogeneità composizionale e della loro tendenza ad agglomerare, una misurazione precisa delle loro dimensioni risulta difficoltosa. A riguardo, i sistemi argillosi presentano la maggiore complessità, in quanto sono costituiti da particelle colloidali (micelle) non facilmente disperdibili per la loro tendenza ad agglomerare a causa delle cariche elettriche superficiali che ne influenzano la stabilità ed il comportamento reologico.
Pertanto nell’eseguire una misura granulometrica di un materiale argilloso disperso in acqua, è necessario adottare opportune modalità di dispersione. Inoltre, sia nelle singole materie che negli impasti, vi è spesso la compresenza di particelle con dimensioni variabili dal micron al millimetro, ma non esiste alcun metodo strumentale in grado di coprire con sufficiente accuratezza un intervallo di misura così ampio. Inoltre le particelle di un materiale ceramico macinato non possono essere considerate sferiche, ma hanno spesso forme irregolari. Ne consegue che molti strumenti di misura ottici della distribuzione granulometrica di sospensioni acquose si basano su conteggi statistici, applicando equazioni correttive del fattore di forma delle particelle (diametro equivalente sferico). Da ultimo, anche le possibili differenze di densità tra particelle di diversa natura mineralogica possono interferire con una corretta determinazione della distribuzione granulometrica, sia nel caso di misure gravimetriche per polveri che nei metodi basati sulla sedimentazione delle sospensioni.
I principali metodi di classificazione granulometrica di materie prime e impasti ceramici sono elencati nella Tab. 5.1, in funzione delle dimensioni minime e massime rilevabili.
Metodo
Intervallo di applicazione [µm]
Setacciatura > 50
Micro setacciatura / filtrazione
0,2 ÷ 50
Microscopio ottico 25 ÷ 2500
SEM - Microscopio elettronico 0,5 ÷ 1000
AFM - Microscopio a forza atomica 0,001 ÷ 5
Sedimentazione 1 ÷ 50
Elutriazione 2 ÷ 50
Centrifugazione
0,05 ÷ 5
Dispersione di raggi X 0,05 ÷ 100
Diffrazione laser 0,05 ÷ 200
Permeabilità ai gas 0,1 ÷ 300
Tab. 5.1 Principali metodi di classificazione dimensionale delle particelle
Nella pratica produttiva, la classificazione delle polveri viene espressa sulla base delle caratteristiche dei setacci stabilite da normative nazionali ed internazionali (lSO internazionale, BS inglese, UNl italiana, DlN tedesca o NF francese); particolarmente utilizzata in tutto il mondo è la classificazione in Mesh ASTM (USA), la cui equivalenza dimensionale è riportata in Tab. 5.2. Nel Paragrafo 12.11 è invece riportata una tabella comparativa dei setacci secondo le diverse normative.
Risulta evidente che i limiti di applicabilità dei metodi di setacciatura sono idonei soltanto per le frazioni più grossolane (> 50 µm). D’altro canto, i metodi idrodinamici, storicamente utilizzati per la determinazione delle particelle fini, sono complessi e richiedono tempi lunghi. Per queste ragioni, le migliori tecniche per l’analisi granulometrica (vedi Paragrafo 6.5) sono oggi rappresentate dalle strumentazioni che impiegano radiazioni incidenti di ridotta lunghezza d’onda (raggi X o luce visibile laser) per rilevare le particelle durante la loro sedimentazione.
Considerando la legge di Stoke, espressa dell’equazione: �������� = 2 9 (��������1 ��������2 ) �������� ���������������� 2
dove v = velocità di caduta di una particella (m/s)
μ = viscosità del mezzo sospendente in Pa∙s (acqua = 1 mPa∙s a 20 °C)
ρ1 = densità della particella (kg/m3)
ρ2 = densità del mezzo sospendente (kg/m3)
g = accelerazione di gravità (9,81 m/s2)
r = raggio della particella, assunta sferica (m).
è infatti possibile correlare la velocità di sedimentazione con le dimensioni medie delle particelle stesse e calcolare la loro distribuzione.
A titolo di esempio, applicando l’equazione di Stoke ad una particella di materiale ceramico di densità 2,5 g/cm3 e di raggio 1 µm, è facile calcolare che la sua velocità di caduta in acqua a 20 °C è pari a 12 mm/ora; questo spiega come i tempi richiesti da un’indagine granulometrica idrodinamica risultino piuttosto lunghi, tanto più se è richiesta la determinazione di frazioni ultrafini inferiori a 1 µm.
Tab. 5.2 Classificazione Mesh ASTM
Graficamente, la distribuzione granulometrica di particelle in sospensione viene rappresentata tramite due curve tipiche (Fig. 5.1): una curva (o un istogramma) di distribuzione differenziale ed una curva cumulativa. In ascissa è riportato il diametro delle particelle, solitamente in scala logaritmica; in ordinata la percentuale relativa in massa corrispondente ad ogni determinato diametro (curva di frequenza) e la somma percentuale in massa delle particelle fino a quel determinato diametro (curva cumulativa).
Fig. 5.1 Curve di distribuzione granulometrica cumulativa (rossa) e differenziale (verde)
Dalla curva cumulativa sopra riportata si evince ad esempio che il 50% delle particelle ha un diametro inferiore ai 10 µm (D50) e dalla curva di distribuzione relativa che la frazione presente con maggior frequenza (moda) è a circa 20 µm in corrispondenza del massimo del picco.
Dal punto di vista produttivo, la pratica industriale ceramica prevede una serie di semplici controlli granulometrici, necessari per assicurare la qualità dei semilavorati, ovvero:
- controllo dimensionale delle materie prime da inviare alla macinazione, in modo da ottimizzare il processo;
- vagliatura del materiale grossolano in uscita dalla macinazione, e suo riciclo in ingresso al mulino;
- controllo granulometrico con un setaccio di riferimento (63 o 45 µm) delle barbottine di impasti e smalti al fine di verificare la correttezza e la costanza dei parametri tecnologici.
Relativamente al processo di macinazione ed omogeneizzazione dell’impasto, è opportuno evidenziare la forte correlazione tra dimensioni delle particelle e caratteristiche del prodotto finito, in quanto la distribuzione granulometrica ha una grande influenza sulla reattività dei singoli componenti durante la fase di cottura.
Infatti il grado di macinazione di una miscela di materie prime condiziona le trasformazioni chimico-fisiche alle alte temperature, le variazioni dimensionali, la densità, la porosità, il peso specifico, la durezza, ecc.
A tale riguardo, il semplice controllo granulometrico di una barbottina di impasto o di smalto, effettuato con una singola setacciatura (il convenzionale residuo a 63 o 45 µm), è solamente una verifica della costanza produttiva di un ben determinato processo di macinazione, ma non è in generale rappresentativo della distribuzione granulometrica totale. È infatti ben evidente come le curve granulometriche rappresentate in Fig. 5.2 presentino un identico residuo (circa 2% a 63 µm), ma siano nettamente diverse come distribuzione. In
particolare l’impasto A mostra una curva gaussiana centrata a 20 µm, mentre l’impasto B presenta una distribuzione bimodale con il massimo valore a circa 0,5 µm ed un diametro medio intorno a 9 µm. Ne consegue che il comportamento tecnologico dei due impasti, pur con un identico valore del residuo a 63 µm, sarà totalmente differente in quanto il prodotto B è molto più fine del prodotto A.
Fig. 5.2 Analisi granulometrica di due impasti con uguale residuo ma differente distribuzione
Infatti la variazione granulometrica determina una diversa superficie specifica delle polveri ed ha un’immediata influenza sulle superfici di contatto fra le particelle e quindi sull’area reattiva che, per reazioni solido-solido o solido-liquido viscoso ad alta temperatura, è di primaria importanza nei processi di sinterizzazione.
5-02 IT
Nella Fig. 5.3 è schematizzata una matrice di impasto ceramico, composta da particelle più grossolane di feldspati (F) e quarzo (Q) contenute in una dispersione di particelle argillose (struttura simile a mattoni).
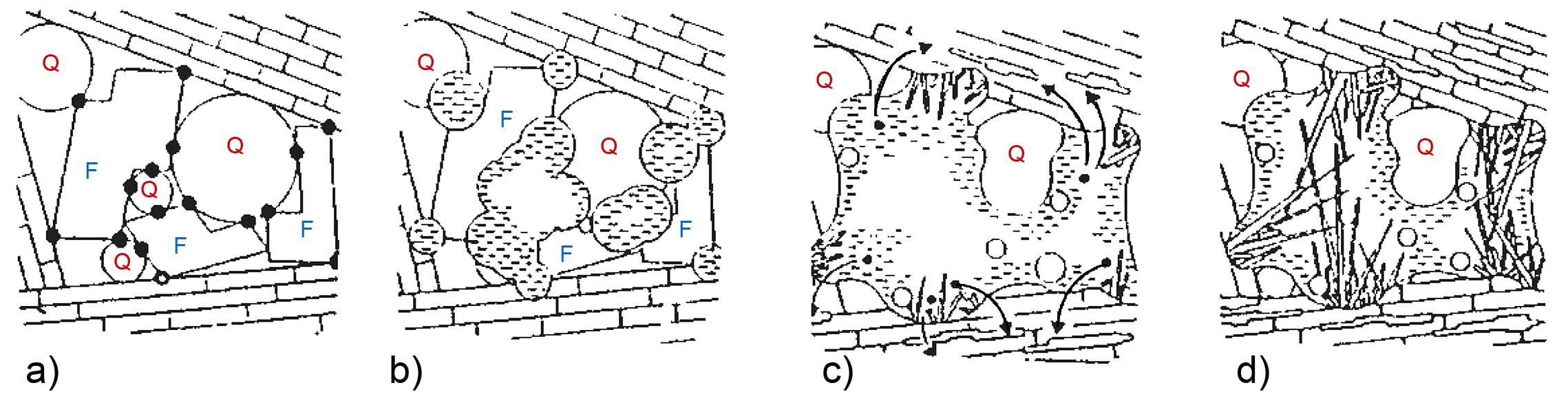
Fig. 5.3 Ipotesi di progressiva fusione-reazione
Il processo di sinterizzazione ha inizio nei punti di contatto delle particelle (a), dove si esplica inizialmente l’azione fondente dei feldspati (punti neri); a seguito della reattività iniziale, che coinvolge più facilmente le particelle più piccole e maggiormente a contatto con i fondenti, si forma una fase liquida (b) che ingloba gradualmente la struttura argillosa circostante (c). Con il progredire del processo termico si ha poi la formazione di nuove fasi cristalline che determinano le proprietà finali del prodotto ceramico (d).
Per quanto concerne l’importanza dei parametri granulometrici nella produzione ceramica, oltre alla preparazione delle barbottine in sospensione acquosa, bisogna considerare anche le dimensioni delle polveri di impasto da utilizzare nei processi di formatura.
A riguardo, il processo di fabbricazione delle piastrelle ceramiche prevede la pressatura di polveri a basso contenuto di umidità (5 ÷ 8%), provenienti sia da impianti di macinazione a secco che soprattutto dall’essiccamento a spruzzo (atomizzazione) di barbottine ottenute per macinazione a umido. Nel primo caso si hanno particelle solide (dense, di forma irregolare e con spigoli vivi) opportunamente bagnate e granulate; nel secondo si ottengono agglomerati di particelle di forma sferoidale, internamente cave, con elevata scorrevolezza e dimensioni comprese tra 125 e 800 µm.
In entrambi i casi le polveri devono poter essere facilmente trasferite dai silos ai sistemi di alimentazione e caricamento delle presse, distribuendosi omogeneamente negli alveoli degli stampi o sul nastro di trasporto della Continua+; conseguentemente la distribuzione delle dimensioni dei granuli ha grande importanza nell’ottenimento di un pezzo ceramico pressato (“verde”) compatto, a densità omogenea ed esente da difetti strutturali.
Le differenze fra i due tipi di macinazione, a secco e a umido, e le caratteristiche delle polveri risultanti sono trattate diffusamente nel Volume II riguardante i processi di produzione.
I minerali argillosi presentano spazi interparticellari e cariche elettrostatiche in grado di interagire con le molecole di acqua che, a causa della forte elettronegatività dell’ossigeno rispetto all’idrogeno, sono di fatto piccoli dipoli, con parziale carica negativa sull’atomo di ossigeno e parziale carica positiva sugli atomi di idrogeno:


Quando le argille vengono mescolate all’acqua, si passa dapprima ad uno stato di pasta plastica (con circa 17% in peso di acqua), per poi ottenere sospensioni (con circa il 30% in peso di acqua) con caratteristiche reologiche estremamente differenziate, che possono essere considerate a tutti gli effetti dispersioni colloidali per le dimensioni sub-micrometriche delle particelle argillose elementari.
Le interazioni elettrostatiche interlaminari sono ulteriormente influenzate dalle irregolarità strutturali o chimiche presenti nella superficie delle particelle, come ad esempio:
- raggruppamenti ionici distorti;
- legami superficiali rotti e non saturati, nei piani di scorrimento o di sfaldamento;
- cationi a carica non saturata, che creano scompensi di cariche nel reticolo cristallino. Infatti, se in un piano basale del reticolo cristallino in equilibrio elettrico avviene una frattura, la superficie generata si trova in condizioni di carenza o eccesso di carica, situazione che può determinare differenti scenari attrattivi o repulsivi nei confronti delle molecole di acqua e soprattutto tra le particelle stesse.
5.4 Meccanismi di flocculazione e deflocculazione di particelle argillose
Nella Fig. 5.4 sono schematizzati i cristalli della caolinite che ai bordi presentano cariche positive (A) e negative (B), nello stato deflocculato (C), con flocculazione bordo-faccia (D), con flocculazione faccia-faccia (E), con flocculazione prodotta da ioni Ca2+ (F) e allo stato deflocculato con ioni Na+ (G).
Fig. 5.5 Distribuzione delle cariche intorno ad una particella di argilla in sospensione
Con riferimento alla Fig. 5.5, si osserva che la superficie delle particelle è caricata negativamente per la presenza dei gruppi OH , che attraggono ioni idrogeno e cationi formando un secondo strato positivo. Quest’ultimo a sua volta crea un forte legame elettrostatico con il dipolo delle molecole d’acqua circostanti, generando uno strato di acqua legata che risulta molto più densificato rispetto all’acqua libera.
Fig. 5-05 IT
Tale primo strato di molecole di acqua, orientate con il dipolo negativo verso le cariche positive e attirate dalla superficie della particella, fa parte del cosiddetto “doppio strato”, ed ha uno spessore variabile fra 7 e 60 Å circa. Il doppio strato può essere considerato una sorta di “condensatore” molecolare, caratterizzato da un potenziale zeta, che determina la stabilità della sospensione delle particelle colloidali, dette micelle.
Relativamente alle sospensioni acquose di polveri di impasti ceramici, i principali fenomeni di natura elettrostatica che è necessario controllare, o meglio guidare, sono la flocculazione ed il suo contrario, la deflocculazione. Con la flocculazione, da una sospensione omogenea di particelle in acqua, si provoca una rapida sedimentazione di “fiocchi”, aggregati di particelle a maggiore densità, poi facilmente separabili dal sistema acquoso; la deflocculazione invece favorisce, tramite opportuni additivi, la dispersione di eventuali aggregati e mantiene le particelle in sospensione anche per tempi lunghi ed in assenza di agitazione. Nella pratica della macinazione ad umido di impasti argillosi, quest’ultima esigenza è, ovviamente, la più sentita.
Entrambi gli effetti sono ottenuti tramite un’opportuna perturbazione del doppio strato elettrico delle particelle colloidali di argilla. In particolare, la flocculazione si ottiene introducendo cationi bi- o tri-valenti, di piccole dimensioni, quindi ad alta densità di carica, in grado di alterare fortemente l’equilibrio elettrostatico delle micelle in sospensione, mentre la deflocculazione è solitamente ottenuta tramite l’addizione di composti ionici, di cationi monovalenti (Na+ o NH4+), organici o inorganici, preferibilmente polimerici (i cosiddetti deflocculanti), che “isolano” una particella dall’altra, favorendone la repulsione.
Grazie alla loro carica negativa le particelle argillose hanno, infatti, la capacità di attrarre nel doppio strato i cationi carichi positivamente, lasciando la restante parte polimerica come barriera alla flocculazione. La forza con cui i cationi vengono attratti è funzione di un bilancio fra la loro carica ed il loro volume idrodinamico, soprattutto considerando quello che assumono dopo idratazione. La tendenza dei cationi ad essere adsorbiti è pertanto la seguente: H+ > Fe3+ > Al3+ > Ba2+ > Sr2+ > Ca2+ > Mg2+ > NH4+ > K+ > Na+ > Li+ quindi cationi interessanti per stabilizzare una sospensione sono, ad esempio, sodio e potassio, mentre cationi ad alta densità di carica (trivalenti, bivalenti o anche monovalenti come il piccolo ione H+) portano alla distruzione del doppio strato elettrico e quindi alla flocculazione. In tal caso il problema viene risolto introducendo un eccesso di cationi a bassa mobilità elettroforetica, che spostano l’equilibrio della reazione di scambio: (Argillan−·Xn+) + nY+ ⇔ (Argillan−·nY+) + Xn+ dove X = catione flocculante e Y = catione deflocculante. In tal modo un’argilla sodica può essere ottenuta da un’argilla calcica aggiungendo un opportuno eccesso di ioni sodio e, eventualmente, sottraendo gli ioni calcio a mano a mano che si formano, ad esempio per precipitazione.
Nella pratica industriale bisogna anche considerare la tempo-dipendenza dei fenomeni di idratazione dei minerali argillosi, della solubilizzazione dei sali presenti e delle reazioni di scambio ionico, tutti fattori che influenzano le caratteristiche di lavorabilità delle sospensioni.
Il termine reologia, dal greco rheo (scorrere) e lógos (studio), significa appunto lo studio dello scorrimento, ovvero della deformazione dei corpi solidi o fluidi sotto l’azione di forze esterne. Un esempio specifico nel settore ceramico è la reologia delle sospensioni argillose (barbottine), ma in modo similare si può anche studiare la reologia delle polveri. È evidente come queste tematiche siano di enorme interesse industriale, perché riguardano le principali fasi dei cicli produttivi di tutti i manufatti ceramici, indipendentemente dalla tecnologia adottata (colaggio, estrusione o pressatura di polveri).
Una particolare attenzione sulle caratteristiche reologiche viene richiesta nella macinazione ad umido delle materie prime per la produzione di impasti o smalti, oltre che nell’applicazione degli smalti stessi. A riguardo, può risultare difficoltoso per il tecnico ceramista tradurre in termini rigorosi il vocabolario spesso figurato, gergale, utilizzato tradizionalmente per descrivere fenomeni di natura reologica. Alcune espressioni tipiche sono ad esempio: “lo smalto fila”, “è bobboso”, “è piombato nella vasca”, “si è snervato”, oppure ancora, “il mulino è imballonato”, “la barbottina è gelata” e così via. Questo modo di esprimersi molto empirico nasconde, in realtà, una certa consapevolezza degli effetti determinati dalle proprietà reologiche dei semilavorati nei processi produttivi frutto di conoscenze costruite sperimentalmente, spesso non razionalizzate in modo da poter essere spiegate. Oggi, viceversa, le esigenze di qualità e flessibilità delle linee produttive non possono più basarsi su conoscenze empiriche. Gli smalti e le barbottine di impasto devono rispettare precisi standard produttivi, con caratteristiche ben definite e misurabili attraverso opportuni sistemi di controllo di processo.
Tra i parametri che spiegano il comportamento reologico, il più significativo è certamente il concetto di viscosità. Questo termine, infatti, è entrato nel linguaggio comune ed è intuitivamente associato alla maggiore o minore facilità di scorrimento di un fluido, ovvero allo sforzo necessario affinché questo raggiunga una determinata velocità. Minore è lo sforzo per muovere un fluido, minore sarà la sua viscosità. Si introduce quindi il concetto di sforzo applicato ad un fluido parallelamente alla sua superficie, ovvero la grandezza fisica nota come sforzo di taglio tangenziale. È altresì intuitivo che una massa fluida sottoposta a una forza non si muove uniformemente come un corpo solido ma presenta al suo interno differenze di velocità. Ad esempio, il flusso di un fluido all’interno di una tubazione sarà massimo al centro e minimo a contatto con la parete. La variazione di velocità degli strati di un fluido in funzione della loro posizione viene espressa come gradiente di velocità. Esistono poi altre due importanti grandezze che caratterizzano il comportamento di un fluido: la prima è lo sforzo iniziale minimo necessario a mettere in moto un fluido, definito come limite di scorrimento; la seconda grandezza, tipicamente riferibile alle sospensioni argillose ed associata alla tempo-dipendenza della loro viscosità, è la tissotropia
A seguire si riporta una trattazione dettagliata con le formule fisiche delle grandezze reologiche sopra citate.
Per calcolare la viscosità di un fluido è necessario misurare le grandezze fisiche che la determinano. Una di queste è appunto il gradiente di velocità. Se in una massa fluida in movimento misuriamo la velocità massima (Vmax) e minima (Vmin) corrispondenti a due diverse posizioni a distanza (L), la differenza tra le due velocità diviso per la distanza si definisce gradiente di velocità (D):
Le dimensioni fisiche di D sono:
cioè l’inverso del tempo, pertanto il gradiente di velocità viene solitamente espresso in secondi reciproci (s–1).
Questa grandezza è direttamente correlata al maggiore o minore attrito interno al fluido. Infatti, se a parità di condizioni sperimentali, un fluido presenta un elevato gradiente di velocità, significa che le sue molecole producono un basso attrito interno.
Il secondo fattore per il calcolo della viscosità è lo “sforzo di taglio” al quale il fluido è sottoposto.
Tornando all’esempio del fluido che scorre in una tubazione, il suo moto richiede l’applicazione di una forza per vincere dapprima l’attrito statico poi l’attrito dinamico sia tra fluido e parete che all’interno del fluido stesso.
Si tratta di forze applicate tangenzialmente (Ft) alla unità di superficie (A) del fluido che ne causano lo scorrimento.
Questa grandezza viene definita sforzo di taglio e si indica solitamente con la lettera greca t (tau):
Dimensionalmente t risulta una pressione, anche se la forza applicata non è perpendicolare bensì parallela alla superficie, e come tale si esprime in Pascal o in unità equivalenti:
5.4.4
Considerando le due grandezze fisiche appena descritte, si comprende chiaramente come il moto di una massa fluida venga generato dall’applicazione di uno sforzo di taglio, che determina velocità di scorrimento decrescenti all’aumentare della distanza, ovvero un gradiente di velocità, funzione dell’attrito interno al fluido stesso. Da un punto di vista fisico la viscosità rappresenta una misura di questo attrito, essendo definita come il rapporto fra lo sforzo di taglio applicato ed il corrispondente gradiente di velocità generato.
Questa grandezza si indica generalmente con la lettera greca η (eta), secondo la formula:
Le sue dimensioni sono quelle di una pressione per un tempo:
Dalla formula si evince che il valore della viscosità è direttamente proporzionale allo sforzo di taglio necessario per generare nel fluido un determinato gradiente di velocità; pertanto quanto maggiore è la viscosità di fluido, tanto maggiore sarà lo sforzo da applicare per produrre la variazione di velocità desiderata.
L’unità di misura della viscosità nel Sistema Internazionale è il Pascal per secondo (Pa∙s); viene comunque ancora utilizzata l’unità Poise (P) secondo la relazione:
1 Pa⋅s = 10 P
Di fatto, però, queste unità di misura sono troppo grandi per molte applicazioni pratiche, per cui si preferisce usare i loro sottomultipli:
milliPascal per secondo = mPa·s = centiPoise = cP
In campo ceramico, l’intervallo di viscosità passa da 1 ÷ 10 cP per smalti applicati a disco o con airless, aumentando fino a 100 ÷ 1000 cP per smalti applicati a velo o per le barbottine da atomizzazione. L’intervallo dei gradienti di velocità ai quali vengono sottoposte le sospensioni ceramiche è ancora più ampio, in quanto si passa da 1 ÷ 10 s–1 nelle vasche munite di agitatore lento, a 100 ÷ 1000 s–1 nelle pompe, per arrivare a 1000 ÷ 10000 s–1 all’interno degli ugelli dell’atomizzatore. Inoltre nella fase di applicazione degli smalti, il fluido può subire repentinamente elevate variazioni del gradiente di velocità, in quanto passa dal moto generato dal dispositivo applicatore ad un valore prossimo a zero sulla superficie della piastrella.
A fronte di intervalli così ampi del gradiente di velocità, si può facilmente dedurre come la viscosità di una sospensione ceramica non possa avere un valore costante, e per tale motivo risulta complesso prevederne il comportamento reologico nelle diverse condizioni di lavoro. Solamente alcune tipologie di fluidi, come l’acqua o l’olio, presentano un comportamento ideale, cosiddetto “newtoniano”, dove il rapporto tra sforzo di taglio e gradiente di velocità rimane costante per qualsiasi valore del gradiente di velocità. Solo in tal caso il comportamento del fluido può essere descritto da un unico valore di η, proprio perché la viscosità rimane costante per tutte le velocità di scorrimento e dipende solo dalla temperatura.
Per i fluidi newtoniani, riportando in grafico i valori di t (sforzo di taglio) misurati al variare di D (gradiente di velocità), si ottiene una retta (vedi Fig. 5.6) definita dalla relazione:
�������� = �������� �������� = tan ��������
Fig. 5.6 Reogramma tipico di un fluido a comportamento newtoniano
La viscosità è il rapporto tra le due grandezze, ovvero la pendenza della retta, cioè la tangente dell’angolo α; maggiore è l’inclinazione della retta più alta è la viscosità.
Questo tipo di rappresentazione grafica (reogramma) è estremamente utile per valutare il comportamento di un fluido che, in questo caso, continuerà a scorrere con velocità decrescente al diminuire dello sforzo di taglio fino a fermarsi quando lo sforzo di taglio si azzera.
Fig. 5-06
Una dimostrazione pratica di un comportamento newtoniano è il versamento di una goccia d’olio sul pavimento: lo scorrimento sarà più o meno lento a seconda della viscosità dell’olio, ma continuerà fino a quando lo sforzo di taglio prodotto dal peso del liquido non verrà controbilanciato dalla tensione superficiale. Dal momento che l’olio ha una tensione superficiale molto bassa, la goccia si spanderà letteralmente “a macchia d’olio”.
L’esperienza quotidiana nella produzione ceramica insegna che le sospensioni ceramiche si comportano in modo molto diverso da un fluido newtoniano come l’olio.
Il principale scostamento dal comportamento ideale risiede nella mancanza di scorrimento di una sospensione ceramica fino a quando lo sforzo applicato non supera un determinato valore di soglia o, allo stesso modo, il moto del fluido si arresta non appena lo sforzo di taglio scende sotto tale valore.
Il valore di soglia dello sforzo di taglio che deve essere superato per iniziare lo scorrimento viene definito limite di scorrimento (yield point), è indicato con t0 (tau zero) e la sua unità di misura è il Pascal.
Il reogramma (t in funzione di D) di un fluido con limite di scorrimento è riportato in Fig. 5.7, dove risulta evidente che lo scorrimento inizia solo dopo aver applicato uno sforzo superiore a t0. Un fluido con questo comportamento è detto fluido di Bingham.
Fig. 5.7 Reogramma di un fluido con limite di scorrimento iniziale t0 (fluido di Bingham)
Le sospensioni ceramiche presentano spesso un limite di scorrimento che ne determina alcune loro caratteristiche, come ad esempio la stesura degli smalti o, nel caso di applicazioni su superfici verticali, l’assenza di colature.
5-07
Tra i comportamenti reologici che si discostano da quelli ideali (newtoniani o di Bingham), riveste un particolare interesse in campo ceramico la tempo-dipendenza. È noto, infatti, che le sospensioni ceramiche, in particolare in presenza di elevate percentuali di argille, possono assumere una consistenza semi-solida se rimangono a riposo per un certo tempo, per poi ritornare fluide quando vengono agitate. Questa caratteristica è particolarmente accentuata in presenza di alune tipologie di argille molto plastiche, come le bentoniti. Questa loro proprietà viene ad esempio sfruttata nell’industria petrolifera per sigillare i pozzi di petrolio: il pozzo viene riempito con una barbottina fluida facilmente pompabile, che, lasciata a riposo, diventa praticamente rigida e costituisce così un’ottima chiusura. Il grande vantaggio di questa tecnica è che il pozzo può essere facilmente riattivato, semplicemente introducendo un getto d’acqua che rende nuovamente fluida la bentonite.
Tale comportamento è determinato dal fatto che, in stato di riposo, la struttura del fluido si modifica in funzione del tempo: tra le particelle di argilla in sospensione si formano legami tridimensionali che rendono rigida la struttura. Questi legami vengono distrutti dalla applicazione di uno sforzo di taglio e necessitano di un certo tempo per riformarsi quando il fluido torna in stato di quiete. In questi sistemi argillosi, anche la viscosità cambia ovviamente in funzione del tempo.
La maggior parte delle barbottine di impasto o di smalto si comporta in questo modo, ma, di solito, la dipendenza della viscosità dal tempo non crea grossi problemi, perché si manifesta a regimi di scorrimento molto bassi, mentre le sospensioni ceramiche vengono generalmente mantenute in agitazione a gradienti di velocità maggiori.
Viceversa il limite di scorrimento, al di sotto del quale non vi è movimento del fluido, è indipendente dal tempo. Tuttavia, molto spesso, una sospensione che manifesta un limite di scorrimento, ha anche la tendenza ad essere tempo-dipendente.
La differenza è che il limite di scorrimento rimane una caratteristica del fluido anche quando è stato sottoposto ad agitazione, mentre, nel caso di un fluido tempo-dipendente, il valore di t o può crescere enormemente se il fluido è lasciato a riposo. Si pone quindi anche il problema di come misurare correttamente il limite di scorrimento.
A riguardo, una possibile metodologia di preparazione del campione per una analisi reologica prevede di sottoporre la sospensione ad un elevato gradiente di velocità per un periodo di tempo sufficiente a demolire la struttura tridimensionale eventualmente formatasi, per poi riportare il gradiente di velocità a zero e procedere con la registrazione del reogramma a gradiente crescente e ritorno.
Idealmente la curva t-D di ritorno (D decrescente) dovrebbe essere sovrapponibile a quella di andata (D crescente). Al contrario, in presenza di un comportamento dipendente del tempo, il reogramma ottenuto al diminuire di D non ripercorre il tracciato registrato all’aumentare di D.
Una ulteriore modalità per valutare la tempo-dipendenza consiste nell’effettuare misure di viscosità al variare del tempo mantenendo il campione di misura a valore di D costante e costruendo quindi un grafico di η rispetto al tempo.
Nella pratica industriale, può succedere che una sospensione tenda a indurire in brevissimo tempo, ostacolando, ad esempio, lo scarico di una barbottina da un mulino discontinuo. In altri casi le sospensioni tendono ad aumentare progressivamente la loro viscosità anche se vengono mantenute in lenta agitazione. In tale situazione, la barbottina si lascia scaricare, pompare e setacciare benissimo ma, una volta introdotta in vasca, tende a flocculare (“gelare”) perché la velocità dell’agitatore non è sufficiente ad impedire la formazione di una struttura tridimensionale. Se non si interviene in tempo con opportuni additivi fluidificanti, la situazione può diventare veramente difficile da gestire perché tutto il contenuto della vasca diventa rigido e soltanto la sezione interessata dalle pale dell’agitatore rimane fluida. A questo punto l’aggiunta di fluidificante diventa difficile perché non si riesce a distribuirlo efficacemente. L’unica cosa da fare è diluire il fluidificante in acqua e aggiungerlo lentamente alla barbottina, facendo in modo di creare un ricircolo tra il fondo e la superficie della vasca, utilizzando una pompa.
Così facendo, lentamente il fluidificante produce il suo effetto, ma sono necessarie diverse ore per ottenere la completa omogeneizzazione. In questo lasso di tempo la barbottina assume un aspetto orribile, in quanto le zone raggiunte dal fluidificante tornano fluide, mentre persistono ammassi galleggianti non ancora fluidificati che sono ancora solidi. Quando una vasca si trova in questa situazione in gergo si dice che la barbottina “ha fatto le rane”, ma è comunque una situazione temporanea che, correttamente gestita, può essere risolta.
Un ultimo parametro, correlato alla tempo-dipendenza e frequente causa di comportamenti anomali è la tissotropia. Si tratta della tendenza delle sospensioni a modificare la loro viscosità in funzione del regime di scorrimento al quale erano sottoposte in precedenza. Nelle sospensioni tissotropiche la viscosità risulta più bassa se il fluido proviene da uno stato di maggiore agitazione, condizione che è spesso associata alla presenza anche di un limite di scorrimento. In pratica, riportando in grafico i valori di t e D, con gradiente di velocità prima crescente e poi decrescente, si ottiene un reogramma con isteresi (Fig. 5.8). Questo effetto di isteresi rappresenta la tissotropia, in quanto non esiste una singola grandezza fisica che possa rappresentare questo comportamento. L’unico modo per quantificare un comportamento tissotropico è quello di effettuare una serie di misure a gradiente di velocità crescente e poi decrescente e quindi riportare in grafico i valori ottenuti. La tissotropia è quindi rappresentata dall’area racchiusa dalle due curve di andata e ritorno. Chiaramente i risultati ottenuti dipendono in maniera strettissima dalla procedura seguita nell’analisi, per cui è necessario operare secondo un metodo standardizzato per garantire una ripetibilità delle misure. Inoltre, siccome la tissotropia è spesso associata alla presenza di un limite di scorrimento e di una tempo-dipendenza, è anche opportuno stabilire un metodo di condizionamento del campione prima di eseguire la misura, in modo da eliminare qualsiasi "effetto memoria".
In ceramica questi particolari effetti reologici sono provocati dalla presenza in sospensione di particelle cariche elettrostaticamente con una forma tale da potersi allineare secondo la direttrice di scorrimento fluido (come, ad esempio, foglietti, bastoncini o filamenti). Partendo da uno stato iniziale parzialmente aggregato, all’aumentare del gradiente di velocità, progressivamente le particelle si allineano e, per tale motivo, quando il gradiente di velocità viene diminuito, le barbottine ceramiche scorrono più facilmente rispetto all’andata (caso (a) di Fig. 5.8). Esistono anche altre tipologie di materiali con comportamenti opposti, dove l’agitazione favorisce la reazione tra le particelle aumentando la viscosità (caso (b) di Fig. 5.8).
Nell’applicazione degli smalti, si è solitamente interessati a valutare il comportamento di una sospensione che proviene da stati di elevata sollecitazione e che rallenta progressivamente fino a fermarsi (come una goccia lanciata da un ugello airless). Il tratto di curva che interessa di più nel reogramma di Fig. 5.8, è quindi quello con gradiente di velocità decrescente che, nel caso degli smalti, risulta quasi perfettamente lineare. Il comportamento reologico della sospensione può quindi essere descritto efficacemente in termini di limite di scorrimento e viscosità plastica (tratto di viscosità costante sopra il limite di scorrimento).
Una eccessiva tissotropia può invece generare problemi applicativi, soprattutto quando è necessaria una bassa viscosità con un certo limite di scorrimento. Inoltre, una elevata tissotropia non permette un buon controllo della viscosità, proprio perché questa varia in funzione della agitazione della sospensione.
Per tale motivo è generalmente opportuno ridurre al minimo la tissotropia di una sospensione ceramica, modificando i rapporti tra i componenti argillosi ed ottimizzando il contenuto e la tipologia dei deflocculanti. In merito a questi ultimi è preferibile utilizzare miscele di diversi additivi chimici con effetti alternativi e/o sinergici.
(a) comportamento tissotropico (es. caolino in acqua)
τ (c) comportamento reopessico (es. ossido di vanadio in acqua)
τ (e) comportamento tisso-reopessico
D
τ (b) comportamento antitissotropico (es. idrossido di magnesio in acqua)
D
D
τ (d) comportamento antireopessico
D
τ (g) Comportamento tissoantitissotropico
D
τ (f) comportamento antireopessico antitissotropico
D
D
τ (h) comportamento antireo-reopessico
D
Fig. 5.8 Reogrammi che mostrano isteresi e corrispondente comportamento
Le barbottine a base di argille presentano un comportamento reologico non newtoniano, in quanto sono classificabili come fluidi plastici e pseudoplastici di tipo tissotropico. Una stessa argilla può determinare sia un comportamento plastico che pseudoplastico in funzione della percentuale di solido nella barbottina.
Generalmente nel settore ceramico la presenza di tissotropia e di un limite di scorrimento, conseguenza del comportamento plastico, sono indesiderabili. Tuttavia, nel ciclo tecnologico di produzione delle barbottine argillose, gli effetti tissotropici hanno una modesta influenza, con possibili interferenze sulle operazioni di setacciatura o nel caso in cui la barbottina rimanga nelle vasche di stoccaggio per tempi molto elevati.
Come descritto nel Paragrafo 5.3, per ottenere una buona reologia delle barbottine, risultano di primaria importanza tutte le grandezze che influenzano direttamente il doppio strato elettrico delle micelle argillose ed in particolare: lo scambio cationico, l’area di contatto tra solido e liquido (dipendente dalla superficie specifica delle particelle), la presenza di sali solubili nell’argilla (soprattutto solfati e cloruri di calcio). Eventuali azioni correttive possono prevedere una diminuzione della percentuale argillosa nella barbottina e/o una ottimizzazione della quantità e tipologia di deflocculante aggiunto. Ovviamente lo stesso processo di macinazione è strettamente correlato al comportamento reologico, in quanto determina la granulometria e la temperatura della barbottina. Una macinazione più spinta aumenta la solubilità dei sali, la superficie di contatto tra acqua e solido, e conseguentemente lo scambio cationico. Ciò comporta un decremento della fluidità della sospensione con aumento della viscosità apparente, della tissotropia e del limite di scorrimento. Questi effetti possono essere ulteriormente accentuati dall’aumento di temperatura che si verifica con tempi di macinazione molto elevati, come nel caso del grès porcellanato. L’aumento di temperatura, anche oltre i 60 °C, determina una positiva iniziale diminuzione della viscosità ma anche un possibile aumento indesiderato della tissotropia e del limite di scorrimento, favorito dalle cinetiche di dissoluzione dei sali e dal conseguente scambio cationico.
Ovviamente una elevata percentuale di argilla nella barbottina peggiora il comportamento reologico in quanto tutti i minerali argillosi hanno la tendenza a generare sistemi tissotropici. Relativamente all’impiego dei deflocculanti per ottimizzare la reologia dei sistemi acqua-argilla, i principali prodotti di uso ceramico sono: il tripolifosfato di sodio, l’esametafosfato di sodio, il silicato di sodio o il poliacrilato di sodio. Questi composti deflocculanti vengono normalmente introdotti in percentuali da 0,1 a 1%, ma bisogna tener presente che concentrazioni troppe elevate possono causare una sovra-deflocculazione, che destabilizza la barbottina e provoca la comparsa di un limite di scorrimento con un aumento della tissotropia. In generale, con riferimento all’uso di un singolo deflocculante, si può affermare che i fosfati e, in particolare il tripolifosfato sodico, forniscono buoni risultati con tutte le tipologie di argille, permettendo di controllare in modo efficace la loro tissotropia. L’utilizzo dei silicati dà normalmente buoni risultati sulle argille magre e/o ad alto contenuto di caolino, ma in ogni caso il controllo della tissotropia risulta più difficoltoso. Gli additivi polimerici di tipo organico risultano generalmente buoni disperdenti e stabilizzanti, ma sono meno efficaci nella riduzione della viscosità apparente. Infine lo studio di opportune miscele di vari deflocculanti,
con diversi meccanismi di azione, può consentire l’ottimizzazione delle proprietà reologiche anche in presenza di sistemi argillosi particolarmente critici.
Come già illustrato nel Capitolo 3, i minerali argillosi più comuni presenti all’interno delle argille comunemente utilizzate nel settore ceramico sono: il caolino, la illite, le smectiti (montmorillonite, nontronite) e le cloriti [1].
Le caratteristiche reologiche di una determinata materia prima argillosa sono pertanto dipendenti dalla tipologia e dalla quantità dei minerali argillosi in essa presenti.
Di seguito vengono illustrate brevemente le caratteristiche reologiche dei minerali argillosi sopra elencati, premettendo che le considerazioni sono riferite ai minerali puri e pertanto hanno unicamente una valenza di indirizzo nel caso di argille reali di uso industriale.
Caolino: la sua struttura mineralogica non permette la presenza di acqua interlamellare, quindi un caolino non presenta una apprezzabile capacità di scambio di cationi. Inoltre, i caolini, solitamente molto puri, non contengono sali solubili. Questi fattori fanno sì che normalmente il caolino non manifesti un comportamento reologico di tipo plastico; pertanto, non si osservano limiti di scorrimento se non operando in condizioni reologiche particolarmente sfavorevoli, come alte concentrazioni, deflocculanti non adatti o macinazioni molto spinte (tenendo comunque presente che, fra i vari minerali argillosi, il caolino è quello che risente meno della macinazione). Il pH dei caolini è normalmente tra 5 e 6; questo fa sì che una deflocculazione ottimale si possa facilmente ottenere con l’uso di composti basici (silicati di sodio, carbonato di sodio o anche idrossido di sodio) che spostano il pH nell’intervallo da 7 a 9. Anche i fosfati ed i polimeri organici salificati agiscono in modo ottimale sul sistema acqua-caolino. Come premesso, i comportamenti descritti sono riferiti a caolini puri, ma le considerazioni effettuate possono essere estrapolate anche alle argille caolinitiche sia bianche che rosse, a condizione che risultino poco contaminate da sali solubili o da minerali argillosi di altra natura.
Illite: per quanto riguarda le caratteristiche reologiche della illite, si può fare riferimento solamente a sistemi argillosi contenenti percentuali più o meno elevate di questo minerale, in quanto non esistono argille totalmente illitiche.
Valutando ad esempio un’argilla di tipo illitico-caolinitica, rispetto a un caolino, si osserva a parità di condizioni un aumento del comportamento plastico del sistema, con una tissotropia più accentuata ed una maggiore viscosità apparente.
Viceversa, se consideriamo una argilla illitico-montmorillonitica per confronto con una montmorillonite, notiamo che il valore della viscosità apparente risulta meno elevato, così come il limite di scorrimento e la tissotropia del sistema.
In definitiva si può affermare che una illite risulta essere un minerale argilloso con un comportamento reologico meno favorevole rispetto a un caolino, in quanto presenta limite di scorrimento e tissotropia. Queste caratteristiche vengono accentuate dalle condizioni di macinazione, essendo l’illite più sensibile della caolinite alla riduzione delle dimensioni delle particelle. I deflocculanti più efficaci risultano il tripolifosfato e il metasilicato di sodio, mentre sono poco efficaci quei prodotti che agiscono solo sul pH come il carbonato e l’idrossido di sodio. Questo è dovuto al fatto che il pH naturale dell’illite risulta meno acido del caolino.
Montmorillonite: dal punto di vista reologico questo minerale è sicuramente quello che presenta le caratteristiche più negative, almeno per quanto riguarda le barbottine per uso ceramico. Infatti, in altri settori, come ad esempio nel campo dei fanghi per trivellazione, la montmorillonite è impiegata proprio per le sue proprietà reologiche.
Le caratteristiche principali della montmorillonite sono una elevata tissotropia e la presenza di un limite di scorrimento particolarmente alto, ovverosia un marcato comportamento plastico della sua sospensione acquosa. Sia la viscosità plastica che quella apparente risultano elevate anche a basse percentuali di contenuto solido della barbottina.
Una certa differenziazione nel comportamento di questi minerali argillosi si può notare in funzione del tipo di cationi presenti negli strati. Infatti, in linea di massima, vi sono tre principali tipi di montmorilloniti: acide, sodiche e calciche. Le acide sono montmorilloniti ottenute per trattamento di montmorilloniti sodo-calciche e risultano estremamente difficili da deflocculare a meno di non utilizzare deflocculanti che provocano un forte innalzamento del pH. Le sodiche viceversa sono quelle più facilmente deflocculabili e possono essere fluidificate con tripolifosfato e poliacrilati. La calcio-montmorillonite è il minerale che generalmente è presente nelle argille per uso ceramico; la sua deflocculazione risulta più difficile del tipo sodico e richiede generalmente l’uso di miscele a base di fosfati o poliacrilati. In ogni caso la presenza di questo minerale deve essere attentamente controllata ed è buona norma limitarne la percentuale al di sotto del 6% per impasti magri e sotto al 4% per impasti plastici.
Clorite: rispetto ai minerali descritti in precedenza, definire le caratteristiche reologiche di una clorite risulta estremamente difficile. Questo è dovuto al fatto che pur essendo la clorite uno dei minerali argillosi più comuni, considerata la sua genesi, è sempre associata a illite e/o smectite. Pertanto, risulta estremamente difficile discriminare il comportamento reologico della clorite da questi minerali. Si può affermare solamente che, considerando la sua struttura e la compresenza di altri minerali argillosi plastici, un’argilla cloritica presenta generalmente scarse caratteristiche reologiche con tissotropia e limite di scorrimento.
A conclusione di questa breve disanima sulle caratteristiche reologiche delle argille, occorre sottolineare come solo le prove di deflocculazione effettuate in laboratorio permettono di quantificare il comportamento reologico di un’argilla, mentre i dati mineralogici e chimici forniscono solamente una stima indicativa.
Gli impasti ceramici sono costituiti essenzialmente da una frazione argillosa plastica e da una frazione inerte-fondente “dura” a base quarzoso-feldspatica, oltre che da componenti in minor quantità come: chamotte, carbonati, recuperi, fanghi, ossidi coloranti, sbiancanti, ecc. L’insieme delle materie prime, opportunamente dosate, viene caricato in mulini a tamburo con i necessari quantitativi di acqua e di deflocculante, quindi macinato fino ad ottenere una barbottina ad un residuo prefissato.
La frazione argillosa presenta naturalmente un intervallo granulometrico molto inferiore ai residui di macinazione richiesti e quindi subisce soltanto l’azione disgregante dell’acqua fino a formare una fase finemente dispersa, mentre i materiali duri subiscono un’azione molitoria per impatto e rotolamento dei corpi macinanti.
Sulla base della costituzione mineralogica e chimica di un impasto, si possono pertanto suddividere i vari componenti in funzione del loro comportamento reologico:
- frazione argillosa colloidale, i cui effetti sono stati trattati nel Paragrafo 5.3;
- materiali inerti o fondenti praticamente insolubili in acqua, quali feldspati, quarzo, silicato di zirconio, allumina;
- materiali solubili in quantità apprezzabili: sali alcalini e alcalino terrosi presenti nelle materie prime, nell’acqua industriale e nelle eventuali aggiunte di scarti e fanghi.
I prodotti insolubili non plastici intervengono nella reologia delle barbottine solo come “numero di particelle per unità di volume” che aumenta con il procedere della macinazione generando particelle più fini caratterizzate da una maggior superficie specifica. Si tratta di corpuscoli sostanzialmente inerti dal punto di vista chimico fisico, ma attivi dal punto di vista reologico in quanto aumentano la densità di particelle in uno spazio già molto affollato di particelle e sali solubili, e quindi incrementano la resistenza allo scorrimento, cioè la viscosità della barbottina.
I sali solubili possono essere costituiti da cationi flocculanti, ad alta carica come Ca2+, Mg2+, Fe2+, Fe3+, Al3+, Zn2+ e/o da cationi monovalenti (Na+ e K+), oltre che da anioni, quali: solfati, cloruri, nitrati, borati, carbonati e bicarbonati. Possono provenire dalle argille, dai fanghi di recupero e soprattutto dall’acqua di macinazione.
Gli effetti degli ioni flocculanti sono ben evidenti: essi contribuiscono alla distruzione del doppio strato elettrico (riduzione del potenziale zeta) esistente fra le micelle, diminuendone le forze repulsive. D’altra parte, anche la presenza degli altri ioni comporta una elevata concentrazione di cariche, con un deciso aumento della forza ionica della soluzione acquosa, che influenza negativamente il potenziale zeta ed ostacola il processo di deflocculazione, anche a basse concentrazioni di cationi flocculanti.
In estrema sintesi, riguardo all’importanza relativa dei tre principali componenti gli impasti ceramici, si può affermare che è la frazione argillosa plastica a rivestire il ruolo principale nel comportamento reologico della barbottina, fermo restando l’influenza dei sali solubili spesso determinante. Viceversa, si può considerare l’effetto dei materiali inerti non plastici decisamente secondario rispetto alle altre due tipologie di componenti.
Prove di macinazione condotte in laboratorio utilizzando acqua deionizzata hanno confermato quanto sia deleteria la presenza di ioni solubili; infatti, a parità delle altre variabili, la viscosità di barbottine preparate con acqua deionizzata è nettamente inferiore rispetto ai test eseguiti con acque potabili o industriali.
Purtroppo, le acque utilizzate per la macinazione degli impasti sono spesso le peggiori dell’intero stabilimento, contenendo i lavaggi ed i recuperi degli altri reparti, quindi smalti, fanghi e sostanze organiche di vario genere. Un’alternativa non può neppure essere l’utilizzo di acqua depurata, poiché si è riscontrato che i flocculanti impiegati nella chiarificazione (cloruro ferrico, cloruro di alluminio, calce, poliacrilammidi), causano effetti ancora peggiori, dal punto di vista reologico, rispetto alle acque di recupero non trattate.
Una ulteriore osservazione sull’acqua di macinazione riguarda il pH: come già osservato esiste un intervallo di pH ottimale per la deflocculazione di sospensione argillose, di norma compreso fra 8 ÷ 9. È evidente che acque i cui valori di pH si discostino notevolmente da questo intervallo contribuiranno a rendere più difficoltoso il processo.
In ambito produttivo risulta pertanto utile un controllo periodico dell’acqua di macinazione. Più che una vera e propria analisi chimica, non eseguibile agevolmente all’interno della maggior parte delle aziende ceramiche, si possono suggerire alcuni test la cui esecuzione è sufficientemente rapida e non richiede strumentazioni di costo elevato:
- pH: la misura di questa grandezza si effettua con un pH-metro;
- conducibilità elettrica: misurata con un conduttimetro (esistono in commercio semplici strumenti portatili), fornisce una indicazione approssimata della concentrazione ionica totale;
- durezza totale: si determina con cartine reattive o per titolazione ed è correlata alla quantità di ioni Ca2+ e Mg2+, con proprietà flocculanti;
- COD (Chemical Oxygen Demand): è una misura delle sostanze organiche ossidabili presenti nell’acqua. Anche questa determinazione, se pure più complessa delle altre, si effettua per titolazione.
Il controllo periodico e frequente di questi parametri permette di avere una sufficiente controllo della qualità dell’acqua di macinazione, di correlare variazioni di questi parametri ad eventuali peggioramenti delle caratteristiche reologiche delle barbottine e di poter fissare empiricamente limiti di tolleranza, superati i quali occorre intervenire ad esempio diluendo con acque non riciclate.
L’intero processo di preparazione dell’impasto ceramico può essere brevemente schematizzato come segue:
- dosaggio delle materie prime principali e ausiliarie, dell’acqua, del deflocculante e caricamento dei mulini;
- omogeneizzazione dei componenti, che avviene negli stadi iniziali di rotazione all’interno dei mulini con bilanciamento delle masse da macinare (questa parte del processo è pressoché inefficace in termini di macinazione ed è di durata estremamente variabile);
- macinazione vera e propria: durante questa fase si verifica una riduzione della granulometria delle particelle solide e ad un affinamento della curva granulometrica verso valori via via inferiori;
- scarico dei mulini;
- setacciatura della barbottina, effettuata con batterie di vibrosetacci;
- sosta in vasca a lenta agitazione;
- travaso in una piccola vasca di servizio, pompaggio ad alta pressione ed atomizzazione.
Durante la fase di dosaggio ed omogeneizzazione, caratterizzata da valori di densità estremamente variabili, avviene anche la dissoluzione del deflocculante (se questo è stato introdotto allo stato solido) e comunque la sua dispersione all’interno della fase acquosa. Spesso, un buon andamento di questa fase iniziale del processo determina l’efficacia dell’intera macinazione, in quanto le eventuali problematiche che si presentano in questo primo periodo si possono trascinare lungo tutto l’arco del processo, creando a volte seri problemi allo scarico e/o macinazioni insufficienti.
Un tipico esempio è “l’impallonamento” dei mulini, riscontrabile in sistemi ad elevato contenuto di frazioni plastiche: il movimento rotatorio genera masse sferoidali compatte (“palloni” o “clay balls”) di dimensioni estremamente variabili, che sono costituite da argilla plastica e che, nei casi peggiori, inglobano anche i corpi macinanti. Se queste masse sono piccole tendono a galleggiare sulla fase dispersa, mentre se aumentano di dimensioni a causa del rotolamento (a effetto valanga), devono essere frammentate meccanicamente a mulino fermo. In ogni caso il materiale “impallonato” non viene macinato ed il residuo finale risulterà troppo alto e con una composizione di impasto modificata.
I motivi per cui si verificano tali fenomeni sono verosimilmente riconducibili alla scarsa bagnabilità di alcune materie prime, ad un caricamento troppo irregolare che può portare a stratificazioni, ad argille eccessivamente umide e plastiche, oppure ad accumuli di deflocculante in zone preferenziali che si traducono in un ritardo nella sua azione disperdente.
Per contrastare l’insorgere di tali fenomeni indesiderati si può intervenire migliorando la premiscelazione di tutti i componenti dell’impasto, addizionando l’acqua contemporaneamente alle materie prime e soprattutto utilizzando deflocculanti in soluzione o già disciolti nell’acqua di macinazione. Una combinazione di interventi come quelli descritti riduce il tempo necessario per l’iniziale fase di dispersione ed omogeneizza rapidamente il sistema, consentendo una efficace azione dei corpi macinanti, con una sensibile riduzione del tempo totale di macinazione.
Se questi interventi non sono risolutivi è possibile che la scarsa disperdibilità degli agglomerati argillosi sia dovuta a corpi macinanti non corretti; in tal caso è necessario rivedere la carica in termini qualitativi, quantitativi e granulometrici.
Dal momento in cui si forma una barbottina sufficientemente omogenea e a densità costante, diventano importanti le caratteristiche reologiche del sistema, una sospensione tendenzialmente instabile, pseudoplastica con limite di scorrimento e tissotropica.
Esaminando il processo di macinazione propriamente detto, la variazione nel tempo di viscosità e residuo (preso come indice di finezza granulometrica) può essere rappresentata come in Fig. 5.9.
viscosità η residuo R A) B)
Fig. 5.9 Andamento nel tempo di residuo (A) e viscosità (B) durante la macinazione
Il residuo di macinazione (curva A) decresce fino a stabilizzarsi; da questo punto in poi un ulteriore tempo di macinazione determina modesti aumenti della finezza e della reattività dell’impasto ma con un notevole consumo dei corpi macinanti.
Fig. 5-09 IT R
La viscosità (curva B) ha invece un deciso calo iniziale, seguito da un periodo più o meno lungo di assestamento su valori pressoché costanti, per poi risalire nella fase finale al prolungarsi del tempo di macinazione.
Il calo iniziale della viscosità è dovuto all’azione del deflocculante, più o meno marcata, secondo i meccanismi precedentemente descritti; la fase a viscosità costante è da imputare al fatto che la frazione argillosa, maggiormente interessata alle interazioni con il deflocculante, si trova già in un sistema omogeneo ad una granulometria fine. L’aumento finale della viscosità è invece imputabile a diversi fattori:
- la riduzione granulometrica della frazione inerte determina un aumento della concentrazione di particelle ad elevata superficie specifica;
- la temperatura del sistema può aumentare oltre il valore considerato ottimale (40 ÷ 50 °C);
- il tempo di macinazione, la temperatura e gli stress fisico-meccanici possono portare ad un parziale deterioramento del deflocculante oltre a interferire negativamente sui meccanismi di scambio ionico.
Durante la fase di macinazione, in cui il sistema è sottoposto continuamente ad elevati sforzi di taglio, ha importanza soprattutto la viscosità, che deve essere sufficientemente contenuta per consentire il movimento agevole dei corpi macinanti, ma non troppo bassa
per non provocare l’eccessivo consumo degli stessi, cosa che si tradurrebbe in uno scarso effetto macinante. Ciò risulta particolarmente vero per chi utilizza corpi ad elevata densità, dove casi di macinazione viscosa hanno portato a notevoli riduzioni dei tempi di lavorazione.
Le eventuali difficoltà di scarico vengono semplicemente risolte aggiungendo piccole quantità di deflocculante in modo da abbassare il limite di scorrimento e favorire il deflusso rapido della barbottina. Queste operazioni risultano facilitate dall’uso di deflocculanti liquidi ad alta efficienza e di sistemi di dosaggio automatici o semiautomatici.
Lo svuotamento o scarico dei mulini discontinui è un’operazione che richiede tempi mediamente compresi fra 30 e 90 min; la barbottina è sottoposta a bassi sforzi di taglio ed acquistano importanza grandezze reologiche come limite di scorrimento e tissotropia, piuttosto che la viscosità propriamente detta.
È in questa fase che barbottine tissotropiche e ad alto limite di scorrimento tendono ad assumere una struttura pseudo-solida (gelificazione), a formare croste superficiali, a lasciare residui quantitativamente importanti sui corpi macinanti e sul rivestimento dei mulini, rendendo lo scarico complessivamente lungo e difficoltoso. Ovviamente questi fenomeni sono pressocché assenti nella barbottina in movimento mentre aumentano progressivamente nel tempo nei sistemi statici o a lento scorrimento.
Considerazioni a parte meritano i sistemi che a fine macinazione raggiungono e superano i 70 ÷ 80 °C; questi, oltre a provocare viscosità più alte, mostrano spesso la formazione di croste solide superficiali dovute ad un rapido aumento di densità locale per evaporazione dell’acqua. Inoltre, eccessive temperature a fine macinazione possono essere sintomo di problemi o irregolarità nella granulometria dei corpi macinanti o del rapporto di carica del mulino.
La fase immediatamente successiva allo scarico è la setacciatura compiuta con setacci a vibrazione. Ovviamente una veloce setacciatura è favorita da un basso limite di scorrimento; ciò risulta tanto più importante per la tendenza ad utilizzare setacci a maglie sempre più fini, allo scopo di trattenere il più possibile eventuali impurezze causa di difetti sul prodotto finito.
Spesso la setacciatura rappresenta lo stadio lento dell’intero processo e molte aziende hanno equipaggiato i reparti di macinazione con batterie di setacci molto numerose.
A questo punto del processo, la barbottina di impasto sosta in vasche munite di lenti agitatori verticali a pale. Il tempo medio di stazionamento è estremamente variabile, e dipende dal dimensionamento dell’impianto: può andare molto orientativamente da 6 ÷ 8 ore a 12 ÷ 15 giorni.
In queste condizioni (sforzi di taglio molto bassi e tempi prolungati) diventano importanti i fenomeni tempo dipendenti come la tissotropia, fenomeni esasperati quando le vasche sono piene e si vengono a creare zone praticamente statiche.
È allora possibile osservare la formazione di “croste” superficiali, talora molto consistenti, che spesso sono un segnale di inizio di “gelificazione” o “ispessimento” dell’intera massa, fenomeno ben conosciuto e temuto dai tecnici di produzione, in quanto fonte di grandi difficoltà di gestione. In questi casi, la soluzione suggerita prevede l’addizione in vasca di deflocculante liquido, meglio se pre-diluito in acqua (in questo modo si contribuisce anche ad abbassare la densità della sospensione). Occorre però attendere, a volte anche diverse ore, affinché l’additivo agisca sull’intera massa.
Come ulteriore osservazione, la naturale scarsa stabilità delle sospensioni ceramiche rende sconsigliabili soste inutilmente prolungate prima dell’atomizzazione. Se i problemi di difficoltà di scarico e setacciatura, e/o gli aumenti di viscosità in vasca dovuti a fenomeni tissotropici sono ricorrenti è invece opportuno effettuare un accurato studio reologico dell’intero processo.
Le cause che portano ad aumenti di viscosità e ad altri fenomeni indesiderati possono essere imputabili a:
1. materie prime
2. acqua di macinazione
3. aggiunte accessorie
4. deflocculante
5. granulometria.
In particolare:
1) Variazione di una o più materie prime, in particolare di quelle argillose, tanto più probabile quanto più di frequente viene effettuato il rifornimento delle stesse. Anche marcate variazioni di umidità delle componenti argillose possono portare a variazioni di viscosità, ma sono facilmente individuabili perché sempre accompagnate da variazioni di densità della barbottina. Se invece è mutata la composizione mineralogica e/o chimica di alcuni componenti dell’impasto, risulta difficile individuare il materiale responsabile della variazione, a meno di accurate analisi chimiche e diffrattometriche, comunque non sempre risolutive. È anche possibile rendersi conto di eventuali variazioni di plasticità attraverso ripetute misure di modulo di rottura in crudo o di ritiro sul pezzo crudo e cotto. Oltre al controllo di alcuni parametri fondamentali (assorbimento, ritiro, colore in cotto, presenza di carbonati, ecc.) è quindi opportuno anche verificare il comportamento reologico delle materie prime. Infine una variabilità delle viscosità può dipendere anche da errori di formulazione o pesatura, ovvero da una maggior o minor presenza dei componenti plastici nella miscela che alimenta i mulini.
2) Variazione della composizione dell’acqua con particolare riferimento al suo contenuto salino (fenomeno già preso in considerazione in precedenza).
3a) Aggiunte (o aumento) di additivi plastificanti o tenacizzanti. L’introduzione o variazioni di questi prodotti nell’impasto vanno sempre precedute da prove di laboratorio.
3b) Aggiunte di fanghi, smalti e recuperi vari provenienti dalle smalterie, i cui effetti andrebbero valutati preventivamente.
3c) Aggiunte (o aumento) di calce esausta proveniente dai filtri dei forni. Poiché lo ione Ca2+ causa la flocculazione delle barbottine, il dosaggio di calce esausta va valutato accuratamente. Per tale motivo diverse aziende hanno rinunciato ad un suo recupero in macinazione, giudicandolo antieconomico rispetto ad altre soluzioni (ad es. conferimento a ditte specializzate).
4a) Diminuzione di deflocculante dovuto a dosaggio errato.
4.b) Diminuzione di deflocculante dovuto ad un forte assorbimento d’acqua dello stesso. È un fenomeno abbastanza raro e comunque solitamente ben visibile ad occhio nudo.
4.c) Deflocculante non adatto alla tipologia di impasto per una fornitura non corretta. Un prodotto diverso può ovviamente dimostrarsi non adeguato per lo specifico impasto utilizzato.
5a) Variazione della granulometria, ovvero aumento della frazione fine o iperfine per un incremento del tempo di macinazione (con conseguente diminuzione del residuo). Ovviamente un deciso aumento del tempo di macinazione porta ad un aumento di viscosità.
5b) Variazione della granulometria dovuta ad aggiunte (o aumento) dei recuperi di polveri dalle presse, a granulometria molto fine. Anche in questi casi è sempre consigliabile effettuare preventive verifiche di laboratorio.
A seguire, la barbottina stoccata e setacciata viene prelevata dalla vasca di servizio ed inviata all’atomizzatore ad una pressione di 15 ÷ 30 bar mediante l’azione di pompe alternative o centrifughe.
L’atomizzazione è la fase dell’intero processo tuttora meno investigata dal punto di vista reologico, anche perché gli stress fisici cui il sistema argilla/acqua è sottoposto sono difficilmente riproducibili dalle strumentazioni di laboratorio. Si può comunque affermare che le barbottine di impasto, sottoposte ad elevatissimi gradienti di scorrimento, mostrano un comportamento newtoniano in un regime laminare, almeno fino all’uscita degli ugelli dove, anche con artifici meccanici, si passa rapidamente ad un regime turbolento per formare le goccioline che, per azione del calore, si trasformano in granuli semi-essiccati. Senza questo rapido essiccamento, i getti di barbottina emessi dagli ugelli verrebbero proiettati contro la superficie superiore della camera, con la formazione di spesse incrostazioni e, nei casi più gravi, compromettendo la riuscita dell’intero processo.
Le caratteristiche come densità (peso specifico) e viscosità della barbottina, correlate al suo comportamento reologico, influenzano il processo di essiccamento a spruzzo e la qualità del prodotto atomizzato, come evidenziato nella Tab. 5.3.
Variabili (valori in aumento)
% acqua nella barbottina
Temperatura barbottina
Peso specifico barbottina
Viscosità barbottina





Tab. 5.3 Effetto di peso specifico e viscosità della barbottina sul prodotto atomizzato
Con il termine deflocculante si intende generalmente una sostanza che, aggiunta in piccola quantità ad un sistema costituito da una fase disperdente e da particelle colloidali, determina una maggior fluidità della dispersione (diminuzione della viscosità apparente) ed una sua stabilizzazione.
Per fase disperdente si intende la fase fluida preponderante nella quale sono disperse particelle solide o liquide non miscibili, che rappresentano quindi la fase dispersa. Nelle comuni sospensioni ceramiche la fase disperdente è acqua, mentre la fase dispersa è costituita da materie prime per impasti o smalti, con presenza di particelle argillose allo stato colloidale.
Per fluidificante si intende una sostanza capace di rendere più scorrevole un fluido, ovvero di abbassarne la viscosità. Al contrario, un addensante è un agente che va ad aumentare la consistenza di una massa fluida.
Nel linguaggio comune ceramico “deflocculante” è spesso sinonimo di “fluidificante”, termine però che implica unicamente una diminuzione della viscosità, rispetto all’azione più complessa dei composti deflocculanti che invece garantiscono anche la stabilità della dispersione, evitando il rapido aggregarsi delle particelle e la conseguente precipitazione dei solidi sospesi.
Pertanto, con il termine “deflocculante” si comprende sia l’azione di dispersione che quella di fluidificazione; il deflocculante certamente agisce anche come fluidificante, ma non tutti i fluidificanti esplicano anche la funzione deflocculante.
I principali additivi deflocculanti utilizzati sono riassunti nella Fig. 5.4:
INORGANICI
Carbonato di sodio
Na2CO3
Silicato di sodio
Na2O · nSiO2
Esametafosfato (NaPO3)6
Tripolifosfato sodico
Na5P3O10
Carbonato di bario BaCO3
ORGANICI
Acidi umici
Tannini
Sali monovalenti di poliacrilati X+ n (CH2CHCOO-)n
Sali monovalenti dell’acido ossalico X+2(COO-)2
Tab. 5.4 Principali additivi reologici
I meccanismi fondamentali che determinano l’azione deflocculante sono i seguenti:
1. spostamento del pH verso valori basici immettendo nel sistema acqua-solido ioni OH– attraverso l’aggiunta di elettroliti basici con cationi monovalenti, tenendo presente che un eventuale eccesso provoca sovra-deflocculazione;
2. sostituzione dei cationi che conferiscono un eccesso di cariche positive al doppio strato diffuso con ioni monovalenti (Na+, K+, Li+, NH4+);
3. aumento della carica negativa esistente sulle particelle argillose per adsorbimento di di anioni (l’adsorbimento è preferenziale per gli anioni con valenza più elevata dotati di un forte campo elettrico);
4. aumento della carica negativa totale del sistema solido-liquido immettendo un colloide non ionico a comportamento elettronegativo;
5. aggiunta di un colloide protettore che preserva le particelle sospese dall’azione dei flocculanti;
6. eliminazione dei flocculanti presenti:
a. per insolubilizzazione dello ione flocculante
Na2CO3 + Ca2+ → CaCO3↓ + 2Na+
BaCO3 + Ca2+ → CaCO3↓ + Ba2+
BaCO3 + SO42– → BaSO4↓ + CO32–
b. per formazione di complessi di coordinazione che sequestrano i cationi flocculanti (Ca2+, Fe3+).
I deflocculanti più efficaci, anche in combinazione tra loro, agiscono secondo più meccanismi tra quelli sopra elencati.
I deflocculanti sono normalmente divisi in inorganici ed organici. Gli inorganici sono elettroliti (basi monovalenti, elettroliti basici, carbonati, silicati e fosfati di sodio). Gli organici possono essere elettroliti o non elettroliti.
Sia che si aggiungano sostanze organiche od inorganiche, bisogna valutare eventuali loro effetti collaterali indesiderabili, ed in particolare la loro influenza sulla velocità di disidratazione dell’impasto, sulla plasticità, sul ritiro e sulla resistenza meccanica.
Va inoltre considerata la possibile influenza del deflocculante sulla tissotropia e gli effetti che possono manifestarsi nelle successive fasi di formatura (adesività agli stampi, aggressività chimica, ecc.).
I deflocculanti inorganici sono più sensibili alla natura ed alla quantità degli ioni già presenti nella sospensione, non vengono eliminati nelle fasi di essiccamento e cottura, e generalmente non manifestano effetti tissotropici.
A parità di concentrazione, i deflocculanti organici presentano una maggiore efficacia, una minore sensibilità alla interferenza di altri ioni ed un maggior potere stabilizzante. Di solito sono tissotropici e di costo più elevato. In cottura subiscono una ossidazione termica, generando sostanzialmente acqua e CO2
La scelta del deflocculante più adatto va condotta, caso per caso, con le necessarie prove reologiche. I problemi più complessi possono essere risolti impiegando sostanze inorganiche ed organiche in miscele compatibili fra di loro per sfruttarne l’effetto sinergico.
Carbonato sodico Na2CO3
Argilla · Ca2+ + Na2CO3 → Argilla · 2Na+ + CaCO3↓
Ca(HCO3)2 + Na2CO3 → CaCO3↓ + 2NaOH + 2CO2
Queste reazioni specifiche rendono il carbonato sodico un ottimo deflocculante quanto più sono presenti ioni Ca2+, in quanto la proprietà di precipitare il calcio favorisce la deflocculazione. Solo nel caso di formatura per colaggio il carbonato di sodio può presentare controindicazioni perché influisce negativamente sulla durata delle forme in gesso (solfato di calcio), per la seguente reazione:
CaSO4 + Na2CO3 → CaCO3↓ + Na2SO4
La formazione del sale Na2SO4 riduce il potere deflocculante del carbonato sodico e crea danni al gesso degli stampi.
Il carbonato sodico Na2CO3 fonde a circa 850 °C, aumenta la plasticità e la resistenza a secco degli impasti ma ne riduce la velocità di essiccamento.
Silicato sodico Na2O · nSiO2
Il silicato di sodio si idrolizza facilmente con reazione alcalina (pH > 11) generando ioni Na+ e OH con separazione di silice colloidale:
Na2O · nSiO2 + H2O → nSiO2 + 2Na+ + 2OH
Il silicato non presenta la stessa aggressività del carbonato verso il gesso ed è dotato di proprietà leganti. In presenza di solfati ha un potere deflocculante ridotto per la formazione di Na2SO4.
La miscela carbonato/silicato (Na2CO3 + Na2O·nSiO2) ha il vantaggio di agire secondo ben quattro dei sei possibili meccanismi descritti nel paragrafo precedente e precisamente 1), 2), 5) e 6); quindi esplica un miglior potere deflocculante rispetto ai singoli componenti e riduce le problematiche di aggressività chimica del solo Na2CO3. Si tratta di soluzioni stabili, che lasciano separare silice gelatinosa e vengono aggiunte in concentrazioni di 0,2 ÷ 0,6% in peso.
Fosfati, polifosfati
I composti fosfatici che presentano un potere deflocculante più marcato sono l’esametafosfato sodico (NaPO3)6 ed i polifosfati, in particolare il tripolifosfato sodico (Na5P3O10). Il loro meccanismo di azione sfrutta il fatto che l’anione fosfato viene preferenzialmente adsorbito dalla particella d’argilla, aumentandone la carica negativa; il conseguente aumento del potenziale zeta determina un maggior effetto disperdente.
I fosfati hanno anche la funzione di sequestrare ioni flocculanti grazie alla formazione di complessi di coordinazione. Un esempio è quello dell’anione esametafosfato [P6O18]6– che forma con gli ioni Ca2+, Mg2+, Fe3+ sali complessi come ad esempio: [CaP6O18]4− e [Ca2P6O18]2−
Il notevole potere deflocculante dell’esametafosfato sodico è quindi da attribuirsi al forte adsorbimento anionico, al sequestro dei cationi flocculanti, oltre che all’azione di scambio del catione Na+
Analogamente si comportano i polifosfati, tra i quali il più usato è il tripolifosfato sodico (Na5P3O10), identificati dalla formula generale:
Mn+2PnO3n+1
dove M = ione monovalente nelle seguenti combinazioni:
n = 1
n = 2
n = 3
n = 4
M3PO4 (ortofosfati)
M4P2O7 (pirofosfati)
M5P3O10 (tripolifosfati)
M6P4O13 (tetrapolifosfati)
Per n > 4 si formano composti a struttura vetrosa.
Anche i polifosfati sequestrano i cationi flocculanti sotto forma di complessi e liberano cationi monovalenti deflocculanti. Avendo poi una struttura a catena, sono efficaci come disperdenti sui materiali argillosi anche in basse concentrazioni, solitamente < 0,4%.
Da notare però che le sospensioni deflocculate con polifosfati si degradano col tempo e riscaldandole aumentano la viscosità.
Carbonato di bario BaCO3
La presenza di anioni SO42− ostacola i processi di deflocculazione per diminuzione del potenziale zeta, in quanto questo anione, a differenza dello ione fosfato, viene adsorbito con facilità dalla particella argillosa in sostituzione degli ioni ioni OH-, soprattutto se SO42− è presente sotto forma di elettrolita forte (ad es. Na2SO4), ad alta solubilità.
Per favorire la deflocculazione è pertanto necessario allontanare i gruppi SO42−, soprattutto in presenza di Ca2+, Mg2+ e Fe3+, cationi flocculanti per eccellenza.
Come sopra descritto, il carbonato di sodio Na2CO3 reagisce con i solfati; ad esempio, con il CaSO4 forma CaCO3 allontanando il catione flocculante Ca2+ ma non elimina gli anioni SO42− che restano sempre come Na2SO4 (solubile in H2O) anche in presenza di silicati o fosfati sodici:
CaSO4 + Na2SiO3 → CaSiO3 + Na2SO4
3CaSO4 + 2Na3PO4 → Ca3(PO4)2 + 3Na2SO4
Per rendere insolubile Na2SO4 può essere utile aggiungere carbonato di bario che, in questo caso specifico, si comporta da deflocculante:
Na2SO4 + BaCO3 → BaSO4↓ + Na2CO3↓
Al posto del carbonato BaCO3, sale quasi insolubile, si potrebbe impiegare il cloruro BaCl2 solubile, ma ciò è possibile solo attraverso un dosaggio stechiometrico dei solfati presenti nella barbottina, in quanto lo ione Ba2+ in eccesso si comporterebbe da pericoloso flocculante, presentando inoltre una tossicità elevata. Per questi motivi si preferisce comunque utilizzare il carbonato BaCO3, nonostante la sua limitata solubilità.
Il carbonato di bario BaCO3 va aggiunto prima dei deflocculanti in quantità da 0,02 a 0,1%.
I deflocculanti organici sono anch’essi elettroliti o polielettroliti sotto forma di sali sodici o ammonici, che agiscono secondo gli stessi meccanismi riportati per i deflocculanti inorganici. Il loro anione funge da colloide protettore, favorendo ulteriormente il potere deflocculante dei sali monovalenti e del pH moderatamente basico.
Acidi umici e derivati
Vengono estratti da humus, torbe e ligniti. Sono derivati di acidi ossicarbossilici a struttura molecolare complessa caratterizzata da anelli condensati, con le seguenti proprietà: natura colloidale del loro radicale anionico, solubilità in ambiente alcalino anche diluito, insolubilità dei loro sali con metalli bi- e tri-valenti.
Si ritiene che gli acidi umici, avviluppando le particelle di argilla, formino con queste un complesso argillo-umico, inibendo così l’azione dei cationi flocculanti e generando un sistema colloidale molto stabile.
Un deflocculante a base di umati in commercio è ottenuto trattando la lignite con soda.
Composti del tannino
Di origine vegetale come gli acidi umici, sono composti dell’acido tannico e dell’acido gallatannico (miscela di esteri del glucosio e dell’acido gallico), appartenenti al vasto gruppo degli acidi polifenolici, composti organici che presentano nella loro molecola contemporaneamente più gruppi carbossilici ed idrossifenolici. Per trattamento termico dei tannini, si ottiene anche il pirogallolo.
I sali alcalini di tali composti (tannato sodico, gallato sodico, pirogallati) sono tutti buoni deflocculanti analogamente ai derivati umici, presentando anch’essi un comportamento colloidale con adsorbimento sulla superficie delle particelle disperse e conseguente diminuzione delle interazioni elettrostatiche flocculanti e della viscosità.
Industrialmente i tannini sono anche utilizzati nel processo di concia per le loro proprietà di creare forti legami a idrogeno con il collagene, rendendo imputrescibili le pelli animali.
Poliacrilati
Derivano dall’acido acrilico
CH2=CHCOOH, acido carbossilico insaturo con acidità leggermente superiore a quella dell’acido acetico (CH3COOH), ed in particolare dai suoi sali di Na+ ed NH4+
L’acido acrilico, grazie al doppio legame etilenico, polimerizza facilmente formando le resine acriliche. Interessanti sono i composti del tipo:
CH2=CHCOOR
dove R = catione alcalino o radicale alchilico.
se R = Na estere sodico
se R = CH3 estere metilico
se R = C2H5 estere etilico
Sono altresì importanti altri monomeri acrilici come:
CH2=CHCN acrilonitrile
CH2=CHCONH2 acrilammide
La loro polimerizzazione avviene per azione del calore, della luce e dei perossidi (es. perossido di benzoile) formando poliesteri, poliacrilonitrili e poliammidi:
L’acido poliacrilico si scioglie abbastanza lentamente in acqua, mentre i suoi sali alcalini sono facilmente solubili. I poliesteri alchilici ed i poliacrilonitrili sono insolubili.
Il poliacrilonitrile sottoposto a parziale idrolisi alcalina dà luogo a polielettroliti con proprietà flocculanti o disperdenti in funzione della lunghezza della catena polimerica (quelli a massa molecolare maggiore si comportano come flocculanti).
L’azione deflocculante dei polielettroliti acrilici avviene attraverso l’adsorbimento dell’anione polimerico sulla superficie delle particelle argillose, garantendo una eccellente azione disperdente ed una ottima stabilità anche nel tempo.
Pag. 5-37
In molti casi i deflocculanti acrilici sono superiori rispetto a quelli inorganici tradizionali (silicato, carbonato, NaOH, polifosfati). Inoltre risultano meno sensibili alla sovra-deflocculazione ed ai cationi interferenti.
In Fig. 5.10 è riportato un confronto tra deflocculanti acrilici ed altri deflocculanti quali il silicato ed il tripolifosfato di sodio, che evidenzia la variazione di viscosità di una barbottina in funzione delle quantità aggiunte.
Fig. 5.10 Effetto di vari deflocculanti sulla viscosità di una barbottina di impasto bianco
Fig. 5-10 IT
Miscele costituite da fluidificanti organici ed inorganici possono anche migliorare la resistenza meccanica in crudo degli impasti, ma resta comunque prioritario privilegiare la stabilità della sospensione bilanciando opportunamente i componenti in funzione delle loro caratteristiche elettrolitiche.
Derivati ammonici
Sostituendo all’ammoniaca (NH3) uno o più radicali organici si ottiene una ammina (RnNH3-n). Le ammine sono sostanze basiche che si dissociano in acqua. Le ammine alifatiche sono più dissociate in soluzione acquosa rispetto all’ammoniaca ed hanno quindi un grado di basicità maggiore. Inferiore risulta la basicità delle ammine aromatiche.
Molte sostanze quali l’etilammina, la dietilammina, la polivinilammina, la piperidina sono buoni deflocculanti, ma sono utilizzate raramente.
Ossalati
Derivano dall’acido ossalico (COOH)2, un acido abbastanza forte che può formare facilmente complessi salini con cationi ad alta valenza. Come deflocculanti si possono usare l’ossalato sodico e quello ammonico, entrambi solubili in acqua a differenza degli altri ossalati che sono insolubili.
Risultano di interesse perché fanno precipitare completamente gli ioni Ca2+ e l’anione ossalato può anche essere adsorbito dalla superficie delle particelle d’argilla.
Solitamente non vengono però utilizzati da soli, ma insieme ad altri composti con maggiore azione deflocculante.
Altri composti organici deflocculanti
Con proprietà deflocculanti sono da ricordare i sali sodici degli acidi alchil-naftalen-solfonici polimerizzati (pH 8 ÷ 10,5), il citrato di litio ed i derivati sodici della carbossilmetilcellulosa (CMC), un elettrolita con anione colloidale (vedi Fig. 5.11).
Carbossimetilcellulosa
Fig. 5.11 Formula di struttura della cellulosa (in alto) e della CMC (in basso)
L’anione della carbossimetilcellulosa viene adsorbito in modo irreversibile alla superficie delle particelle minerali aumentandone la loro carica negativa e la capacità di dispersione.
Fig. 5-11 IT
Si ottiene un notevole effetto deflocculante tanto maggiore quanto più piccolo è il grado di polimerizzazione.
La CMC viene usata in ceramica soprattutto come legante, tenendo però in considerazione anche le sue proprietà deflocculanti.
Il processo tecnologico che prevede la macinazione ad umido in mulini a tamburo e la successiva atomizzazione della barbottina rappresenta a tutt’oggi la miglior soluzione per ottenere impasti complessi (multicomponente), omogenei, privi di impurità e con caratteristiche granulometriche ottimali per la successiva pressatura e cottura in forni a ciclo rapido. Il processo di macinazione ad umido implica una grande quantità di acqua (30 ÷ 40% della massa totale di barbottina), che viene poi ridotta a 5 ÷ 8% nella fase di atomizzazione. L’uso degli additivi reologici, deflocculanti o fluidificanti, è quindi legato essenzialmente alla necessità di effettuare la macinazione con la minima percentuale di acqua possibile, compatibilmente con valori di viscosità tali da rendere agevoli i vari stadi del processo (svuotamento dei mulini, sosta in vasca a lenta agitazione, setacciatura, pompaggio e atomizzazione). Il criterio è quindi economico-produttivo: un abbassamento del tenore di acqua, a parità di altre condizioni, porta contemporaneamente a minori volumi di acqua da evaporare e ad una maggiore produttività dell’impianto, a tutto vantaggio della sostenibilità del processo. Chiaramente i benefici di un minor contenuto d’acqua vanno commisurati al costo del deflocculante.
I principali deflocculanti per impasto sono i seguenti:
- sodio carbonato
- sodio silicato
- polifosfati di sodio
- composti organici naturali (acidi umici e umati, tannini, ligninsulfonati)
- composti organici sintetici (poliacrilati, polimetacrilati e derivati).
Le loro caratteristiche sono state descritte in dettaglio nei paragrafi precedenti, mentre di seguito si riportano alcune considerazioni specifiche sul loro impiego negli impasti.
Sodio carbonato: è usato molto raramente negli impasti per atomizzazione, solamente per impasti molto poveri di frazioni argillose plastiche (es. bicottura di pasta bianca), in ragione del suo scarso effetto fluidificante.
Sodio silicato: è un colloide-protettore delle micelle argillose in miscele prive di ioni polivalenti; è quindi molto indicato per barbottine costituite da argille poco plastiche che non contengano composti organici. Il silicato di sodio in forma liquida è la base principale di molte miscele di deflocculanti, presentando un basso costo ed una rapida azione nel processo di macinazione. È anche utilizzato spesso nelle barbottine per il colaggio dei sanitari in combinazione con sodio carbonato che elimina i cationi di calcio dalla barbottina.
Polifosfati e poliacrilati: sono di larghissimo uso negli impasti, in particolare il tripolifosfato di sodio. È difficile valutare a priori quale sia il prodotto migliore per una specifica composizione di impasto; pertanto, è sempre opportuno eseguire prove reologiche comparative in laboratorio prima di passare alla lavorazione industriale.
Composti organici naturali: sono utilizzati molto raramente come additivi per impasti. Un effetto collaterale comune a questa tipologia di deflocculanti è il fenomeno della sovra-deflocculazione, ovvero di un aumento della viscosità apparente dovuto ad un eccesso di sostanza attiva. Per quanto poi riguarda l’introduzione negli impasti di additivi organici naturali, e in minor misura anche sintetici, bisogna tener presente la loro tendenza ad accentuare il fenomeno del “cuore nero”, ovvero di una insufficiente ossidazione in cottura della parte centrale del supporto ceramico. Viceversa è possibile riscontrare un aumento di tenacità in crudo dei pezzi pressati, analogamente all’utilizzo sia dei deflocculanti organici che inorganici (silicati o polifosfati di sodio).
Relativamente al settore degli smalti, l’uso degli additivi reologici non è tanto governato da considerazioni economico produttive, quanto dalla necessità di lavorare con sospensioni aventi precise caratteristiche chimico-fisiche, idonee all’applicazione dello smalto con differenti tecnologie applicative, che implicano macchine basate su principi di funzionamento estremamente differenziati.
Pertanto, in questo paragrafo specifico, si è preferito suddividere gli additivi più utilizzati negli smalti, non in base alla loro natura chimica, ma in base alla funzionalità richiesta, ovvero: - deflocculanti - sospensivanti - collanti.
Deflocculanti. L’effetto principale esercitato sulla barbottina di smalto è un abbassamento della viscosità apparente. Questi prodotti possono essere addizionati preferenzialmente in macinazione o in linea di smaltatura (quest’ultima operazione risulta, però, agevole solo per prodotti liquidi).
Le classi principali di prodotto sono quelle già viste per gli impasti: polifosfati (tripolifosfato ed esametafosfato) di sodio e derivati sodici o ammonici dell’acido poliacrilico, polimetacrilico o copolimeri a struttura più complessa.
I vantaggi di una macinazione condotta ad alta densità, cioè con alta percentuale di solido, sono i seguenti:
- riduzione dei tempi di macinazione
- migliore distribuzione granulometrica
- maggiore versatilità nelle successive applicazioni.
Le percentuali di utilizzo degli additivi deflocculanti sono assai variabili, dipendentemente dall’efficienza del prodotto utilizzato e dalla specificità delle fritte e delle materie prime impiegate nella formulazione dello smalto; in via del tutto generale variano da 0,03% a 0,4%.
Le aggiunte in linea hanno principalmente l’obiettivo di una correzione istantanea della viscosità, al fine di permettere una regolazione ottimale dei macchinari e soddisfare specifiche esigenze degli effetti estetici da realizzare.
Effetti secondari o collaterali:
- ritardo di asciugamento dello smalto
- miglioramento della stesura, soprattutto per applicazioni con airless
- rallentamento della velocità di sedimentazione (nel breve periodo)
- riduzione della plasticità e tissotropia per sospensioni di smalti ad alta densità, per applicazioni a velo (vela/campana).
Effetti da sovradosaggio:
- viscosità troppo bassa con scorrevolezza eccessiva che può portare ad irregolarità di applicazione (es. colature sui bordi)
- formazione di sedimenti estremamente tenaci (cementazione) nel medio o lungo periodo
- sovra-deflocculazione.
Sospensivanti. La loro azione si esplica mediante interazioni elettrostatiche (nel caso di sali solubili), aumento della viscosità (nel caso di CMC) o maggiore contenuto di colloidi protettori.
Il loro effetto, in ogni caso, è una diminuzione della velocità di sedimentazione, che a sua volta dipende da una serie di fattori, tra cui: il peso specifico e la distribuzione granulometrica dei solidi sospesi, la densità e la viscosità della barbottina, la presenza di colloidi.
I prodotti sospensivanti maggiormente utilizzati sono:
- sodio cloruro
- miscele di elettroliti in soluzione
- derivati cellulosici (CMC ad alta viscosità)
- argille bentonitiche
- silice colloidale.
Collanti. Esistono colle di vario genere utilizzate per migliorare l’adesione degli smalti: metilcellulose, amidi eterificati ed esterificati, ecc. Tra questi, i prodotti più comunemente impiegati sono le carbossimetilcellulose (CMC). Se ne sfruttano le proprietà leganti, in quanto migliorano la coesione delle particelle di smalto crudo, l’adesione di questo al supporto, la capacità di regolare l’asciugatura (rallentando l’evaporazione della fase liquida) e la stesura degli smalti.
Gli effetti reologici dipendono strettamente dal tipo di CMC usata, classificabile sulla base della viscosità di una soluzione al 2% in acqua:
- CMC a bassa viscosità (circa 5 ÷ 50 mPa·s)
- CMC a media viscosità (circa 100 ÷ 1.000 mPa·s)
- CMC ad alta viscosità (circa 1.000 ÷ 10.000 mPa·s ed oltre)
I rispettivi comportamenti reologici, ad una percentuale di uso normale, del 0,1 ÷ 0,2%, possono essere così riassunti:
- una CMC a bassa viscosità provoca un lieve calo di viscosità nella sospensione e può dar luogo a fenomeni di sedimentazione
- una CMC a media viscosità ha scarso effetto sulla viscosità ed un leggero effetto sospensivante
- una CMC ad alta viscosità provoca un aumento di viscosità e del potere sospensivante.
Alcuni possibili effetti dovuti ad un sovra-dosaggio possono essere:
- aumento eccessivo di viscosità
- allungamento dei tempi di asciugatura
- assorbimento irregolare dello smalto da parte del supporto.
Alcuni andamenti reologici esemplificativi sono riportati nelle figure successive; le curve di viscosità sono state ottenute dopo aver sottoposto lo smalto ad un elevato gradiente di velocità (D) per un tempo prestabilito.
In Fig. 5.12 si mostra il comportamento reologico di uno smalto da bicottura per applicazione a campana con viscosità (pendenza della curva) elevata e limite di scorrimento nullo.
Fig. 5.12 Comportamento reologico di uno smalto da bicottura per applicazione a campana
La Fig. 5.13 mostra invece il comportamento reologico di uno smalto per applicazione airless; rispetto allo smalto per campana si nota infatti una minore viscosità e la presenza di un limite di scorrimento.
5-12
Fig. 5.13 Comportamento reologico di uno smalto per applicazione airless
Fig. 5-13
La Fig. 5.14 mostra l’effetto dell’aggiunta di piccoli quantitativi di sodio tripolifosfato (NaTPF) e di carbossimetilcellulosa (CMC) su uno smalto con elevato limite di scorrimento.
senza additivo
0.05% TPF 0.05% CMC30
Fig. 5.14 Effetto dell’aggiunta di piccoli quantitativi di NaTPF e CMC
La Fig. 5.15 mostra l’aumento di viscosità di uno smalto causato dall’aggiunta di CMC ad elevato peso molecolare.
Fig. 5-14 IT
senza additivo
Fig. 5.15 Effetto dell’aggiunta di CMC ad elevato peso molecolare
Fig. 5-15 IT
La Fig. 5.16 mostra invece l’effetto di aggiunte di caolino e bentonite: la bentonite aumenta notevolmente viscosità e limite di scorrimento, il caolino invece produce solo un piccolo aumento del limite di scorrimento.
Fig. 5.16 Effetto delle aggiunte di caolino e bentonite
La Fig. 5.17 mostra infine l’effetto della densità apparente sul comportamento reologico di uno smalto. All’aumentare della densità aumentano la viscosità (pendenza) ed il limite di scorrimento (scostamento dall’origine).
Fig. 5-16 IT
1850 g/L
1800 g/L
Fig. 5-17 NEW 1700 g/L
Fig. 5.17 Effetto della densità apparente di uno smalto
Da ultimo, è opportuno segnalare alcuni effetti indesiderati, non reologici, collegabili alla presenza dei seguenti composti:
A. polifosfati
B. sodio
C. cloro
D. sali solubili
E. altri prodotti inorganici non volatili (SiO2, Al2O3)
F. sostanze organiche di vario tipo.
A. I polifosfati portano, durante il processo di cottura, alla formazione di vetri a base di P2O5, insolubili nei vetri a base di SiO2; questo può causare difetti come microbolle e puntinature.
B. Il sodio entra a far parte dei vetri, esercitando una marcata azione fondente ed originando fenomeni di “puntinatura” da sovracottura.
C. La decomposizione termica dei cloruri dà luogo alla formazione di cloro gassoso, che può deteriorare le parti metalliche ed i rivestimenti dei forni per la formazione di condense chimicamente aggressive, oltre che risultare una emissione inquinante per l’ambiente.
D. La presenza di sali solubili può comportare la loro migrazione, soprattutto durante la fase di essiccamento, verso i bordi delle piastrelle, con conseguente formazione di macchie, aloni, spillature e ribolliture localizzate.
E. Qualunque composto inorganico non volatile alla temperatura di cottura entra a far parte dello smalto, contribuendo in misura più o meno rilevante alla modifica della sua composizione e quindi ne altera le caratteristiche di fusibilità e aspetto superficiale.
F. Le sostanze organiche si decompongono per riscaldamento, dando origine a composti gassosi che fuoriescono dalla superficie smaltata. Se il ciclo di cottura non è adeguato per la quantità e qualità delle sostanze organiche presenti, possono sorgere difetti da degasazione e puntinatura nera. Un altro inconveniente dovuto alla presenza di taluni prodotti organici (amidi, CMC ed altri derivati cellulosici) è la loro decomposizione biologica ad opera di microorganismi (muffe e batteri), che avviene in particolare nelle sospensioni acquose. Gli effetti sulle barbottine di smalto sono una variazione importante della viscosità ed una perdita del potere collante, con conseguenti spillature e sfondini sulla superficie smaltata, fino a microfratture e fessurazioni sui bordi. Il problema si presenta con smalti contenenti CMC, applicati alcuni giorni dopo la macinazione. In questi casi sono utili alcuni additivi ad effetto “antibatterico” che permettono di mantenere i parametri reologici e le proprietà leganti per periodi più lunghi.
[1] C. Klein e C. Hurlburt, Manual of mineralogy, Wiley & Sons, 1988.
[2] R. Grimshaw, The chemistry and physics of clays, Benn, 1971.
[3] Società Ceramica Italiana, Reologia ceramica applicata, Faenza Ed., 1990.
[4] Società Ceramica Italiana, Reologia ceramica applicata (teoria e pratica), SALA, 2006.
[5] P. Pozzi e C. Galassi, La reologia dei materiali ceramici tradizionali, Faenza Ed., 1994.
DEFLOCCULANTE
Con il termine “deflocculante” si intende un additivo in grado di impedire o contrastare i fenomeni di flocculazione di una sospensione ceramica.
DISPERDENTE
Con il termine “disperdente” si intende un additivo in grado di migliorare la separazione tra le particelle di una sospensione e prevenirne l’aggregazione.
FLOCCULAZIONE
Processo chimico-fisico che dà luogo ad una sedimentazione solida nelle sospensioni colloidali, come le barbottine di impasti e smalti ceramici. In tal caso la flocculazione può essere determinata da una diminuzione del pH o dalla presenza di cationi ad alta carica (Fe3+, Al3+, Ca2+, ecc.), fattori che interferiscono negativamente con il doppio strato elettrico delle micelle argillose.
FLUIDIFICANTE
Con il termine “fluidificante” si intende un additivo in grado di abbassare la viscosità di una sospensione ceramica.
GRADIENTE
Con il termine gradiente si indica generalmente la variazione che una grandezza fisica subisce da un punto all’altro dello spazio lungo una certa direzione. Ad esempio, si parla di gradiente termico per esprimere la variazione della temperatura lungo una data direzione. Nella pratica ceramica con il termine gradiente si intende, per estensione, la variazione di uno specifico parametro in una serie di misure effettuate (p.es. gradiente di densità, gradiente di pressatura, gradiente di cottura, ecc.).
Il potenziale zeta (ζ) o potenziale elettrocinetico, è il potenziale generato in seguito alla formazione di un doppio strato elettrico nelle particelle in sospensione. Esso è responsabile dei fenomeni elettrocinetici e della stabilità dei colloidi. Un valore elevato di potenziale zeta conferisce maggiore stabilità ai sistemi colloidali, in quanto si originano repulsioni elettrostatiche che impediscono l’aggregazione delle particelle disperse. Quando il potenziale è basso, le forze attrattive prevalgono sulle repulsioni e può verificarsi la flocculazione.
La superficie specifica è l’area superficiale per unità di massa (espressa in m2/g) di solidi granulari, polveri, materiali porosi o particelle in sospensione. La superficie specifica di un materiale fornisce utili indicazioni sulla sua reattività, come ad esempio la capacità di scambio ionico delle particelle argillose o la cinetica di sinterizzazione delle polveri ceramiche.
TISSOTROPIA
La tissotropia è la proprietà di fluidi pseudo-plastici, come le sospensioni argillose, di aumentare la propria viscosità passando da uno stato di agitazione ad uno stato di quiete. Il fenomeno è determinato dalla formazione di una struttura solida tridimensionale (castello di carte) che conferisce rigidità al fluido e che può essere dispersa nuovamente per agitazione o con l’aggiunta di additivi deflocculanti. Il comportamento contrario alla tissotropia, ovvero quando un fluido aumenta la sua viscosità in presenza di sollecitazioni meccaniche, viene chiamato reopessia.
Grandezza fisica che misura la resistenza di un fluido allo scorrimento ed è associabile ad un coefficiente di attrito interno al fluido stesso. Si calcola dal rapporto tra lo sforzo di taglio ed il gradiente del flusso di scorrimento. La sua unità di misura è il Pa·s (Pascal × secondo) e suoi sottomultipli.
Le proprietà delle argille e delle altre materie prime utilizzate nell’industria ceramica dipendono dalla tipologia e dalla quantità dei minerali di cui sono costituite. Risulta pertanto di primaria importanza identificare la natura di ciascun materiale mediante le varie tecniche di analisi disponibili.
La caratterizzazione di una materia prima ceramica ha quindi l’obiettivo di individuare la sua natura composizionale e le sue proprietà fondamentali, quali:
- composizione chimica (usualmente espressa in ossidi)
- composizione mineralogica e struttura cristallina
- caratteristiche termiche
- granulometria
- comportamento tecnologico.
Soprattutto per le materie prime argillose, una caratterizzazione precisa è però piuttosto difficoltosa in quanto questi materiali di natura sedimentaria non hanno una composizione mineralogica ben definita e solitamente contengono diversi altri minerali, come quarzo, feldspati, miche, carbonati, ossidi di ferro, ecc. Le stesse componenti argillose sono spesso costituite da diverse tipologie di argille (caolinitiche, illitiche, cloritiche o smectiche) che possono essere meglio identificate valutando anche le loro proprietà reologiche, il loro grado di plasticità ed il loro comportamento tecnologico nelle fasi di formatura, essiccamento e cottura.
La caratterizzazione di una materia prima si compone quindi sia di valutazioni indirette delle sue proprietà tecnologiche sia di metodologie analitiche specifiche, come le analisi chimico-mineralogiche e le analisi termiche descritte nel presente capitolo.
Qualunque analisi da eseguire su un materiale disomogeneo, come spesso risultano le materie prime ceramiche, richiede preliminarmente un adeguato campionamento dell’aliquota di materiale necessario, affinché questa sia rappresentativa delle grandi quantità provenienti da una cava e/o immagazzinate in fabbrica.
Partendo dal caso più generale del campionamento in cava, per il quale esistono apposite procedure standardizzate, il campione dovrà essere prelevato in differenti parti del fronte estrattivo e a diverse profondità. Dopo miscelazione e quartatura, qualora il materiale si presenti uniforme, si potrà utilizzare un campione pari a circa l’1% della quantità presa in esame, che dovrà essere maggiore (fino al 5%) nel caso di un materiale disomogeneo. Lo stesso procedimento vale per campionamenti effettuati da navi o autotreni carichi di materiali sfusi.
Il campione preliminare così selezionato andrà poi ulteriormente omogeneizzato e nuovamente quartato tramite formazione di mucchi appiattiti e prelevamento di quarti opposti, sino ad ottenere un campione finale di circa 10 ÷ 12 kg.
Quest’ultimo sarà poi sottoposto ad una moderata macinazione e quartato, sino ad ottenere un campione ideale di circa 3 kg con granulometria media non superiore a 0,5 ÷ 2 mm. Anche l’umidità contenuta dal campione non dovrebbe essere completamente eliminata durante la comminuzione e la quartatura, per evitare alterazioni della microstruttura o perdite di sali solubili. L’eventuale essiccazione e una macinazione fine del campione saranno effettuate soltanto subito prima dell’analisi, ed in funzione di quanto necessario per il tipo di analisi stessa. A riguardo si tenga presente che la fase di macinazione può risultare critica per il possibile inquinamento da parte dei mezzi macinanti, specie se metallici, mentre un essiccamento a temperature elevate può modificare la struttura di taluni minerali.
La determinazione qualitativa e quantitativa della composizione chimica di una materia prima è fondamentale per individuare la tipologia di materiale e per valutarne il contributo in una formulazione di impasto o smalto.
I metodi di analisi chimica di un materiale inorganico sono molteplici ma, tra questi, le moderne tecniche strumentali permettono di ottenere risultati rapidi e accurati, tenendo sempre presente che il prelievo e la preparazione del campione sono determinanti per conseguire risultati corretti e rappresentativi.
Le analisi chimiche dei materiali ceramici vengono normalmente espressi in ossidi ed in particolare, le materie prime da impasto (argille, sabbie e feldspati), sono caratterizzate dai seguenti principali otto elementi:
- SiO2 biossido di silicio (silice)
- Al2O3 triossido di alluminio (allumina)
- TiO2 biossido di titanio
- Fe2O3 triossido di ferro (ossido ferrico)
- CaO ossido di calcio
- MgO ossido di magnesio
- Na2O ossido di sodio
- K2O ossido di potassio.
Gli eventuali composti volatili, derivati ad esempio da carbonio e zolfo o anche gli ossidrili presenti nei reticoli cristallini (acqua di costituzione), vengono espressi complessivamente come perdita al fuoco a 1060 °C.
Ovviamente possono essere presenti anche altri elementi in quantità solitamente basse, alcune frazioni percentuali, come ad esempio litio, boro, bario, stronzio e metalli di transizione (rame, cromo, manganese, ecc.).
I metodi analitici strumentali impiegati per l’analisi chimica quantitativa di materiali ceramici si basano sulle tecniche spettroscopiche di assorbimento o di emissione degli elementi, ovvero sulle radiazioni elettromagnetiche assorbite o emesse da ciascun elemento ad una determinata lunghezza d’onda caratteristica di una sua transizione elettronica. Grazie alle loro prestazioni superiori (precisione, ripetibilità e rapidità delle analisi), le spettroscopie hanno soppiantato gli storici metodi di analisi manuali come la gravimetria, la colorimetria o la complessometria, tutti estremamente laboriosi.
Le principali tecniche analitiche spettroscopiche oggi utilizzate sono pertanto le seguenti:
- Spettrometria ad assorbimento atomico (AAS-GFAAS)
Si basa sull’assorbimento da parte di una determinata specie atomica di una radiazione monocromatica generata da una lampada a catodo cavo.
Il suo principio di funzionamento può essere schematizzato come in Fig. 6.1.
Lampada Atomizzatore Monocromatore Rivelatore
La lampada emette una radiazione specifica per l’elemento di interesse
L’atomizzatore (a fiamma o forno elettrico in grafite) converte il campione in soluzione in atomi liberi che assorbono la radiazione emessa dalla lampada
Il monocromatore seleziona la lunghezza d’onda utilizzata per la misurazione
Il rivelatore misura la luce assorbita dagli atomi liberi proporzionale alla loro concentrazione
Fig. 6.1 Principio di funzionamento della spettroscopia di assorbimento atomico (AAS)
Da sottolineare come questo metodo di misura richieda la solubilizzazione del campione, ovvero la sua mineralizzazione secondo i procedimenti descritti nel Paragrafo 6.4. Tramite questa tecnica è possibile ottenere ottimi risultati analitici per qualsivoglia elemento metallico, sino a risoluzioni molto basse, dell’ordine di frazioni di parti per milione (ppm).
Fig. 6-01 IT
- Spettrometria ad emissione atomica (AES)
È una tecnica simile alla precedente, dalla quale si differenzia per la misurazione della radiazione emessa (non assorbita) da un determinato elemento atomico eccitato. Anche in questo caso la soluzione del campione viene atomizzata ad alta temperatura tramite un arco elettrico o una fiamma. Più recentemente è stata introdotta una sorgente di ionizzazione al plasma accoppiato induttivamente (ICP), nota come torcia al plasma (vedi Fig. 6.2). Questa è costituita da un tubo in quarzo avvolto all’estremità da una bobina di induzione magnetica che genera un plasma di argon (Ar+) ad altissima temperatura (6000 ÷ 10000 °K). Gli atomi del campione da analizzare iniettati nel flusso di argon che attraversa la torcia si ionizzano ed emettono radiazioni elettromagnetiche a lunghezze d’onda caratteristiche. Il vantaggio principale dei sistemi ad emissione rispetto alla spettrometria ad assorbimento risiede nella possibilità di effettuare analisi sequenziali delle specie atomiche costituenti il campione, senza dover modificare la sorgente della radiazione di eccitazione. In entrambi i casi i limiti di rilevazione dei singoli elementi sono molto buoni, permettendo di determinare concentrazione nell’ordine dei ppm ed in taluni casi anche dei ppb (µg/L).

Fig. 6.2 Torcia al plasma di uno spettrometro ICP-AES
- Spettrometria a fluorescenza di raggi X (XRF)
Si basa sulla emissione di radiazioni di fluorescenza caratteristiche degli elementi atomici costituenti il campione quando questo viene irradiato con raggi X ad alta energia. La radiazione X infatti eccita gli elettroni interni agli orbitali degli elementi presenti, provocando una conseguente emissione di luce fluorescente (vedi Fig. 6.3). Si distinguono due sistemi principali per analisi XRF: a dispersione di energia o a dispersione di lunghezza d’onda. Nel primo caso tutte le radiazioni di fluorescenza emesse dal campione vengono raccolte da un rilevatore allo stato solido che le distingue in base al loro livello energetico; nel secondo caso le radiazioni di fluorescenza vengono disperse geometricamente da un cristallo, in modo che i diversi livelli energetici possano essere rilevati singolarmente in funzione dell’angolo di dispersione. Quest’ultima tecnica consente una maggior risoluzione soprattutto con elementi a basso peso atomico, come litio e boro, mentre la XRF a dispersione ha come limite inferiore fluoro e sodio. Inoltre, a differenza dalle altre tecniche spettroscopiche sopra descritte, il campione viene analizzato allo stato solido. Fig. 6-03

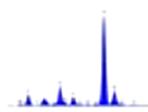
- Fotometria a fiamma
Si tratta di una tecnica analitica semplificata che sfrutta gli stessi principi della spettrometria ad emissione atomica. È particolarmente adatta per misurare la concentrazione di metalli alcalini ed alcalino-terrosi in diverse matrici, in quanto questi metalli raggiungono uno stato eccitato già alle temperature inferiori di una fiamma, emettendo radiazioni elettromagnetiche nella regione dello spettro visibile. Sono infatti ben noti i colori tipici di riconoscimento di alcuni metalli mediante il saggio alla fiamma: rosso (litio), giallo/arancio (sodio), arancione/rosso (calcio), violetto (potassio), verde (bario), verde/azzurro (rame).
- Analizzatori carbonio / zolfo
Utilizzano un fornetto tubolare ceramico con resistenze elettriche per portare il campione a temperature fino a circa 1300 °C, causando la conversione del carbonio e dello zolfo presenti rispettivamente in CO2 e SO2, la cui concentrazione viene determinata da un sistema di misura a infrarossi.
Infine, sono disponibili vari metodi analitici specifici, semplici ed efficaci, per la determinazione del contenuto di sali solubili ed in particolare degli anioni solfato, cloruro, nitrato e carbonato, la cui valutazione è di estrema importanza per stabilire l’idoneità di una materia prima nel processo ceramico.
Una volta selezionato accuratamente il campione, come descritto nel Paragrafo 6.1, fino ad ottenere pochi grammi omogenei, si procede ad una sua essiccazione a bassa temperatura (< 100 °C) per non alterare il contenuto di sostanze volatili termolabili. Si esegue poi la macinazione di laboratorio che deve essere effettuata con mezzi macinanti idonei a raggiungere finezze inferiori a 63 µm, senza ovviamente inquinare il campione. In funzione della durezza del campione, si passa da metodi manuali in mortai di corindone o di agata, a micro-mulini con corpi macinanti in leghe dure, come il carburo di tungsteno.
Pesato con bilancia analitica il quantitativo di campione necessario, si procede con l’eventuale attacco chimico o fusione dello stesso, in modo da ottenere una sua completa omogeneizzazione. Si può ad esempio sciogliere il campione con opportuni reagenti acidi ed ottenere una soluzione acquosa omogenea, oppure vetrificarlo con opportuni fondenti nel caso si debba eseguire l’analisi su su un provino allo stato solido.
Notoriamente i materiali ceramici, essendo costituiti da silicati, alluminati ed ossidi vari, sono piuttosto difficili da solubilizzare; a riguardo esiste una vastissima letteratura scientifica sulle metodologie di attacco chimico a caldo con miscele di acido fluoridrico (HF) e altri acidi minerali, quali: acido nitrico (HNO3), acido cloridrico (HCl), acido solforico (H2SO4). La dissoluzione in acidi del campione (mineralizzazione) può anche essere eseguita in appositi contenitori plastici ad alta resistenza (ad esempio in Teflon).
I principali metodi di attacco e solubilizzazione acida delle materie prime di interesse ceramico sono pertanto:
- Attacchi acidi a caldo in contenitori aperti, tramite l’utilizzo di miscele acide-ossidanti, a base di HCl, HNO3 ed HClO4; la necessità di disgregare la matrice silicatica rende poi quasi sempre indispensabile anche l’uso di HF (da cui la impossibilità di utilizzare la normale vetreria in vetro borosilicato). L’utilizzo di contenitori aperti, congiuntamente all’uso di solubilizzanti acidi ad alta temperatura, può favorire la perdita di componenti volatili.
- Attacchi acidi a caldo in contenitori chiusi e quindi anche ad elevate pressioni. Questi sistemi, sempre più diffusi per la rapidità dell’attacco e dissoluzione, impiegano normalmente contenitori in Teflon e sistemi di riscaldamento programmabili a microonde. - Fusioni alcaline con formazione di una perla vetrosa, che può essere utilizzata direttamente se la tecnica di analisi prevede l’uso di un campione solido (come la XRF) o che può essere facilmente solubilizzata con acido cloridrico. Per la vetrificazione esiste una vasta gamma di fondenti alcalini, utilizzabili in funzione della temperatura di fusione desiderata e delle necessità analitiche; in tutti i casi, il composto fondente comporta l’aggiunta di almeno un elemento, che pertanto non è più quantificabile nel campione incognito. La Tab. 6.1 riporta una serie di composti fondenti con la relativa temperatura di vetrificazione e, tra questi, i più utilizzati per l’analisi delle materie prime ceramiche sono: NaKCO3 (carbonato sodico-potassico), NaOH (idrossido di sodio), LiBO2 (metaborato di litio) e Li2B4O7 (tetraborato di litio).
B2O3
K2Cr2O7
(1)
LiOH (2)
(4)
(1) Dopo decomposizione e trasformazione del bisolfato in pirosolfato K2S2O7)
(2) Normalmente introdotto nella forma idrata
(3) Come PbO dopo decomposizione e fuoriuscita di anidride carbonica a 315°C
(4) Come PbO dopo decomposizione a 500°C circa
Tab. 6.1 Temperature di fusione di composti fondenti
La fusione viene preferibilmente effettuata mediante una perlatrice che garantisce condizioni operative standardizzate, utilizzando contenitori di platino; la perla di vetro ottenuta può essere sottoposta direttamente a lettura strumentale (XRF) o essere solubilizzata per la lettura tramite spettroscopia di assorbimento o di emissione.
Qualunque sia la tecnica strumentale utilizzata, è ovviamente necessario predisporre opportune curve di calibrazione nell’intervallo composizionale dei campioni incogniti, tramite l’utilizzo di soluzioni o provini solidi a concentrazione nota.
Indubbiamente l’analisi elementare fornisce risultati precisi sulla natura chimica dei materiali, tuttavia in molti casi non è sufficiente a spiegare il comportamento tecnologico di una materia prima ceramica, che dipende anche dalle forme mineralogiche presenti, dalla microstruttura e dalle proprietà fisiche.
Questo tipo di indagine permette di determinare le singole fasi cristalline presenti in una materia prima ceramica, ovverosia la sua composizione mineralogica, la cui valutazione è di primaria importanza per definire il comportamento tecnologico della materia prima stessa e il suo contributo in un impasto.
Una valutazione preliminare sulla natura mineralogica di una materia prima può già essere effettuata con un esame visivo del campione tal quale e, in modo più approfondito, con l’ausilio di un microscopio ottico (in luce riflessa, trasmessa, polarizzata ed a prismi di Nicol incrociati). In particolare, esaminando provini in strato sottile inglobati in una resina, con la necessaria preparazione tecnico-scientifica, è possibile riconoscere i diversi minerali presenti e la loro conformazione strutturale attraverso le proprietà ottiche come l’indice di rifrazione (Fig. 6.4).

Fig. 6.4 Cristalli di muscovite (Ms) e di quarzo (Qtz) in un granito, osservati con luce polarizzata (//) e con prismi di Nicol incrociati (+) [1]
La tecnica principalmente utilizzata per la caratterizzazione mineralogica è indubbiamente la diffrattometria a raggi X (XRD), eseguibile su singoli cristalli o, più comunemente per le materie prime ceramiche, su polveri mediante l’utilizzo di un diffrattometro dedicato (Fig. 6.5).
Sorgente raggi X
Fig. 6.5 Schema di funzionamento del diffrattometro a raggi X (XRD)
Fig. 6-05 IT
di diffrazione misurato sul goniometro
Fig. 6.6 Principio di funzionamento della diffrattometria a raggi X (XRD)
La radiazione X incidente sul campione, opportunamente filtrata per ottenere la monocromaticità, interagisce con il reticolo cristallino dello stesso (Fig. 6.6), dando luogo a riflessioni in funzione della distanza tra i piani reticolari (d) e dell’angolo di incidenza (θ) tramite l’equazione di Bragg:
6-06 IT
n λ = 2d sin θ
dove n = numero intero positivo e λ = lunghezza d’onda della radiazione incidente.
Tra le possibili sorgenti di raggi X, la più utilizzata è quella generata da un anticatodo in rame, filtrata con nichel per ottenere la sola lunghezza d’onda λ (CuKα) = 1,541 Å.
L’analisi diffrattometrica consiste pertanto nel registrare i picchi di risposta dei composti cristallini presenti nel campione, in funzione dell’angolo 2θ (diffrattogramma).
In Fig. 6.7 si riporta a titolo di esempio lo spettro di diffrazione di un quarzo.
Fig. 6.7 Diffrattogramma di un quarzo
6-07
Come si può osservare, il picco principale si trova a 26,6 °(2θ) e corrisponde ad una distanza reticolare di 3,34 Ångstrom (Å).
Per tutte le sostanze cristalline sono disponibili tavole o database con le distanze reticolari caratteristiche e le intensità dei relativi picchi di diffrazione, come negli esempi riportati nella Fig. 6.8 e Fig. 6.9 riferiti a quarzo (SiO2) e caolinite (Al2Si2O5(OH)4).
Fig. 6.8 Distanze reticolari caratteristiche del quarzo e intensità relativa dei picchi [2]
Fig. 6.9 Distanze reticolari caratteristiche della caolinite e intensità relativa dei picchi [2]
Utilizzando i database dei parametri cristalIografici, i software specifici per l’analisi diffrattometrica permettono sia l’identificazione qualitativa dei minerali cristallini presenti in una determinata materia prima sia la loro misurazione quantitativa con opportuni protocolli di calibrazione. A riguardo, il procedimento tuttora più utilizzato è il metodo di Rietveld, sviluppato già nel 1969 dal cristallografo olandese H. Rietveld ed ovviamente ottimizzato grazie
alle potenzialità di calcolo dei moderni processori.
Per quanto concerne la preparazione del campione, normalmente l’analisi si esegue su un campione in polvere ottenuto per macinazione, evitando di alterarne le caratteristiche microstrutturali, in particolar modo se argilloso, e favorendo l’omogeneizzazione e l’orientamento casuale di tutti i piani cristallini presenti. Oggi l’analisi diffrattometrica a raggi X può essere considerata una tecnica routinaria, semplice e rapida, che permette di identificare i composti cristallini presenti anche in matrici relativamente complesse, come quelle delle materie prime naturali. Inoltre, in combinazione con l’analisi chimica quantitativa, è possibile stabilire con buona approssimazione la composizione espressa in composti (analisi razionale). Ad esempio, una scheda di caratterizzazione di un impasto ceramico riporta solitamente sia la composizione chimica in ossidi, sia la composizione mineralogica (quarzo, caolinite, illite, calcite, dolomite, feldspato sodico, ecc.) come evidenziato nella Fig. 6.10.
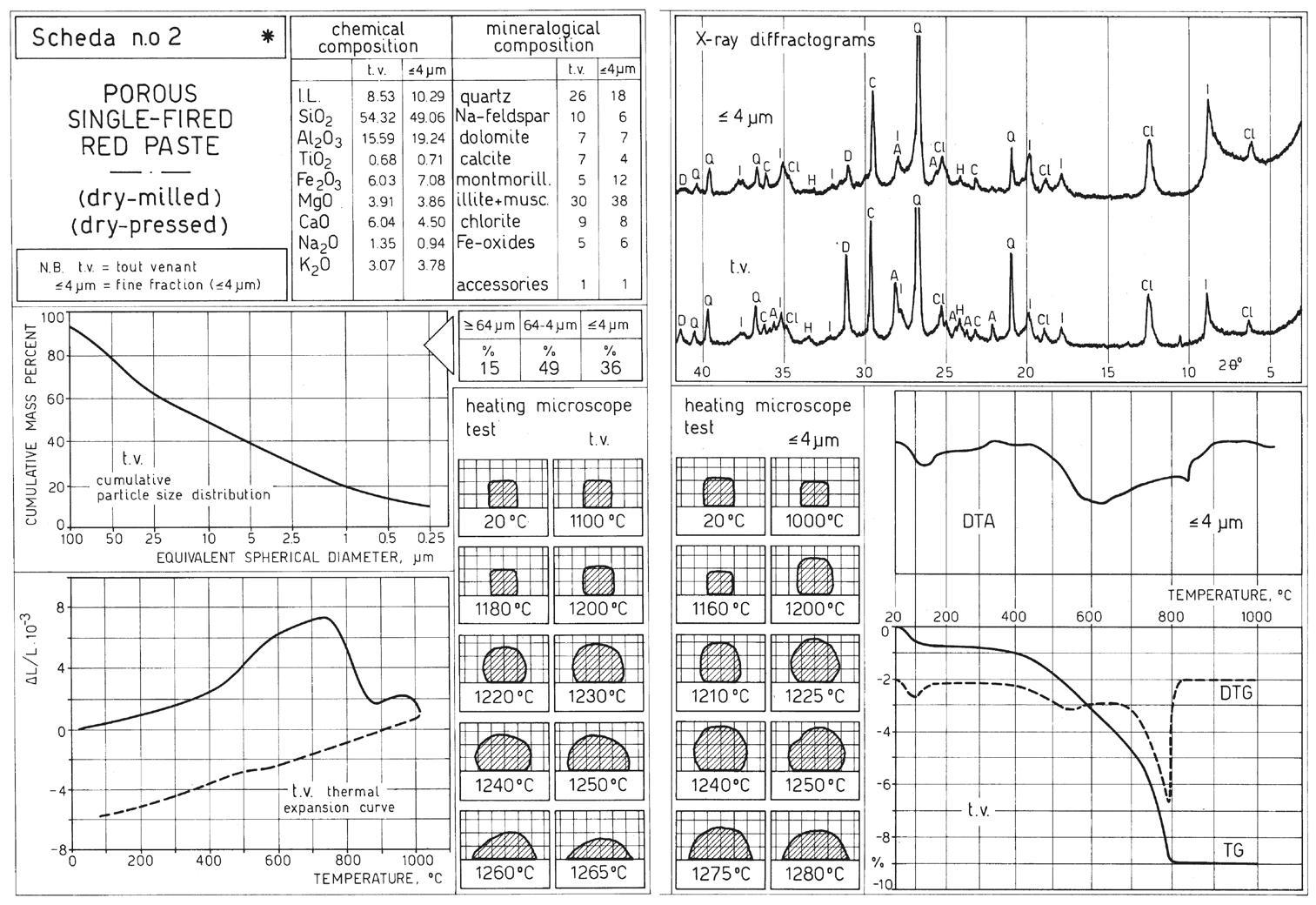
Fig. 6.10 Esempio di scheda-impasto con analisi chimica e mineralogica quantitativa
Le possibili analisi dei parametri fisici di un materiale che variano in funzione dell’aumento e/o diminuzione della temperatura, sono molteplici. Sottoponendo un campione ceramico ad uno specifico gradiente termico, è infatti possibile registrare: la perdita di peso, il calore emesso o assorbito, le variazioni dimensionali, la deformazione, la fusibilità, l’emissione di sostanze gassose, ecc.
Per effettuare queste misure si utilizzano tecniche strumentali specifiche, come ad esempio:
- analisi termodifferenziale (DTA)
- calorimetria differenziale a scansione (DSC)
- analisi termogravimetrica (TGA)
- analisi dei gas emessi (EGA)
- analisi dimensionale (Dilatometria)
- analisi termomeccanica (TMA)
- analisi della piroplasticità (Flessometria)
- analisi della fusibilità.
Queste tipologie di analisi termiche hanno il principale scopo di valutare il comportamento di una singola materia prima o di un impasto ceramico nei processi di essiccamento e cottura. Rappresentano però anche validi metodi per acquisire informazioni sulla composizione chimica-mineralogica di un materiale ceramico, essendo i parametri misurati strettamente correlati alla sua natura chimica ed alla struttura cristallina. Si possono infatti individuare le trasformazioni di fase di vari minerali (ad es. la transizione quarzo α quarzo β) oppure riconoscere la presenza di minerali soggetti a decomposizione termica come carbonati, solfati, solfuri, ecc.
Spesso è possibile anche combinare alcune di queste analisi in un’unica strumentazione, ad esempio TGA+DTA oppure TGA+EGA, in modo da ottenere con un’unica misura una maggior quantità di informazioni sul campione.
L’analisi DTA si basa sulla misura delle differenze di temperatura tra il campione da esaminare ed un materiale di riferimento inerte (es. caolino calcinato), sottoposti simultaneamente a riscaldamento fino ad alte temperature (es. 1200 °C).
Se nel materiale incognito avvengono reazioni endotermiche (con assorbimento di calore) o esotermiche (con emissione di calore), si produce una differenza di temperatura rispetto al riferimento e lo strumento di misura registrerà graficamente un picco negativo (endo-) o positivo (eso-). La posizione dei picchi rispetto all’asse delle temperature consente una determinazione qualitativa di molti minerali e di altri composti soggetti a decomposizioni termiche. Ad esempio, i minerali argillosi presentano un caratteristico picco endotermico intorno ai 600 °C associato alla perdita dell’acqua di costituzione, mentre la calcite (CaCO3) mostra un forte picco endotermico a circa 900 °C, dovuto alla sua trasformazione in ossido di calcio con emissione della CO2
La Fig. 6.11 riporta schematicamente le analisi DTA di alcuni minerali argillosi di interesse ceramico.
Caolinite
Metahalloysite
Na-montmorillonite
Ca-montmorillonite
Fig. 6.11 Analisi DTA di minerali argillosi
Fig. 6-11 IT
Nel caso poi venga eseguita una analisi simultanea TG+DTA è possibile correlare le trasformazioni termiche con l’eventuale perdita di massa associata, come evidenziato in Fig. 6.12 dove la reazione termica di deidratazione a ca. 600 °C di un caolino corrisponde ad una perdita in massa per l’eliminazione dell’acqua di costituzione, mentre il picco esotermico a ca. 1000 °C non è associato ad alcuna variazione di peso in quanto si tratta di una cristallizzazione di mullite primaria.
Fig. 6.12 Curve TG, DTG (derivata della TG) e DTA di un caolino
Fig. 6-12
L’analisi DSC è analoga alla DTA ma, rispetto a quest’ultima, permette di misurare la quantità di calore assorbita o rilasciata da un campione alle rispettive temperature di reazione. Conseguentemente, nota la quantità del materiale sottoposto ad analisi e conoscendo le entalpie teoriche associate ad una determinata reazione, è possibile effettuare anche una analisi composizionale quantitativa.
La Tab. 6.2 riporta alcuni esempi delle entalpie associate a tipiche reazioni di trasformazione di materie prime ceramiche, alle temperature di riferimento; il segno + indica una reazione endotermica (con assorbimento di calore).
(cristallo) −229
(vetro) −203
Li2SiO3 (vetro) −311
Mullite −426
Na2SiO3 (cristallo) −560 kcal/kg °C
Na2SiO3 (vetro) −520 Quarzo α → β +5 573
PbSiO3 (vetro) −3,2 Cristobalite α → β +3 250
Sillimanite −274 Tridimite α → β +0,7 117
Willemite −31 Quarzo → Cristobalite +30 1470
Wollastonite −171 Quarzo → Tridimite +21 867
Tab. 6.2 Entalpie di trasformazione (espresse in kcal/kg) per composti di interesse ceramico
L’analisi termogravimetrica misura la perdita in peso che subisce un materiale sottoposto a riscaldamento fino ad alta temperatura (es. 1200 °C).
L’esame del diagramma ottenuto, analogamente a quanto avviene per l’analisi termica differenziale a cui la termogravimetria è spesso associata, permette un’analisi qualitativa e quantitativa dei minerali e di altri composti anche organici, soggetti a degradazione termica. A titolo di esempio in Fig. 6.13 si riporta il grafico dell’analisi TG di una dolomite, carbonato di calcio e magnesio dalla formula CaMg(CO3)2
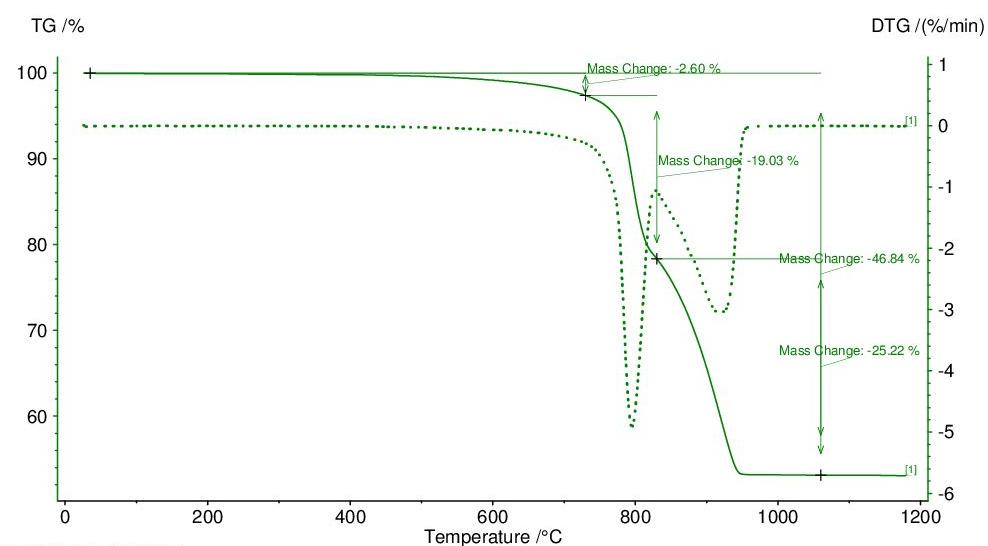
Fig. 6.13 Curva TG di una dolomite
Come si può osservare, la curva della TG presenta due principali perdite di peso in successione, la prima del 19,03% con la massima pendenza a circa 800 °C, come evidenziato dalla derivata DTG (curva tratteggiata), la seconda del 25,22% poco dopo i 900 °C. Tali perdite di peso corrispondono alla decomposizione della dolomite con emissione di CO2 che avviene in due stadi secondo la reazione:
CaMg(CO3)2 → CaCO3 + MgO + CO2↑ → CaO + MgO + CO2↑
Sulla base dei valori delle perdite in peso rispetto alle percentuali teoriche si può quindi valutare la purezza della dolomite analizzata.
La combinazione della TGA con uno spettrometro di massa (MS) o con la spettroscopia in trasformata di Fourier (FTIR) consente di studiare la natura dei prodotti gassosi sviluppati (EGA, Evolved Gas Analysis).
Questo tipo di analisi risulta di particolare interesse in relazione alla degradazione termica ed all’ossidazione dei composti organici additivati agli impasti (come ad es. deflocculanti e tenacizzanti) o all’applicazione superficiale di colle, fissatori ed inchiostri a base solvente.
L’analisi EGA permette infatti di identificare le sostanze volatili emesse, talvolta potenzialmente nocive come nel caso della formaldeide o dell’acroleina.
L’analisi dilatometrica permette di misurare le variazioni dimensionali (espansione e ritiro) di un campione opportunamente preformato (di solito in forma di parallelepipedo o cilindretto di lunghezza 35 ÷ 50 mm), sottoposto ad un riscaldamento con gradiente termico controllato da temperatura ambiente alla temperatura massima prefissata (tipicamente 100, 400, 700, 1000 o 1200 °C).
La misura del comportamento dilatometrico può essere effettuata su provini di materie prime e impasti sia crudi che cotti; nel primo caso l’andamento della curva rivela le trasformazioni strutturali del materiale (transizioni di fase, decomposizioni, fusioni, ecc.), nel secondo caso se ne determina il coefficiente di dilatazione termica lineare α, espresso dalla seguente relazione:
in cui α è il coefficiente di dilatazione termica lineare fra T1 e T2, L0 è la lunghezza iniziale del provino, ∆L è la differenza fra LT1 ed LT2.
Tipicamente per i materiali ceramici i valori di α30-400°C sono compresi tra 50 e 80×10−7°C−1
Nella pratica produttiva si utilizza spesso anche il coefficiente di dilatazione cubica 3α, ricavato moltiplicando per 3 il coefficiente lineare α ottenuto dalla misura dilatometrica, con l’ipotesi che la variazione dimensionale del materiale sia la stessa in tutte le direzioni (ipotesi valida per un materiale perfettamente isotropo).
Il coefficiente di dilatazione è estremamente importante per valutare la bontà dell’accoppiamento di materiali differenti, come lo smalto e il supporto. In tal caso, infatti, un diverso comportamento dilatometrico determina, soprattutto durante il raffreddamento, tensionamenti che nelle piastrelle smaltate possono portare a variazioni della planarità o addirittura alla rottura dello strato di smalto (vedi Fig. 6.14).
αsmalto < αsupporto
αsmalto > αsupporto
compressione trazione
Fig. 6.14 Effetti sulla planarità di un non corretto accordo dilatometrico fra smalto e supporto
Nella pratica produttiva industriale è preferibile ottenere piastrelle con un leggero stato di compressione e, conseguentemente, il coefficiente di dilatazione dello smalto deve essere di poco inferiore a quello del supporto.
Fig. 6-14 IT NEW
La registrazione della curva di dilatazione di un supporto ceramico cotto evidenzia anche la presenza di quarzo libero residuo, in quanto la transizione quarzo α → quarzo β è caratterizzata da un marcato aumento di volume alla temperatura teorica di 573°C (Fig. 6.15).
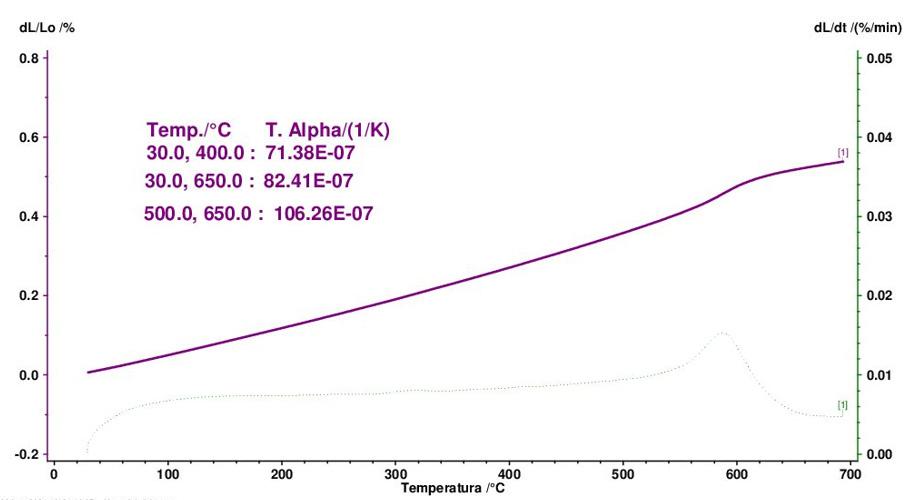
Fig. 6.15 Curva dilatometrica di un supporto cotto con transizione di fase del quarzo
L’entità della variazione dilatometrica dovuta al quarzo può essere quantificata calcolando il coefficiente α tra 500 e 650 °C; un valore basso è particolarmente importante nella produzione di grandi formati per evitare rotture in fase di raffreddamento (sfilo).
L’analisi dilatometrica di un impasto crudo fin oltre la sua temperatura di cottura permette invece di valutare il suo comportamento nella fase di sinterizzazione, ed in particolare stabilità e ritiro.
Si riporta come primo esempio (Fig. 6.16) la curva di dilatazione di un impasto carbonatico da rivestimento (monoporosa).
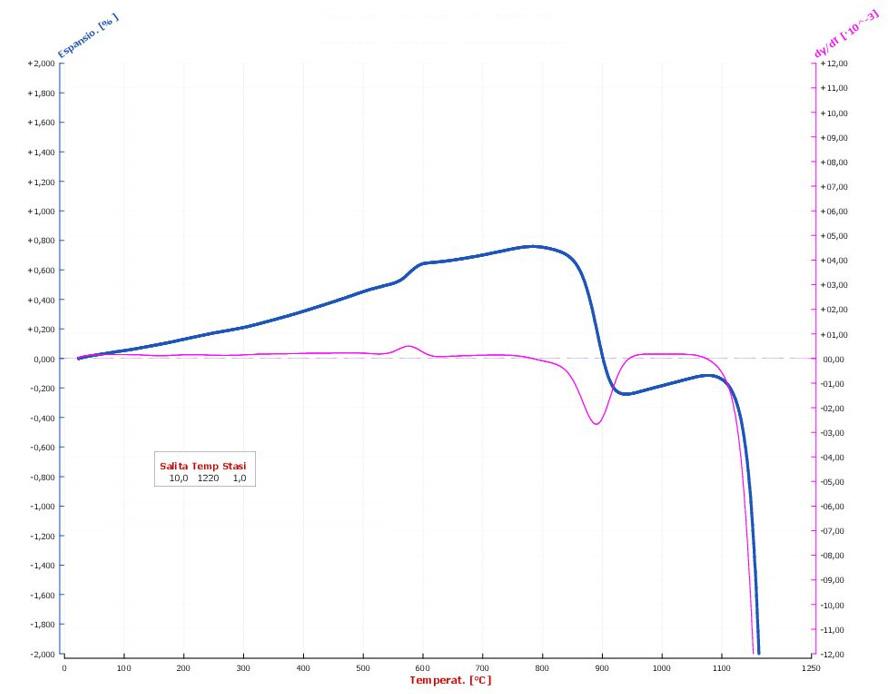
Fig. 6.16 Curva dilatometrica di un supporto crudo di monoporosa
Il grafico mostra una iniziale dilatazione regolare, la transizione del quarzo a 573 °C, una normale dilatazione fino a ca. 850°C dove avviene una brusca contrazione dovuta alla decomposizione dei carbonati presenti in formulazione, un pianerottolo quasi orizzontale corrispondente alla formazione di silicati e silico-alluminati di calcio e magnesio (diopside e gehlenite) ed una rapida fusione finale. Per ottenere un prodotto dimensionalmente stabile è opportuno che la temperatura massima di cottura sia in corrispondenza del plateau.
La Fig. 6.17 riporta invece l’analisi al dilatometro ottico di un impasto crudo da grès porcellanato.
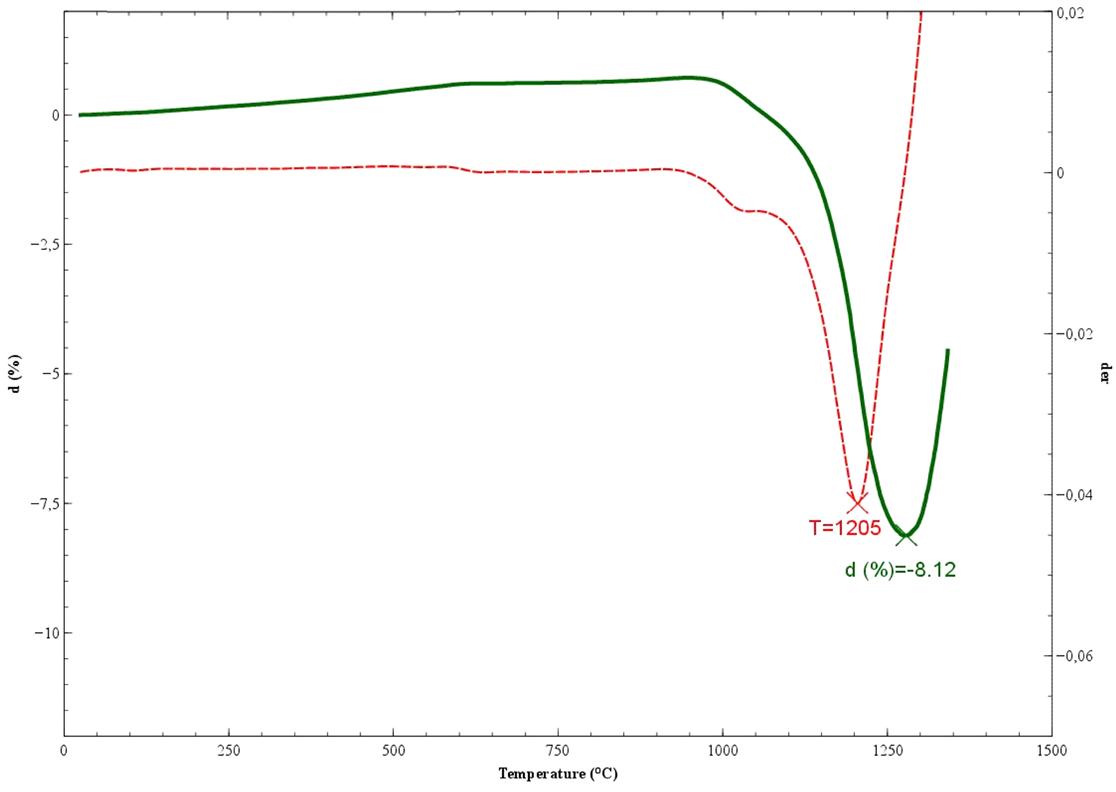
Fig. 6.17 Curva dilatometrica di un supporto crudo di grès porcellanato
Come si può osservare, dopo la fase di bassa dilatazione fino sopra ai 1000 °C, inizia immediatamente la greificazione con la massima velocità di ritiro a 1205 °C (picco della derivata), successivamente alla quale il materiale va in espansione per l’incipiente fusione. È possibile anche valutare la stabilità dell’impasto alla greificazione, mantenendo il campione per un certo tempo alla temperatura massima desiderata. La Fig. 6.18 mostra, ad esempio, che il campione mantenuto a 1205 °C, la temperatura di massima sinterizzazione registrata in precedenza, presenta una ottima stabilità dimensionale con un ritiro di 8,3%.
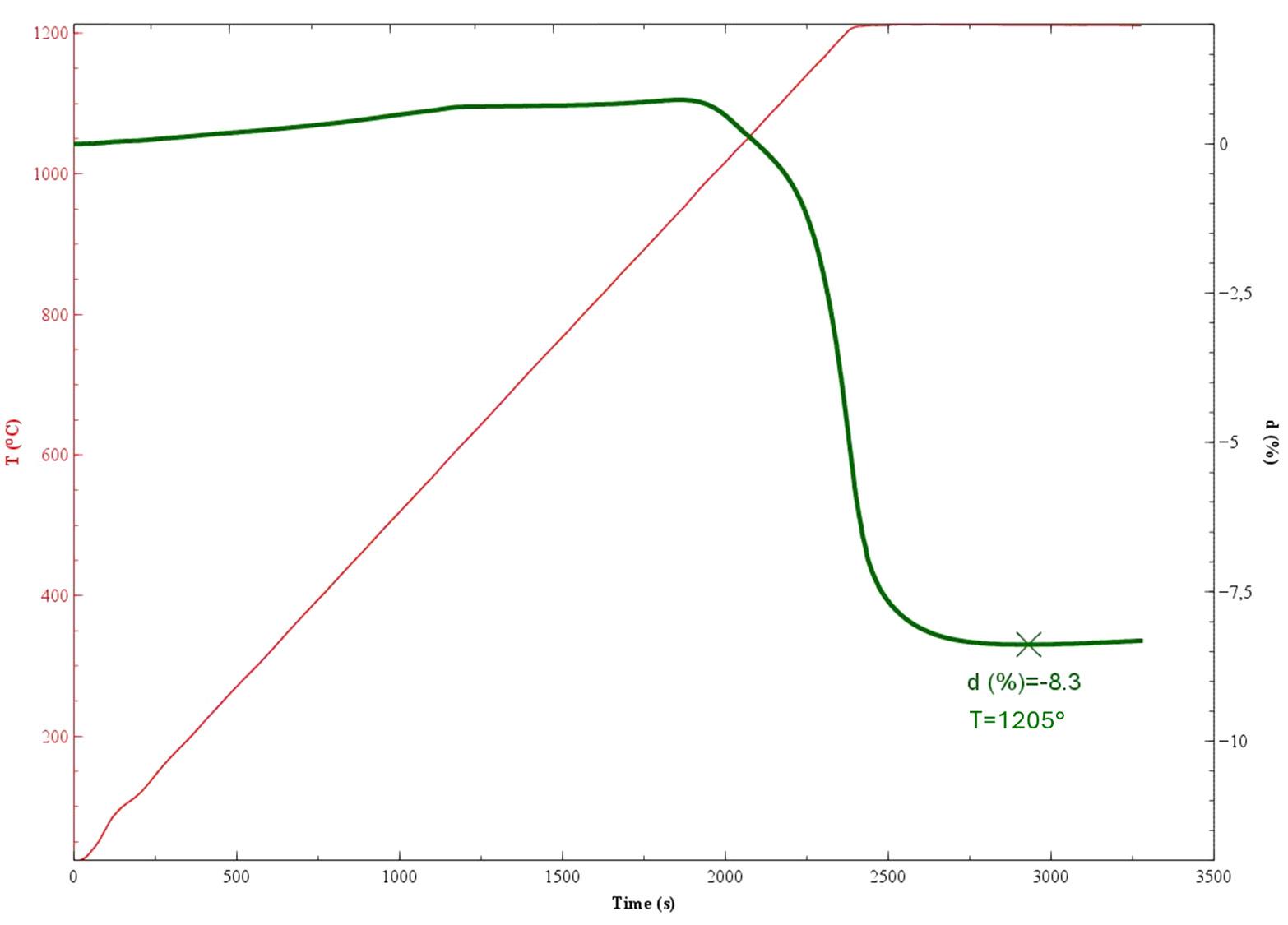
Fig. 6.18 Curva dilatometrica con permanenza a 1205 °C
L’analisi termomeccanica viene utilizzata per studiare il comportamento dei materiali sottoposti ad uno sforzo a temperature variabili, tramite un trasduttore di spostamento induttivo. Dalle misure è possibile ricavare i coefficienti di dilatazione lineare, le temperature di transizione di fase, le temperature di rammollimento e sinterizzazione, le variazioni di densità, ecc. Si tratta comunque di una analisi piuttosto sofisticata, poco applicata ai materiali ceramici tradizionali.
L’analisi termica al flessimetro ottico viene effettuata per determinare la deformazione piroplastica di un impasto ceramico greificabile, tipicamente per grès porcellanato. Dalla piastrella cruda viene ricavata una “barretta” che viene sottoposta ad un ciclo termico di riscaldamento con permanenza di 4 min alla temperatura massima di sinterizzazione (Fig. 6.19).

Fig. 6.19 Flessimetro ottico per l’analisi della piroplasticità
Durante la fase di greificazione dell’impasto ceramico si ha la formazione di fasi viscose ed il campione si deforma curvandosi sotto l’azione del proprio peso. Una fotocamera ottica ad alta risoluzione misura la flessione del pezzo nel suo punto mediano. Il valore di deformazione piroplastica è espresso dalla percentuale di flessione del campione rispetto alla distanza dei supporti. Per non incorrere in problematiche di deformazione in cottura, specialmente nel caso di lastre ceramiche a spessore sottile e/o con un passo rulli del forno inadeguato, è opportuno che il valore di deformazione piroplastica sia inferiore al 2,5% (Fig. 6.20).
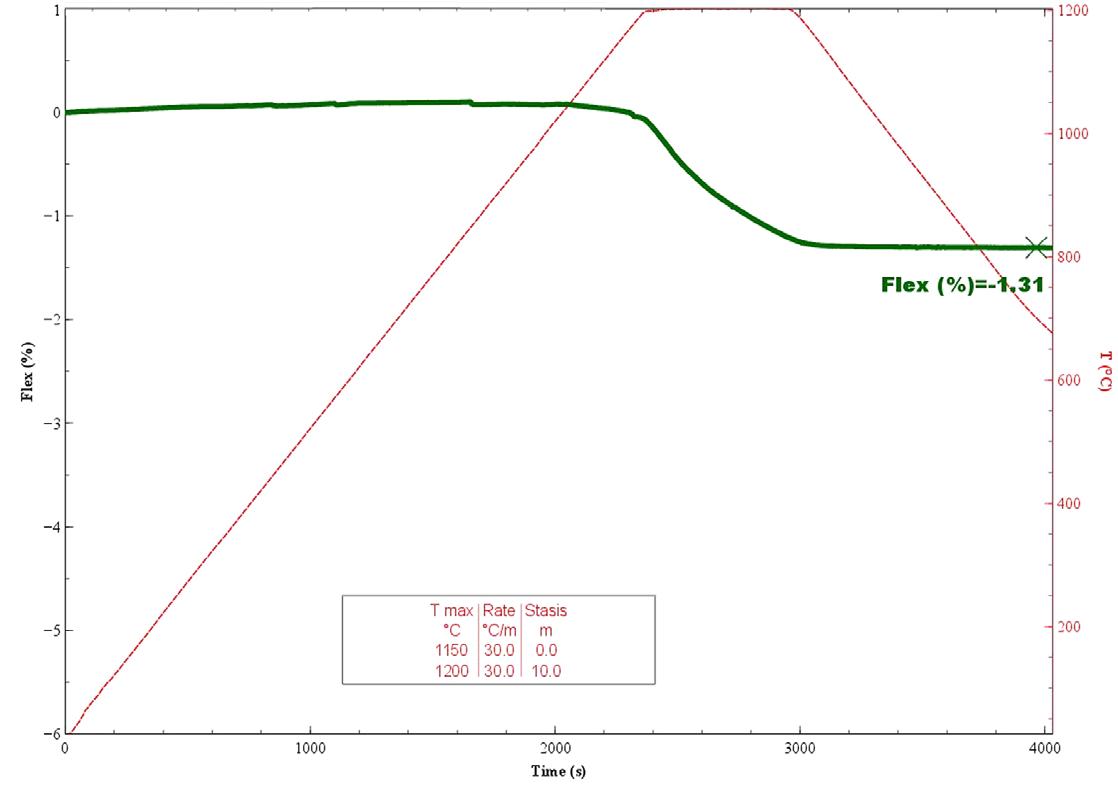
Fig. 6.20 Misura della deformazione al flessimetro ottico
In ceramica l’analisi della fusibilità di materie prime, impasti, fritte e smalti viene generalmente eseguita con il microscopio riscaldante. L’analisi consiste nell’osservazione diretta di un piccolo provino del materiale crudo pressato, posto all’interno di un fornetto al quale viene imposto un incremento di temperatura con un gradiente termico rappresentativo del ciclo di cottura industriale (es. 50 °C/min). L’apparecchio consente simultaneamente, tramite un sistema di lenti e una telecamera, la visione diretta a monitor e la registrazione dell’immagine del campione sottoposto a riscaldamento. Tale prova permette di rilevare le temperature di inizio ritiro, di rammollimento, di fusione e l’angolo di contatto col supporto refrattario una volta raggiunta la fusione (Fig. 6.21).
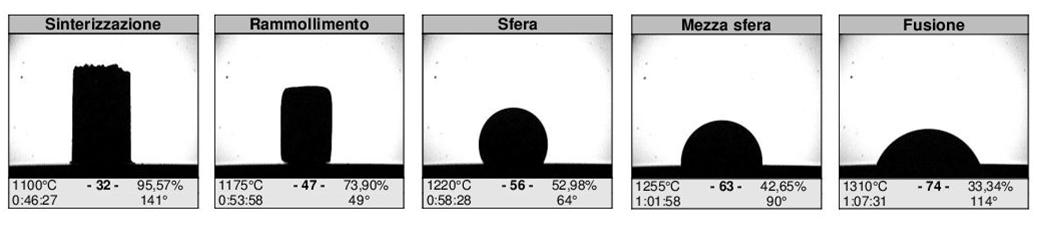
L’analisi strumentale della distribuzione granulometrica delle singole particelle in sospensione è di particolare interesse per i materiali argillosi e per gli impasti macinati a umido, in quanto permette di determinare anche le frazioni più fini, inferiori al micrometro, caratteristiche dei composti colloidali come le argille (vedi Paragrafo 5.2).
Esistono fondamentalmente due tecniche strumentali per la determinazione del diametro di particelle in sospensione, una basata sulla diffrazione della luce (light scattering) secondo la teoria di Fraunhofer, l’altra associata alla velocità di sedimentazione secondo la legge di Stokes.
Nel caso di strumenti di tipo light scattering (Fig. 6.22), alcune gocce di sospensione vengono iniettate e mantenute in ricircolo all’interno di una cella di misura attraversata da una radiazione laser solitamente nel visibile (es. λ=520 nm).
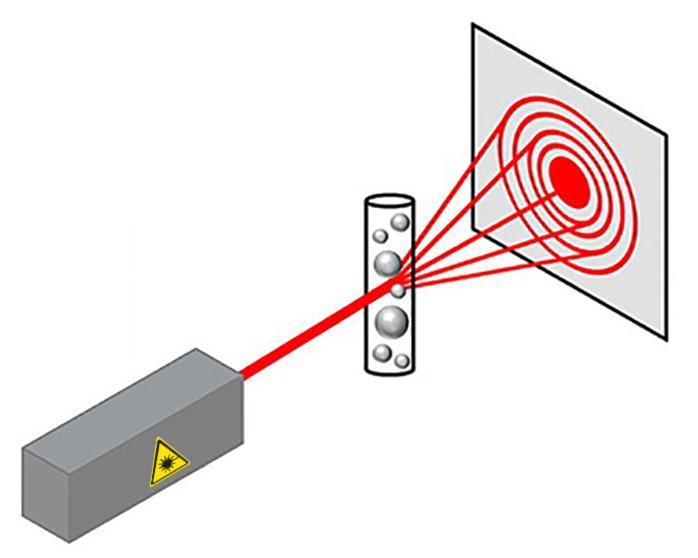
Fig. 6.22 Principio di misura granulometrica per diffrazione laser
Analogamente, nella misura per sedimentazione, una piccola quantità di sospensione viene immessa in una cella verticale e, tramite un fascio di raggi X che scansiona le particelle dal basso verso l’alto, viene misurato il loro diametro sulla base della velocità (spazio/tempo) di precipitazione (Fig. 6.23).
Fig. 6-22
Fig. 6.23 Principio di misura granulometrica per sedimentazione con raggi X (fonte: Micromeritics US)
Con entrambe le tecniche è possibile ricavare le curve di distribuzione granulometrica differenziale e cumulativa, in un ampio intervallo dimensionale, da 0,01 µm ad alcuni millimetri (light scattering) e da 0,1 a 300 µm (X-ray sedimentation). Qualora la granulometria del campione da analizzare comprenda frazioni più grossolane rispetto all’intervallo di misura dello strumento, è necessario setacciare la sospensione ed alimentare solamente la porzione passante, mantenendo comunque la possibilità di inserire la percentuale tolta nel computo della curva di distribuzione granulometrica.
La tecnologia della diffrazione laser viene anche utilizzata per la determinazione della distribuzione granulometrica di polveri fini allo stato secco. Il campione in polvere viene alimentato e disperso in un circuito di aria compressa, quindi analizzato tramite light scattering. L’intervallo di misurazione può andare da 0,1 a 8750 µm cambiando opportunamente le distanze focali (Fig. 6.24).
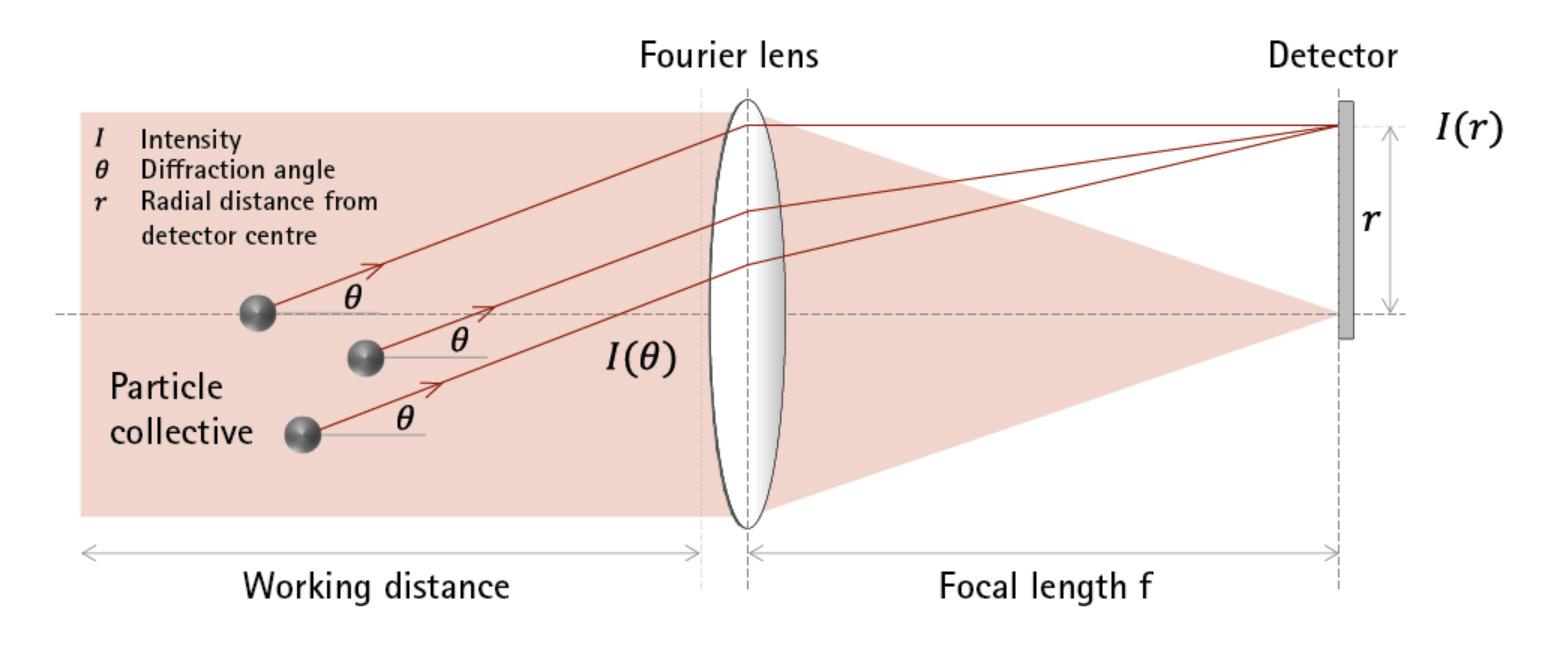
La prassi di effettuare sistematicamente semplici controlli tecnologici su materie prime e impasti consente di mantenere una buona qualità dei semilavorati e del prodotto finito in tutto il processo produttivo.
Le caratteristiche dei materiali in arrivo che devono essere determinate con regolarità sono le seguenti:
- aspetto
- umidità
- residuo ai setacci
- contenuto di carbonati
Aspetto del materiale in arrivo
Si annotano forma, dimensioni (pezzatura) e colore del materiale in arrivo con l’obiettivo di rilevare immediatamente eventuali anomalie nella fornitura.
Umidità naturale del campione
Si pesa un campione di materiale umido (Pu), si essicca a peso costante in stufa da laboratorio a 110 °C per circa 24 h e si pesa il campione secco (Ps). La differenza delle due pesate rapportata al peso del campione umido moltiplicato per 100 esprime l’umidità percentuale del campione:
Per una maggior rapidità di analisi vengono più convenientemente utilizzate termobilance costituite da una unità di pesatura ed una unità di riscaldamento elettrico, che automaticamente forniscono il valore percentuale dell’umidità del campione.
Residuo ai setacci
Si pesano 100 g del campione secco e si trattano in un agitatore da laboratorio per circa 30 min con 1000 mL di acqua, eventualmente addizionata con 1 g di sodio tripolifosfato o sodio poliacrilato, finché le particelle siano completamente disgregate e disperse.
Si setaccia la barbottina su vagli da 1.000, 2.500 e 10.000 maglie/cm2 (equivalenti ad una luce di passaggio di 180, 125 e 63 µm). Si essiccano i residui ottenuti nei vari setacci fino a peso costante e si procede alla loro pesatura. Questi valori danno direttamente la percentuale dei residui ai vari setacci.
Anche per le materie prime non plastiche si può effettuare una determinazione a secco della granulometria, vagliando circa 200 g di materiale secco su una pila di setacci vibranti (Fig. 6.25) aventi ad esempio le seguenti reti:

Fig. 6.25 Vibrosetaccio per la misura della granulometria di polveri
Nel caso invece dell’analisi granulometrica di polveri di impasto atomizzato, la sequenza di setacci utilizzati è generalmente la seguente:
Il parametro principale da mantenere sotto controllo è la percentuale di polvere fine inferiore a 125 µm che, preferibilmente, deve essere inferiore al 5%.
Contenuto di carbonati (espresso in % CaCO3)
La determinazione viene eseguita con un semplice calcimetro Pizzarelli o Dietrich-Frühling (Fig. 6.26). Un campione di 1 g del materiale essiccato a 110 °C viene posto nell’apposito contenitore, nel quale viene versato, attraverso un rubinetto, acido cloridrico diluito (1:1).
Qualora il materiale contenga carbonati di qualunque tipo si libera CO2 che, esercitando una pressione proporzionale alla quantità dei carbonati presenti, abbasserà la colonna di acqua del cilindro graduato sul quale si legge direttamente il valore percentuale dei carbonati (sotto forma di CaCO3), contenuti nella materia prima. Noto il volume di CO2 sviluppato VCO2, e la temperatura assoluta T, il calcolo da applicare è ����������������2 % = 53621 × ������������������������2 [
] �������� [�������� ]
Se si assume che la CO2 sia stata sviluppata soltanto da CaCO3, allora vale la relazione:
× CO2%

[1] Università di Parma, «Atlas of Italian Rocks», [Online]. Available: https://www.rocks. unipr.it/.
[2] ICDD, Mineral Powder Diffraction File Databook, 1993.
[3] A. Ravaglioli e G. Pollifrone, Materie prime ceramiche - Vol.3 (metodi attuali di indagine di laboratorio e industriali), Faenza Ed., 1979.
[4] A. Bresciani e B. Spinelli, «Porcelain tile pyroplastic deformation during firing and post-firing variations in planarity», Ceramic Forum Int.l 89, 2012.
[5] E. Sánchez et al., «Firing deformation in large size porcelain tiles. effect of compositional and process variables», in Qualicer, 2018.
[6] R. C. MacKenzie, Differential Thermal Analysis - Vol.1 (Fundamental Aspects), Academic Press, 1970.
[7] W. Smykatz-Kloss, Differential Thermal Analysis. Application and Results in Mineralogy, Springer-Verlag, 1974.
È una tecnica di analisi per lo studio dei composti cristallini basata sull’interazione dei loro piani reticolari con una radiazione X. Per la diffrattometria di polveri si utilizza solitamente la radiazione del rame CuKα a lunghezza d’onda di 1,541 Å (8,04 keV) che viene riflessa dai piani cristallini con un angolo determinato dalla distanza caratteristica di ciascun piano cristallino (legge di Bragg).
PPM / PPB
Con ppm si intende “parti per milione” (dall’inglese parts per million) ovvero la concentrazione di una specie (elemento, composto, ecc.) espressa in milionesimi (1/106 = 10−6) rispetto all’unità. Viene usata per concentrazioni molto basse.
Per analogia, con ppb si intende “parti per miliardo” (dall’inglese parts per billion) ovvero la concentrazione espressa in miliardesimi (1/109 = 10−9) rispetto all’unità. Viene usata per concentrazioni bassissime.
Valgono pertanto le seguenti equivalenze:
1% = 0,01 = 10.000 ppm
1‰ = 0,001 = 1.000 ppm
1 ppm = 1.000 ppb
Procedimento standard di campionamento di materiali in polvere, che prevede una serie di passaggi mirati ad ottenere un campione, da sottoporre ad analisi, con le stesse caratteristiche di composizione del lotto iniziale e che sia il più omogeneo possibile.
Ottenere un campione di polvere che abbia le stesse caratteristiche granulometriche dell’intero lotto non è semplice; fenomeni di segregazione e stratificazione possono rendere molto diverse le composizioni in superficie rispetto a quelle più interne. La quartatura permette, con una successione di semplici azioni, di passare dal prelievo iniziale ad un suo sottoinsieme rappresentativo della composizione media.
Un reticolo cristallino (o reticolo di Bravais) è costituito dalla ripetizione ordinata di una singola unità strutturale detta cella elementare. Le celle elementari sono poliedri formati da atomi o ioni disposti ai loro vertici, nelle facce o al centro. Bravais dimostrò che nello spazio tridimensionale sono possibili soltanto 14 tipi di celle elementari che generano i ben noti sistemi cristallini dei minerali (cubico, tetragonale, ortorombico, monoclino, triclino, trigonale, esagonale).
Indagine chimico-fisica basata sulla misura dello spettro elettromagnetico o di singole radiazioni luminose emesse o assorbite da un determinato materiale e pertanto caratteristiche della sua composizione.
Spettroscopie di assorbimento sono ad esempio la spettroscopia infrarossa, la spettroscopia UV/visibile o la spettroscopia di assorbimento atomico.
La spettroscopia di emissione atomica prevede invece di portare gli atomi in uno stato elettronico eccitato (tramite altissime temperature, irradiamento con RX o bombardamento elettronico) in modo che questi emettano radiazioni di decadimento a più bassa energia, dette radiazioni di fluorescenza.
Nei precedenti capitoli sono state descritte le principali caratteristiche delle singole materie prime utilizzate per impasti e smalti ceramici che, insieme alle condizioni di processo, determinano le proprietà delle piastrelle, intese come prodotti finiti con determinate funzioni tecniche ed estetiche. Infatti, a seconda della destinazione d’uso, la piastrella dovrà presentare adeguate caratteristiche meccaniche, fisiche, chimiche ed anche estetiche, che rendono il materiale ceramico idoneo al suo impiego in edilizia.
Ad esempio una piastrella per pavimentazioni deve possedere una elevata resistenza ai carichi, all’abrasione ed alla sporcabilità. Inoltre le piastrelle devono garantire precise caratteristiche geometriche, riferite alle loro dimensioni (calibri) ed alla loro planarità, anche in relazione a possibili variazioni di umidità e, più in generale, delle condizioni climatiche ambientali.
Tutte queste proprietà delle piastrelle ceramiche sono definite e regolamentate dalle normative tecniche internazionali, che verranno sinteticamente illustrate nei prossimi paragrafi.
La varietà delle piastrelle ceramiche e dei loro utilizzi ha portato negli anni ad una normativa internazionale continuamente aggiornata dalla ISO (International Organization for Standardization) e, nello specifico, dal Comitato Tecnico ISO/TC 189 “Ceramic Tiles”, a partire dal 1985.
La principale norma ISO di riferimento è la ISO 13006 che stabilisce le definizioni, la classificazione, le caratteristiche e le modalità di marcatura per le piastrelle ceramiche. I metodi di misura dei requisiti tecnici prescritti sono invece definiti nelle diverse parti della norma ISO 10545.
Secondo la ISO 13006 [1], con piastrella ceramica si intende una lastra sottile composta da argille e/o altre materie prime inorganiche, generalmente utilizzata come rivestimento di pavimenti e pareti, solitamente formata mediante estrusione (A) o pressatura (B) a temperatura ambiente, ma anche mediante altri processi (C), quindi essiccata e successivamente cotta a temperature sufficienti per sviluppare le proprietà richieste. Le piastrelle possono essere smaltate (GL) o non smaltate (UGL), sono incombustibili e non vengono alterate dalla luce.
Le piastrelle di ceramica sono classificate in gruppi in base al metodo di fabbricazione e al loro assorbimento d’acqua. Questa suddivisione non considera quindi l’utilizzo che verrà fatto dei vari prodotti ceramici.
I metodi di fabbricazione individuati dalla norma sono essenzialmente due:
- metodo A: piastrelle estruse, ovvero formate allo stato plastico mediante un estrusore;
- metodo B: piastrelle pressate a secco, per le quali il materiale ceramico è finemente macinato e la polvere ottenuta viene formata ad elevate pressioni di compattazione.
Con assorbimento d’acqua (AA) la norma intende la quantità di acqua che può impregnare la piastrella, espressa come % di massa d’acqua rispetto alla massa secca. La misura dell’assorbimento d’acqua è normata dalla ISO 10545-3. In funzione della quantità di acqua assorbita dalla piastrella, la norma individua tre Gruppi:
- Gruppo I: basso assorbimento di acqua (AA ≤ 3%)
- Gruppo II: medio assorbimento di acqua (3% < AA ≤ 10%)
- Gruppo III: alto assorbimento di acqua (AA > 10%)
Il Gruppo I è ulteriormente suddiviso in due sottogruppi:
- Gruppo Ia: AA ≤ 0,5%
- Gruppo Ib: 0,5% < AA ≤ 3%
Anche il Gruppo II è ulteriormente suddiviso in due sottogruppi:
- Gruppo IIa: 3% < AA ≤ 6%
- Gruppo IIb: 6% < AA ≤ 10%
Alla luce di questa classificazione, considerando solamente le piastrelle pressate (Metodo B), i vari Gruppi risultanti sono quelli richiamati in Tab. 7.1.
Metodo di formatura
Tipologia di prodotto
Grès porcellanato (UGL/GL)
Tab. 7.1
Monocottura vetrificata (UGL/GL)
Pavimento semi-vetrificato (GL)
Pavimento poroso (GL)
Classificazione ISO 13006 delle piastrelle pressate a secco
Rivestimento poroso (GL)
Una volta definita la suddivisione in Gruppi, la ISO 13006 individua poi una serie di caratteristiche dimensionali, fisiche e chimiche che identificano il singolo prodotto ceramico, specificando inoltre quali di queste caratteristiche sono rilevanti in funzione dell’uso (interno, esterno) e della destinazione (pavimento, rivestimento). Per ogni caratteristica misurabile è indicata la rispettiva norma ISO 10545 di riferimento che descrive i metodi di prova (vedi Tab. 7.2).
Infine, per ogni Gruppo, la norma prescrive in dettagliate tabelle i requisiti di accettabilità in relazione ad ognuna delle caratteristiche misurate.
Caratteristiche
Dimensioni e qualità della
Lunghezza e larghezza
(curvatura e svergolamento)
Proprietà fisiche Assorbimento
dovuta all’umidità
al cavillo (GL)
al gelo
Proprietà chimiche
Resistenza a basse concentrazioni di acidi e basi
Resistenza ad alte concentrazioni di acidi e basi
Resistenza ai detergenti per la casa e ai sali per piscine
Resistenza alle macchie
Cessione di piombo e cadmio (GL)
GL = piastrelle smalatate (glazed)
UGL = piastrelle non smaltate (unglazed)
Tab. 7.2 Caratteristiche secondo la norma ISO 13006
10545-13
La norma ISO 10545 [2] definisce in maniera sistematica le varie caratteristiche (dimensionali, fisiche, chimiche) che devono essere verificate per attestare la conformità del prodotto ceramico. Per ogni caratteristica (o gruppo di caratteristiche) la norma prevede una parte dedicata che illustra dettagliatamente la metodica di verifica; in Tab. 7.3 sono elencate le parti attualmente in vigore, mentre altre sono tuttora in fase di elaborazione.
La Parte 1 descrive i criteri di campionamento e di accettazione delle piastrelle da testare secondo le parti successive.
Norma Titolo
ISO 10545-1
ISO 10545-2
ISO 10545-3
ISO 10545-4
ISO 10545-5
ISO 10545-6
ISO 10545-7
ISO 10545-8
ISO 10545-9
ISO 10545-10
ISO 10545-11
Campionamento e criteri di accettazione
Determinazione delle caratteristiche dimensionali e della qualità della superficie
Determinazione dell’assorbimento di acqua, della porosità apparente, della densità relativa apparente e della densità apparente
Determinazione della resistenza a flessione e della forza di rottura
Determinazione della resistenza all’urto mediante misurazione del coefficiente di restituzione
Determinazione della resistenza all’abrasione profonda per piastrelle non smaltate
Determinazione della resistenza all’abrasione superficiale per piastrelle smaltate
Determinazione della dilatazione termica lineare
Determinazione della resistenza agli sbalzi termici
Determinazione della espansione dovuta all’umidità
Determinazione della resistenza al cavillo per piastrelle smaltate
ISO 10545-12 Determinazione della resistenza al gelo
ISO 10545-13 Determinazione della resistenza chimica
ISO 10545-14 Determinazione della resistenza alle macchie
ISO 10545-15 Determinazione del piombo e del cadmio ceduto dalle piastrelle
ISO 10545-16 Determinazione di piccole differenze di colore
ISO 10545-18 Determinazione del valore di riflettanza della luce (LRV)
ISO 10545-20 Determinazione della flessione delle piastrelle di ceramica per il calcolo del loro raggio di curvatura
Tab. 7.3 Parti della norma ISO 10545 attualmente in vigore (2024)
Nelle successive tabelle (Tab. 7.4, Tab. 7.5 e Tab. 7.6) sono riportati i valori delle principali caratteristiche per i due Gruppi estremi (BIa e BIII) tra le classi di piastrelle ceramiche prodotte industrialmente:
- Gruppo BIa: AA ≤ 0,5% (prodotti in grès porcellanato tecnici o smaltati)
- Gruppo BIII: AA > 10% (prodotti porosi da rivestimento smaltati)
Per tutti gli altri gruppi, la norma ISO 13006 prevede valori intermedi tra questi due estremi. Relativamente alle caratteristiche dimensionali della Tab. 7.4, la norma richiede di adottare il valore minore tra i due parametri indicati (tolleranza percentuale ed assoluta). Per esempio, nel caso di una piastrella di 600 × 600 mm, la tolleranza percentuale sulla lunghezza risulterebbe ± 3.6 mm, mentre quella assoluta è ± 2.0 mm; si adotta quindi quest’ultimo intervallo, più restrittivo.
Tab. 7.4 Criteri di accettabilità per le caratteristiche dimensionali e metodi di prova relativi per piastrelle con lato > 15 cm
all’urto test disponibile 10545-5
Resistenza all’abrasione profonda (UGL) ≤ 175 mm3 - 10545-6
Resistenza all’abrasione superficiale (GL) Classe PEI Classe PEI 10545-7
Dilatazione termica lineare test disponibile 10545-8
Resistenza agli sbalzi termici test disponibile 10545-9
Espansione dovuta all’umidità test disponibile 10545-10
Resistenza al cavillo (GL) resistente 10545-11
Resistenza al gelo resistente test disponibile 10545-12
Piccole differenze di colore
∆E < 0.75 (GL) ∆E < 1 (UGL) 10545-16
Tab. 7.5 Criteri di accettabilità per le caratteristiche fisiche e metodi di prova relativi per piastrelle con spessore ≥ 7,5 mm
Caratteristiche chimiche
Resistenza a basse concentrazioni di acidi e basi Classe B min. Classe B min. 10545-13
Resistenza ad alte concentrazioni di acidi e basi Classe B min. Classe B min. 10545-13
Resistenza ai detergenti per la casa e ai sali per piscine Classe B min. Classe B min. 10545-13
Resistenza alle macchie Classe 3 min. Classe 3 min. 10545-14
Cessione di piombo e cadmio (GL) - - 10545-15
Tab. 7.6 Criteri di accettabilità per le caratteristiche chimiche e metodi di prova relativi
La norma ISO 17889, introdotta nel 2021, affronta in modo puntuale, con una serie di valutazioni oggettive, il tema della sostenibilità dei prodotti ceramici e dei materiali per la loro posa; la Parte 1 di questa norma è specifica per le piastrelle ceramiche [3].
Scopo della norma è identificare i requisiti ambientali, economici e sociali, al fine di:
- promuovere lo sviluppo e l’uso di piastrelle ceramiche sostenibili;
- orientare tutti gli stakeholder alla responsabilità ambientale lungo tutta la filiera delle piastrelle e dei materiali di posa;
- fornire una risorsa verificabile per le specifiche del prodotto "piastrella" e consentire ai professionisti della progettazione, agli appaltatori e ai consumatori di identificare piastrelle e materiali di installazione sostenibili;
- aumentare il valore delle piastrelle e dei materiali di posa sostenibili lungo tutta la catena di fornitura creando maggiore consapevolezza e domanda del mercato.
La norma fornisce un sistema di valutazione della sostenibilità lungo tutto il loro ciclo di vita (criteri LCA) utilizzando una serie di indicatori qualitativi e quantitativi. Vengono così specificate due categorie di requisiti:
- Requisiti obbligatori (tipo M = mandatory): del tipo vero/falso (pass/fail), la cui conformità è un prerequisito per un prodotto valutato come sostenibile. Se un prodotto ceramico non è conforme a tutti i requisiti obbligatori non può essere classificato come sostenibile.
- Requisiti volontari (tipo V = voluntary): requisiti ai quali un prodotto può rispondere a diversi livelli, assegnati attraverso un rating opportunamente calcolato. Tutti i requisiti volontari multirating richiedono un livello minimo obbligatorio del 100%. Il livello di conformità ai requisiti multirating contribuisce alla valutazione finale del prodotto.
In totale la norma identifica 38 requisiti, così suddivisi:
- 15 requisiti obbligatori (tipo M)
- 23 requisiti volontari (tipo V, suddivisi in V1, V2, V3)
I requisiti sono inoltre così suddivisi:
- 27 requisiti riguardano la sostenibilità ambientale: materie prime, fabbricazione, distribuzione e installazione, utilizzo, fine vita, marcatura ambientale del prodotto.
- 10 requisiti riguardano la sostenibilità sociale: salute e sicurezza nella produzione, durante l’installazione e durante l’uso del prodotto.
- 1 requisito è relativo alla sostenibilità economica: rispetto delle normative di settore e dichiarazioni del produttore.
Per ogni requisito volontario è fornita una griglia di valutazione basata su parametri oggettivi misurabili, ovviamente differenti in funzione del requisito. La valutazione del livello di sostenibilità va da 100% (più basso) a 130% (più alto).
I requisiti volontari hanno un peso diverso a seconda della difficoltà di raggiungimento, il costo o gli sforzi per l’implementazione, ecc. I requisiti V1 richiedono generalmente la conferma dell’esistenza di un determinato impianto o servizio, mentre i requisiti V2 vanno più in profondità, occupandosi dei sistemi di gestione e organizzazione della fabbrica. I requisiti V3 riguardano invece le prestazioni di sostenibilità misurabili come le emissioni inquinanti o il riciclo dell’acqua o dei rifiuti. Vista la difficoltà crescente, alle tre classi V è stato associato un peso diverso:
- V1 = requisiti di tipo vero/falso: peso = 1
- V2 = requisiti manageriali: peso = 3
- V3 = requisiti quantitativi: peso = 6
Il calcolo del rating finale avviene solamente se sono stati superati tutti i requisiti M (obbligatori). Se sì, si procede al calcolo delle seguenti tre medie aritmetiche per i requisiti V1, V2, V3 (che possono avere solamente questi valori: 100%, 110%, 120%, 130%):
Il rating finale di sostenibilità S ra è quindi calcolato come media ponderata delle medie aritmetiche di cui sopra, utilizzando i pesi indicati, secondo la seguente formula:
(6 × ����������������������������������������3 )]
+ 3 + 6
Ovviamente S ra avrà un valore compreso tra 100% e 130%.
Sulla base della S ra così ottenuta, ogni singolo prodotto sarà ritenuto conforme alla norma ISO 17889 se sono rispettati i seguenti criteri:
- tutti i requisiti obbligatori (M) devono essere soddisfatti
- S ra deve essere maggiore di 117,5%.
Per i dettagli sui singoli requisiti e la griglia di valutazione si rimanda direttamente alla consultazione dell’Allegato A della norma ISO 17889-1.
Una delle proprietà fondamentali della ceramica, oltre alle caratteristiche estetiche, è indubbiamente la sua resistenza meccanica. A riguardo, negli ultimi tempi lo sviluppo di lastre in grande formato e la progressiva diminuzione degli spessori hanno ulteriormente evidenziato l’importanza di una adeguata resistenza meccanica del prodotto, sia durante il processo produttivo, che nella sua commercializzazione (immagazzinamento, trasporto e messa in opera). È quindi importante comprendere le correlazioni esistenti tra resistenza meccanica e dimensioni/spessore, in funzione di una corretta progettazione del prodotto ceramico.
Una lastra o piastrella ceramica si rompe prevalentemente quando è sollecitata a flessione. La sollecitazione può nascere ad esempio per effetto di un carico concentrato in corrispondenza di un vuoto nel sottofondo su sui è appoggiata (caso del pavimento) ovvero per azione del vento o di altra azione dinamica (caso di parete esterna con facciata ventilata).
Dalla scienza delle costruzioni è noto come la resistenza meccanica a flessione Wf di una trave a sezione rettangolare aumenta col quadrato dello spessore h, secondo la relazione
dove b è la larghezza e h è lo spessore della sezione. Se quindi si riduce lo spessore h della lastra (come avviene dei prodotti ceramici sottili) si ha un sensibile indebolimento della resistenza meccanica, con possibili problemi di integrità e sicurezza.
Ad esempio, se si suppone di dimezzare lo spessore della lastra, passando da 9,6 mm a 4,8 mm, le principali grandezze cambiano come indicato in Tab. 7.7:
supponendo b = 1 mm
Tab. 7.7 Confronto tra spessore normale e sottile
Il costo di produzione è calcolato ipotizzando il contesto Italia (2024) per prezzi di energia, gas e materie prime. Chiaramente ridurre lo spessore impatta principalmente sui costi di materia prima (per il supporto) e di energia (soprattutto termica). I costi per materiali indiretti (imballi, consumabili, ecc.), personale e Capex si riducono in minore misura.
Dall’esempio di Tab. 7.7 si vede come, a fronte di una riduzione del 50% dello spessore (e quindi della massa), la resistenza meccanica a flessione cala grandemente (−75%), mentre il costo di produzione si riduce solo del 27%.
Fig. 7.1 Relazione tra resistenza e spessore nelle piastrelle ceramiche
Fig. 7-01 IT
Nel grafico di Fig. 7.1 sono stati riportati gli andamenti della forza di rottura (prova a flessione) in relazione allo spessore (mm), per tre diversi materiali (grès porcellanato) aventi carico di rottura (MOR) variabile da 40 a 60 MPa. Il campo colorato rappresenta la zona non accettabile secondo quanto prescritto dalla norma ISO 13006 per il Gruppo BIa (prodotti in grès porcellanato) nel caso di prodotti aventi spessore inferiore a 7,5 mm (spessori sottili). Le curve dei vari materiali corrispondono al caso in cui la larghezza del campione è pari alla distanza tra gli appoggi (L = b) e sono descritte dalla relazione �������� = 2 3 ������������������������ ∙ ℎ2
Come si evince dal grafico, lo spessore h minimo per soddisfare la ISO 13006 deve essere nell’intorno di 4,6 mm, precisamente tra 4,2 mm (per MOR 60 MPa, impasto di grande qualità) e 5,1 mm (MOR = 40 MPa). Piastrelle e lastre con spessori inferiori a 4 mm non si possono quindi considerare conformi all’attuale normativa.1
In tempi recenti, per sopperire alla perdita di resistenza meccanica, soprattutto per grandi formati a spessore sottile, si è proceduto ad irrobustire le lastre con un rinforzo posteriore, accoppiando stuoie di varia natura (fibre di vetro, carbonio, plastica) interponendo uno strato di resina (poliuretanica, epossidica, ecc.). L’aggiunta di questi materiali permette, dal punto di vista strutturale, un aumento della resistenza meccanica in funzione dello spessore e del modulo elastico, secondo la relazione: ℎ2 = ℎ1
dove h1, E1 sono spessore e modulo elastico della ceramica e h2, E2 sono quelli del ma-
1 Al momento di andare in stampa (2025) è in elaborazione presso il TC189 ISO una norma specifica per grandi formati e spessori sottili; tuttavia, per ora restano in vigori i valori prescritti dalla norma ISO13006.
teriale di rinforzo. Dalla relazione appena esposta risulta che, per esempio, se si dimezza lo spessore della piastrella ed in luogo della ceramica si applica un nuovo materiale con spessore metà rispetto alla ceramica tolta, allora il suo modulo elastico E2 deve essere 4 volte quello della ceramica, per eguagliare la resistenza iniziale.
Nella Tab. 7.8 sono riportati alcuni esempi di materiali di rinforzo con indicazione del modulo elastico e del costo (valori indicativi). Per il grès porcellanato si è considerato il solo costo diretto di produzione (valori medi Italia, 2021):
Materiale di rinforzo
Modulo elastico [GPa]
Costo per mm di spessore [€/mq]
Grès porcellanato 60 0,40
Poliuretano (90 Shore A) 10 4,0
Tab. 7.8 Alcuni materiali per rinforzo posteriore
Come si evince dalla tabella, il grès porcellanato è di gran lunga il materiale a minor costo, con prestazioni meccaniche più che accettabili. Tra i materiali di rinforzo, la fibra di carbonio e l’acciaio garantirebbero un sensibile miglioramento strutturale ma il loro costo è decisamente troppo elevato. Oltretutto bisogna considerare anche il costo aggiuntivo per l’incollaggio del rinforzo al prodotto ceramico.
Viceversa la fibra di vetro, oggi ampiamente utilizzata per il rinforzo delle lastre a spessore sottile, ha un modulo elastico paragonabile a quello del grès porcellanato e non può quindi incrementarne la resistenza meccanica.
L’unico beneficio certo dell’applicazione di un rinforzo posteriore in fibra di vetro su piastrelle e lastre ceramiche è l’aumento di resistenza ai carichi impulsivi (urti, sollevamento, ecc.) e la funzione di sicurezza (anticaduta) assicurata dalla rete.
Per tale motivo, nel caso di applicazioni particolari (facciate ventilate, arredamento, ecc.) dove le lastre vengono supportate in pochi punti e non incollate sull’intera superficie posteriore, l’utilizzo di reti e stuoie rinforzanti diventa indispensabile. Per i sistemi e le linee di applicazione di tali stuoie si veda il Paragrafo 7.5 del Volume II.
Tra le proprietà finali richieste ad un supporto ceramico, sia esso smaltato o meno, si possono considerare di primaria importanza la resistenza meccanica e le caratteristiche geometriche (calibro e planarità). Queste proprietà devono presentare valori minimi e tolleranze in funzione della tipologia di prodotto che si intende realizzare, ma esse sole non sono sufficienti per definire le specifiche necessarie alla formulazione di un buon impasto. Vi sono, infatti, tanti altri vincoli da rispettare, quali: la sufficiente plasticità per la formatura e la lavorazione dei pezzi crudi, la essiccabilità a cicli rapidi, l’idoneità al processo di cottura, ecc. Qualora le caratteristiche dell’impasto non soddisfino queste necessità, il prodotto finito potrebbe presentare svariati difetti, come laminazioni e crepe, incompatibilità con la superficie smaltata, cuore nero, post-espansione, ecc.
Per quanto riguarda specificatamente la resistenza meccanica, come esposto nei paragrafi precedenti, le norme ne fissano i valori minimi da rispettare (Fig. 7.2). Più precisamente, per ciascuna tipologia di prodotto, la normativa ISO definisce i limiti per il carico di rottura (MOR, espresso in N/mm2, in ordinata nel grafico di Fig. 7.2) e anche per lo sforzo di rottura (valore normalizzato espresso in N, riportato all’interno delle barre di Fig. 7.2), in modo da assicurare la resistenza meccanica del prodotto indipendentemente da spessore e formato. Infatti, mentre il modulo di rottura è una grandezza propria di ogni materiale (indipendente dalle dimensioni del campione), il carico effettivo di rottura indica il comportamento in esercizio del pezzo specifico e dipende, oltre che dal materiale, anche da dimensioni e spessore della piastrella.
Fig. 7.2 Requisiti di resistenza meccanica secondo le norme ISO
Fig. 7-02 IT
Per estremizzare, un prodotto sottile greificato presenta un MOR molto elevato (conseguenza della qualità del materiale), ma il suo sforzo di rottura potrebbe essere inferiore a quello di una piastrella da rivestimento a spessore normale. Possiamo ad esempio analizzare il comportamento di una piastrella cruda, realizzata con lo stesso spessore in formato piccolo (es. 30 × 30 cm) e molto grande (es. 120 × 120 cm). In tal caso il modulo MOR del
materiale è lo stesso (stessa composizione, pressione di compattazione ecc.), ma la forza effettivamente misurata nella prova di rottura a flessione, e quindi la resistenza alle sollecitazioni sulla linea di produzione, risulterà inferiore per il formato maggiore (e infatti i sistemi di trasporto per grandi formati devono garantire attenzioni e accuratezze superiori).
Pertanto, i requisiti di resistenza meccanica che le normative impongono ai prodotti finiti fissano valori minimi di MOR da 15 N/mm2 (rivestimento - Gruppo BIII) a 35 N/mm2 (grès porcellanato - Gruppo BIa), mentre i valori di sforzo di rottura, normalizzato rispetto alle dimensioni, vanno rispettivamente da 600 a 1300 N.
Soddisfare questi requisiti di resistenza meccanica è, peraltro, abbastanza facile tanto che uno stesso impasto potrebbe essere utilizzato per più di un gruppo di prodotti; ma così non è se si considera l’insieme delle proprietà fondamentali del materiale ceramico.
In primo luogo, occorre considerare il comportamento dell’impasto alla greificazione: le curve di greificazione (espressione di assorbimento di acqua, ritiro e carico di rottura in funzione della temperatura massima di cottura) relative alle diverse tipologie di prodotti (rivestimento in pasta rossa e bianca, pavimento rosso e chiaro, grès porcellanato) mostrano quanto marcate siano le differenze.
Per quanto riguarda l’assorbimento d’acqua (si veda Fig. 7.3), gli impasti da rivestimento presentano elevate porosità in un intervallo piuttosto ampio alle basse temperature, per poi greificare troppo rapidamente per poter essere utilizzati come pavimento. Un impasto destinato alla greificazione totale (AA = 0%), come il grès porcellanato, presenta, invece, un intervallo di stabilità molto ampio alle basse porosità, a differenza dei più tradizionali impasti da pavimento smaltato (AA = 3 ÷ 6%).
Questi comportamenti non solo devono essere conosciuti per ottimizzare i processi di lavorazione, ma possono essere spiegati, predetti e modificati in funzione delle caratteristiche chimico-fisiche delle materie prime e delle formulazioni di impasto.
Assorbimento
Grès porcellanato
Pavimento (rosso)
Rivestimento (rosso)
Rivestimento (bianco)
Pavimento (bianco)
Fig. 7.3 Curve di greificazione: assorbimento acqua
Fig. 7-03 IT NEW
Anche le curve di ritiro mostrano la grande diversità di comportamento che sussiste tra i vari impasti (vedi Fig. 7.4). A riguardo, si riscontra generalmente una maggiore stabilita degli impasti bianchi rispetto alle paste rosse, più argillose e composizionalmente meno bilanciate
nei loro componenti, sia nei prodotti greificabili che in quelli porosi. Per quanto concerne i materiali a supporto poroso, utilizzati per rivestimento, va in particolare sottolineato il ritiro contenuto (< 1%) alle basse temperature di cottura, prerogativa necessaria per garantire la massima uniformità di calibro, trattandosi di piastrelle normalmente non rettificate meccanicamente.
Ritiro [%]
Pavimento (rosso)
Pavimento (bianco)
Grès porcellanato
(rosso)
(bianco)
7.4 Curve di greificazione: ritiro
Fig. 7-04 IT NEW
La resistenza meccanica (in Fig. 7.5) è in buona parte legata ai comportamenti manifestati in sinterizzazione. Di conseguenza si possono osservare valori più elevati per gli impasti rossi, che hanno maggiore tendenza alla greificazione, ad eccezione del grès porcellanato, la cui composizione è appositamente studiata per sviluppare le fasi vetrose.
Grès porcellanato
Pavimento (rosso)
Rivestimento (rosso)
Pavimento (bianco)
Rivestimento (bianco)
Fig. 7.5 Curve di greificazione: carico di rottura MOR
Fig. 7-05 IT
Assorbimento d’acqua, ritiro e carico di rottura rappresentano i fattori macroscopici che devono essere sistematicamente verificati e controllati durante il processo produttivo. A riguardo, un’analisi più approfondita delle modificazioni strutturali che avvengono durante la cottura rende ancor più evidenti le differenze tra impasti porosi e greificati
Nei prodotti porosi (Fig. 7.6) si osserva come gli impasti da rivestimento siano caratterizzati dalla presenza di carbonati di calcio e/o magnesio, che si decompongono al di sopra degli 800 °C, liberando gli ossidi corrispondenti.
Dalle successive reazioni di questi ossidi con la restante matrice ceramica si formano, a temperature relativamente basse (tra 1000 e 1140 °C), nuovi composti cristallini, silico-alluminati di Ca e Mg, che garantiscono una discreta stabilità di cottura e lo sviluppo di una sufficiente resistenza meccanica, prima di dar luogo ad una fusione incontrollabile (oltre 1140 °C).
Assorbimento Acqua
AssorbimentoRitiro
Decomposizione dei carbonati
Presenza di CaO libero
Formazione di fasi cristalline (gehlenite, diopside, wollastonite, anortite)
Formazione incontrollata di fase vetrosa
Degasazioni attraverso lo smalto
Tendenza alla post-espansione
Aumento della resistenza meccanica
Stabilità dimensionale
Aumento eccessivo del ritiro
Deformazione indesiderata
Fig. 7.6 Comportamento in cottura degli impasti porosi rossi
Fig. 7-06 IT NEW
Le numerose specie cristalline che possono essere sviluppate da un sistema CaO, Al2O3, SiO2 sono riportate nel diagramma ternario di Fig. 7.7. In particolare si evidenzia la formazione di gehlenite, pseudowollastonite ed anortite che, in associazione tra loro, possono determinare abbassamenti della temperatura di fusione favorendo, attraverso la formazione di fasi liquide, ulteriori reazioni tra gli ossidi presenti.
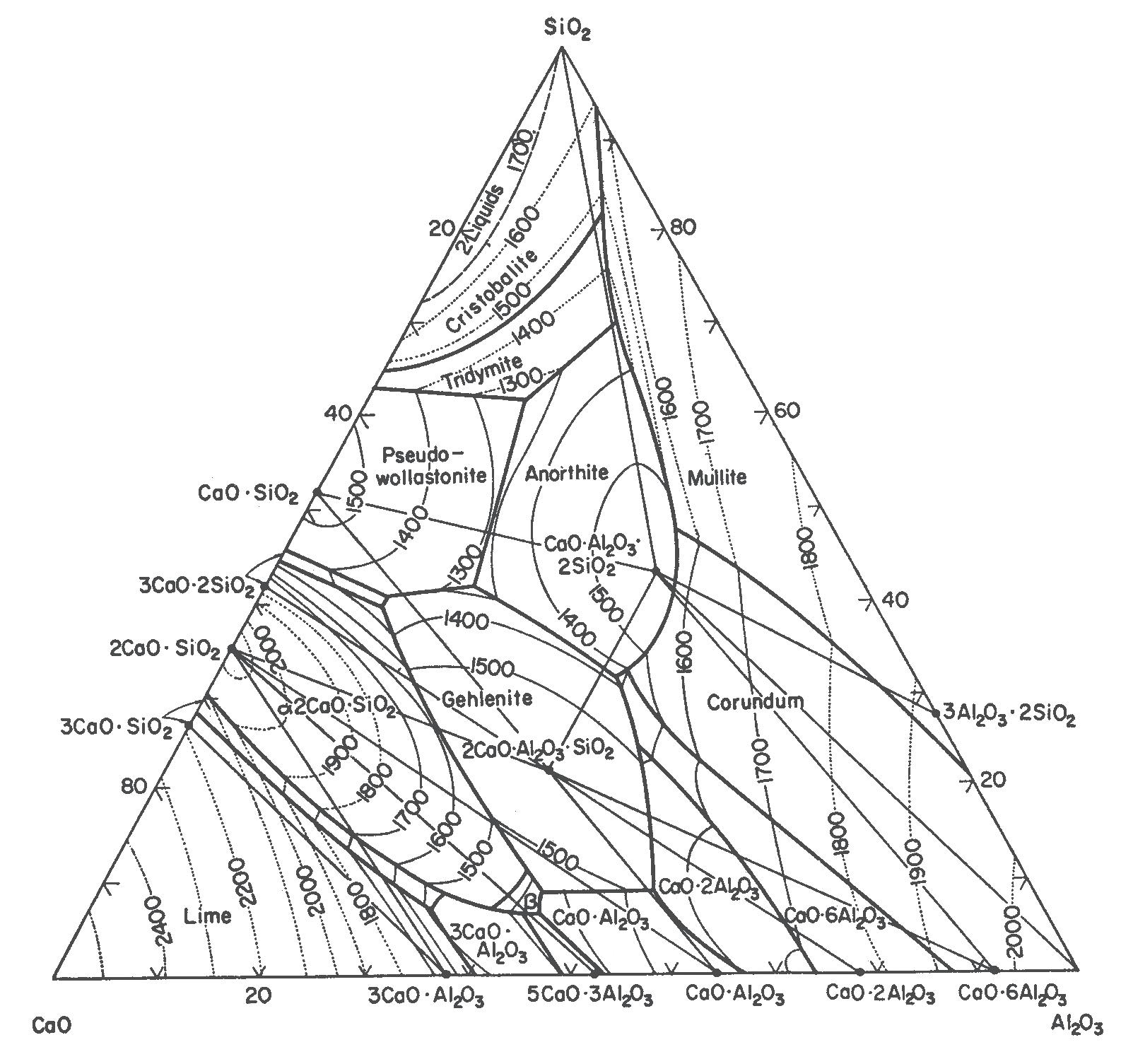
Fig. 7.7 Diagramma di fase per silico-alluminati di calcio
Composto
Quarzo SiO2 170 1713
Albite
Na2O·Al2O3·6SiO2 78 1118
Ortoclasio K2O·Al2O3·6SiO2 50 1170 (*)
Anortite CaO·Al2O3·2SiO2 50 1553
Gehlenite 2CaO·Al2O3·SiO2 81 1593
p-Wollastonite CaO·SiO2 70 1544
Diopside CaO·MgO·2SiO2 75 1392
Fosterite 2MgO·SiO2 94 1900
Spinello MgO·Al2O3 85 2135
Mullite 3Al2O3·2SiO2 48 1880 (*) Fusione incongruente
Tab. 7.9 Valori di α e Tfusione per composti presenti dopo cottura
La Tab. 7.9 riporta alcune proprietà dei composti cristallini già presenti nelle composizioni di partenza e di altri, cosiddetti di neoformazione, che si generano nel processo di cottura e contribuiscono a determinare le proprietà strutturali finali dei prodotti porosi. Si notino i
coefficienti di dilatazione, tendenzialmente elevati, dei nuovi composti contenenti calcio e magnesio che permettono di contenere il ritiro dei pezzi, congiuntamente all’aumento delle proprietà meccaniche dovuto ai nuovi cristalli formatisi.
Fig. 7.8 Curva dilatometrica di impasto poroso
La Fig. 7.8 mostra, infatti, come la stabilità dimensionale nella zona di cottura (intervallo 1000 ÷ 1100 °C) permetta dopo raffreddamento di ottenere un ritiro finale molto contenuto (inferiore a 0,5%).
Fig. 7-08 IT
Le temperature teoriche di fusione dei composti puri sono comunque molto elevate, e la loro formazione da reazioni solido-solido o solido-liquido è certamente difficoltosa; pertanto, le percentuali che si rilevano nei prodotti cotti, particolarmente a ciclo rapido, sono generalmente basse. Ciò è particolarmente vero per la mullite, che difficilmente ha modo di formarsi nei prodotti da rivestimento, in considerazione delle basse temperature di cottura. I diffrattogrammi a raggi X sulle polveri di un impasto poroso cotto, come nell’esempio di Fig. 7.9, mostrano dunque ancora la presenza dei minerali cristallini originari, soprattutto quarzo (Q) e feldspato (F), oltre ad una moderata formazione di nuove fasi (N).
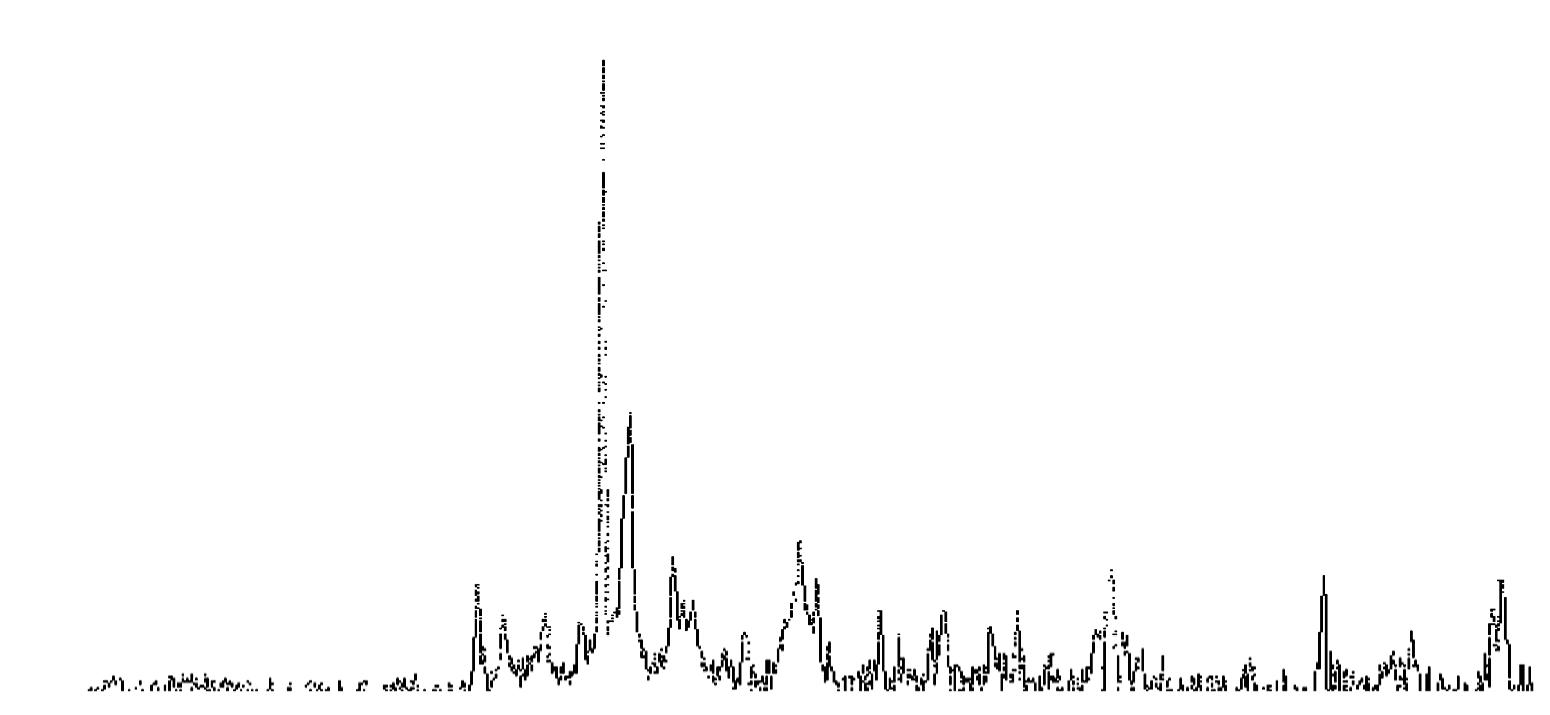
Fig. 7.9 Diffrattogramma RX di un supporto poroso
Cicli di cottura troppo rapidi, specie se associati a formulazioni non correttamente bilanciate nei componenti e nelle loro granulometrie, non danno tempo sufficiente per lo sviluppo di fasi cristalline stabili. Il rapido raffreddamento dopo le zone di massima temperatura determina un "congelamento" della struttura in fasi metastabili, la cui permanenza nel tempo è comunque ben superiore al ciclo di vita di un prodotto ceramico.
Fig. 7-09
Viceversa, se si considerano i meccanismi di formazione dei prodotti greificati, basati sulla azione fondente dei composti di sodio e potassio, si nota che, rispetto ai rivestimenti (costituiti da sistemi calcio-magnesio) le temperature di reazione si spostano a livelli termici più elevati (Fig. 7.10). Di conseguenza, le caratteristiche meccaniche a bassa temperatura dei prodotti greificabili sono generalmente più scadenti di quelle dei prodotti porosi per rivestimento. Inoltre, i composti intermedi di neoformazione che iniziano a svilupparsi in una matrice non ancora sufficientemente legata, fanno sì che impasti greificabili sotto-cotti presentino una maggior tendenza ad espandere (nel tempo) con l’umidità rispetto ai prodotti da rivestimento. È quindi sconsigliabile utilizzare come rivestimento impasti greificabili cotti a bassa temperatura, anche qualora non vi siano particolari esigenze di controllo del ritiro, ovvero di calibro del prodotto finale.
di cottura
Assorbimento Acqua
Trasformazioni di fase metacaolino-spinello
Parziale distruzione cristalli (albite, ortoclasio, quarzo)
Formazione di fase vetrosa
Formazione di nuovi cristalli (mullite)
Aumento della resistenza meccanica
Aumento di ingelività, impermeabilità, ritiro, deformazione
Aumento della resistenza meccanica
Fig. 7.10 Comportamento in cottura degli impasti totalmente greificabili
Fig. 7-10 IT
Il diagramma di fase Na2O, K2O, Al2O3, SiO2 (Fig. 7.11), riferito alle combinazioni tra silico-alluminati di sodio e potassio con la silice, mostra come in determinate regioni composizionali sia possibile lo sviluppo di fasi liquide a temperature decisamente basse: 1118 °C per il feldspato sodico (albite) e 1170 °C per il feldspato potassico (ortoclasio), che presenta una fusione incongruente, separando leucite (KAlSi2O6). Ovviamente i diagrammi di fase si riferiscono a sistemi in equilibrio termodinamico e non danno informazioni sulle cinetiche di reazione, né quindi sul tempo necessario alla greificazione degli impasti che, particolarmente nei cicli rapidi di cottura, devono avvenire nel breve intervallo di permanenza alle massime temperature.
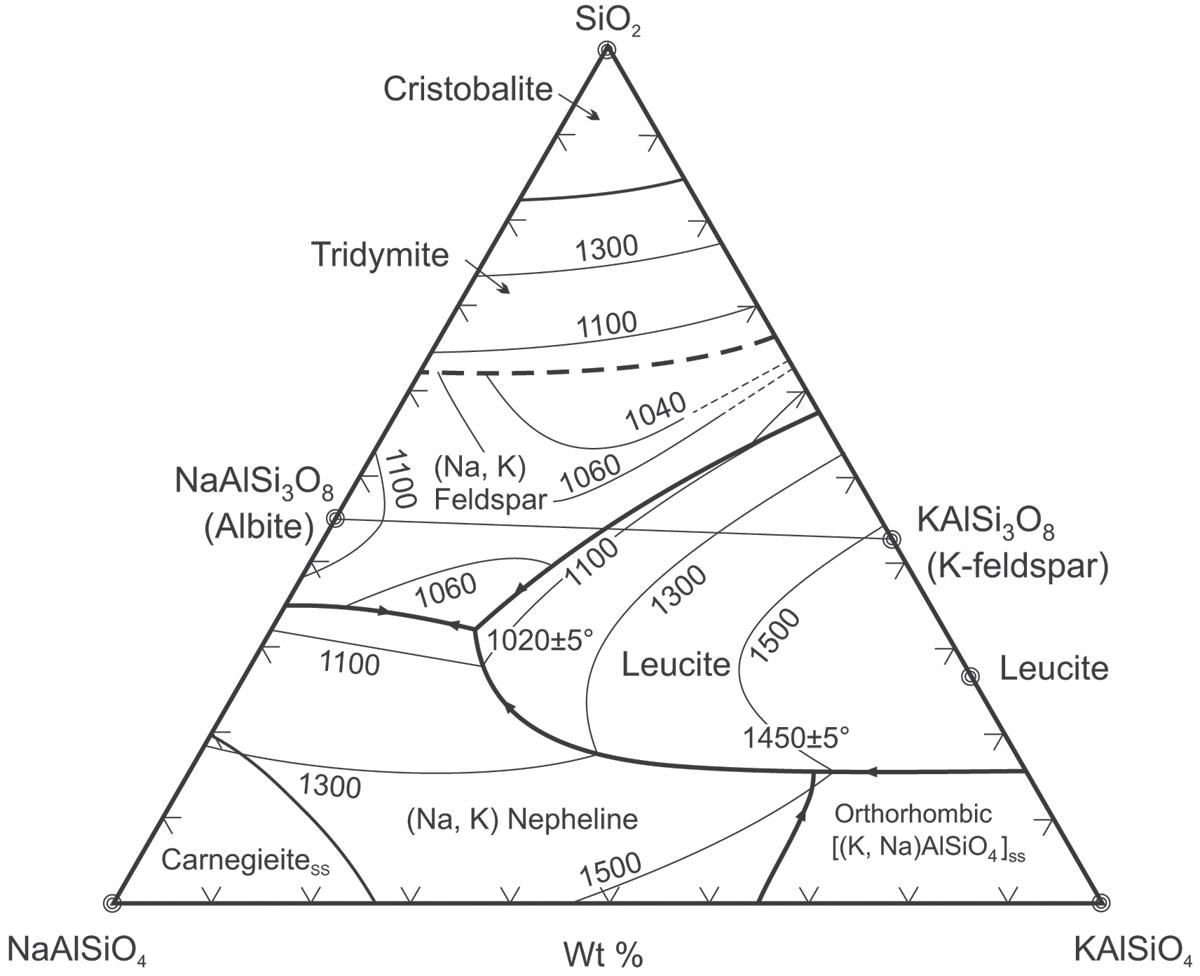
Fig. 7.11 Diagramma di fase per i feldspati sodico-potassici
Inoltre, si deve tenere presente che la velocità di reazione di composti allo stato solido è fortemente dipendente dalla superficie specifica disponibile, e quindi dalla loro granulometria. Ad esempio, una quantità di materiale a densità 2,5 g/cm3 presenta una superficie di reazione di 24 cm2/g, se le particelle hanno un diametro medio di 1 mm, mentre, riducendo le dimensioni delle particelle a 50 µm, la sua superficie di reazione diventa 480 cm2/g. In ogni caso, difficilmente si riuscirà a completare la reazione di una particella solida cristallina all’interno di un impasto ceramico, ma si manifesteranno prevalentemente reazioni di “cementazione” tra i singoli grani, favorite tra l’altro dalla presenza di imperfezioni dei reticoli cristallini e di impurità che, anche in piccola quantità, possono fungere da catalizzatori o dare origine ad eutettici basso fondenti.
La curva di dilatazione di un impasto greificabile crudo (Fig. 7.12) evidenzia un processo di vetrificazione dapprima graduale poi repentino: la temperatura ottimale di cottura è ben individuabile attraverso la derivata (in arancio) della curva di dilatazione, che presenta un picco minimo in corrispondenza della massima velocità di ritiro (valori di dilatazione negativi).
A questa temperatura il tempo necessario alla vetrificazione sarà minimo, mentre temperature superiori rappresentano uno spreco di energia e possono dar luogo a strutture meno dense, per l’incipiente espansione.
Fig. 7.12 Curva dilatometrica di un supporto crudo greificabile
Associando le curve di dilatazione termica in riscaldamento ed in raffreddamento di un supporto greificabile (Fig. 7.13), risulta ben evidente la sua differenza di comportamento rispetto all’impasto poroso di Fig. 7.8.
Fig. 7-12
Fig. 7.13 Curva dilatometrica di impasto greificabile
Fig. 7-13 IT Intervallo di cottura
In questo caso la cottura avviene in un tratto già in forte contrazione, determinando così anche un elevato ritiro finale del materiale in uscita forno. Inoltre, piccole variazioni del livello termico di cottura possono facilmente generare calibri diversi, ovvero piastrelle con dimensioni variabili nell’intorno di un valore medio di riferimento. In particolare, nel grès porcellanato questa problematica è stata superata tramite lavorazioni meccaniche di squadratura e rettifica, ottenendo così piastrelle e lastre con una geometria perfettamente definita, idonee ad essere posate con fughe distanziatrici minime.
In conclusione, sulla base delle richieste di mercato, è possibile definire le migliori condizioni produttive per garantire le proprietà ottimali per le differenti tipologie di piastrelle ceramiche.
Attualmente le principali classi di prodotto sono:
- rivestimento
- pavimento
- grès porcellanato
- lastre di grande formato.
I prossimi capitoli sono dedicati alla descrizione dettagliata di ciascuna tipologia.
[1] ISO, International Organization for Standardization, «ISO 13006 Ceramic tiles – Definitions, classification, characteristics and marking», 2018.
[2] ISO, International Organization for Standardization, «ISO 10545 Ceramic tiles – Part 1-20», 2014-2022.
[3] ISO, International Organization for Standardization, «ISO 17889-1 Ceramic tiling systems – Sustainability for ceramic tiles and installation materials», 2021.
[4] L. Cini, Guida all’interpretazione dei diagrammi di stato in fase condensata, Faenza Ed., 1973.
Il modulo di rottura, anche detto carico di rottura, è la principale caratteristica di un materiale e indica la sua capacità di resistere alle sollecitazioni meccaniche. Maggiore il suo valore, maggiore la resistenza del materiale. Non dipende dalle dimensioni e dallo spessore della piastrella, ma solo dalle caratteristiche fisiche del materiale. Ha le dimensioni di una forza (N) diviso una superficie (mm2), come la pressione. Per le piastrelle ceramiche, secondo ISO 10545-4, si misura mediante prova di flessione a tre punti.
Il carico di rottura, anche chiamato MOR dall’inglese Modulus of Rupture, è calcolato con la relazione
dove: F forza di rottura [N]
L distanza dei rulli di supporto [mm]
b larghezza della piastrella [mm]
h spessore minimo della piastrella [mm]
In passato il carico MOR veniva espresso in kgforza/cm2, ma tale unità di misura è stata sostituita da N/mm2 (equivalente a MPa). L’equivalenza tra le due unità vale circa
Per es. un carico MOR di 500 kg/cm2 corrisponde approssimativamente a 50 N/mm2.
Lo sforzo di rottura è la forza (misurata in N) a cui si rompe il campione durante la prova di flessione, opportunamente corretta per tenere conto della geometria della prova attraverso il fattore di forma L/b. Lo sforzo di rottura vale pertanto
dove: F forza di rottura [N]
L distanza dei rulli di supporto [mm]
b larghezza della piastrella [mm]
Lo sforzo di rottura corrisponde numericamente alla forza di rottura (letta sulla macchina di prova) quando il campione è quadrato (L = b).
A differenza del carico MOR, lo sforzo S è fortemente influenzato da dimensioni e spessore del campione.
Importante parametro fisico, che fornisce una stima della porosità aperta del corpo ceramico in esame. Viene normalmente misurato per differenza di massa tra il campione dopo immersione in acqua e il campione prima dell’immersione (l’impregnazione con acqua viene effettuata sotto vuoto). L’assorbimento di acqua viene espresso in % ed è calcolato mediante la relazione
���������������� = ��������2 ��������1 ��������1 × 100 [%]
dove: m1 massa del campione asciutto (cotto) [kg] m2 massa del campione umido [kg]
Per un solido, indica la capacità di resistere ad una sollecitazione di flessione. Per una piastrella avente sezione rettangolare, il modulo vale ���������������� = ��������ℎ2 6 [mm3 ]
dove: b larghezza della piastrella [mm] h spessore della piastrella [mm]
Dalla definizione discende che la resistenza alla flessione è direttamente proporzionale alla larghezza ed al quadrato dello spessore. Quindi, per esempio, un raddoppio dello spessore comporta un aumento della resistenza a flessione del quadruplo.
Le fasi storiche di sviluppo dell’industria ceramica in Italia sono state determinanti nel favorire la continua evoluzione dei processi e dei materiali ceramici da rivestimento. Particolarmente significativo è stato il passaggio dalla bicottura tradizionale alla bicottura e monocottura rapide, con la realizzazione di piastrelle a dimensioni sempre maggiori caratterizzate da un più elevato contenuto estetico e decorativo.
Questa evoluzione è avvenuta soprattutto durante l’ultimo ventennio del secolo scorso, come naturale prosieguo delle esperienze maturate nel settore della monocottura da pavimento. Agli inizi degli anni ‘80 infatti, in concomitanza con la prima crisi energetica mondiale, si è sviluppato e adottato il processo della monocottura anche per i materiali ceramici porosi.
L’uso delle tecnologie di cottura mono- e bi-rapida si è quindi diffuso nel mondo e la scelta tra i due processi è stata determinata da diversi fattori, quali: il livello tecnico-professionale degli operatori, il costo dell’energia e della manodopera, le materie prime disponibili, le esigenze merceologiche e di qualità, gli aspetti culturali caratteristici di ciascuna specifica area geografica [1].
La Fig. 8.1 mostra la distribuzione complessiva della produzione ceramica mondiale suddivisa tra le categorie: rivestimento, pavimento in monocottura e grès porcellanato (principalmente da pavimento ma che, in funzione della tipologia, può essere impiegato anche nel rivestimento). Come si può notare, i prodotti da rivestimento porosi (in mono e bicottura) rappresentano circa un terzo del totale (pari a circa 5 ÷ 6 miliardi m2 annui).
Rivestimento
Pavimento (monocottura)
Grès porcellanato
Fig. 8.1 Produzione mondiale in funzione delle tipologie produttive (stime 2019)
La Fig. 8.2 mostra invece come le diverse aree produttive del mondo presentino quote differenti tra pavimento e rivestimento; questo in relazione ad aspetti logistici (presenza di materie prime domestiche idonee al rivestimento) e culturali. Rimane comunque ben evidente la predominanza del pavimento sul rivestimento.
Fig. 8.01 IT
Fig. 8.2 Suddivisione tra piastrelle per pavimento e rivestimento nelle principali zone produttive mondiali (stima 2019)
Fig. 8.02 IT
Infine, la Fig. 8.3 mostra una ulteriore segmentazione delle piastrelle da rivestimento prodotte nella zona geografica / paese, in base alla suddivisione tra pasta rossa e pasta bianca. La tendenza generale è a favore della pasta rossa, ad esclusione della Cina ed in misura minore della stessa Europa. Anche in questo caso, la preferenza tra pasta rossa e bianca deriva dalle materie prime disponibili, oltre che da aspetti culturali e di mercato.
Fig. 8.3 Suddivisione delle piastrelle da rivestimento (pasta bianca o rossa) per le principali zone produttive mondiali (stima 2019)
8.03 IT
All’interno della classificazione generale delle “piastrelle da rivestimento” sono compresi prodotti ottenuti sia con il processo di bicottura che di monocottura.
Secondo la norma internazionale ISO 13006 [2] le piastrelle ceramiche vengono classificate in vari gruppi a seconda del metodo di formatura e del valore di assorbimento d’acqua del prodotto finito (vedi Tab. 8.1).
Le piastrelle da rivestimento poroso, avendo un assorbimento d’acqua superiore al 10%, sono comprese nel gruppo BIII.
Metodo di formatura
Tipologia di prodotto
Grès porcellanato
Monocottura greificata
Pavimento semi-greificato
Pavimento poroso
Rivestimento poroso
Tab. 8.1 Classificazione ISO 13006 delle piastrelle pressate a secco [2]
In generale, i materiali da rivestimento presentano le seguenti caratteristiche:
- massima stabilità dimensionale in cottura, con valori di contrazione praticamente nulli (< 1%);
- porosità compresa tra il 12% ed il 18% (espressa come assorbimento d’acqua);
- valori del carico di rottura in cotto compresi tra 20 ÷ 25 N/mm2
Le proprietà sopra riportate sono da ritenersi indicative ma sono comunque utili per inquadrare il prodotto sotto il profilo merceologico, in relazione alla sua destinazione d’uso.
Le specifiche contemplate dalla norma ISO 10545 [3] per il gruppo BIII riguardano in particolare le caratteristiche dimensionali, fisiche e chimiche dei prodotti.
Nella Tab. 8.2 viene riportato un quadro riassuntivo riguardo a questi ultimi due aspetti prescindendo dalle caratteristiche dimensionali. Nella stessa tabella sono indicati i valori prescritti dalle norme e, per confronto, le caratteristiche medie dei prodotti commercializzati.
Caratteristiche Norma Valori prescritti dalla norma Valori reali di mercato
Assorbimento d’acqua
di rottura MOR
Sforzo di rottura
Resistenza abrasione
Coefficiente dilatazione
Resistenza sbalzi termici
Espansione all’umidità
Resistenza cavillo
Resistenza al gelo
Resistenza ai prodotti chimici
Resistenza agli acidi e alle basi
Resistenza macchie
Estrazione Pb e Cd dai vetri
Differenza di colore
10545-3
ISO 10545-4 > 600 N (spess. ≥7.5 mm) > 200 N (spess. <7.5 mm) > 700 N (spess. ≥7.5 mm) > 300 N (spess. <7.5 mm)
ISO 10545-6/-7 dichiarata dal produttore dichiarata dal produttore
ISO 10545-8 test disponibile 6,5 ÷ 7,5 × 10-6
ISO 10545-9 test disponibile conforme
ISO 10545-10 test disponibile < 0,06 %
ISO 10545-11 richiesta conforme
ISO 10545-12 test disponibile dichiarata dal produttore
ISO 10545-13 classe GB min classe GB min
ISO 10545-13 test disponibile dichiarata dal produttore
ISO 10545-14 classe 3 min classe 3 min
ISO 10545-15 test disponibile dichiarata dal produttore
ISO 10545-16 test disponibile dichiarata dal produttore
Tab. 8.2 Caratteristiche tecniche dei rivestimenti porosi secondo la normativa ISO 13006 [2] rispetto ai valori di mercato
Nell’evoluzione degli smalti progressivamente adeguati alle diverse tecnologie di cottura (tradizionale, bi-rapida, monoporosa), le caratteristiche tecniche di resistenza alla macchia e agli agenti chimici sono rimaste pressoché inalterate. Relativamente agli smalti bianchi ed alle cristalline, i vincoli e le possibilità offerte dalle nuove tecnologie di cottura rapida hanno richiesto diverse formulazioni di fritte e smalti, passando da prodotti “basso fondenti-viscosi” a composti “alto-fondenti” con fusione eutettica. Quest’evoluzione composizionale ha interessato sia gli smalti della bicottura rapida che, in particolare, della monoporosa [4].
Formati
In passato, i formati prodotti con il processo di cottura tradizionale erano principalmente il 15×15 ed il 20×20 cm. Con l’impiego della bicottura rapida ed anche della monocottura porosa le dimensioni si sono assestate su formati più grandi e rettangolari, quali 25×33, 33×45, 40×80 cm ed oltre.
I formati più diffusi risultano comunque quelli medi (quali: 20×30, 25×40, 30×60 cm), facilmente realizzabili nei forni monostrato e ben gestibili tramite i sistemi di automazione e movimentazione adottati nei nuovi impianti di cottura rapida.
Tuttora limitata risulta invece la produzione di grandi formati porosi che implica indubbiamente maggiori difficoltà tecniche, soprattutto per quanto riguarda il controllo della planarità.
Pezzi speciali
Sono prodotti che presentano rilievi realizzati principalmente in pressatura (listelli, inserti, torelli, ecc.), fabbricati a piccoli lotti in linee dedicate con presse di limitata potenza e stampi speciali. Nel caso di forme complicate o con variazioni di spessore molto pronunciate, rispetto alla monoporosa si preferisce la tecnologia della bicottura.
Aspetto superficiale
La caratteristica superficiale tipica delle piastrelle da rivestimento nella maggioranza dei casi è sempre stata quella della brillantezza. Conseguentemente, l’aspetto degli smalti è prevalentemente lucido, trasparente (con solo cristallina) oppure opaco (bianco), mentre risultano meno diffuse le superfici matt.
Tra queste rientrano le tipologie definite “anticate”, con effetti gessosi simili ad intonaci invecchiati, messi in risalto attraverso le tecniche di strutturazione delle superfici.
Tuttavia, la percentuale più elevata della produzione di piastrelle da rivestimento è costituita da superfici smaltate lucide con decori ispirati alle pietre naturali, soprattutto marmi.
Caratteristiche generali
Le composizioni degli impasti per piastrelle da rivestimento hanno subito un’evoluzione nel tempo adattandosi fondamentalmente ai cicli delle cotture rapide. Particolare attenzione è stata rivolta nell’adeguare le caratteristiche del supporto per far fronte alle problematiche derivanti dalla cottura rapida, soprattutto se simultanea di supporto e smalto.
Nella monoporosa, infatti, si possono facilmente sviluppare interferenze tra la degasazione di alcune materie prime del supporto ed il vetro in fusione, con la conseguente formazione di difetti superficiali nello smalto stesso. Pertanto l’evoluzione delle composizioni degli impasti, da quelle utilizzate nella cottura tradizionale, poi adattate alla bicottura rapida ed infine alla monocottura porosa, ha comportato le seguenti variazioni:
- diminuzione della percentuale di minerali argillosi;
- introduzione di percentuali più elevate di materie prime inerti e complementari (feldspati, sabbie feldspatiche, quarzo);
- contenimento dei minerali che sviluppano in cottura fasi gassose ad alta temperatura (calcite e/o dolomite), particolarmente nel caso della monocottura porosa.
Perseguendo questi obiettivi è stato comunque possibile utilizzare materie prime già note, variando unicamente il loro intervallo composizionale.
Nel caso della monoporosa è inoltre preferibile l’impiego di materie prime caratterizzate da elevata purezza e granulometria tendenzialmente fine, in modo da ridurre l’insorgenza di eventuali difetti provocati da componenti reattivi o inquinanti.
Per quanto concerne i prodotti finiti, di norma le piastrelle da rivestimento devono possedere una elevata stabilità dimensionale. A riguardo, la formazione di nuovi composti cristallini, quali wollastonite, gehlenite, anortite e diopside, oltre ad assicurare stabilità dimensionale, condizionano e stabilizzano anche altre caratteristiche come il valore di post-espansione all’umidità del supporto poroso, il coefficiente di dilatazione e la resistenza meccanica.
Caratteristiche delle materie prime per impasto
Sulla base di una sommaria classificazione commerciale, gli impasti idonei a produrre piastrelle da rivestimento possono essere distinti in “rossi” e “bianchi”. In entrambi i casi le materie prime usate sono costituite da due tipologie fondamentali, quali: - materiali argillosi; - materiali complementari (feldspati, sabbie feldspatiche, quarzo, calciti).
Nelle Tab. 8.3 e Tab. 8.4 sono riportate le composizioni chimiche e le caratteristiche fisiche delle materie prime usate più frequentemente nelle formulazioni di impasti sia per bicottura rapida che per monoporosa.
Argilla semiplastica
Argilla
Argilla caolinitica
Sabbia feldspatica
Carbonato di calcio
Tab. 8.3 Composizione delle materie prime per prodotti da rivestimento (monocottura porosa per interni)
Caratteristiche
Argilla (A) Argilla (B) Argilla (C) Argilla (D)
Carico di rottura (in crudo) N/mm2 0,6÷0,8 0,8÷1,0 1,0÷1,5 0,4÷0,6
Carico di rottura (in essiccato) N/mm2
Dopo cottura a 1100 °C
Porosità % 8÷12 2÷6 5÷10 15÷20
Ritiro % 2÷3 4÷6 3÷5 2÷4
Carico di rottura (in cotto) N/mm2 18,0÷20,0 20,0÷30,0 20,0÷30,0 8,0÷12,0
Tab. 8.4 Caratteristiche fisiche delle argille di Tab. 8.3
Di seguito vengono riportate alcune note in merito alle caratteristiche generali e mineralogiche dei materiali utilizzati.
Materie prime plastiche
Argille carbonatiche-marnose: le associazioni mineralogiche presenti possono essere illitico-cloritiche ed eventualmente illitico-caolinitiche (quest’ultima in quantità inferiore). La quantità di calcite presente nella matrice argillosa può essere variabile e raggiungere valori anche molto elevati. Queste argille contribuiscono in parte a conferire plasticità all’impasto. Generalmente dopo cottura risultano di un colore beige-arancio per la presenza di minerali ferrosi.
Argille plastiche greificabili cuocenti rosso: sono caratterizzate dall’assenza quasi totale di carbonati. I minerali argillosi possono essere di natura illitico-cloritica. Hanno la funzione di conferire plasticità all’impasto, assicurando l’ottenimento di valori elevati del carico di rottura in verde e secco ed anche dopo cottura in quanto contribuiscono alla greificazione.
Argille ball clay: cuocenti generalmente bianco. La matrice argillosa risulta di tipo caolinitico, con poca illite. Il tessuto granulometrico tendenzialmente è fine e pertanto si possono prevedere comportamenti plastici significativi. Dopo cottura questi materiali evidenziano caratteristiche di resistenza meccanica e porosità idonee per tutte le tipologie di prodotti ceramici.
Argille caolinitiche (china clay): il comportamento è generalmente refrattario; la modesta plasticità dipende essenzialmente dalla granulometria intrinseca del materiale. Il loro utilizzo va limitato a percentuali non troppo elevate.
Risulta evidente come l’impiego delle argille carbonatiche e/o greificabili sia finalizzato all’ottenimento di un supporto color beige-arancio, mentre l’utilizzo delle ball clay e china clay permette di ottenere supporti bianchi.
Materie prime complementari
Sabbie feldspatiche, feldspati e quarzo: vengono introdotti nella composizione come inerti e/o smagranti per facilitare la degasazione dei composti volatili che si sviluppano durante la cottura. Nel caso di utilizzo di feldspati è preferito il tipo potassico, perché meno reattivo del sodico. L’utilizzo di feldspati contribuisce anche all’abbassamento del coefficiente di dilatazione complessivo della massa ceramica. Anche il quarzo risulta un materiale fondamentale per regolare il coefficiente di dilatazione, aumentandolo proporzionalmente alla quantità presente. È inoltre importante sottolineare come la reattività del quarzo libero verso gli ossidi alcalino-terrosi (CaO e MgO) sia modesta, in considerazione dello stato granulometrico piuttosto grossolano e del ciclo rapido di cottura.
Calcite e dolomite: sono materiali fondamentali negli impasti da rivestimento. Le percentuali presenti possono essere variabili da 8% a 12%. Particolare importante risulta la loro granulometria naturale nonché lo stato granulometrico dopo macinazione. Infatti, granulometrie molto fini favoriscono sia le reazioni di decarbonatazione che le successive reazioni con i composti residui dei materiali argillosi alterati termicamente, in particolare la silice amorfa e l’allumina, generando composti cristallini di neoformazione a temperature superiori a 900 °C. Di estrema importanza risulta la cinetica della decarbonatazione e, quindi, della completa espulsione della fase gassosa (CO2), prima del rammollimento del vetro di superficie (smalto). Il completarsi delle reazioni di sintesi tra gli ossidi alcalino-terrosi (di calcio e magnesio), silice ed allumina, assume indubbiamente un ruolo importante nel definire le caratteristiche fisico-meccaniche del pezzo ceramico dopo cottura (resistenza meccanica, coefficiente di dilatazione ecc.). È evidente altresì come i rapporti quantitativi ottimali fra materiali argillosi, calcite, feldspati, quarzo siano dipendenti dalla natura mineralogica e dalla granulometrica intrinseca delle argille.
Chamotte: polvere ottenuta dalla macinazione di prodotti ceramici cotti (solitamente scarti), a volte utilizzata nella composizione come materiale complementare ad effetto smagrante e stabilizzante.
A titolo indicativo nella Fig. 8.4 sono riportati gli intervalli composizionali di impasti porosi bianchi e rossi, realizzabili sia in monoporosa che in bicottura, ed una terza rappresentazione di composizioni costituite da sole argille rosse. Quest’ultima tipologia di formulazione viene utilizzata prevalentemente in bicottura ed è abbastanza diffusa in alcune aree del mondo (ad esempio nel Sudamerica) dove vi è disponibilità di materiali argillosi che per loro natura già contengono i minerali complementari necessari.
Impasto poroso bianco
Argille plastiche
30 ÷ 40%
Argille caolinitiche 20 ÷ 30%
Sabbie feldspatiche 10 ÷ 15%
Feldspato
Quarzo
Calcite
Impasto poroso rosso
Argille carbonatiche
5 ÷ 10%
5 ÷ 10%
7 ÷ 10%
50 ÷ 60%
Argille greificabili 10 ÷ 20%
Sabbie feldspatiche 15 ÷ 20%
Feldspato
5 ÷ 10%
Impasto poroso rosso (solo argille)
Argille carbonatiche
65 ÷ 75%
Argille greificabili 15 ÷ 25% Chamotte 5 ÷ 10%
8.4 Possibili composizioni per impasti porosi bianchi e rossi
8.04 IT NEW
Come descritto, i materiali da rivestimento possono essere ottenuti con i processi di bicottura o di monocottura, entrambi con cicli rapidi. Inoltre, le composizioni degli impasti possono essere sviluppate per la produzione di prodotti con supporto bianco o rosso.
Le paste rosse sono costituite prevalentemente da argille più o meno carbonatiche ad alto contenuto di ferro. Gli altri materiali componenti l’impasto possono essere sabbie feldspatiche, feldspati e quarziti e, quando necessario, calciti e/o dolomiti. Gli impasti cuocenti bianco contengono invece miscele più equilibrate e ponderate di argille chiare, calcite, sabbie feldspatiche, feldspati e quarzo.
La differenza più rilevante tra le composizioni bianche e rosse consiste nelle quantità e tipologia di argille utilizzate, mentre la differenza più significativa fra impasti da bicottura e da monocottura porosa risiede nella percentuale di calcite e/o dolomite più elevata per la bicottura (15 ÷ 18%); per quest’ultima possono essere usate anche materie prime di purezza leggermente inferiore [5].
La Tab. 8.5 riporta, a titolo esemplificativo, diverse composizioni chimiche di impasti per monocottura porosa e bicottura rapida, cuocenti rosso e bianco.
Tab. 8.5 Esempi di composizioni per monoporosa e bicottura
Caratteristiche del prodotto
Le caratteristiche finali di una massa ceramica per rivestimento sono influenzate in modo fondamentale dai composti di neoformazione che si sviluppano durante la cottura. La loro formazione è da attribuirsi alla reattività degli ossidi di calcio e magnesio provenienti rispettivamente dalla decomposizione della calcite e/o della dolomite durante cottura, con la silice e l’allumina derivante dalla distruzione dei reticoli argillosi. I principali composti di neoformazione sono la gehlenite (2CaO·Al2O3·SiO2), il diopside (CaO·MgO·2SiO2), l’anortite (CaO·Al2O3·2SiO2) e la wollastonite (CaO·SiO2). In particolare, caratteristiche quali la resistenza meccanica, il coefficiente di dilatazione termica e l’espansione all’umidità dipendono dalla
quantità e qualità di questi composti. È evidente come nei cicli rapidi di cottura la possibilità di formare nuovi composti, partendo da fasi allo stato solido scarsamente reattive, sia fortemente dipendente dalla granulometria dei materiali allo stato naturale e dal grado di macinazione del semilavorato.
L’evoluzione delle reazioni di decomposizione e di sintesi, che si sviluppano in un impasto di monoporosa sottoposto a cottura rapida, è riportata in Fig. 8.5; si evidenzia chiaramente la dinamica delle reazioni di decomposizione dei minerali di partenza e la formazione di nuovi composti.
Fig. 8.5 Evoluzione delle reazioni di decomposizione e di sintesi che si sviluppano durante la cottura di un impasto da monoporosa
8.05 IT
Materie prime per smalti
Gli smalti più frequentemente utilizzati nel rivestimento sono brillanti, del tipo trasparente (cristalline) ed opaco (fondamentalmente bianco). Questi smalti risultano composti da fritte diverse dipendendo dal tipo di tecnologia, cioè bicottura rapida e monoporosa.
Congiuntamente alle fritte, la cui percentuale di utilizzo è nell’ordine del 90 ÷ 95%, vengono introdotte nella miscela anche piccole percentuali (5 ÷ 10%) di materiali plastificanti quali caolino e ball clay, nonché additivi organici per regolare la reologia nella fase di applicazione e aggancio al supporto (colle).
Bicottura rapida
Gli smalti adatti a questa tecnologia, il cui ciclo di cottura è nell’ordine dei 30 ÷ 50 min, dispongono di tempi di maturazione del vetro estremamente brevi (2 ÷ 4 min). Di conseguenza le fritte evidenziano punti di rammollimento a temperature moderate ed una bassa viscosità in fusione alla massima temperatura di cottura (approssimativamente 1050 ÷ 1130 °C).
Per conseguire questi obiettivi si utilizzano nella maggioranza dei casi fondenti alcalino-borici. Al fine di ottimizzare ulteriormente i valori di viscosità ai valori della temperatura massima di cottura, le fritte più attuali presentano nella formulazione elevati quantitativi di CaO e ZnO, a scapito principalmente dell’ossido di sodio.
Monoporosa
La necessità di usare nelle composizioni d’impasto per rivestimento minerali carbonatici utili per introdurre ossido di calcio e magnesio ha comportato notevoli problemi di carattere tecnologico nello studio degli smalti per la monocottura porosa.
Le emissioni di gas (CO2), derivanti dall’uso di calcite e/o dolomite, in un arco di temperature tra 750 ÷ 950 °C (corrispondenti alla zona di maturazione degli smalti tradizionali) sono state uno dei problemi maggiori per lo studio e lo sviluppo di questo processo. L’esigenza di ottenere temperature di rammollimento degli smalti più elevate di 1000 °C ha richiesto nuove formulazioni alto-fondenti, basate su composizioni a fusione eutettica. Questo è stato possibile diminuendo ossidi quali B2O3 e Na2O ed introducendo CaO, MgO, ZnO e K2O come elementi attivi a provocare la “fusione eutettica” ad alta temperatura.
Per chiarire meglio questo comportamento vengono proposti in Fig. 8.6 i grafici di rammollimento al microscopio riscaldante di due campioni di fritta A e B. Il primo campione è uno smalto per bicottura tradizionale che evidenzia un rammollimento anticipato di circa 60 ÷ 70 °C rispetto a quello B. Da ciò si deduce che il campione A non è adatto alla monocottura porosa, a causa della sua tendenza ad impermeabilizzare la superficie troppo precocemente rispetto alla degasazione dei carbonati presenti nel supporto. Sempre nella Fig. 8.6 sono riportate le differenti composizioni chimiche relative alle fritte A e B, usate rispettivamente in bicottura tradizionale e monocottura porosa.
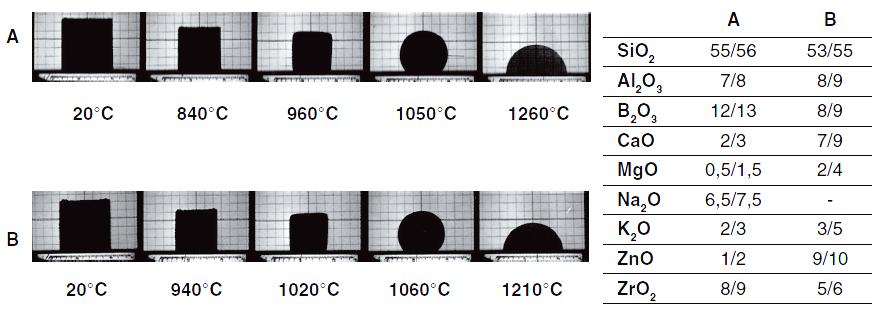
Fig. 8.6 Immagini al microscopio riscaldante per due fritte con diverso punto di rammollimento
Oltre alla temperatura di rammollimento, altri fattori molto importanti, utili per definire la storia termica di una fritta per monoporosa, sono ad esempio la tensione superficiale e la viscosità a caldo del vetro medesimo. Una bassa tensione superficiale favorisce l’eliminazione delle eventuali bolle di gas presenti nel vetro durante la cottura. Bassi valori di viscosità a caldo favoriscono invece una migliore stesura dello smalto, migliorando anche la sua bagnabilità verso le superfici con le quali viene a contatto (ingobbio e/o supporto).
Altri fattori importanti nelle caratteristiche di uno smalto sono la reattività con l’ingobbio e/o con il supporto, ed il valore del coefficiente di dilatazione. Senza alcun dubbio, la compatibilità dilatometrica con il supporto e l’ingobbio è un fattore imprescindibile al fine di poter controllare la planarità delle piastrelle.
Ingobbi
Con il termine ingobbio si intende una composizione più o meno vetrosa che normalmente viene applicata sul supporto prima dello smalto. L’applicazione dell’ingobbio risulta praticamente indispensabile nella monoporosa e nella bicottura. Tra le funzioni principali dell’ingobbio è opportuno richiamare le seguenti:
- inibizione di eventuali reazioni dello smalto con impurezze cromofore dell’impasto;
- avvicinamento dell’accordo dilatometrico tra supporto e smalto;
- diminuzione del costo dello smalto applicato (l’uso dell’ingobbio permette l’applicazione di una quantità di smalto inferiore)
- protezione dello smalto trasparente (cristallina) da possibili macchie di umidità che possono attraversare il supporto poroso nelle piastrelle posate in opera.
Gli ingobbi normalmente utilizzati per monoporosa e bicottura rapida sono costituiti da una percentuale di fritte (30 ÷ 40%), argille di tipo ball clay, silicato di zirconio ed eventualmente feldspato e quarzo. Le funzioni specifiche dei singoli costituenti possono essere così riassunte:
- le fritte contribuiscono a formare la matrice vetrosa;
- le argille (ball clay) apportano le necessarie caratteristiche plastiche all’ingobbio;
- il silicato di zirconio migliora il grado di bianco e l’opacità;
- il quarzo ed il feldspato permettono di controllare sia la fusibilità della miscela che il coefficiente di dilatazione.
Le caratteristiche finali di un ingobbio per monoporosa in genere consistono in: - assenza di sviluppo di una fase vetrosa apprezzabile sino a 1000 °C e quindi un’inerzia chimica sino a tale temperatura; - grado di bianco elevato (coprenza); - impermeabilità (non macchiabilità) a soluzioni acquose/colorate che possono venire a contatto con il supporto cotto; - adesione al supporto cotto e formazione di un interstrato fra supporto e smalto, fortemente integrato.
A titolo di esempio, nella Fig. 8.7 è riportata una macrofotografia in sezione che evidenzia la funzione dell’ingobbio nel contenere l’inquinamento generato da eventuali impurezze presenti nel supporto.

8.7 Funzione dell’ingobbio nel contenimento delle impurezze presenti nel supporto
Congiuntamente alla natura chimica ed alle associazioni mineralogiche delle materie prime, le caratteristiche finali di un prodotto da rivestimento dipendono fortemente dai parametri tecnologici adottati nel corso del processo di lavorazione.
Nel definire quindi le condizioni di lavoro in un determinato processo è molto importante valutare le relazioni che intercorrono tra gli aspetti tecnologici di natura chimico-fisica ed i parametri di lavorazione nelle varie fasi di processo, in particolare:
- macinazione
- atomizzazione
- pressatura
- essiccamento
- cottura biscotto
- smaltatura
- cottura monoporosa
- cottura vetrato.
Di seguito si riporta una trattazione dettagliata per ciascuna fase del processo.
Macinazione
Ha come obiettivo la comminuzione ed omogeneizzazione delle materie prime in ingresso allo stabilimento ceramico, fino ad ottenere granulometrie finali dei semilavorati ottimali e costanti.
Nel caso di impasti per monocottura porosa il grado di macinazione delle materie prime, congiuntamente ad altri fattori di ordine chimico e fisico, può influenzare la velocità di decomposizione dei carbonati in cottura e quindi condizionare sensibilmente la temperatura in cui non si riscontrano più emissioni di gas (CO2) dal supporto. Allo stesso tempo il grado di macinazione può condizionare la reattività tra i vari componenti in cottura e favorire quindi il formarsi di composti di neoformazione in maniera più o meno completa.
Una consistente reattività favorisce la formazione di composti cristallini e contribuisce ad aumentare le caratteristiche meccaniche del materiale cotto. Il grafico riportato in Fig. 8.8 evidenzia chiaramente le variazioni del carico di rottura che si possono riscontrare in materiali cotti ottenuti partendo da composizioni con valori di residuo e grado di macinazione differenti.
Fig. 8.8 Andamento del carico di rottura (in cotto) e del contenuto di calcite nel residuo in funzione del residuo di macinazione
8.08 IT
Nello stesso grafico è anche mostrato come varia la percentuale di calcite presente nel residuo. In linea di massima, il residuo dopo macinazione, nel caso di impasti compositi sia di monoporosa che di bicottura risulta nell’ordine del 4 ÷ 6% (residuo a 63 µm = 230 mesh). Nel caso di impasti cuocenti rosso, costituiti principalmente da materiali argillosi, il residuo può diminuire sino a 3 ÷ 4%.
Atomizzazione
Tale processo è finalizzato all’evaporazione parziale dell’acqua contenuta nella barbottina congiuntamente alla formazione di granuli sferoidali. La distribuzione granulometrica di un impasto per rivestimento non risulta particolarmente differente rispetto alle altre tipologie di impasti atomizzati per monocottura da pavimento o grès porcellanato.
Nella Fig. 8.9 sono mostrate le fasce granulometriche tipiche per impasti da monoporosa e bicottura. Non si riscontrano differenze sostanziali tra i due semilavorati. Si può notare in entrambi i casi una forte concentrazione di particelle, circa 70 ÷ 80%, nella fascia compresa tra 180 e 425 µm.
Monoporosa Bicottura
Pressatura
Fig. 8.9 Distribuzione granulometrica dell’atomizzato per impasti da monoporosa e bicottura (valori indicativi)
L’obiettivo della formatura tramite pressatura a secco è quello di ottenere in crudo la massima densificazione delle polveri possibile, compatibilmente con i problemi di “cuore nero” e di degasazione che possono manifestarsi in cottura. Forze di pressatura differenti esercitate sulle polveri determinano una disomogeneità di densità apparente nel pezzo pressato e, conseguentemente, differenti valori di contrazione e porosità dopo cottura.
Fig. 8.09 IT
Nel caso di prodotti porosi la variazione della densità apparente in crudo non comporta però cambiamenti sostanziali della contrazione in quanto i valori di ritiro per questi prodotti sono molto bassi (inferiori a 1%). Viceversa, differenze di densità apparente nel medesimo pezzo pressato (imputate ad errori di caricamento delle polveri) possono portare alla formazione di aree con valori di porosità differente, e quindi a problemi di stesura ed assorbimento degli smalti applicati.
Valori di densità apparente elevati (superiori a 2,1 ÷ 2,2 g/cm3) possono rendere più difficoltosa la fuoriuscita dei gas dal supporto durante la cottura e causare quindi problemi di “sobbollitura” dello smalto (in monoporosa).
Il grafico della Fig. 8.10 riporta la relazione tra la densità apparente in essiccato e la temperatura ottimale di rammollimento della fritta, al fine di evitare problemi di decarbonatazione. Nel grafico vengono riportate tre diverse fritte (A, B, C) aventi temperatura di rammollimento crescente; le aree rosse indicano zone a rischio di difetti nello smalto. La curva mostra la correlazione tra pressione di compattazione e densità apparente (in essiccato).
Generalmente la pressione di formatura per impasti da rivestimento è nell’ordine di 200 ÷ 250 daN/cm2 (1 daN/cm2 equivale a circa 1 kgF/cm2); per gli impasti cuocenti rosso, tendenzialmente più plastici, la pressione può risultare anche inferiore, fino a soli 150 daN/cm2.
Densità apparente in essiccato [g/cm3]
Fig. 8.10 Correlazione tra densità in essiccato e temperatura ottimale di rammollimento della fritta
Essiccamento
Questa operazione è considerata oggi apparentemente semplice, in quanto i fenomeni fisici che si verificano durante l’evaporazione dell’umidità residua dell’impasto (4 ÷ 7%) sono sufficientemente noti e controllabili. In questa fase, contemporaneamente all’evaporazione dell’acqua residua, si riscontra un aumento della resistenza meccanica del pezzo ceramico, attribuibile ad un avvicinamento delle particelle e quindi al formarsi di legami più forti tra esse. Nel caso della monoporosa, la resistenza meccanica dopo essiccamento deve essere elevata; infatti, per sopportare le sollecitazioni meccaniche nella fase di smaltatura e stoccaggio, il carico di rottura (MOR) dovrà essere superiore a 2,5 N/mm2.
Con i cicli di essiccamento attualmente utilizzati, per non incorrere in problemi di crepe e piccole fessurazioni nel perimetro delle piastrelle, è buona regola contenere i movimenti dimensionali nella fase di essiccamento in valori di ritiro compresi tra lo 0 ÷ 0,3%.
Cottura biscotto
In questa fase, le curve di cottura e la temperatura d’esercizio dei forni devono permettere e favorire l’evolversi delle reazioni fra i diversi componenti fino ad ottenere le caratteristiche finali del supporto, quali: porosità, resistenza meccanica, coefficiente di dilatazione, ecc. È evidente, in base a quanto riportato finora, che le reazioni di sinterizzazione di una massa ceramica non dipendono solamente dalla natura chimico-fisica dell’impasto in questione ma anche dal grado di macinazione, dalla densità apparente del pressato ed infine da ciclo e temperatura di cottura. Nel grafico di Fig. 8.11 si può osservare l’influenza della densità apparente (in essiccato) sui valori di assorbimento d’acqua (parametro indicativo della porosità aperta) e del ritiro dopo cottura.
Ritiro
Assorbimento d'acqua
Ritiro Assorbimento
Densità apparente [g/cm3]
Fig. 8.11 Influenza della densità apparente sull’assorbimento d’acqua (porosità) e sulla contrazione dopo cottura
Fig. 8.11 IT
Il grafico riportato nella Fig. 8.12 evidenzia invece l’andamento del coefficiente di dilatazione termica lineare α (tra 30 e 400 °C) nel caso di campioni cotti a differenti temperature.
Fig. 8.12 Andamento del coefficiente di dilatazione termica lineare α con la temperatura di cottura
I dati delle Fig. 8.11 e Fig. 8.12 si riferiscono a misure effettuate su un impasto cuocente rosso.
Fig. 8.12 IT NEW
Per la ceramica da rivestimento, le tipologie di prodotti più commercializzate presentano smalti brillanti, in particolare cristalline trasparenti e bianchi lucidi opacizzati. Più limitata è invece la commercializzazione di prodotti con superfici diverse (rustiche o matt).
I dispositivi più frequentemente utilizzati per l’applicazione di smalti da rivestimento sono sistemi a campana e a vela. Questi dispositivi permettono di ottenere superfici lisce e speculari tramite l’applicazione sul supporto crudo o biscottato di un velo continuo di smalto a spessore controllato con velocità di caduta costante.
Attualmente per ottimizzare l’applicazione degli smalti e per inibire le problematiche relative alla smaltatura dei supporti crudi (in monoporosa) si tende a ridurre il più possibile il quantitativo di acqua nelle barbottine di smalto. Il comportamento reologico di tali sospensioni si allontana pertanto da quello ideale per assumere caratteristiche plastiche e spesso anche tissotropiche. Le caratteristiche reologiche ottimali di uno smalto standard tradizionale, nel caso di applicazione a campana o vela sono:
- densità elevata
- limite di scorrimento basso
- viscosità costante
- tissotropia molto bassa.
Nella Fig. 8.13 vengono proposti due reogrammi relativi a sospensioni di smalti aventi valori differenti di contenuto d’acqua (e quindi di densità).
acqua 27%
acqua 28,5%
Fig. 8.13 Variazione della viscosità per smalti con differente contenuto d’acqua
Il diagramma riporta la variazione del gradiente di velocità D (1/s) in funzione dello sforzo di taglio t (Pa), il cui rapporto esprime la viscosità apparente. Si nota come piccole variazioni del contenuto d’acqua causano una marcata variazione delle caratteristiche reologiche.
Fig. 8.13 IT
Nella Tab. 8.6 sono riportate le caratteristiche reologiche (viscosità, limite di scorrimento e tissotropia) di differenti sospensioni di smalti utilizzate industrialmente.
Sospensione di
Ingobbio 1
2
Tab. 8.6 Caratteristiche reologiche per differenti sospensioni di smalto
Al fine di minimizzare le problematiche relative all’impiego di smalti ad altissima viscosità, in tempi recenti l’applicazione a campana è stata ottimizzata per evitare la formazione di bolle d’aria e la rigatura in genere.
Durante la smaltatura dei supporti biscottati si riscontra la diffusione dell’acqua dello smalto nel supporto per capillarità. Questa determina l’assorbimento, facilita l’aderenza al supporto e condiziona la stesura dello smalto, così come il suo tempo di essiccamento. È pertanto necessario controllare la velocità di assorbimento del biscotto, proprietà che dipende dalla composizione di base del supporto, dal grado di cottura del biscotto, dalla densità e dalla viscosità dello smalto.
Nel caso della smaltatura della monoporosa le variabili di processo da controllare sono molteplici e particolarmente restrittive sono le tolleranze accettabili riguardo a:
- caratteristiche reologiche dello smalto
- corretto funzionamento dei dispositivi d’applicazione
- caratteristiche delle piastrelle dopo essiccamento (resistenza meccanica, umidità residua e temperatura delle piastrelle).
Altri aspetti importanti sono la setacciatura degli smalti ed il trattamento per mezzo di deferrizzatori. Per la setacciatura sono ormai universalmente impiegati vibrosetacci circolari ad elevata efficienza. La separazione magnetica delle particelle cromofore inquinanti viene invece eseguita con deferrizzatori che possono essere di diversi tipi, quali:
- barre magnetiche cilindriche
- a nido d’ape
- a rulli.
L’efficacia dei deferrizzatori è legata alla portata ed alla densità degli smalti.
Cottura monoporosa
Questa fase della produzione è particolarmente critica perché in essa si sviluppano tutte le reazioni che determinano le caratteristiche del prodotto finito.
La dinamica della cottura di questi prodotti risulta particolare in quanto il supporto contiene materiali carbonatici la cui decomposizione deve essere compatibile con le caratteristiche dello smalto. Quest’ultimo deve presentare un comportamento “eutettico”, ovverosia evidenziare una iniziale refrattarietà e quindi presentare una discreta permeabilità ai gas sino ad una temperatura compresa tra 1000 ÷ 1050 °C, per poi fondere bruscamente. A tale riguardo si evidenziano in dettaglio i comportamenti tecnologici, relativi sia all’impasto che allo smalto, associati ad una curva di cottura tipica della monoporosa (vedi Fig. 8.14).
di cottura [min]
Fig. 8.14 Curva di cottura tipica per monocottura porosa
Il primo tratto A della curva (fino a 800 °C) corrisponde alla fase di preriscaldo del materiale ed alla distruzione dei materiali argillosi. Nella zona B tra 800 ÷ 950 °C avviene la decomposizione dei carbonati con la diffusione all’esterno della CO2. È quindi importante che in questo intervallo termico lo smalto mantenga una certa porosità per favorire l’espulsione dei gas. Nel tratto C tra 950 ÷ 1100 °C si sviluppano le reazioni di sintesi tra gli ossidi alcalino-terrosi (CaO e MgO) provenienti dalla decomposizione dei carbonati, con le fasi amorfe residuali derivanti dalla degradazione termica delle argille.
Fig. 8.14 IT
La sintesi di questi composti di neoformazione è di fondamentale importanza per regolare e definire le caratteristiche fisico-meccaniche del prodotto finale. Nella zona D alla massima temperatura si completa la sinterizzazione del supporto e allo stesso tempo si ottimizza la stesura e la fusione dello smalto. Il tratto E corrisponde alla fase di raffreddamento rapido della piastrella sino alla temperatura di circa 600 °C. In seguito si ha una fase di raffreddamento lento per contenere le tensioni associate alla trasformazione del quarzo libero residuale, ancora presente nel supporto cotto.
Nel grafico di Fig. 8.14 sono riportate tre curve di temperatura, che si riferiscono: alla piastrella, alla zona superiore e alla zona inferiore rispetto al piano dei rulli. Infatti, gradienti di temperatura differenti tra la faccia superiore e inferiore della piastrella possono, entro certi limiti, condizionare e controllare la sua planarità. Diventa così possibile, sfruttando le caratteristiche di espansione e ritiro del materiale, indurre tensioni interne tra le superfici superiore ed inferiore della piastrella che ne modificano la planarità.
Cottura vetrato
Nel processo della bicottura vengono solitamente utilizzate due tipologie di fritte classificabili come “tradizionali” o “a composizione eutettica”.
La temperatura di cottura delle fritte tradizionali può essere nell’ordine di 1020 ÷ 1050 °C, mentre per le fritte di tipo eutettico, alto-fondenti, si raggiungono 1080 ÷ 1130 °C. In entrambi i casi, tuttavia, la cottura del materiale richiede regolazioni e curve differenti da quelle usate nel caso della monoporosa.
Nella fattispecie le curve di cottura e le temperature di esercizio dei forni devono permettere l’evoluzione della fusione del vetro, che rispettivamente risultano di 1030 ÷ 1050 °C per le fritte tradizionali e 1080 ÷ 1130 °C per quelle a composizione eutettica. In entrambi i casi la massima temperatura viene mantenuta per alcuni minuti per favorire la stesura e la brillantezza del vetro.
Indipendentemente dal tipo di fritta usata, dopo cottura si procede ad una fase di raffreddamento rapido fino a circa 600 °C. Raggiunta tale temperatura, la discesa fino a 500 °C deve essere molto lenta per attenuare le tensioni associate alla trasformazione di fase da quarzo β a quarzo α (a 573 °C) che possono provocare anche la rottura del pezzo (sfilo da raffreddamento).
I cicli di cottura adottati oscillano tra 30 e 50 min, a seconda del formato delle piastrelle. Nella Fig. 8.15 sono riportate le curve di cottura rispettivamente del biscotto e del vetrato.
Caratteristiche prodotto finito
Per valutare complessivamente le caratteristiche dei prodotti da rivestimento, nella Fig. 8.16 è riportato un grafico in cui sono rappresentate in modo qualitativo le variazioni delle caratteristiche di carico di rottura (in cotto), assorbimento d’acqua e ritiro per diverse temperature di cottura. Nello stesso grafico si è indicato anche l’intervallo di temperatura ottimale (4) per la cottura.
Fig. 8.16 Carico di rottura (1), assorbimento (2) e ritiro (3) in funzione della temperatura di cottura; l’area (4) rappresenta l’intervallo di cottura ottimale per prodotti da rivestimento
Fig. 8.16 IT NEW2
Nella pratica produttiva, le proprietà meccaniche delle piastrelle porose variano entro intervalli abbastanza ampi [6]. Generalmente i prodotti in pasta chiara, sia in monoporosa che in bicottura, sono contraddistinti da valori più elevati del carico di rottura, in parte per la loro porosità mediamente inferiore, ma soprattutto per la differente microstruttura rispetto agli impasti rossi. A tale riguardo i parametri da considerare sono la distribuzione dimensionale dei pori, la finezza granulometrica (peggiore nel caso di paste rosse macinate a secco) ed il rapporto tra silicati di neoformazione e componenti inerti residui (quarzo, feldspato potassico, ecc.).
La dinamicità del settore ceramico è stata determinante nel favorire la continua evoluzione dei processi di produzione. A partire dagli anni ‘70 del secolo scorso si è assistito ad una trasformazione tecnologico-impiantistica con il passaggio dal tradizionale processo di cottura in forni a tunnel ai forni a rulli, che permettono cicli di cottura molto più rapidi su formati diversificati con conseguente aumento della produttività.
L’adozione di queste tecnologie ha richiesto nuovi layout e l’elevato grado di automazione raggiunto ha permesso anche una forte riduzione dei costi industriali. Infatti, già negli anni ‘80 è avvenuta una progressiva evoluzione dei processi produttivi con particolare riferimento a:
- diffusione della macinazione ad umido
- introduzione del processo di atomizzazione
- diffusione delle presse idrauliche
- adozione degli essiccatoi rapidi
- diffusione dei forni a rulli.
L’introduzione dei processi di cottura rapida da rivestimento non è stata comunque così immediata e radicale come per il pavimento. Questo perché la tecnologia della bicottura tradizionale non poteva essere facilmente convertita a cicli rapidi sia per le limitazioni impiantistiche del periodo, che soprattutto per l’elevato valore estetico delle piastrelle allora prodotte.
Infatti, solamente la disponibilità commerciale di smalti eutettici alto-fondenti ha reso possibile l’applicazione delle nuove tecnologie produttive alle piastrelle da rivestimento, con l’ottenimento di superfici smaltate ad elevato valore tecnico ed estetico. Gli anni ‘90 hanno così consolidato l’affermarsi dei processi produttivi della monoporosa e della bicottura rapida, permettendo importanti perfezionamenti tecnologici e una notevole riduzione dei costi industriali.
Gli impianti attuali presentano ulteriori livelli di complessità, dipendentemente dalle loro dimensioni, dal grado di flessibilità delle linee e, conseguentemente, dalle automazioni introdotte e dai sistemi di controllo addottati.
Nella Fig. 8.17 si riportano i diagrammi a blocchi relativi ai processi di monoporosa e bicottura rapida, dove vengono evidenziate le varie fasi di lavorazione, prevedendo l’uso della macinazione in continuo ormai affermatasi anche in impianti di medio-piccole dimensioni. Tra le varie fasi di lavorazione illustrate, si nota nel caso della monoporosa la possibilità di stoccare il materiale dopo smaltatura e cottura, come pure la possibilità di indirizzare direttamente il materiale dall’uscita forno alla scelta. Nel caso della bicottura è invece previsto lo stoccaggio del biscotto e dello smaltato in aree dedicate.
La pressatura è effettuata con presse idrauliche di adeguato tonnellaggio, l’essiccamento con essiccatoi rapidi oggi prevalentemente orizzontali, la cottura rapida con forni a rulli, la scelta con macchine completamente automatizzate.
La movimentazione dei carrelli di stoccaggio delle piastrelle crude e cotte è anch’essa totalmente automatizzata grazie all’impiego di veicoli a guida autonoma.
Materie prime
Dosaggio
Macinazione continua odiscontinua
Atomizzazione
Stoccaggio polveri atomizzate
Pressatura
Essiccamento
Smaltatura e decorazione
Stoccaggio crudo
Cottura
Stoccaggio cotto
Scelta e imballo
Magazzino
Preparazione smalti
Materie prime
Dosaggio
Macinazione continua odiscontinua
Atomizzazione
Stoccaggio polveri atomizzate
Pressatura
Essiccamento
Cottura biscotto
Stoccaggio biscotto
Smaltatura e decorazione
Stoccaggio smaltato
Cottura vetrato
Stoccaggio cotto
Scelta e imballo
Magazzino
Preparazione smalti
Fig. 8.17 Diagramma a blocchi del processo di monoporosa (sinistra) e bicottura (destra).
Gli effetti della rapida evoluzione tecnologico-impiantistica avvenuta negli ultimi decenni si possono riassumere in:
- aumento della produttività (per addetto)
- diminuzione dell’area occupata dall’impianto
- diminuzione dei costi industriali del prodotto.
Nella Fig. 8.18 è riportato un moderno layout per un impianto idoneo alla produzione di piastrelle da rivestimento con il processo di bicottura rapida su forno bicanale (piano inferiore: biscotto, piano superiore: vetrato). A destra si vede l’area di dosaggio delle materie prime e di preparazione impasto (macinazione e atomizzazione). Segue l’area di formatura con pressatura ed essiccamento rapido (in essiccatoio orizzontale multipiano). Le piastrelle essiccate entrano direttamente nel piano inferiore del forno bicanale per il primo trattamento termico (biscottatura). Il prodotto biscottato in uscita dal forno viene convogliato (a ritroso) sulla linea di smaltatura e decorazione digitale e, tramite un tratto di trasporto inclinato, reinserito nel piano superiore del forno bicanale per la cottura dello smalto. All’uscita dal forno il prodotto vetrato viene indirizzato verso la linea di squadratura (solo per i formati più grandi) e quindi alla linea di scelta e confezionamento.
dosaggio e preparazione impasto
preparazione smalti
pressatura ed essiccamento
smaltatura e decorazione
8.18 IT
cottura biscotto e vetrato (in forno bicanale )
squadratura (grandi formati)
scelta e confezionamento
Fig. 8.18 Schema di impianto per la produzione di rivestimento in bicottura rapida (le parti in rosso rappresentano un eventuale raddoppio produttivo)
Di seguito vengono sinteticamente illustrate le macchine caratteristiche che compongono un impianto tipico per la produzione di piastrelle da rivestimento con tecnologia di macinazione a umido.
È comunque possibile utilizzare anche il processo di macinazione a secco e successiva rigranulazione delle polveri, in particolare nel caso della bicottura ottenuta da composizioni d’impasto molto ricche in materiale argilloso e/o di materiali aventi caratteristiche morfologiche similari tra loro. Meno frequente e comunque più difficoltosa risulta invece la produzione di monoporosa con impianti che prevedono questo tipo di macinazione.
Dosaggio materie prime
Il dosaggio può essere eseguito con macchine ed impianti che presentano livelli di automazione differenti. Nel caso del processo di macinazione discontinua per il dosaggio delle materie prime si possono utilizzare sistemi che prevedono l’impiego di cassoni pesatori singoli, dotati di celle di carico deformabili. Nei processi di macinazione continua è generalmente previsto un sistema di pesatura diretta su nastro gestita da un processore.
La miscela dosata viene stoccata temporaneamente in un silo di precarica per essere poi immessa nel mulino continuo. In alcuni casi, quando le caratteristiche delle argille lo richiedono, si può prevedere la scioglitura preventiva di una parte dei materiali argillosi. In questo caso la sospensione argillosa, che contiene già tutto o parte del fluidificante, viene immessa, dopo opportuno dosaggio, direttamente all’ingresso del mulino.
La macinazione ad umido dell’impasto può essere effettuata con il processo di tipo sia continuo che discontinuo. La scelta del tipo di macinazione dipende da diversi criteri quali ad esempio:
- caratteristiche delle materie prime
- dimensione dell’impianto
- professionalità delle maestranze.
Dal punto di vista tecnologico i vantaggi più immediati della macinazione continua sono:
- maggior costanza delle caratteristiche della barbottina
- aumento della densità
- migliori caratteristiche reologiche
- automazione totale del processo.
Dal punto di vista tecnico-organizzativo si riscontra quindi una migliore razionalità nella gestione dell’impianto. Inoltre, sotto l’aspetto economico, la macinazione continua comporta un risparmio diretto sul personale impiegato ed indiretto sull’energia necessaria all’evaporazione dell’acqua delle barbottine all’atomizzatore in quanto è possibile gestire barbottine aventi maggiore contenuto di solido rispetto al sistema di macinazione discontinuo.
Allo scarico del mulino viene effettuato un controllo della barbottina prima con un setaccio grossolano, poi con una batteria di setacci più fini. Il residuo di setacciatura viene immesso nell’acqua predosata destinata al carico del mulino, mentre la barbottina filtrata viene inviata ad una vasca di raccolta munita di agitatore, dalla quale può essere direttamente prelevata ed inviata all’atomizzatore.
Qualora le caratteristiche fisiche delle materie prime lo permettano (ad es. con bassi valori di residuo a 63 µm) si può prevedere la scioglitura di una parte delle argille ed even-
tualmente dello scarto crudo in un turbo-dissolutore gestito in parallelo al mulino, evitando così il passaggio di questi materiali nel mulino stesso.
In alternativa, si può anche prevedere una scioglitura preliminare delle materie prime argillose che saranno successivamente inviate alla macinazione nel mulino con gli altri componenti dell’impasto. Questa opzione si dimostra particolarmente interessante soprattutto nei casi in cui gli impasti siano costituiti da una frazione rilevante di argille fortemente plastiche e con umidità naturale relativamente alta (superiore al 18 ÷ 20%).
L’atomizzazione è il processo nel quale si attua la pressoché totale evaporazione dell’acqua contenuta in una barbottina. La diminuzione del contenuto d’acqua da valori iniziali fino al 40% viene effettuata mediante un processo di essiccamento a spruzzo in controcorrente di aria calda. L’operazione si effettua iniettando la barbottina dal basso verso l’alto mediante pompe ad elevata pressione (16 ÷ 22 bar) e successiva nebulizzazione all’interno della camera cilindrica d’essiccamento dell’atomizzatore, tramite ugelli di varie dimensioni omogeneamente distanziati.
Il processo di macinazione e atomizzazione svolge anche la funzione di “digestore” delle acque di lavaggio, dei fanghi di varia provenienza e di tutte le polveri di filtraggio e pulizia dell’impianto, in quanto vengono smaltite in modo controllato nella barbottina stessa dell’impasto ceramico.
L’atomizzatore è solitamente dotato di filtri a secco o ad umido per abbattere, entro i limiti richiesti dalle norme di legge vigenti nei diversi paesi, le polveri più fini che altrimenti si disperderebbero nell’atmosfera con il vapor d’acqua in uscita dal camino.
Se ritenuto opportuno, la macchina può essere alimentata anche con aria calda proveniente da sistemi di cogenerazione al fine di diminuire i costi energetici d’esercizio.
Con l’atomizzatore, il semilavorato (barbottina) viene trasformato in polvere a granulometria ed umidità controllata (normalmente 5,5 ÷ 6,5%), idonea ad essere convogliata attraverso nastri trasportatori nei silos di stoccaggio.
Pressatura
Questa fase del processo risulta centrale nella tecnologia produttiva dei materiali ceramici in quanto conferisce la forma al manufatto da realizzare. Normalmente, nell’operazione di formatura, si cerca di ottenere il massimo addensamento delle particelle in crudo, compatibilmente con i problemi di degasazione e di “cuore nero” in cottura. Le pressioni di formatura generalmente adottate per i prodotti porosi da rivestimento oscillano intorno ai 250 ± 50 daN/cm2.
Le presse idrauliche attualmente disponibili risultano particolarmente adatte alla formatura di questi materiali e allo stesso tempo permettono di garantire elevate affidabilità e ridurre al minimo l’assorbimento d’energia.
Vanno anche considerati alcuni importanti accessori che sono parte integrante della pressa: i carrelli di alimentazione polveri e gli stampi.
I carrelli, specifici dispositivi preposti all’alimentazione delle polveri nello stampo, permettono un omogeneo caricamento dell’alveolo del medesimo.
Gli stampi generalmente utilizzati nella formatura di questo prodotto sono del tipo a punzoni entranti, mentre per formati maggiori del 30×60 cm si preferisce l’impiego di stampi a formatura superiore (SFS); i punzoni sono normalmente rivestiti in gomma per diminuirne
la sporcabilità e quindi la frequenza di pulizia. Il punzone “lato marca” può essere del tipo tradizionale (rigido) oppure di tipo isostatico per ottimizzare l’omogeneità di pressatura e quindi ottenere valori di densità apparente uniformi nei vari punti della piastrella.
Essiccamento
L’essiccamento è la fase di lavorazione in cui si ha la eliminazione dell’umidità residua di pressatura nelle piastrelle appena formate. Questa operazione può essere effettuata mediante essiccatoi verticali o, preferibilmente, orizzontali. Le piastrelle in uscita dalle presse sono raccolte da linee a rulli ed inviate agli essiccatoi.
Nel caso di essiccatoi verticali i cicli di essiccamento risultano di 35 ÷ 70 min, mentre per gli orizzontali i cicli possono essere molto più rapidi (6 ÷ 20 min). Ovviamente, la durata del ciclo dipende dal tipo d’impasto, dal formato e dallo spessore dei pezzi.
In particolare per la monoporosa è indispensabile ottenere all’uscita dell’essiccatoio una temperatura uniforme della piastrella. A tale riguardo, gli essiccatoi orizzontali, disponendo di un’elevata circolazione dei flussi d’aria nei singoli moduli e alla elevata automazione, mantengono costante la temperatura di superficie delle piastrelle in uscita, garantendo minime tolleranze anche in presenza di fermate delle linee di smaltatura a valle.
Le linee di smaltatura idonee alla produzione di monoporosa e bicottura possono ritenersi sostanzialmente uguali, a parte alcuni dispositivi complementari quali gli sbavatori che dipendono dalla natura del materiale (“biscottato” o “crudo”). Generalmente a parità di applicazioni, le linee della monoporosa sono più lunghe. Come accennato, le tipologie più frequenti di piastrelle da rivestimento presentano superfici lisce e speculari, per ottenere le quali i dispositivi più idonei per l’applicazione dell’ingobbio e dello smalto risultano essere i gruppi a “campana” e “vela”. Inoltre, sono utilizzati sistemi specifici per la nebulizzazione (cabine airless) idonei per realizzare un’omogenea distribuzione dell’acqua ed eventualmente degli ingobbi.
Riguardo alla decorazione, considerando le tipologie ed i disegni oggi richiesti, vengono utilizzate quasi esclusivamente stampanti digitali a getto d’inchiostro. Queste macchine si sono rapidamente diffuse poiché oltre alla rapidità nei cambi di prodotto da realizzare (file grafico) garantiscono una elevata definizione dell’immagine stampata e non necessitano di continui interventi da parte degli operatori. Esistono diversi modelli di macchine digitali la cui distinzione è basata essenzialmente sulla larghezza di stampa e sul numero di barre colore utilizzabili. Tra le caratteristiche peculiari della decorazione digitale vi è indubbiamente l’assenza di contatto con la piastrella da decorare.
Attualmente si sta assistendo ad una evoluzione dell’automazione delle linee di smaltatura e decorazione, grazie all’impiego di dispositivi per il controllo dinamico del processo e di opportune piattaforme di gestione dei dati.
La sempre maggiore comprensione del comportamento di materie prime, impasti, ingobbi e smalti, oltre all’esperienza acquisita hanno semplificato e standardizzato il processo di cottura.
Le macchine termiche sono oggi dotate di opportune strumentazioni che permettono di effettuare regolazioni e controlli della temperatura molto accurati.
Le larghezze dei forni sono aumentate con i formati e con le maggiori produttività richieste; di pari passo sono migliorati i sistemi di combustione, idonei a mantenere la temperatura costante ed omogenea in tutta la sezione del forno. Oltre ai bruciatori ad alta velocità sono anche disponibili bruciatori in grado di distribuire la fiamma in zone specifiche della sezione del forno. Ciò consente una distribuzione termica ottimale relativamente ai materiali da cuocere specialmente nelle zone più critiche delle curve di cottura.
L’adozione della cottura rapida su rulli, mono o bi-canale, ha facilitato la movimentazione delle piastrelle favorendo la realizzazione di formati di maggiori dimensioni con elevata flessibilità produttiva.
La precisione nella predisposizione e misurazione delle curve di cottura, congiuntamente al mantenimento delle condizioni di esercizio del forno, è assicurata da sistemi di controllo elettronico che garantiscono il mantenimento della temperatura prestabilita entro limiti molto ristretti. Il sistema di controllo del forno esegue inoltre la supervisione e la memorizzazione dei dati di processo e provvede autonomamente alla sua gestione.
Scelta
L’operazione della scelta, pur non intervenendo sulle caratteristiche del prodotto, costituisce la fase conclusiva del ciclo di lavorazione. Anche in questo reparto si è sviluppata la completa automazione delle operazioni fino all’inscatolamento ed alla successiva pallettizzazione. La geometria delle piastrelle (dimensione e planarità) può essere verificata con sistemi di visione elettronica, indirizzando poi il prodotto verso le specifiche uscite. Il numero di queste dipende dal criterio di classificazione sulla base dei valori di planarità e tono del prodotto. Ad esempio, si possono prevedere sette posizioni di scarico (uscite) considerando due toni e tre classi di scelta. L’intervento dell’operatore, negli impianti più automatizzati, è limitato all’analisi dei difetti estetici ed alla codifica delle piastrelle per definirne la classe di appartenenza.
Talvolta le operazioni di scelta sono precedute dalla squadratura a secco dei lati delle piastrelle, in particolare nel caso di formati medio-grandi (ad es. 35×70 cm o superiori).
Movimentazione e stoccaggio
Oltre ai tradizionali impianti di movimentazione e stoccaggio con veicoli a guida automatica si stanno sempre più diffondendo gli impianti di movimentazione con stoccaggio minimo alimentando in diretta al forno. Nel caso della bicottura possono essere previsti sistemi di stoccaggio del supporto su panconi. Il prelievo delle piastrelle viene eseguito con dispositivi muniti di ventose o di piani aspirati che poi trasferiscono le stesse alla linea di smaltatura. Altre soluzioni prevedono l’impiego di box di stoccaggio, nei quali vengono accumulate le piastrelle. Questi sono poi convogliati alle linee di smaltatura attraverso carrelli trasportatori che si muovono su superfici piane guidati da un raggio laser. Lo stesso sistema di movimentazione tramite LGV (laser guided vehicle) può essere utilizzato nel caso della monocottura per movimentare i carri contenitori delle piastrelle sia crude che cotte. In entrambi i casi una stazione computerizzata sovrintende e controlla i flussi dell’intero sistema di trasporto, monitorando in tempo reale i flussi produttivi da e per lo stoccaggio delle piastrelle.
La monocottura porosa e la bicottura rapida hanno indubbiamente raggiunto valenze tecniche ed estetiche superiori a quelle della bicottura tradizionale. Dal punto di vista produttivo, la monoporosa e la bicottura rapida possono ritenersi due tecnologie alternative e complementari. La definizione dell’impianto ideale per ogni specifica esigenza dipende da fattori tecnici, economici, culturali e geografici.
L’aumento delle dimensioni dei formati, con effetti di superficie sempre più simili ai marmi e alle pietre naturali, ha incrementato l’utilizzo di queste tipologie di prodotti ceramici come materiali da rivestimento.
[1] A. Brusa e L. Contoli, «The choice between monoporous and fast double firing», Ceramic World Review, vol. 4, 1992.
[2] ISO, International Organization for Standardization, «ISO 13006 Ceramic tiles – Definitions, classification, characteristics and marking», 2018.
[3] ISO, International Organization for Standardization, «ISO 10545 Ceramic tiles – Part 1-20», 2014-2022.
[4] G. Biffi, La monocottura rapida porosa, Faenza Ed., 1987.
[5] A. Bertoni, C. Siligardi e M. Montorsi, «Bicottura e monoporosa a confronto», Ceramica Informazione, vol. 467, pp. 135-143, 2008.
[6] Dondi M. et al., «Proprietà meccaniche delle piastrelle ceramiche a supporto poroso», Ceramica Informazione, vol. 402, p. 985, 2000.
BICOTTURA
Prodotto ceramico a supporto poroso, costituito da un impasto carbonatico rosso o bianco, sottoposto ad una prima cottura a ciclo lento o rapido (biscotto), quindi smaltato e decorato, poi ricotto (da cui il nome bicottura) in forno a rulli a ciclo rapido.
BI-RAPIDA
Tecnologia o prodotto appartenente alla bicottura, che prevede l’impiego di una cottura rapida in forno a rulli sia per il biscotto che per il vetrato.
EUTETTICO
Si definisce eutettico una specifica miscela composta da due o più componenti che presenta una temperatura di fusione inferiore alle temperature di fusione dei singoli componenti puri; quindi la miscela è più “facile da fondere” rispetto ai singoli materiali. Solitamente si identificano come eutettici le composizioni con le minime temperature di fusione.
MONOCOTTURA
Prodotto ceramico ottenuto con una cottura unica del supporto smaltato, solitamente riferito ad un supporto semi-greificato, costituito da un impasto non carbonatico rosso o bianco, smaltato e decorato in crudo, quindi cotto in forno a rulli a ciclo rapido.
MONOPOROSA
Prodotto ceramico a supporto poroso, costituito da un impasto carbonatico rosso o bianco, smaltato e decorato in crudo, quindi cotto in forno a rulli a ciclo rapido.
L’impiego della ceramica come pavimento ha conosciuto un grande impulso a partire dagli anni ‘50 del secolo scorso, con l’avvento sul mercato del “cottoforte”, tipico prodotto italiano ottenuto con la tecnologia tradizionale della bicottura, le cui caratteristiche tecniche ne permettevano l’uso anche come pavimento.
La sempre maggior richiesta del mercato, congiuntamente all’introduzione delle presse automatiche e dei forni a rulli, hanno fatto sì che, a partire dagli anni ‘60 siano apparsi i primi impianti con cottura rapida, soprattutto grazie alla tecnologia della monocottura che ha rappresentato una vera e propria rivoluzione produttiva.
Con il termine “monocottura” si indica comunemente un processo produttivo che prevede la cottura contemporanea (quindi “mono”) del supporto e dello smalto in un’unica soluzione, in contrasto con la “bi-cottura” tradizionalmente utilizzata nei processi ceramici.
Per metonimia il termine “monocottura” si è poi trasferito al prodotto ceramico stesso, ovvero ha identificato la piastrella smaltata da pavimento, a supporto greificato, ottenuta attraverso il processo di monocottura.
Nel periodo compreso tra gli anni ‘70 e ‘80 sono accaduti tre eventi molto importanti per il settore ceramico:
- la diffusione dell’automazione industriale
- l’affermarsi della tecnologia di cottura rapida
- la crisi energetica.
Certamente l’introduzione della tecnologia della monocottura, con l’utilizzo di cicli rapidi di cottura ha portato in quel periodo storico a mutamenti sostanziali, sia della tecnologia ceramica che delle macchine termiche, in particolare dei forni [1] [2].
Parallelamente sono state introdotte numerose soluzioni innovative in tutto il ciclo produttivo e, tra queste, nuovi sistemi di movimentazione delle piastrelle. Si è infatti passati da forni a tunnel che prevedevano il trasporto delle piastrelle impilate su carrelli rotabili a forni a rulli monostrato, con piastrelle dapprima supportate da piastre refrattarie, fino ad arrivare ai forni attuali dove le piastrelle avanzano direttamente sui rulli ceramici, che fungono essi stessi da supporto dei pezzi.
Gli stessi concetti termodinamici di cottura applicati ai nuovi forni a rulli risultarono totalmente differenti da quelli utilizzati nei forni a tunnel. Le nuove teorie relative alla cottura rapida delle piastrelle si rifacevano a modelli elaborati dal prof. M. Korach [3] che furono poi applicati ai forni a rulli industriali.
Il forno monostrato a rulli si rivelò inoltre la macchina termica a minor consumo specifico. Anche per questo motivo, la monocottura rapida si impose all’attenzione dei produttori quasi contemporaneamente alla crisi energetica degli anni ‘70.
Oltre al risparmio energetico, conseguente all’eliminazione di una seconda fase di cottura, vi sono state altre condizioni che hanno contribuito alla diffusione della monocottura greificata: - la semplificazione dei sistemi di smaltatura per prodotti greificati - la possibilità di produrre formati più grandi
- l’adozione nell’impiantistica di sistemi automatici di movimentazione dei pezzi.
Inoltre, l’introduzione della monocottura greificata nel mercato ha portato ad un miglioramento delle caratteristiche fisico-meccaniche del materiale ceramico, incrementandone allo stesso tempo le caratteristiche estetiche.
Qualità tecnica e contenuto estetico, congiuntamente a una riduzione dei costi, sono stati i fattori determinanti per l’affermazione di questo prodotto da pavimento.
La Fig. 9.1 mostra la distribuzione complessiva della produzione ceramica mondiale suddivisa tra le categorie: rivestimento, pavimento in monocottura e grès porcellanato (prodotto ad elevate prestazioni che viene utilizzato prevalentemente come pavimento ma anche per rivestimenti). Come si può notare, i prodotti da pavimento in monocottura rappresentano circa il 12% del totale (pari a circa 2 miliardi m2 annui).
Rivestimento
Pavimento (monocottura)
Grès porcellanato
Fig. 9.1 Produzione mondiale in funzione delle tipologie produttive (stime 2019)
La Fig. 9.2 mostra invece come le diverse aree produttive del mondo presentino quote differenti tra pavimento e rivestimento; questo in relazione ad aspetti logistici, come la presenza di materie prime domestiche idonee all’una o all’altra tecnologia, ed a fattori culturali. Risulta comunque evidente la netta predominanza del pavimento sul rivestimento.
Fig. 9.01 IT
Fig. 9.2 Suddivisione tra piastrelle da pavimento e rivestimento per le principali zone produttive mondiali (stime 2019)
A seguire, la Fig. 9.3 mostra un’ulteriore segmentazione delle piastrelle da pavimento prodotte nelle principali zone geografiche / paesi, in base alla suddivisione tra pasta rossa (più economica) e pasta bianca (più pregiata). La tendenza generale è decisamente a favore della
Fig. 9.02 IT
pasta bianca, nonostante esistano ancora importanti produzioni in pasta rossa in Spagna, in Medio Oriente e nel continente Sud-americano. Anche in questo caso, la preferenza tra pasta rossa e bianca deriva da contingenze locali, aspetti culturali e di mercato.
Fig. 9.3 Suddivisione delle piastrelle da pavimento (pasta bianca o rossa) per le principali zone produttive mondiali (stime 2019)
9.03 IT
La definizione del prodotto “monocottura greificata” è di per sé stessa esplicativa delle sue caratteristiche. Infatti, nel gergo ceramico, il termine “grès” indica un materiale con una struttura “compatta”, non porosa, ovvero greificata. A riguardo le norme internazionali prevedono differenti livelli di porosità espressi come assorbimento d’acqua, per differenti gruppi di prodotti.
Secondo la norma internazionale ISO 13006 [4] le piastrelle ceramiche vengono classificate in vari gruppi a seconda del metodo di formatura (estrusione, pressatura o colaggio) e del valore di assorbimento d’acqua del prodotto finito (vedi Tab. 9.1).
Le piastrelle in monocottura greificata ottenute per pressatura ricadono nei gruppi BIb (assorbimento compreso tra 0,5 e 3%) e BIIa (assorbimento tra 3 e 6%).
pressatura a secco
Tipologia di prodotto Grès porcellanato
Monocottura vetrificata
Pavimento semi-vetrificato
Pavimento poroso Rivestimento poroso
Tab. 9.1 Classificazione ISO 13006 delle piastrelle pressate a secco; in evidenza i gruppi di riferimento dei prodotti da pavimento [4]
La differenza tra la monocottura BIIa rispetto alla BIb risiede quindi nella maggior greificazione di quest’ultima, che per tale motivo presenta anche caratteristiche di antigelività che la rendono idonea per applicazioni in ambienti esterni.
Viceversa il gruppo BIIb si riferisce ad un pavimento con supporto poroso, con caratteristiche tecniche inferiori, che trova comunque uno spazio commerciale come prodotto utilizzabile tanto per pavimento quanto per rivestimento.
Le specifiche contemplate dalla norma ISO 10545 [5] riguardano le caratteristiche dimensionali, fisiche e ceramiche dei prodotti ceramici.
10545
105456 <
ISO 105457 dichiarata dal produttore dichiarata dal produttore dichiarata dal produttore
ISO 105458 test disponibile test disponibile test disponibile
ISO 105459 test disponibile test disponibile test disponibile
ISO 1054510 test disponibile test disponibile test disponibile
ISO 1054511 richiesta richiesta richiesta
ISO 1054512 r ichiesta (per uso esterno) dichiarata dal produttore dichiarata dal produttore
ISO 1054513 classe GB min / UB min classe GB min / UB min classe GB min
ISO 1054513 test disponibile test disponibile test disponibile
ISO 1054514 classe 3 min classe 3 min classe 3 min
ISO 1054516 test disponibile test disponibile test disponibile
Caratteristiche
Sforzo di rottura
Resistenza abrasione profonda
Resistenza abrasione superf iciale
Coefficiente dilatazione
Resistenza sbalzi termici
Espansione all’umidità
Resistenza cavillo
Resistenza al gelo
Resistenza ai prodotti chimici
Resistenza agli acidi e alle basi
Resistenza macchie
Differenza di colore
Tab. 9.2 Caratteristiche tecniche dei pavimenti secondo la normativa ISO 13006 [4]
Nella Tab. 9.2 viene riportato un quadro riassuntivo delle caratteristiche fisico-ceramiche per i gruppi BIb, BIIa e BIIb (con porosità maggiore: 6 ÷ 10%).
L’insieme di tutte le caratteristiche esposte conferma infatti che questi materiali, in grado di sopportare usure e sollecitazioni meccaniche, sono particolarmente idonei ad essere utilizzati come pavimentazioni.
È inoltre opportuno accennare alle caratteristiche degli smalti che costituiscono la superficie di lavoro del prodotto in esercizio e che, nella storia della monocottura greificata, hanno conosciuto diverse fasi evolutive.
In una prima fase, gli smalti ricalcavano le caratteristiche dei prodotti ottenuti mediante cottura tradizionale. Successivamente, negli anni ‘80 e ‘90, in linea con le superiori caratteristiche tecniche del supporto, si sono imposti smalti (graniglie, sinterizzati, devetrificati) con maggiore durezza e resistenza all’abrasione della superficie. Questi smalti venivano generalmente applicati con tecniche miste (a secco e ad umido). L’aspetto dei prodotti era piuttosto semplice, basato sull’accoppiamento di colori e formati, in quanto se ne privilegiava la valenza tecnica rispetto a quella estetica.
Attualmente, seguendo una tendenza che ormai perdura da diversi anni, gli effetti materici delle superfici smaltate si rifanno alle pietre naturali, con particolare diffusione di superfici dall’aspetto invecchiato, chiamato appunto genere “anticato”. In ogni caso, si può affermare che l’evoluzione delle tecnologie di decorazione e l’ulteriore ottimizzazione delle caratteristiche tecniche permette di ottenere comunque ottimi risultati estetici.
Nella maggioranza dei casi i valori di resistenza all’abrasione delle piastrelle smaltate di questa tipologia possono essere classificati nella Classe 4 o in alcune tipologie nella Classe 5 (la più elevata), secondo il metodo di prova definito dalla norma ISO 10545-7, conosciuto anche come indice PEI (dal Porcelain Enamel Institute - USA).
In merito alle caratteristiche del supporto, si riscontra una sempre maggior tendenza ad avvicinarsi alle caratteristiche porosimetriche del grès porcellanato, con valori di assorbimento d’acqua nell’ordine di 1%. I prodotti da pavimento si evolvono pertanto dalle caratteristiche della tradizionale monocottura (gruppo BIIa: 3% < AA < 6%) verso prodotti aventi porosità inferiori a 1%, compresi quindi nel gruppo BIb.
Formati
Storicamente il formato principale del pavimento era il 30×30 cm, mentre negli ultimi tempi si è passati a formati medio grandi come 60×60 e 80×80 cm. Più modesta risulta invece la produzione dei formati rettangolari per pavimento in monocottura smaltata.
Aspetto superficiale
Particolarmente in Italia, si è affermata la tendenza a produrre piastrelle da pavimento con superfici matt satinate, con un effetto “anticato” o simile alle pietre naturali. Queste tipologie di prodotti, oltre agli smalti utilizzati, sono valorizzate dalle tecniche di strutturazione della superficie e, talvolta, anche del perimetro della piastrella.
Originariamente, le linee di smaltatura per ottenere questi effetti risultavano abbastanza complesse essendo costituite da diverse attrezzature e sistemi applicativi (dischi, aerografi, macchine serigrafiche, spazzolatrici, ecc.) mentre in tempi più recenti il numero di applicazioni si è notevolmente ridotto.
Se invece si considera la produzione mondiale di monocottura, la percentuale maggiore è caratterizzata da smalti ad effetto lucido, limitando l’utilizzo di finiture matt a funzioni antiscivolo e di resistenza all’abrasione.
A riguardo, negli ultimi tempi, si assiste ad un forte incremento delle superfici smaltate con graniglie, generalmente applicate a secco. Tali effetti riprendono i motivi classici dei marmi e delle pietre naturali, conferendo micro-strutturazioni alle superfici perfettamente sincronizzate con le decorazioni digitali.
Secondo una classificazione tecnico-commerciale gli impasti di monocottura greificata possono essere distinti in rossi e bianchi. In entrambi i casi, le materie prime usate sono costituite da due tipologie fondamentali: - materiali argillosi; - materiali complementari (feldspati, quarzi, pegmatiti, ecc.).
Nel caso degli impasti rossi, i minerali argillosi sono solitamente di natura illitico-cloritica ed illitico-caolinitica. Per gli impasti bianchi invece, la natura delle argille è di tipo ball clay o china clay, rispettivamente di natura illitico-caolinitica e caolinitica.
La seconda tipologia di minerali, a carattere fondente e/o inerte, comprende rocce quali: feldspatoidi, feldspati, sabbie feldspatiche e quarzo. È evidente che i rapporti quantitativi fra materiali argillosi, fondenti, e quarzo sono dipendenti dalla natura mineralogica intrinseca e dalla granulometria delle argille, oltre che dalle caratteristiche dei fondenti utilizzati.
Gli impasti bianchi a loro volta sono suddivisi in due tipologie, “potassico” e “sodico”, a seconda della predominanza dei rispettivi ossidi nella formulazione.
Nelle Tab. 9.3 e Tab. 9.4 sono riportate le composizioni chimiche e le caratteristiche fisiche delle materie prime usate più frequentemente nelle formulazioni di impasti per la produzione di monocottura bianca e rossa da pavimento. Materia prima
Argilla bianca plastica (A) 55÷65 22÷29 0,5÷1 0,5÷1 0,5÷1 0,1÷0,5 1÷2
Argilla bianca semi plastica (B)
Argilla rossa plastica (C)
Feldspato potassico
Feldspato sodico
Tab. 9.3 Analisi chimica in ossidi delle materie prime per prodotti da pavimento (monocottura bianca e rossa)
Caratteristiche
di rottura (in crudo)
Carico di rottura (in essiccato)
Dopo cottura a 1100 °C
di rottura (in cotto)
Tab. 9.4 Caratteristiche fisiche delle argille
Le materie prime di tipo ball clay e alle argille rosse greificabili, rispettivamente usate per impasti bianchi e rossi, hanno il compito di conferire ai semilavorati le necessarie caratteristiche in crudo, quali plasticità e resistenza meccanica in verde ed essiccato.
Le argille di tipo china clay sono da ritenersi secondarie per le caratteristiche in crudo, mentre sono fondamentali per l’elevato apporto di allumina, necessario per controllare la greificazione in cotto.
I feldspati, i feldspatoidi, le sabbie feldspatiche sono gli elementi fondenti, che contribuiscono con la formazione di fasi vetrose alla densificazione del materiale durante la cottura.
Infine, il quarzo è l’elemento equilibratore delle fasi vetrose in quanto ne regola la viscosità e contiene il ritiro del materiale in cottura.
Come già descritto, le composizioni per monocottura da pavimento possono essere costituite da impasti bianchi o rossi [6]. Questi ultimi, a differenza di quelli bianchi, sono composti prevalentemente da argille ad alto contenuto di ferro e da modeste percentuali di materiali complementari, quali feldspati e quarziti.
Gli impasti cuocenti bianco risultano invece miscele più articolate e ponderate di diverse materie prime quali: argille esenti da ossidi di ferro, feldspati, feldspatoidi, quarzo, ecc.
La Fig. 9.4 riporta gli intervalli composizionali per impasti cuocenti bianco e rosso.
Monocottura greificata (bianca)
Argille plastiche
10 ÷ 15%
Argille semi plastiche 15 ÷ 25%
Argille caolinitiche 10 ÷ 20%
Feldspatoidi
25 ÷ 35%
Feldspato 5 ÷ 10%
Quarzo
10 ÷ 15%
Talco - Dolomite 1 ÷ 2%
Monocottura greificata (rossa)
Argille rosse greificabili
Feldspatoidi
70 ÷ 80%
15 ÷ 20%
Quarzo 10 ÷ 20%
Fig. 9.4 Composizioni degli impasti per monocottura greificata bianca e rossa
Gli impasti bianchi per la natura delle materie prime usate presentano in cottura valori di stabilità dimensionale e porosimetrica in generale più ampia rispetto a quelli rossi, grazie al maggior contenuto di allumina ed alla minor presenza di ossidi di ferro. Di conseguenza, i diagrammi di greificazione sono sostanzialmente differenti tra impasti rossi e bianchi (vedi Fig. 9.5).
L’aumento della contrazione lineare (ritiro) e la diminuzione dell’assorbimento d’acqua si sviluppano in forma meno graduale ed a temperature inferiori nel grès rosso rispetto al grès bianco. Infatti, nel primo caso si ha un forte sviluppo di fase liquida, generata principalmente da minerali illitici ferrosi che conferiscono basse viscosità al sistema. Tale comportamento è valido soprattutto per gli impasti rossi costituiti principalmente da minerali argillosi (75 ÷ 80%). Nel caso invece di impasti compositi, in cui le argille rosse vengono miscelate con feldspati e quarzo, il comportamento in cottura (variazioni di ritiro e porosità) risulta più simile a quello degli impasti chiari.
9.04 IT
Intervallo di cottura
Ritiro impasto rosso
Intervallo di cottura
Ritiro impasto bianco
impasto bianco
impasto rosso
Fig. 9.5 Diagramma di greificazione di impasti bianchi e rossi
La Tab. 9.5 mostra diverse formulazioni d’impasto da monocottura greificata così suddivise: sette composizioni di impasto cuocente chiaro, di cui tre ottenute con fondente di tipo prevalentemente sodico (A) e quattro con fondenti di tipo potassico (B), e quattro composizioni di monogreificata rossa (C).
Fig. 9.05 IT
Tab. 9.5 Esempi di composizioni di impasti greificabili bianchi e rossi
Materie prime per smalti
Le caratteristiche fisiche degli smalti, quali resistenza all’usura, resistenza agli acidi ed alle basi, condizionano e determinano le prestazioni del prodotto finito nei differenti ambienti di utilizzo. Di conseguenza si sono sviluppati e si stanno tuttora ricercando superfici vetrose con proprietà fisico-meccaniche e chimiche migliorative.
Le caratteristiche di uno smalto dipendono essenzialmente dalle materie prime con cui lo stesso è formulato ed anche dalla temperatura di cottura.
La formulazione di uno smalto si basa sull’utilizzo di fritte di differente natura (a media e alta viscosità) e di fritte a fusione eutettica (calcio e zinco), in percentuale variabile.
Assieme alle fritte vengono introdotti negli smalti altri materiali naturali e sintetici con caratteristiche specifiche, in funzione della qualità superficiale (tessitura) e delle proprietà dei vetri che si vogliono ottenere.
Alcuni di questi componenti non frittati come i feldspati, la nefelina ed il quarzo contribuiscono congiuntamente con le fritte a formare la matrice vetrosa dello smalto [7].
Altri invece come la sabbia di zirconio o il corindone, dissolvendosi solo parzialmente nel vetro, contribuiscono a migliorare la resistenza all’abrasione e agiscono come opacificanti (silicato di zirconio) o come mattizzanti (ossido di alluminio).
Altri componenti che si usano nella formulazione degli smalti sono: - anatasio (biossido di titanio) che oltre ad essere un mattizzante esercita un effetto positivo sulle proprietà meccaniche e chimiche del vetro; - wollastonite e carbonato di calcio e magnesio, minerali a base alcalino-terrosa che agiscono come mattizzanti e possono contribuire a formare parte della matrice vetrosa.
Oltre che dalla natura chimica e mineralogica delle materie prime, le caratteristiche finali di un prodotto dipendono in modo rilevante anche dai parametri tecnologici adottati nel corso del processo di lavorazione.
Nel definire quindi le condizioni di lavoro in un determinato processo è molto importante valutare le relazioni che intercorrono tra gli aspetti tecnologici, di natura chimico fisica, ed i parametri di lavorazione nelle varie fasi del processo, quali:
- macinazione
- atomizzazione
- pressatura
- essiccamento
- smaltatura
- cottura
che vengono di seguito analizzate in dettaglio.
Macinazione
Ha come obiettivo la comminuzione ed omogeneizzazione delle materie prime in ingresso allo stabilimento ceramico, al fine di ottenere granulometrie finali dei semilavorati costanti.
Nel caso di impasti greificati il grado di macinazione delle materie prime, congiuntamente ad altri fattori di ordine chimico e fisico possono influenzare il grado di greificazione del materiale e quindi condizionare sensibilmente i valori di ritiro e porosità.
Il grafico riportato in Fig. 9.6 evidenzia chiaramente le variazioni di ritiro che si riscontrano nel prodotto finale, utilizzando come semilavorato barbottine di partenza uguali a livello composizionale, però con residui di macinazione differenti. Il grafico mette in risalto anche l’entità di queste variazioni nel caso di cottura del prodotto a differenti temperature.
Residuo 2,4%
Residuo 3,7%
Residuo 6,4%
Fig. 9.6 Ritiro di cottura in funzione del residuo di macinazione (setaccio a 63 µm)
Fig. 9.06 IT
In linea di massima il residuo dopo macinazione, per impasti greificati compositi, risulta nell’ordine del 7 ÷ 10% (setaccio 63 µm = 230 mesh). Nel caso di impasti rossi costituiti principalmente da materiali argillosi, tale valore può diminuire fino al 4 ÷ 6%.
Atomizzazione
Tale processo è finalizzato all’evaporazione parziale dell’acqua contenuta nella barbottina congiuntamente alla formazione di granuli sferoidali.
La distribuzione granulometrica di un impasto per monocottura greificata non risulta particolarmente differente dagli altri semilavorati atomizzati idonei ad ottenere ad esempio il grès porcellanato o i rivestimenti porosi.
Nella Fig. 9.7 viene proposto un grafico rappresentativo delle fasce granulometriche tipiche per un impasto da monocottura greificata. Come si può notare si riscontra una forte concentrazione di particelle (oltre 60% del totale) nella fascia compresa tra 250 ÷ 425 µm.
Fig. 9.7 Granulometria di un atomizzato per monocottura greificata (valori indicativi)
Pressatura
L’obiettivo della formatura tramite pressatura è quello di ottenere in crudo la massima densificazione delle polveri, compatibilmente con i problemi di “cuore nero” o di degasazione che possono manifestarsi in cottura. È chiaro che differenti pressioni specifiche di formatura esercitate sulle polveri, portano ad ottenere valori distinti di densità apparente del pezzo pressato e, conseguentemente valori diversi della contrazione e della porosità dopo cottura. Con riferimento ad un impasto cuocente bianco, i grafici riportati in Fig. 9.8 evidenziano l’andamento del ritiro e della porosità (AA) al variare della pressione di formatura e della temperatura di cottura.
Fig. 9.07 IT NEW
Generalmente la pressione di formatura è nell’ordine di 250 ÷ 350 daN/cm2 per gli impasti bianchi; per quelli cuocenti rosso, tendenzialmente più plastici, la pressione può risultare anche inferiore, nell’ordine dei 200 daN/cm2 (1 daN/cm2 equivale a circa 1 kgF/cm2).
Fig. 9.8 Variazione del ritiro e dell’assorbimento d’acqua in funzione della pressione di formatura e della temperatura di cottura
Essiccamento
Fig. 9.08 IT
Questa operazione è considerata oggi apparentemente semplice in quanto i fenomeni fisici che si verificano durante l’evaporazione dell’umidità residua dell’impasto (4 ÷ 7%) sono ormai noti e controllabili.
Tuttavia, è bene considerare che impasti prevalentemente caolinitici, tendono ad evidenziare comportamenti dimensionali in espansione dopo essiccamento, dimostrando modesti valori di resistenza meccanica sull’essiccato e ciò può comportare la formazione di crepe nei pezzi qualora la velocità di essiccamento sia troppo rapida.
Con i cicli oggi in uso è opportuno che le variazioni dimensionali dopo essiccamento siano tendenti a 0% o preferibilmente in leggera contrazione, fino a valori massimi dell’ordine di 0,3%; la resistenza meccanica dei pezzi essiccati deve invece essere superiore ad almeno 2,5 N/mm2
Smaltatura
Le numerose tipologie di smalti oggi presenti nei prodotti commercializzati dalle aziende ceramiche sono riconducibili alle seguenti famiglie:
- brillanti
- matt (devetrificati)
- rustici.
Per la smaltatura vengono impiegate tecniche di applicazione sia a secco che ad umido.
Nell’applicazione a umido vengono usate cabine airless, smaltatrici a campana o a vela. Nel caso invece dell’applicazione per via secca (graniglie di opportune fasce granulometriche) vengono utilizzati i granigliatori.
L’uso degli smalti richiede quindi flessibilità nelle metodologie di lavoro e adeguate configurazioni delle linee di smaltatura. La combinazione di diverse tipologie di smalti da applicare nel medesimo prodotto richiede conoscenze specifiche per ciascun semilavorato.
Nei prodotti da pavimento, vi sono tipologie che richiedono applicazioni semplici, con il dosaggio dello smalto tramite cabina airless in basse quantità (da 100 a 500 g/m2), mentre altre tipologie richiedono applicazioni più complesse, con quantità di smalto fino a 2 kg/m2 In tal caso gli smalti possono essere applicati sia ad umido (con densità da 1,2 a 1,9 kg/L) che a secco con granulometrie variabili. Spesso sono anche necessarie applicazioni di collanti e riservanti organici.
Notevoli quindi sono le sollecitazioni a cui viene sottoposto il supporto crudo, che deve essere capace di assorbire elevati quantitativi di acqua e contemporaneamente non deve flettere eccessivamente per non compromettere le successive operazioni di decorazione digitale e le fasi di movimentazione fino alla cottura.
È importante sottolineare come la tecnologia di decoro digitale abbia permesso di decorare le piastrelle “non a contatto” riducendo le difettologie ed ottenendo definizioni grafiche di elevato grado estetico. Inoltre i limitati quantitativi dei colori applicati con le stampanti digitali e la loro elevata efficienza hanno portato ad una riduzione dei costi per metro quadrato di prodotto.
Per mantenere una buona resistenza all’usura dei prodotti da pavimento si è quindi diffusa l’applicazione di smalti protettivi trasparenti ad effetto lucido o matt subito dopo la decorazione digitale.
Cottura
L’utilizzo di curve di cottura ottimali permette il corretto sviluppo delle reazioni di fusione e cristallizzazione dei componenti l’impasto e lo smalto. Tali reazioni contribuiscono all’ottenimento dei parametri di assorbimento e contrazione del supporto, con incremento delle caratteristiche tecniche e dei contenuti estetici della superficie smaltata. Come noto, le condizioni di greificazione di una massa ceramica dipendono non solo dalla natura chimico-fisica dell’impasto in questione, ma anche dal grado di macinazione, dalla densità apparente del pressato e dalla temperatura massima di cottura.
Densità [g/cm3] 1,82 1,92 2,02
Fig. 9.9 Influenza della densità (in verde) su ritiro e assorbimento dopo cottura per un impasto greificato rosso
Nel grafico di Fig. 9.9 si evidenzia l’influenza della densità apparente (in crudo) sui valori di porosità (assorbimento d’acqua) e ritiro dopo cottura di un impasto rosso greificabile.
Assorbimento Acqua AA Fig. 9.09 IT
Nel valutare le caratteristiche dell’impasto risulta molto importante analizzare la stabilità dimensionale e le variazioni della porosità nel caso di cotture effettuate a differenti cicli e gradienti di temperatura.
La Fig. 9.10 riassume l’andamento del carico di rottura, del ritiro e dell’assorbimento d’acqua al variare della temperatura di cottura per un impasto da monocottura greificata. Nello stesso grafico viene evidenziato l’intervallo (4) relativo alla temperatura ottimale di cottura.
Fig. 9.10 Carico di rottura (1), assorbimento (2) e ritiro (3) in funzione della temperatura di cottura; l’area (4) rappresenta l’intervallo di cottura ottimale per prodotti greificati
La dinamicità del settore ceramico è stata determinante nel favorire la continua evoluzione dei processi di produzione. Negli anni ‘70 è avvenuta la trasformazione tecnologico-impiantistica con il passaggio dei processi produttivi dalla bicottura alla monocottura. L’adozione di questa tecnologia, con cicli rapidi in forni a rulli, ha permesso di rivoluzionare i lay-out delle fabbriche ceramiche.
La riduzione dei tempi di cottura determinata dall’impiego dei forni a rulli monostrato ha comportato l’introduzione di una maggiore automazione, con una conseguente riduzione dei costi diretti industriali.
Parallelamente si è assistito ad una evoluzione dei processi produttivi nelle diverse fasi di lavorazione, attraverso le seguenti innovazioni:
- diffusione della macinazione ad umido discontinua
- introduzione del processo di atomizzazione
- diffusione delle presse idrauliche
- adozione degli essiccatoi rapidi
- diffusione dei forni a rulli.
Già negli anni ‘90 queste tecnologie si sono consolidate con l’aumento delle capacità produttive, determinato dalla forte domanda del mercato, e hanno permesso una diminuzione dei costi di fabbricazione pur incrementando la qualità estetica delle piastrelle ceramiche.
Gli impianti oggi possono presentare diversi livelli di sofisticazione, dipendendo appunto dalle automazioni introdotte, e dai sistemi di controllo addottati (supervisore di reparto, supervisione di impianto, ecc.).
I fattori più importanti da considerare per valutare il grado di complessità di un impianto sono in genere:
- il livello tecnologico e culturale locale
- le dimensioni dell’impianto (produttività)
- la flessibilità richiesta all’impianto.
Nella Fig. 9.11 si riporta un diagramma a blocchi relativo al processo di monocottura greificata, dove vengono evidenziate le varie fasi di lavorazione. Il processo indicato prevede l’uso della macinazione in continuo, oramai diffusa anche in impianti di dimensioni medio-piccole. La pressatura è effettuata con presse idrauliche ad alto tonnellaggio, l’essiccamento con essiccatoi rapidi automatizzati (verticali o orizzontali), la cottura rapida in forni a rulli, la scelta con macchine completamente automatizzate.
L’adozione di dispositivi elettronici ha permesso di automatizzare completamente la movimentazione dei carrelli di stoccaggio piastrelle crude e cotte, con l’impiego di sistemi laser a guida autonoma.
Materie prime
Dosaggio
Macinazione continua o discontinua
Atomizzazione
Stoccaggio polveri atomizzate
Pressatura
Essiccamento
Smaltatura e decorazione
Stoccaggio crudo
Cottura
Stoccaggio cotto
Scelta e imballo
Magazzino
Fig. 9.11 Diagramma a blocchi del processo di monocottura
Dalle considerazioni sopra riportate appaiono evidenti gli effetti della rapida evoluzione tecnologico-impiantistica negli ultimi decenni che si possono riassumere in:
Fig. 9.11 IT
- aumento della produttività (per addetto)
- diminuzione dell’area occupata dall’impianto
- diminuzione dei costi industriali del prodotto.
Analizzando le varie fasi di lavorazione si può osservare lo stoccaggio sia del materiale crudo dopo la smaltatura che del cotto, come pure la possibilità di indirizzare direttamente il materiale crudo al forno o il cotto alla scelta.
Nella Fig. 9.12 è riportato il layout di un moderno impianto per monocottura greificata che addotta il processo di macinazione continua. All’estrema destra si vede la zona di dosaggio materie prime e preparazione impasto (macinazione e atomizzazione). Seguono quattro linee di pressatura, che alimentano due essiccatoi orizzontali multipiano, collegati in diretta con le due linee di smalteria e decorazione digitale. Le linee di smalteria sono a loro a volta direttamente connesse (senza stoccaggio intermedio) a due forni monostrato a rulli di grande lunghezza (oltre 150 m) e larghezza (2950 mm). Il prodotto cotto viene stoccato in un magazzino intermedio, inviato alle linee di squadratura a secco (quando necessario) e infine alla scelta ed al confezionamento.
dosaggio e preparazione impasto
squadratura (grandi formati)
cottura (forni monostrato) smaltatura e decorazione
scelta e confezionamento
stoccaggio (prodotto cotto)
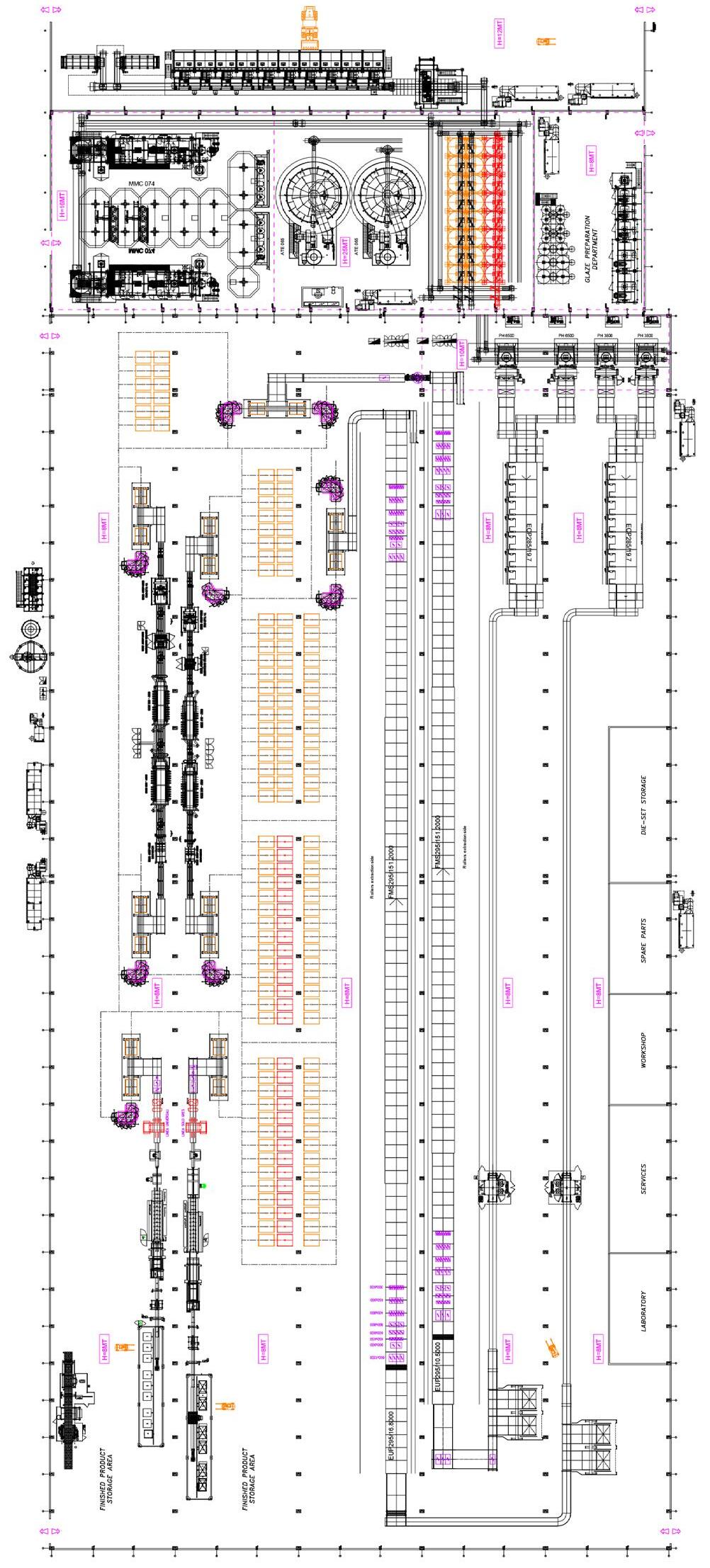
preparazione smalti
pressatura ed essiccamento
Fig. 9.12 Impianto per la produzione di piastrelle in monocottura greificata
Di seguito vengono sinteticamente descritte le macchine caratteristiche che compongono un impianto tipico per la produzione di piastrelle in monocottura greificata, che contempla nelle sue fasi di lavorazione il processo di macinazione ad umido e l’atomizzazione. Non sono state considerate le macchine per la macinazione a secco e rigranulazione delle polveri, talvolta utilizzate per produrre piastrelle da pavimento soprattutto in pasta rossa, ma generalmente meno diffuse.
Dosaggio materie prime
Il dosaggio può essere eseguito con macchine ed impianti che presentano livelli di automazione differenti. Nel caso del processo di macinazione discontinua, per il dosaggio delle materie prime si possono prevedere sistemi costituiti da cassoni pesatori singoli, dotati di celle di carico deformabili.
Nei processi di macinazione continua è generalmente previsto un sistema di pesatura dinamico su nastro gestito da un processore. La miscela dosata viene stoccata temporaneamente in un silo di precarica per essere poi immessa nel mulino continuo.
In alcuni casi, quando le caratteristiche delle argille lo richiedono, si può prevedere la scioglitura preventiva di una parte dei materiali argillosi. In questo caso la sospensione argillosa, che contiene già in toto o in parte il fluidificante, può essere indirizzata, dopo opportuno dosaggio, all’ingresso del mulino.
La macinazione ad umido dell’impasto può essere effettuata con il processo di tipo continuo o discontinuo. La scelta del tipo di macinazione dipende da diversi fattori quali ad esempio:
- dimensione dell’impianto
- professionalità delle maestranze
- caratteristiche delle materie prime.
Dal punto di vista tecnologico i vantaggi più immediati della macinazione continua sono:
- maggior costanza delle caratteristiche della barbottina
- aumento della densità
- migliori caratteristiche reologiche
- automazione totale del processo.
Dal punto di vista tecnico-organizzativo si riscontra quindi una migliore razionalità nella gestione dell’impianto. Riguardo all’aspetto economico, la macinazione continua comporta un risparmio diretto sul personale impiegato ed indiretto sull’energia necessaria all’evaporazione dell’acqua delle barbottine all’atomizzatore, in quanto è possibile gestire con questo processo barbottine aventi maggiore contenuto di solido rispetto al sistema di macinazione discontinuo.
Allo scarico del mulino viene effettuato un controllo della barbottina prima con un setaccio grossolano, poi con una batteria di setacci più fini che garantiscono il taglio granulometrico desiderato. Il residuo di setacciatura viene immesso nell’acqua predosata destinata al carico del mulino, mentre la barbottina filtrata viene inviata ad una vasca di raccolta munita di agitatore, dalla quale può essere direttamente prelevata ed inviata all’atomizzatore.
Qualora le caratteristiche fisiche delle materie prime lo permettano (bassi valori di residuo a 63 micron) si può prevedere la scioglitura di una parte delle argille ed eventualmente dello
scarto crudo in un turbo dissolutore gestito in parallelo al mulino evitando il passaggio di questi materiali dal mulino stesso. ln alternativa, si può anche prevedere una scioglitura preliminare delle materie prime argillose che saranno successivamente inviate alla macinazione nel mulino con gli altri componenti dell’impasto. Questa opzione si dimostra particolarmente interessante soprattutto nei casi in cui gli impasti siano costituiti da una frazione rilevante di argille fortemente plastiche e con umidità naturale relativamente alta (superiore a 18 ÷ 20%).
L’atomizzazione è il processo nel quale avviene la pressoché totale evaporazione dell’acqua contenuta in una barbottina. La diminuzione del contenuto d’acqua sino a valori del 4 ÷ 7% viene effettuata mediante il processo di essiccamento a spruzzo. L’operazione si effettua iniettando la barbottina dal basso verso l’alto mediante pompe ad elevata pressione (16 ÷ 22 bar) e successiva nebulizzazione all’interno della camera cilindrica d’essiccamento dell’atomizzatore, impiegando ugelli di varie dimensioni opportunamente distanziati tra loro. Il processo di macinazione e atomizzazione svolge anche la funzione di “digestore” delle acque di lavaggio, dei fanghi di varia provenienza e di tutte le polveri di filtraggio e pulizia dell’impianto, in quanto vengono smaltite in modo controllato nella barbottina stessa dell’impasto ceramico.
L’atomizzatore è solitamente dotato di filtri a secco o ad umido per abbattere, entro i limiti richiesti dalle norme di legge vigenti nei diversi paesi, le polveri più fini che altrimenti si disperderebbero in atmosfera assieme al vapor d’acqua in uscita dal camino.
Se ritenuto opportuno, la macchina può essere alimentata anche con aria calda proveniente da sistemi di cogenerazione al fine di diminuire i costi energetici d’esercizio.
Con l’atomizzatore la barbottina viene trasformata in polvere a granulometria ed umidità controllata (es. 6%), idonea ad essere convogliata, attraverso nastri trasportatori nei silos di stoccaggio.
Pressatura
Questa fase del processo risulta molto importante nella tecnologia produttiva dei materiali ceramici in quanto conferisce la forma al manufatto da realizzare. Normalmente nell’operazione di formatura, si cerca di ottenere il massimo addensamento delle particelle in crudo, compatibilmente con i problemi di degasazione e di “cuore nero”. Le pressioni di formatura generalmente adottate per i prodotti da pavimento vanno da 200 a 350 daN/cm2, o anche maggiori. Le presse idrauliche attualmente disponibili risultano particolarmente adatte alla formatura di questi materiali e allo stesso tempo permettono di garantire elevate affidabilità e ridurre al minimo l’assorbimento d’energia.
Vanno anche considerati alcuni importanti accessori che sono parte integrante della pressa: gli stampi e i carrelli di alimentazione polveri.
Gli stampi generalmente utilizzati nella formatura di questo prodotto (monogreificata) sono del tipo a punzoni entranti, ma per formati maggiori del 60x60 cm spesso si preferisce l’impiego di stampi a formatura superiore (SFS); i punzoni sono normalmente rivestiti in gomma per diminuire la sporcabilità e quindi la frequenza di pulizia. L’utilizzo della gomma è anche particolarmente idoneo per ottenere effetti superficiali di tipo strutturato. Il punzone “lato marca” può essere del tipo tradizionale (rigido) oppure di tipo isostatico per ottimizzare l’omogeneità di pressatura e quindi ottenere valori di densità apparente uniformi nei vari punti della piastrella.
Nella formatura dei materiali ceramici greificati, oltre alla forza di pressatura è fondamentale un caricamento omogeneo delle polveri all’interno dell’alveolo dello stampo; i caricamenti anomali generano infatti problemi di geometria nella piastrella (ortogonalità e calibro). Questa problematica viene risolta con l’utilizzo di specifici dispositivi preposti all’alimentazione delle polveri nello stampo, in sinergia con i tamponi isostatici, permettendo una notevole riduzione dei difetti geometrici nelle piastrelle soggette a ritiro in cottura (per approfondimenti si veda il Paragrafo 3.2 del Volume II).
Essiccamento
L’essiccamento è la fase di lavorazione in cui si ha la eliminazione dell’umidità residua di pressatura nelle piastrelle appena formate. Questa operazione può essere effettuata mediante essiccatoi verticali e orizzontali. Le piastrelle in uscita dalle presse sono raccolte da linee a rulli ed inviate agli essiccatoi.
Nel caso di essiccatoi verticali i cicli d’essiccamento risultano nell’ordine di 35 ÷ 70 min, nel caso invece degli orizzontali i cicli possono essere molto più rapidi, da 6 a 20 min. In entrambi i casi però la durata del ciclo dipende dal tipo d’impasto, dalla dimensione e dallo spessore dei pezzi.
Smaltatura e decorazione
Le esigenze del mercato mondiale della piastrella, sempre più sofisticate ed articolate, richiedono macchine ed attrezzature per realizzare molteplici applicazioni di smalto e decoro. Parallelamente si riscontra la necessità di limitare le attività manuali nelle operazioni di smaltatura e decorazione, a favore di linee totalmente automatizzate.
A riguardo, un elevato grado di flessibilità dalle linee di smaltatura è stato certamente ottenuto grazie alla decorazione digitale che ha permesso rapidi cambi dei prodotti da realizzare. Dall’iniziale sviluppo di linee di smaltatura molto lunghe, si è passati quindi ad una netta semplificazione impiantistica, mantenendo comunque la possibilità di realizzare una vasta gamma di prodotti.
Ad esempio, per ottenere effetti materici caratteristici, di tipo anticato, rustico o pietra, sono necessarie le seguenti applicazioni: smaltobbio con cabina airless, decoro digitale e smalto protettivo con cabina airless. Invece, per ottenere superfici brillanti con stesura ottimale, i dispositivi più idonei all’applicazione dell’ingobbio e dello smalto risultano tuttora essere i sistemi a campana e a vela per formati di larghezza fino al 1200 mm.
Riguardo alla decorazione, considerando le tipologie grafiche oggi richieste, si impiegano quasi unicamente stampanti digitali a getto d’inchiostro. Queste si sono rapidamente diffuse poiché, oltre alla rapidità nei cambi di prodotto da realizzare, garantiscono una alta definizione dell’immagine stampata e necessitano di interventi di controllo ed assistenza limitati. Esistono diversi modelli di macchine digitali la cui distinzione è basata essenzialmente sulla larghezza di stampa e sul numero di barre colore utilizzabili. Tra le caratteristiche più interessanti della decorazione digitale vi è indubbiamente la stampa non a contatto dell’immagine da riprodurre sul pezzo da decorare e la elevata definizione.
Attualmente si sta assistendo ad una evoluzione dei concetti di automazione delle linee di smaltatura, grazie all’impiego di dispositivi per il controllo dinamico del processo e di opportune piattaforme di gestione elettronica dei dati.
La sempre maggiore comprensione del comportamento di materie prime, impasti, ingobbi e smalti, e l’esperienza acquisita hanno semplificato e standardizzato il processo di cottura. Le macchine termiche sono oggi dotate di apparecchiature sempre più sofisticate che permettono di effettuare regolazioni e controlli della temperatura molto accurati.
Le sezioni dei forni sono aumentate, e di pari passo sono migliorati i sistemi di combustione idonei a mantenere costante ed omogenea la temperatura nell’intera sezione. Oltre ai bruciatori ad alta velocità sono disponibili bruciatori capaci di distribuire la fiamma in zone specifiche della sezione del forno. Ciò consente una distribuzione termica ottimale in relazione ai materiali da cuocere specie nelle zone critiche delle curve di cottura.
L’adozione della cottura rapida su rulli, mono o bi-canale, ha facilitato la movimentazione delle piastrelle favorendo la realizzazione di formati di maggiori dimensioni con elevata flessibilità produttiva.
I cicli e le temperature di cottura adottate risultano compresi in linea di massima tra 35 ÷ 60 min e 1100 ÷ 1200 °C rispettivamente, dipendendo dalla natura delle masse ceramiche da cuocere e dalle dimensioni e spessore delle piastrelle stesse.
Nonostante il preciso controllo delle temperature del forno si possono riscontrare variazioni del ritiro di cottura nelle piastrelle prodotte dovute a molti fattori (materie prime impasto, residuo di macinazione, ecc.) che nel caso di formati grandi rendono difficoltoso mantenere le dimensioni all’interno di intervalli molto stretti. Per questo motivo con impasti a medio-alto ritiro si utilizzano linee di rettifica per squadrare il prodotto dopo la cottura e mantenere la geometria e le dimensioni entro i ristretti limiti richiesti dal mercato.
Nella Fig. 9.13 sono riportati due esempi tipici di curve di cottura. La curva verde risulta normalmente adatta per impasti greificati standard, la curva rossa invece per impasti sensibili al problema del “cuore nero”. In questa seconda curva il materiale permane più tempo nell’intervallo 600 ÷ 1000 °C per consentire la fuoriuscita dei gas derivanti dalla combustione dei composti organici e dalla ossidazione della massa ceramica.
di cottura [min]
Fig. 9.13 Curve di cottura per monocottura greificata: impasti standard (verde) e impasti sensibili al “cuore nero” (rossa)
Fig. 9.13 IT NEW
La precisione nella predisposizione e misurazione delle curve di cottura, congiuntamente al rispetto delle condizioni di esercizio del forno è assicurata da sistemi di controllo elettronico che garantiscono il mantenimento della temperatura prestabilita entro limiti molto ristretti. Il sistema di controllo del forno esegue inoltre la supervisione e la memorizzazione dei dati di processo e provvede autonomamente alla sua gestione.
Scelta
L’operazione della scelta, pur non intervenendo sulle caratteristiche del prodotto, costituisce un momento molto importante del ciclo di lavorazione. Anche in questo reparto è stata sviluppata la completa automazione delle operazioni fino all’inscatolamento e successiva pallettizzazione. La geometria delle piastrelle (calibro e planarità) può essere verificata con sistemi di ispezione elettronici (telecamere, sensori laser, ecc.), indirizzando poi il prodotto verso le specifiche uscite. L’intervento dell’operatore, negli impianti più automatizzati, è limitato all’analisi dei difetti estetici ed alla codifica delle piastrelle per definirne la classe di appartenenza.
Frequentemente, le operazioni di scelta sono precedute dalla squadratura a secco dei lati delle piastrelle, in particolare nel caso di formati medio-grandi (ad es. 60×60 cm o superiori).
Movimentazione e stoccaggio
Oltre ai tradizionali impianti di movimentazione dei box intermedi di stoccaggio con veicoli a guida automatica si stanno sempre più diffondendo gli impianti di movimentazione con stoccaggio minimo alimentando in diretta al forno. Il prelievo delle piastrelle cotte viene poi eseguito con dispositivi muniti di piani aspiranti o ventose che prevedono l’impiego di panconi, sui quali vengono accumulate le piastrelle. Questi sono poi convogliati alle linee di scelta attraverso carrelli trasportatori che si muovono su superfici piane tramite LGV (laser guided vehicle). Una stazione computerizzata sovrintende e controlla i flussi dell’intero sistema di trasporto, monitorando in tempo reale i flussi produttivi da e per lo stoccaggio delle piastrelle.
La piastrella in monocottura per pavimento, dopo decenni di continuo sviluppo ed aggiornamento tecnologico, ha sicuramente raggiunto valenze tecniche ed estetiche di ottimo livello, che possono trovare ancora notevoli potenzialità di miglioramento grazie alle tecnologie digitali.
D’altra parte, le prestazioni tecniche e strutturali del prodotto ceramico sono generalmente superiori alle esigenze richieste dai capitolati edili ed anche i suoi contenuti estetici sono tali da competere con successo con materiali alternativi.
Risulta pertanto evidente che la pavimentazione ceramica unisce aspetti tecnici, estetici ed economici di grande competitività.
[1] T. Emiliani, G. Ortelli e G. Biffi, «Single-firing of single-pressed and glazed wall or floor tiles», in 2nd CIMTEC, 1973.
[2] A. Baudran e R. Ducarre, «Single-firing. Actual problems and solutions», in 2nd CIMTEC, 1973.
[3] Società Italiana per la Ceramica (Assiceram), Scritti di Maurizio Korach, Faenza Ed., 1977.
[4] ISO, International Organization for Standardization, «ISO 13006 Ceramic tiles – Definitions, classification, characteristics and marking», 2018.
[5] ISO, International Organization for Standardization, «ISO 10545 Ceramic tiles – Part 1-20», 2014-2022.
[6] P. G. Burzacchini, «Monocottura e cottura rapida», Ceramica Informazione, n. 230, pp. 283-288, 1985.
[7] N. Tozzi, «The use of miscellaneous feldspar materials in the preparation of glazes for fast single firing», in Tecnargilla International Forum, Rimini, 1989.
Durante la cottura di piastrelle ceramiche, qualora l’impasto contenga percentuali importanti di materia organica, può presentarsi un alone scuro internamente alla piastrella, conosciuto con il nome di “cuore nero”. Tale variazione di colore, più o meno estesa, è determinata da una incompleta ossidazione del materiale ceramico a causa appunto dei composti organici che sottraggono ossigeno. Nel caso dei processi di monocottura rapida, sia per materiali tipo pavimento che rivestimento, questo difetto è accentuato dai cicli di cottura molto rapidi e, nella fase di pressatura, dall’utilizzo di pressioni di formatura molto elevate. Anche l’uso di tenacizzanti nell’impasto, come pure l’applicazione di colle e riservanti, contribuiscono ad esasperare questo difetto, in quanto comportano un incremento del contenuto di materia organica nella massa.
Particolare finitura della superficie ceramica, atta a conferire una apparenza di prodotto usurato dal tempo e, nel caso del pavimento, dal calpestio. L’effetto è ottenuto normalmente con superfici non planari, opportunamente strutturate (con punzoni di pressatura sagomati) e talvolta anche con bordi irregolari, non rettilinei (prodotti “rustici”). Anche le applicazioni di smalti e decori imitano il passare del tempo, con macchie e incrostazioni casuali riprodotte in massa o mediante decorazione digitale.
Metodo per la valutazione della resistenza all’abrasione superficiale di piastrelle smaltate, attraverso una procedura accelerata di prova a strisciamento rotante di corpi duri abrasivi. In funzione del numero di giri effettuati, i campioni vengono classificati dalla Classe 0 (la meno performante) alla Classe 5 (la più elevata) in base alla alterazione della superficie; i materiali idonei per piastrelle da pavimento solitamente ricadono nelle Classi 4 e 5.
Il metodo di prova è definito dalla norma ISO 10545-7, ed è commercialmente conosciuto anche come indice PEI, dal nome del Porcelain Enamel Institute (USA).
Con materiale greificato o “grès” si intende una ceramica con una bassa porosità residua, ovvero caratterizzata dalla formazione in cottura di fasi vetrose (vetrificazione) che densificano il materiale ceramico aumentandone la resistenza meccanica. Ovviamente questo processo di sinterizzazione comporta un ritiro elevato (5 ÷ 8%) con la conseguente generazione di calibri diversi sul prodotto finito in funzione del grado di omogeneità della formatura e del processo di cottura.
Con monocottura greificata si intende quella particolare tipologia di piastrelle smaltate da pavimento, sinterizzate con una unica cottura (supporto e smalto), che presentano un grado di greificazione mediamente elevato (assorbimento d’acqua compreso tra 0,5 ÷ 6%) e conseguentemente caratteristiche meccaniche idonee per pavimentazioni.
Il grès fine porcellanato, o più comunemente grès porcellanato [1] [2], è un prodotto con ottime caratteristiche tecniche in quanto presenta una elevatissima resistenza meccanica con valori di assorbimento d’acqua estremamente contenuti, spesso inferiori allo 0,1%.
Pertanto, nel contesto dell’industria delle piastrelle ceramiche il grès porcellanato ha assunto un’importanza sempre crescente, in relazione alla sua espansione da segmenti di mercato inizialmente ridotti per quantità e specializzazione applicativa, a destinazioni d’uso sempre più diversificate; il risultato è stato uno straordinario incremento dei volumi produttivi. Il prodotto, che in origine veniva considerato unicamente per le sue caratteristiche tecniche, ha invece dimostrato notevoli potenzialità estetiche che hanno consentito la penetrazione in numerosi contesti applicativi.
Sotto il profilo ceramico, il grès porcellanato trae origine da tipologie produttive già in uso nel passato, come il grès chimico o il klinker. Il suo recente sviluppo è stato possibile grazie all’introduzione di composizioni chimico-mineralogiche innovative, adeguate alle moderne tecnologie come la formatura di polveri ad elevate pressioni, le recenti tecniche di smaltatura e decorazione e, non ultima, la cottura rapida in forni a rulli monostrato.
La Fig. 10.1 mostra la distribuzione complessiva della produzione ceramica mondiale suddivisa tra le categorie: rivestimento, pavimento (in monocottura) e grès porcellanato principalmente impiegato per pavimentazione ma utilizzabile anche per rivestimento. Come si può notare, i prodotti in grès porcellanato rappresentano la fetta predominante della produzione mondiale, con circa il 54% del totale (pari ad oltre 8 miliardi di m2 annui).
Rivestimento
Pavimento (monocottura)
Grès porcellanato
Fig. 10.1 Distribuzione della produzione mondiale per tipologie produttive (stime 2019)
Senza dubbio il grès porcellanato, sviluppatosi a partire dai primi anni ‘90 come prodotto tecnico, è diventato il vero market leader tra i materiali ceramici destinati a pavimenti e rivestimenti in tutto il mondo.
Ad ulteriore conferma della qualità espressa dal grès porcellanato, nel grafico successivo (Fig. 10.2) si può constatare come la produzione italiana si sia concentrata quasi esclusivamente su questo materiale di fascia alta che ha raggiunto addirittura il 92% del totale delle piastrelle prodotte (dati 2023).
Fig. 10-01 IT
Rivestimento
Pavimento (monocottura)
Grès porcellanato
Fig. 10.2 Distribuzione della produzione italiana suddivisa per tipologie produttive (dati 2023)
Fig. 10-02 IT
Questo successo di mercato è certamente conseguenza delle ottime qualità intrinseche del prodotto (resistenza meccanica, ingelività, stabilità dimensionale, resistenza all’usura e agli agenti atmosferici), combinata alle valenze estetiche che, nella versione smaltata (divenuta predominante) permettono di qualificare il prodotto in ogni contesto applicativo con una conseguente grande diffusione commerciale.
La grande varietà di prodotti oggi realizzati in grès porcellanato comprende tutte le possibili tipologie estetiche (legni, marmi, pietre, cementi, ecc.) impiegate principalmente nelle pavimentazioni di interni, inclusi spazi prettamente commerciali come supermercati, hotel, aeroporti, ecc.
La possibilità di diversificare gli spessori rispetto ai valori standard di 10 ÷ 12 mm permette inoltre di impiegare il grès porcellanato anche per applicazioni da esterno ad elevato carico (pavimentazioni carrabili, pavimenti galleggianti, ecc.) utilizzando in tal caso prodotti a spessore 20 mm o anche 30 mm.
Viceversa, le versioni a spessore sottile possono trovare impiego come rivestimento delle pareti in abbinamento cromatico con i pavimenti a spessore standard, oppure anche per la ristrutturazione di pavimenti esistenti senza dover rimuovere il vecchio pavimento sottostante. Destinazione d’uso
Rivestimenti smaltati
INDOOR RESIDENZIALE
INDOOR COMMERCIALE
OUTDOOR
Pavimenti smaltati
Pavimenti tecnici
Rivestimenti grande formato
Pavimenti heavy duty
Rivestimenti facciate GRÈS
Tab. 10.1 Suddivisione merceologica dei principali utilizzi delle piastrelle ceramiche
La Tab. 10.1 mostra, in modo puramente indicativo, le attuali segmentazioni del mercato delle piastrelle ceramiche; per ogni destinazione d’uso (interno/esterno e residenziale/commerciale) si sono indicate le tipologie più diffuse, assieme ai formati e spessori di riferimento. Come si evince dalla tabella, il grès porcellanato ha oggi assunto una diffusione globale, spaziando sia nei formati che negli spessori, con molteplici finiture superficiali, e trovando così collocazione in tutti i segmenti di mercato come materiale di assoluta eccellenza.
La denominazione “grès porcellanato” è esplicativa nel richiamare le origini e le caratteristiche del prodotto; “grès”, nella terminologia ceramica, indica infatti un materiale a massa estremamente compatta, costituita da varie fasi cristalline immerse in una matrice vetrosa, mentre l’aggettivo “porcellanato” ha la stessa radice etimologica del termine “porcellana”, il materiale ceramico per eccellenza, noto ed apprezzato da secoli.
Secondo la norma internazionale ISO 13006 [3] le piastrelle ceramiche vengono classificate in vari gruppi a seconda del metodo di formatura e del valore di assorbimento d’acqua del prodotto finito (vedi Tab. 10.2). Le piastrelle in grès porcellanato appartengono al gruppo Bla, che comprende i materiali totalmente greificati, caratterizzati da valori di porosità inferiori allo 0,5%, espressi in termini di assorbimento d’acqua. In realtà i prodotti realizzati e commercializzati presentano in molti casi una porosità nettamente inferiore a quanto previsto dalla normativa, anche minore dello 0,1%.
Tipologia di prodotto Grès porcellanato Monocottura vetrificata
Pavimento semivetrificato
Pavimento poroso Rivestimento poroso
Tab. 10.2 Classificazione ISO 13006 delle piastrelle pressate a secco; in evidenza il gruppo di appartenenza del grès porcellanato [3]
Il grès porcellanato è, fra i materiali ceramici per pavimentazione, quello che meglio si presta ad applicazioni dove è richiesta una elevata resistenza all’usura, in virtù della sua elevata durezza superficiale. Inoltre, possiede ottime caratteristiche di antigelività, di resistenza meccanica a flessione e compressione, di resistenza all’attacco chimico e alle macchie, ecc.
I metodi di prova delle caratteristiche dimensionali, fisiche e chimiche delle piastrelle ceramiche sono definiti dalla norma ISO 10545 [4].
La Tab. 10.3 presenta un quadro esplicativo delle principali caratteristiche, mettendo a confronto i valori minimi prescritti dalle norme ed i valori reali generalmente rilevabili sui prodotti in commercio.
Caratteristiche
Assorbimento d’acqua
Norma Valori prescritti
10545-3
Carico di rottura MOR ISO 10545-4 > 35 N/mm2 > 50 N/mm2
Sforzo di rottura
ISO 10545-4 > 1300 N (spess. ≥7.5 mm) > 700 N (spess. <7.5 mm) > 2000 N (spess. ≥7.5 mm) > 1500 N (spess. <7.5 mm)
Resistenza abrasione profonda (UGL) ISO 10545-6 < 175 mm3 < 150 mm3
Resistenza abrasione superficiale (GL)
ISO 10545-7 dichiarata dal produttore dichiarata dal produttore
Coefficiente dilatazione ISO 10545-8 test disponibile 7,0 × 10-6
Resistenza sbalzi termici ISO 10545-9 test disponibile nessuna alterazione
Espansione all’umidità ISO 10545-10 test disponibile test disponibile
Resistenza cavillo ISO 10545-11 richiesta conforme
Resistenza al gelo ISO 10545-12 richiesta senza difetti visibili
Resistenza ai prodotti chimici
Resistenza agli acidi e alle basi
Resistenza macchie
Estrazione Pb e Cd dai vetri
Differenza di colore
ISO 10545-13 dichiarata dal produttore classe LA
ISO 10545-13 test disponibile dichiarata dal produttore
ISO 10545-14 classe 3 min classe 4
ISO 10545-15 test disponibile dichiarata dal produttore
ISO 10545-16 test disponibile dichiarata dal produttore
Tab. 10.3 Caratteristiche tecniche del grès porcellanato secondo la normativa ISO 13006 [3] rispetto ai valori di mercato
Altre caratteristiche interessanti sono l’igienicità del grès porcellanato (effetto batteriostatico) e la possibilità di rendere il prodotto antistatico; queste proprietà ne consentono l’uso in locali adibiti a servizi informatici, ambienti comunitari, mense, strutture sanitarie (es. sale operatorie).
Indubbiamente il grès porcellanato è di fatto la piastrella ceramica che presenta le migliori caratteristiche tecniche, in quanto è un prodotto fondamentalmente costituito di fasi cristalline ad elevata durezza, sinterizzato ad alta temperatura.
Relativamente alla sola tipologia del grès porcellanato tecnico (minoritaria rispetto al prodotto smaltato) va comunque sottolineato come un materiale ceramico eterofasico presenti una microporosità intrinseca, dovuta all’impossibilità di colmare, anche dopo cottura prolungata, i vuoti esistenti tra le particelle solide cristalline e la fase vetrosa circostante.
Il grès porcellanato rientra in questa categoria di materiali e presenta una microporosità, sia pur minima, che, se non adeguatamente controllata, può dare origine a fenomeni più o meno marcati di sporcabilità del prodotto tecnico (non smaltato). Infatti, anche se i valori di porosità aperta sono molto contenuti (circa 0,1% in termini di assorbimento d’acqua e 0,5% secondo la porosimetria a mercurio) la porosità chiusa interna è stimabile intorno al 6%, con dimensioni dei pori comprese tra 1 e 10 μm. Tale porosità chiusa viene portata alla luce dal processo di levigatura del grès tecnico, con l’asportazione di circa 0,5 ÷ 1 mm di strato superficiale, accentuando la macchiabilità del prodotto. Esistono diverse possibilità di intervento, per ridurre gli effetti o l’incidenza stessa delle microporosità residue sulle prestazioni del prodotto in condizioni di esercizio.
È stato ad esempio dimostrato che la penetrazione di agenti macchianti e la rimozione degli stessi è correlata alla forma e al diametro dei pori. Pori aperti di dimensioni relativamente elevate favoriscono una facile pulizia del prodotto, determinando però un decadimento visivo della superficie. Al contrario pori estremamente piccoli non consentono l’intrusione di sostanze estranee, e pertanto questa condizione è da preferirsi.
A tal fine, opportune regolazioni dei parametri di lavorazione possono incrementare sensibilmente il grado di densificazione del materiale, ad esempio mediante:
- un aumento della superficie specifica dei componenti l’impasto, agendo sul grado di macinazione specie dei materiali duri (quarzo e feldspati);
- un aumento della compattazione delle polveri, con l’impiego di alte pressioni di formatura;
- l’utilizzo di fondenti più energici, compatibilmente con la stabilità chimico-fisica degli impasti e la loro tendenza alla deformazione piroplastica.
Nel caso dei prodotti smaltati, non si riscontrano generalmente problematiche di macchiabilità della superficie in quanto la vetrosità dello smalto protettivo garantisce una totale impermeabilità. Tuttavia una certa attenzione è richiesta per le tipologie di prodotti smaltati lappati, per i quali comunque le microporosità superficiali sono minime rispetto al grès tecnico levigato. A riguardo va sottolineato come la lappatura sia molto più blanda rispetto ad una levigatura profonda e come le mole utilizzate permettano di ottenere una superficie liscia e compatta.
Formati
Storicamente la produzione del grès porcellanato tecnico si è affermata con i formati quadrati di piccole dimensioni, tipicamente 30×30 cm o 33×33 cm, che hanno rappresentato per un lungo periodo uno standard di prodotto.
Con la forte crescita del grès porcellanato smaltato, associata anche allo sviluppo impiantistico in particolare nelle fasi di formatura e decorazione, i formati si sono rapidamente evoluti nelle loro dimensioni massime, acquisendo indubbiamente un maggior pregio estetico ed architettonico.
Oggi la produzione italiana di grès porcellanato registra una prevalenza dei formati rettangolari rispetto ai quadrati, entrambi con dimensioni nettamente maggiori rispetto al passato. Più in dettaglio, il formato rettangolare prevalente è il 60×120 cm mentre il formato quadrato di riferimento è tuttora il 60×60 cm, con quote significative anche di 80×80 cm e 120×120 cm.
Inoltre, grazie allo sviluppo di nuove tecnologie produttive d’avanguardia, si è oramai consolidata anche la produzione di grandissimi formati (160×320 cm ed oltre), con la diffusione del grès porcellanato in segmenti di mercato ad alto valore aggiunto, come valida alternativa tecnica ed estetica rispetto alle lastre in pietra naturale (vedi Capitolo 11).
Da ultimo è importante evidenziare la crescente tendenza a lappare le superfici smaltate del grès porcellanato, con possibilità di realizzare finiture lucide, satinate o matt, che conferiscono in particolare ai grandi formati un aspetto estetico di grande impatto.
Tipologie
La combinazione degli aspetti tecnici e dei contenuti estetici determina, di fatto, la destinazione d’uso del grès porcellanato, come rappresentato schematicamente nella Fig. 10.3 che mostra, in generale, gli attuali settori applicativi di piastrelle e lastre ceramiche.
PAVIMENTI
residenziali commerciali outdoor
RIVESTIMENTI
pareti interne facciate esterne
ARREDI
piani cucina componenti per mobili
Fig. 10.3 Destinazioni d’uso dei prodotti in grès porcellanato È interessante notare come il grès porcellanato, originariamente sviluppatosi come pavimentazione tecnica non smaltata, si sia fortemente espanso nel settore residenziale, sostituendo gran parte della monocottura greificata, grazie al connubio tra le superiori caratteristiche tecniche e l’elevato pregio estetico determinato dalle nuove tecnologie di smaltatura e decorazione digitale.
Fig. 10-03 IT
A dimostrazione della notevole differenziazione dei prodotti oggi presenti nel mercato si riporta una tabella riassuntiva delle principali tipologie associate alle molteplici destinazioni d’uso sopra citate (Fig. 10.4).
PAVIMENTI
residenziali commerciali outdoor pareti interne facciate esterne piani cucina componenti per mobili
Grès porcellanato tecnico (non smaltato)
Tinta unita o sale-pepe Naturale
Tinta unita o sale-pepe Levigato
Grès porcellanato smaltato
Decorato Naturale
Decorato Strutturato
Decorato Lappato
Fig. 10.4 Tipologie di grès porcellanato e loro applicazioni tipiche
10-04 IT
Le materie prime usate per la formulazione di impasti per il grès porcellanato possono essere ricondotte ad alcuni gruppi di minerali specifici, ciascuno dei quali esercita una propria funzione caratteristica; in particolare le materie prime argillose conferiscono plasticità all’impasto, mentre quelle complementari, non plastiche, comprendono i minerali fondenti e quelli a funzione prevalentemente smagrante e strutturale.
Alla prima famiglia appartengono i minerali argillosi di natura illitico-caolinitica o montmorillonitica, con caratteristiche plastiche più o meno marcate in relazione alla propria struttura mineralogica e alla granulometria delle particelle. I minerali fondenti sono invece rappresentati da feldspati e feldspatoidi, talco, euriti, pegmatiti, ecc., mentre i componenti più refrattari, a funzione strutturale, sono il quarzo e le quarziti in genere.
Per tutte le materie prime utilizzate nelle formulazioni per grès porcellanato è richiesta una bassa concentrazione di ossidi coloranti, quali Fe2O3 e TiO2, in modo da evitare eccessivi sviluppi cromatici indesiderati.
I rapporti quantitativi fra i componenti dipendono dalla natura mineralogica e dalla granulometria dei minerali argillosi e, in ultima analisi, dalla loro reattività nei confronti dei minerali fondenti. Le Tab. 10.4 e Tab. 10.5 forniscono un quadro delle caratteristiche chimiche e fisiche delle composizioni e dei prodotti di grès porcellanato.
Tra le materie prime evidenziate in Tab. 10.4 viene riportato il solo feldspato sodico, ma possono essere usati anche feldspati misti sodico-potassici e lo stesso feldspato potassico con aumento della stabilità nella fase di greificazione. Materia
sodico
Quarzo
Dolomite
Tab. 10.4 Composizione delle materie prime per grès porcellanato
L’argilla plastica (B) apporta la necessaria plasticità in verde, quindi la lavorabilità alla pressa e la resistenza meccanica anche dopo essiccamento (si veda Tab. 10.5). Il tipo china clay (A) è un’argilla caolinitica complementare alla precedente che non migliora le caratteristiche in crudo, mentre è fondamentale sotto il profilo chimico per incrementare il contenuto di allumina. Il feldspato (o eventualmente il talco e la dolomite in piccole quantità) è un materiale fondente agli usuali livelli termici di cottura (1200 ÷ 1220 °C). Il quarzo, quando partecipa alla fusione con i feldspati, è un componente equilibratore della viscosità e dei flussi vetrosi;
quando invece non partecipa alle reazioni costituisce la matrice base della fase cristallina presente nel prodotto finito, insieme ad una modesta quantità di mullite, generatasi dalla decomposizione delle caoliniti.
Caratteristiche
Carico di rottura (in crudo) N/mm2 0,4 ÷ 0,6 0,8 ÷ 1,0
Carico di rottura (in essiccato) N/mm2 1,0 ÷ 1,5 3,0 ÷ 4,0
Dopo cottura a 1100 °C
Porosità % 10 ÷ 12 3 ÷ 6
Ritiro % 5 ÷ 7 4 ÷ 6
Carico di rottura (in cotto) N/mm2 12 ÷ 15 25 ÷ 35
Tab. 10.5 Caratteristiche fisiche delle argille di Tab. 10.4
Il grès porcellanato può essere considerato l’evoluzione del “grès chimico” [5], un prodotto ceramico molto resistente realizzato negli anni ‘70 in piccoli formati (5×5 cm, 10×10 cm) con tecnologie oggi in disuso. Le nuove composizioni rese possibili dall’impiego dei forni a rulli per cottura rapida e delle presse idrauliche di elevato tonnellaggio hanno permesso di reinterpretare il prodotto sia sotto il profilo tecnologico che, conseguente, merceologico.
A titolo di esempio, la Fig. 10.5 fornisce un confronto fra una composizione di grès tradizionale adottata nei cicli di lavorazione in forni a tunnel (temperatura di cottura 1200 ÷ 1220 °C, ciclo di 30 ÷ 50 ore) ed una composizione attuale per cicli rapidi di cottura (temperatura 1200 ÷ 1220 °C, ciclo di 40 ÷ 60 min).
La struttura dei materiali cotti è simile nei due casi; tuttavia, nel caso della cottura a ciclo lento si può riscontrare la presenza di mullite in alta percentuale e l’assenza di microporosità (pori chiusi e aperti), proprietà che conferisce al cotto un’elevatissima resistenza alle macchie. La quasi assoluta assenza di porosità e la formazione dei cristalli di mullite sono da attribuirsi principalmente al prolungato tempo di cottura, che favorisce la sinterizzazione e la densificazione del materiale.
La composizione per il grès porcellanato attuale è riferita ad un impasto base per prodotti smaltati. Nel caso sia richiesto un supporto superbianco, oltre alle materie prime menzionate, per aumentare il grado di bianco dell’impasto base vengono aggiunti silicato di zirconio e/o allumina; tali materie prime, che hanno caratteristiche refrattarie, vengono generalmente inserite nella formulazione a parziale sostituzione del quarzo.
Grès tradizionale (cottura in tunnel)
Argilla caolinitica
Argilla plastica
Feldspato
Quarzo
35 ÷ 45%
15 ÷ 20%
25 ÷ 35%
10 ÷ 20%
Grès porcellanato (cottura rapida)
Argilla caolinitica
Argilla plastica
Feldspato
Quarzo
Talco / Dolomite
10 ÷ 20%
20 ÷ 30%
45 ÷ 55%
0 ÷ 10%
0 ÷ 3%
Fig. 10.5 Possibili composizioni di grès porcellanato formulate con le materie di Tab. 10.4
La Tab. 10.6 fornisce le analisi chimiche di alcune composizioni di impasti per grès porcellanato. In particolare: grès chimico tradizionale (A), grès porcellanato base (B1, B2), grès porcellanato superbianco (C1, C2).
Fig. 10-05 IT
Tab. 10.6 Esempi di composizioni in ossidi di impasti da grès porcellanato
Le caratteristiche degli smalti da grès porcellanato, come resistenza all’usura ed ai prodotti chimici, dipendono essenzialmente dalle materie prime con cui gli stessi vengono formulati ed anche dalla temperatura di cottura che in questa tipologia raggiungono i 1180 ÷ 1220 °C.
Di conseguenza si sono sviluppate formulazioni per alte temperature di cottura con proprietà fisico-meccaniche e chimiche differenti, idonee per l’utilizzo del prodotto nei differenti ambienti d’esercizio.
La prima applicazione è solitamente uno smaltobbio, materiale che combina sia le caratteristiche di un engobbio che di uno smalto. La sua formulazione comprende prevalentemente materie prime da impasto (argille, caolini, feldspati, ecc.) di qualità, con piccole aggiunte di silicato di zirconio e di fritta, permettendo di ottenere con una sola applicazione un fondo coprente e vetroso idoneo allo sviluppo del colore degli inchiostri digitali.
Tuttavia, in considerazione delle elevate temperature di cottura, l’impiego delle fritte nello smaltobbio, come pure in molti smalti satinati e matt, è generalmente limitato. Viceversa, per gli smalti dove è richiesta una elevata trasparenza, le percentuali di fritte utilizzate possono raggiungere anche il 60 ÷ 70% della composizione.
Le fritte introdotte negli smalti presentano alte temperature di rammollimento, tipicamente oltre i 1120 ÷ 1140 °C. Inoltre, mentre le fritte lucide trasparenti hanno un elevato contenuto di calcio, magnesio ed in minor misura di zinco, quelle matt hanno solitamente quantitativi elevati di bario, allumina e calcio, e cristallizzano pur rimanendo trasparenti.
L’utilizzo negli smalti di materie prime in elevate percentuali è possibile solo se queste sono di buona purezza con bassi tenori di inquinanti, preferendo materiali con basse perdite al fuoco come feldspati di sodio e potassio, nefelina e wollastonite. L’aggiunta di carbossimetilcellulosa nella sospensione di smalto è indispensabile per favorire l’aggancio nell’interstrato smalto-supporto fino alla cottura.
Gli smalti macinati vengono impiegati per realizzare nel prodotto varie tipologie di effetti: lucido, semilucido, satinato, matt.
La “sbobba trasparente” è realizzata con una parte di smalto macinato a umido e una parte di micrograniglie; viene applicata con airless e permette una successiva lappatura.
Le graniglie per applicazione a secco sono miscele di fritte a granulometria appositamente controllata, che vengono impiegate in quantità elevate consentendo ulteriori lavorazioni della superficie come il “full lappato” a finitura lucida o satinata.
Congiuntamente alla composizione chimica e mineralogica delle materie prime, le caratteristiche finali del prodotto ceramico dipendono fortemente dai parametri tecnologici adottati nel corso del processo di lavorazione.
Grado di macinazione: per favorire le reazioni di vetrificazione e densificazione in cottura, il valore del residuo a 230 mesh (63 µm) della barbottina dopo macinazione deve essere molto contenuto: per il grès tecnico tra 0,5 ÷ 1% con macinazione discontinua e tra 2 ÷ 3% nel caso di mulini continui, mentre per il grès smaltato rispettivamente tra 2 ÷ 3% (MTD) e tra 2,5 ÷ 4,5% (MTC o MMC). A questi valori di residuo corrispondono normalmente diametri medi delle particelle compresi tra 15 ÷ 20 µm. Questo grado di finezza contribuisce ad aumentare la superficie specifica delle particelle componenti la massa ceramica e quindi la loro reattività in cottura.
Densità in crudo: l’obiettivo è quello di raggiungere, in fase di pressatura, il massimo grado di compattazione in crudo delle polveri atomizzate compatibilmente con i problemi di degasazione che si possono manifestare in cottura. La pressione di formatura normalmente utilizzata è di 350 ÷ 420 daN/cm2 (1 daN/cm2 equivale a circa 1 kgF/cm2) e consente di ottenere valori di densità del pezzo pressato di 1,95 ÷ 2,10 g/cm3.
Ciclo e temperatura di cottura: per il compimento del processo, in funzione dei parametri di macinazione e pressatura, la temperatura e il tempo di cottura sono fondamentali per l’ottenimento di un materiale vetrificato a bassissima porosità. Mediamente la cottura rapida del grès porcellanato a spessore standard (10 ÷ 12 mm) richiede cicli di 40 ÷ 60 min e temperature di cottura dell’ordine di 1200 ÷ 1220 °C.
È importante osservare che i parametri di macinazione, pressatura e cottura devono essere analizzati nel loro complesso, considerando le loro interazioni soprattutto nel caso di variazioni contemporanee di più fattori. Ad esempio, per diminuire l’assorbimento d’acqua dopo cottura (AA) si può agire:
- riducendo il residuo di macinazione (materiale più fine, quindi più reattivo)
- aumentando la pressione di compattazione (maggiore densità in crudo, quindi anche in cotto)
- aumentando la massima temperatura di cottura in forno (favorendo le reazioni di sinterizzazione dei componenti).
Nel caso particolare di impasti colorati mediante aggiunta di elevate percentuali di pigmenti, è molto importante uniformare le rispettive curve di greificazione, in quanto alcuni pigmenti possono modificare la fusibilità e spostare il punto ottimale di cottura, rendendo a volte necessario l’aggiustamento della composizione. Ad esempio, in Fig. 10.6 sono mostrate alcune curve di greificazione di impasti colorati con pigmenti diversi; si può notare che in funzione della temperatura i valori di ritiro ed assorbimento differisco anche sensibilmente. Può quindi essere opportuno intervenire sulla composizione e/o sui parametri di processo per minimizzare tali differenze.
Ritiro
Assorbimento
Assorbimento
Fig. 10.6 Curve di greificazione relative ad impasti colorati con diversi pigmenti
Il grafico in Fig. 10.7 raffigura invece l’andamento di resistenza a flessione, ritiro in cottura ed assorbimento d’acqua per diversi intervalli di temperature.
Fig. 10-06 IT
Temperatura di cottura (°C)
Fig. 10.7 Carico di rottura (1), assorbimento (2) e ritiro (3) in funzione della temperatura di cottura; l’area (4) rappresenta l’intervallo di cottura ottimale per grès porcellanato
Fig. 10.7 IT NEW
È quindi importante sottolineare come i requisiti del prodotto finito (assorbimento d’acqua quasi nullo, valori molto elevati di resistenza alla flessione ed alla abrasione profonda, scarsa macchiabilità) siano influenzati sia dalla scelta delle materie prime, sia dalle condizioni di processo (dosaggio, macinazione, pressatura, essiccamento e cottura).
Le migliori caratteristiche del prodotto si ottengono ovviamente con un grado di cottura ottimale. Una cottura insufficiente è riconoscibile dall’elevato assorbimento d’acqua (> 0,5%), mentre una cottura eccessiva si evidenzia misurando la densità del prodotto cotto; una eventuale sua diminuzione indica la formazione di una porosità chiusa (sovracottura).
Rispetto alla produzione di altre tipologie di piastrelle ceramiche, la realizzazione del grès porcellanato introduce alcuni aspetti tecnologici ed impiantistici peculiari:
- la possibile colorazione delle barbottine, in particolare per la produzione di grès porcellanato tecnico non smaltato;
- il dosaggio e la miscelazione delle polveri colorate richiesti per la realizzazione di prodotti decorati nello spessore (demiscelati, venati, ecc.);
- l’introduzione di grani e scaglie nella sola superficie o in tutto lo spessore mediante miscelazione con le polveri di impasto;
- la distribuzione selettiva delle polveri colorate in fase di pressatura nella produzione dei venati;
Nella Fig. 10.8 si riporta un diagramma a blocchi con alcune ipotesi di flusso riferite alle possibili fasi di lavorazione richieste dalle diverse tipologie di prodotti realizzabili.
Materie prime
Dosaggio
Macinazione
Atomizzazione
Stoccaggio polveri atomizzate
Pressatura
Essiccamento
Smaltatura e decorazione
Stoccaggio crudo
Cottura
Stoccaggio cotto
Squadratura
Scelta e imballo
Magazzino
Colorazione a umido
Colorazione a secco
Preparazione smalti
Levigatura / Lappatura
Fig. 10.8 Diagramma a blocchi del processo per la produzione di grès porcellanato
Fig. 10-08 IT NEW
La Fig. 10.9 riporta un esempio di layout produttivo di un impianto versatile, predisposto per la produzione delle principali tipologie di grès porcellanato (smaltate, decorate e lappate). Dopo le fasi iniziali di preparazione degli impasti e degli smalti, l’impianto si sviluppa con le linee di pressatura e l’essiccamento rapido (multipiano orizzontale a rulli), collegate in diretta (senza stoccaggio) con le linee di smaltatura e decorazione digitale. Le due linee di decorazione alimentano direttamente due forni monostrato a rulli, preceduti da essiccatoi pre-forno; il prodotto cotto esce dai forni e viene inviato all’area di stoccaggio mediante veicoli a guida autonoma. Dallo stoccaggio i prodotti sono poi prelevati ed inviati alle eventuali linee di lucidatura/lappatura o direttamente alla squadratura, ed infine alle operazioni di scelta e confezionamento.
Di seguito vengono illustrate in dettaglio le varie fasi di lavorazione necessarie alla produzione di piastrelle in grès porcellanato.
Dosaggio materie prime
In relazione al grado di sofisticazione dell’impianto ed in particolare al tipo di processo di macinazione adottato (discontinuo o continuo), la fase di dosaggio può presentare differenti gradi di automazione.
Per i processi discontinui si può operare con dispositivi meccanici tradizionali a leva o con celle di carico deformabili in dotazione al cassone di contenimento.
Nei processi continui un sistema automatico di controllo gestisce la pesatura e il dosaggio della miscela dei componenti l’impasto, con stoccaggio intermedio nel silo che alimenta il mulino, aggiungendo il deflocculante alla sospensione acquosa contenente i residui di setacciatura, normalmente sottoposti a ricircolo.
In alcuni casi, si può prevedere una scioglitura preventiva di una parte dell’argilla (generalmente la frazione più fine e a maggior plasticità) e, eventualmente, dello scarto crudo dell’impasto base, con produzione di una sospensione argillosa che può essere dosata all’entrata del mulino oppure a valle di quest’ultimo, per essere poi miscelata con la barbottina prodotta dal mulino stesso.
Anche per la produzione del grès porcellanato, l’utilizzo del mulino continuo è diventato prioritario. Generalmente il mulino viene impiegato per produrre la barbottina di impasto base ed eventualmente le composizioni dei superbianchi, se richiesti in grande quantità.
Allo scarico dei mulini sono collocati diversi setacci, il primo dei quali ha il compito di arrestare eventuali corpi solidi grossolani (es. residui dei corpi di macinazione), mentre i successivi hanno la precisa funzione tecnologica di vagliare la barbottina da atomizzare. Il residuo di setacciatura viene trasportato dall’acqua predosata destinata al mulino e quindi riciclato.
stoccaggio (prodotto cotto)
cottura (con essiccatoio preforno)
levigatura lappatura
squadratura
scelta e confezionamento
smaltatura e decorazione
pressatura ed essiccamento
preparazione smalti
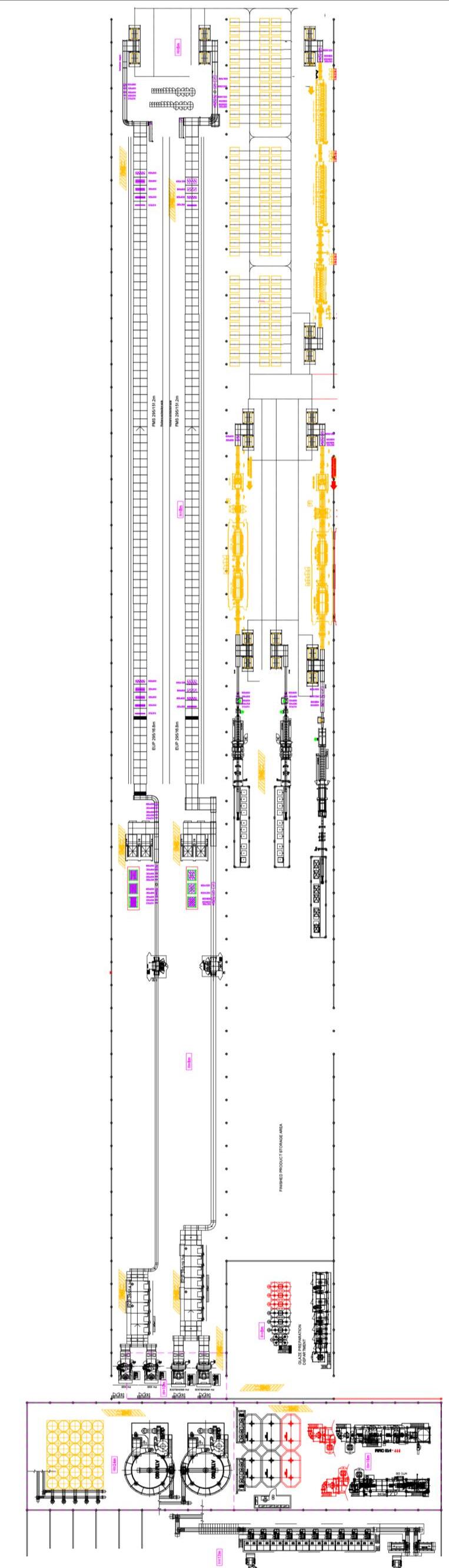
Fig. 1009 IT
dosaggio e preparazione impasto
Fig. 10.9 Esempio di impianto per la produzione di grès porcellanato
Colorazione degli impasti
La colorazione dell’impasto base può essere effettuata per addizione discontinua di “sciroppi” concentrati (per sciroppo si intende una barbottina colorata con una elevata concentrazione di pigmenti). La macinazione di queste sospensioni si effettua in mulini specifici, oppure per dosaggio e miscelazione continua degli sciroppi stessi, per mezzo di dispositivi di misura e regolazione della portata volumetrica o massica. Il sistema volumetrico prevede il dosaggio di sciroppi concentrati provenienti da apposite vasche (ciascuna delle quali dedicata ad un colore), e permette di effettuare la regolazione della portata volumetrica per differenti livelli di concentrazione. I dosatori massici prevedono invece il dosaggio di semilavorati liquidi tenendo conto della loro densità, garantendo pertanto una maggiore accuratezza e stabilità della colorazione. Le unità di dosaggio fanno capo ad un quadro di controllo predisposto all’impostazione ed alla gestione dei parametri del processo di miscelazione.
La specifica tipologia di prodotto che si intende realizzare ed il numero di colori delle polveri atomizzate richieste costituiscono fattori determinanti nel dimensionamento del reparto. Infatti, nonostante l’operazione di atomizzazione sia concettualmente identica per tutti i prodotti ceramici, la complessità del mix di polveri da produrre ed immagazzinare, determinata dalle esigenze estetiche e commerciali, stabilisce il numero di vasche e di sili necessari, oltre a numero e potenzialità degli atomizzatori.
La granulometria ottimale delle polveri atomizzate è del tutto analoga a quella consigliata per le altre tipologie di prodotti. A titolo di esempio, in Fig. 10.10 si riporta un esempio di distribuzione granulometrica per un comune impasto di produzione. Va comunque posta particolare attenzione a non eccedere nel contenuto delle frazioni più grossolane e più fini.
Fig. 10.10 Granulometria di atomizzato per grès porcellanato (valori indicativi)
Fig. 10-10 IT NEW
L’elevata compattazione alla quale viene sottoposto il materiale produce un forte addensamento nel corpo della piastrella, permettendo di contenere il ritiro e ridurre sensibilmente la porosità sul prodotto cotto. Infatti, ad elevate pressioni di formatura corrispondono valori elevati di densità apparente del pressato e quindi ritiri più contenuti, con bassi valori di porosità. La pressione di formatura, che solitamente è di 350 ÷ 420 daN/cm2, non deve essere eccessiva per facilitare l’evaporazione nella fase di essiccamento e per consentire l’ossidazione delle sostanze organiche presenti nell’impasto e l’allontanamento dei gas generati durante la cottura.
Oltre alla pressione di formatura, in fase di compattazione risulta fondamentale l’omogeneità del caricamento delle polveri nell’alveolo dello stampo (o sul nastro di caricamento dei sistemi di compattazione continua). A riguardo, particolarmente critica risulta la gestione di miscele costituite da polveri atomizzate e granuli, che per la loro eterogeneità rendono più difficoltosa la fase di pressatura. Per ottenere un caricamento ottimale delle miscele, i carrelli delle presse possono essere dotati di griglia flottante alveolata. Tali dispositivi permettono di contenere le variabilità nel caricamento degli alveoli riducendo i difetti di superficie (righe di tono, marcate differenze di tono, ecc.) e di geometria delle piastrelle (variazioni di calibro e mancanza di ortogonalità). Un ruolo indispensabile per l’omogeneità delle caratteristiche geometriche delle piastrelle dopo cottura è demandato all’utilizzo degli stampi isostatici.
Essiccamento
L’essiccamento del grès porcellanato non presenta particolari difficoltà nelle normali condizioni di lavoro dell’essiccatoio, né sostanziali differenze nei cicli rispetto alle altre produzioni industriali di piastrelle.
Le macchine termiche utilizzate possono essere essiccatoi del tipo verticale od orizzontale, senza particolari distinzioni riguardo all’aspetto tecnologico, se non per la gestione di grandi formati. Tuttavia, nelle produzioni di grès tecnico colorato non smaltato è preferibile l’essiccatoio orizzontale, per evitare bande di concentrazione dei sali con variazioni di colore dopo la cottura in corrispondenza dei rulli di supporto dell’essiccatoio verticale. Comuni cicli di essiccamento negli essiccatoi verticali sono compresi tra 45 e 65 min, in funzione del formato e dello spessore del pezzo (i formati e gli spessori maggiori richiedono tempi più lunghi). Negli essiccatoi orizzontali i cicli variano da 15 a 30 min, sempre in funzione delle dimensioni delle piastrelle. Per le successive decorazioni digitali sulle linee di smaltatura, è fondamentale che la temperatura all’uscita dell’essiccatoio sia stabilizzata, al fine di minimizzare le differenze di assorbimento e di evaporazione dell’acqua contenuta negli smalti. Se questo non avvenisse, si accentuerebbero i problemi legati ai toni ed alla qualità delle superfici smaltate.
La costante evoluzione estetica dei prodotti, con la richiesta di tipologie sempre più sofisticate, comporta una sempre maggiore disponibilità di linee di smaltatura funzionali e flessibili. Tecnicamente la smaltatura su piastrella calda è indispensabile per far evaporare l’acqua della sospensione di smalto e quindi favorirne il suo essiccamento, riducendo il rischio di crepe e rotture dovute alla diminuzione della resistenza meccanica del supporto bagnato. Le applicazioni più comuni sono quelle eseguite con cabine ad airless mobili; per i formati più piccoli si hanno anche cabine ad airless fissi, mentre per le applicazioni di quantità elevate di smalto si utilizza l’applicazione con “vela” o campana.
L’applicazione dello smaltobbio copre la superficie dei pezzi con una sola applicazione ottenendo un fondo vetroso idoneo allo sviluppo del colore degli inchiostri applicati con la decorazione digitale.
Alcuni effetti a rilievo si ottengono grazie ad una applicazione digitale di un riservante prima dell’applicazione della sospensione acquosa di smaltobbio che viene spostato e lascia scoperta la zona del riservante (idrorepellente).
Dopo la sua applicazione, lo smaltobbio deve asciugarsi terminando l’emissione di vapore dalla superficie dei pezzi per evitare pericolose condense sulle barre delle stampanti digitali; per velocizzare questa fase è buona norma installare ventole supplementari.
Dopo la stampa dei decori digitali in quadricromia e/o con ulteriori colori specifici, si procede all’applicazione di uno o più strati protettivi di varia natura (trasparenti, lucidi, matt, ecc.); nel caso di sbobbe o graniglie ad elevato spessore è poi possibile effettuare dopo la cottura anche lavorazioni di finitura come lappature lucide o satinate.
Infatti, l’applicazione a liquido con airless della sbobba permette di ottenere una superficie smaltata di spessore sufficiente alle operazioni di lappatura, tuttavia la sbobba apporta anche una grande quantità di acqua su pezzi oramai freddi che può addirittura richiedere l’installazione di opportuni sistemi termici di asciugatura, come lampade infrarossi o veri e propri essiccatoi.
A riguardo, la tecnica dell’applicazione a secco di graniglie, sviluppata per far fronte a nuove esigenze estetiche e tecniche, limita notevolmente anche l’impiego di acqua.
Con lo scopo di realizzare decori a spessore, le graniglie di fritta si possono depositare su un supporto smaltato previa applicazione digitale di una colla liquida secondo la grafica desiderata. In questo modo si realizza il fissaggio della graniglia solamente dove è presente la colla ed è sufficiente un flusso di aria per asportare la graniglia non fissata, ottenendo effetti decorativi a rilievo.
Le graniglie sono solitamente impiegate in granulometrie fini, con fasce granulometriche tipiche comprese fra 0.1 ÷ 1 mm.
Una ulteriore applicazione di colla anche sulla graniglia evita poi che una sua frazione possa staccarsi per effetto delle correnti d’aria del forno. Spesso si sostituisce tale applicazione con quella di uno smalto protettivo che, oltre alla finitura superficiale, migliora anche il fissaggio della graniglia.
Ultima applicazione è quella dell’ingobbio refrattario sottomarca che serve a ridurre la sporcatura dei rulli del forno in quanto le elevate temperature di esercizio possono generare attaccature di residui fusi e conseguentemente causare problematiche di planarità del prodotto.
Essiccamento
Molte nuove installazioni prevedono una ridotta zona di movimentazione e stoccaggio del prodotto già smaltato, inviando direttamente il semilavorato appena smaltato alla fase di cottura. In questo caso l’umidità delle piastrelle crude smaltate potrebbe essere troppo alta per le temperature all’entrata del forno e provocare possibili scoppi del materiale. Per evitare tale problema si possono inserire essiccatoi a rulli in fronte al forno, in modo da recuperare aria calda dalla zona di raffreddamento del forno stesso ed abbassare l’umidità del prodotto smaltato da circa 2,5% fin sotto a 1% in ingresso al forno vero e proprio.
Gli obiettivi di questa fase del processo sono: la vetrificazione della massa (con valori di assorbimento quasi nulli) e la stabilità dimensionale nell’intervallo di temperatura di cottura, senza deformazioni o altre variazioni geometriche (curvature, lunette, ecc.).
Il raggiungimento di questi obiettivi è legato a molti fattori, tra i quali ricordiamo i più importanti:
- la reattività tra i componenti dell’impasto
- il grado di macinazione della barbottina
- la pressione di formatura
- la temperatura e il ciclo di cottura.
Il grado di sinterizzazione ottimale per le piastrelle completamente greificate è funzione della reattività dei componenti argillosi e dell’azione sinergica di vetrificazione sviluppata dai feldspati e dalla fase vetrosa che si va gradualmente formando.
La complessa reazione di distruzione dei reticoli argillosi, che comporta la formazione di fasi vetrose fino a produrre un materiale sinterizzato, è controllata dall’energia termica fornita durante la fase di cottura. Gli altri fattori, quali il grado di macinazione della barbottina e la densità del pressato, influenzano la cinetica della reazione, ovvero determinano la velocità alla quale essa avviene.
L’energia termica, necessaria per permettere le reazioni tra i componenti, è correlata alla curva di cottura (Fig. 10.11), progettata in modo da fornire costantemente la quantità di energia sufficiente affinché le reazioni avvengano in modo graduale, in relazione anche al formato delle piastrelle.
T zona inferiore
T zona superiore
Tempo di cottura [min]
Fig. 10.11 Esempio di curva di cottura per grès porcellanato
I cicli di cottura attualmente in uso per il grès porcellanato variano dai 30 min richiesti per la cottura di piastrelle di formato e spessore medio-bassi (es. 60×60 cm, spessore 6 mm) a 65 min necessari per i formati dimensioni maggiori (es. 120×120 cm) a spessore 12 mm, fino a 180 min per formati spessorati di grandi lastre (es. 160×320 cm).
Fig. 10-11 IT
Le temperature massime di cottura variano tra 1180 ÷ 1220 °C in funzione delle caratteristiche di composizione dell’impasto, nonché del grado di macinazione della barbottina, della compattazione delle polveri pressate, ecc.
I moderni forni a rulli permettono il mantenimento delle condizioni di cottura quindi delle temperature prestabilite entro limiti molto contenuti (± 5°C).
La determinazione e predisposizione delle curve, congiuntamente al mantenimento delle condizioni di cottura è assicurata da sistemi di controllo a microprocessore e tramite un computer si effettua la supervisione, la memorizzazione dei dati di processo e la gestione automatizzata del forno.
Scelta
L’operazione di scelta, pur non intervenendo sulle caratteristiche del prodotto, costituisce l’atto finale di tutto il processo produttivo. Infatti, in questa fase il materiale viene selezionato e suddiviso secondo i criteri indicati dalla direzione aziendale sulla base degli obiettivi di mercato. Il prodotto selezionato contribuisce pertanto a determinare l’immagine esterna dell’azienda, oltre che la redditività; per questo motivo i criteri che guidano la scelta devono essere attentamente valutati e adeguati alle specifiche necessità.
L’elevata compattezza del grès porcellanato permette di ottenere, tramite il processo di levigatura, superfici lucidate a specchio, conferendo in tal modo un valore estetico di prestigio al prodotto tecnico non smaltato.
Generalmente la levigatura si effettua ad umido su piastrelle già selezionate in funzione della loro planarità. Le macchine sono composte da diverse stazioni in serie, dotate di teste operatrici rotanti con organi oscillanti e mole di materiale abrasivo. La levigatura consta di tre fasi distinte:
- calibratura della superficie: si effettua con l’utilizzo di utensili diamantati. Questa operazione serve per creare una superficie planare uniformando altresì lo spessore delle piastrelle;
- levigatura: viene realizzata per ridurre progressivamente la rugosità superficiale mediante una successione di utensili abrasivi a grana decrescente;
- lucidatura: è l’ultimo stadio di finitura della superficie per mezzo di abrasivi sempre più fini che permettono di ottenere una superficie a specchio.
Lappatura
Con l’affermarsi del grès porcellanato smaltato, si è sviluppata anche la tecnologia di sola lappatura, più adatta per trattare una superficie di smalto a minor durezza rispetto al grès. In questo caso l’obiettivo è asportare meno materiale possibile, anche solamente 0,1 ÷ 0,2 mm, attraverso una sequenza di abrasivi a grana sempre più fine e feltri per la lucidatura finale, ottenendo così indici di gloss elevatissimi (90 ÷ 100). In alternativa è possibile anche ottenere superfici matt morbide o satinate.
A complemento del processo di lappatura vengono suggeriti e realizzati trattamenti della superficie con materiali a base di silice colloidale per chiudere le microporosità eventualmente apertesi con l’asportazione dello strato più esterno.
In generale, e obbligatoriamente nel caso di levigatura o lappatura, si effettua una rettifica dei bordi con macchine squadratrici-bisellatrici per rifinire il perimetro delle piastrelle. Questa operazione si realizza con apposite macchine rettificatrici, dotate di mandrini squadratori e bisellatori con utensili diamantati funzionanti a secco. Lo spessore di materiale asportato può variare da 4 ÷ 8 mm per ogni lato, in relazione alle dimensioni della piastrella.
La squadratura meccanica dei bordi consente di produrre piastrelle con un solo calibro ed è oggi ampiamente utilizzata anche su gran parte della produzione indipendentemente dal processo di levigatura.
Il grès porcellanato rappresenta oggi la principale categoria di prodotto ceramico grazie alle sue caratteristiche tecniche ed alle molteplici possibilità di arricchimento estetico.
La sua recente evoluzione è indubbiamente legata allo sviluppo dei prodotti smaltati che hanno di fatto sopravanzato la più tradizionale monocottura greificata.
A riguardo il grès porcellanato smaltato, oltre alla miglior qualità tecnica, si è rivelato estremamente versatile in quanto ha consentito lo sviluppo di formati sempre più grandi con la realizzazione di un ampio intervallo di spessori, da pochi millimetri ad oltre due centimetri.
Si è quindi affermato come prodotto ceramico polivalente, in grado di offrire soluzioni per tutte le possibili applicazioni: dai pavimenti residenziali a quelli ad alto transito o outdoor, dai rivestimenti a spessore sottile alle facciate ventilate.
Il comune denominatore è la garanzia di una elevata qualità del prodotto, riconosciuta ed apprezzata da tutto il mercato mondiale.
[1] G. Biffi, Grès porcellanato: tecnologia, produzione, mercato, Faenza Ed., 1993.
[2] G. Biffi, Il grès porcellanato: manuale di fabbricazione e tecniche di impiego, Faenza Ed., 1997.
[3] ISO, International Organization for Standardization, «ISO 13006 Ceramic tiles – Definitions, classification, characteristics and marking», 2018.
[4] ISO, International Organization for Standardization, «ISO 10545 Ceramic tiles – Part 1-20», 2014-2022.
[5] P. G. Burzacchini, G. P. Emiliani e M. Morganti, Dizionario enciclopedico della ceramica, Ed. Polistampa, 2016.
Durante la cottura di piastrelle ceramiche, qualora l’impasto contenga percentuali importanti di materia organica, può presentarsi un alone scuro internamente alla piastrella, conosciuto con il nome di “cuore nero”. Tale variazione di colore, più o meno estesa, è determinata da una incompleta ossidazione del materiale ceramico a causa appunto dei composti organici che sottraggono ossigeno. Nel caso dei processi di monocottura rapida, sia per materiali da pavimento che da rivestimento, questo difetto è accentuato dai cicli di cottura molto rapidi e, nella fase di pressatura, dall’utilizzo di pressioni di formatura molto elevate. Anche l’uso di tenacizzanti nell’impasto, come pure l’applicazione di colle e riservanti, contribuiscono ad accentuare questo difetto, in quanto comportano un incremento del contenuto di materia organica nella massa.
Trattamento della superficie di prodotti in grès porcellanato smaltato, realizzato con macchine lucidatrici dotate di utensili abrasivi. Lo strato di smalto applicato in superficie (solitamente trasparente) deve avere uno spessore sufficiente per permettere la lucidatura completa dell’intera piastrella (full), ed è ottenuto applicando (sopra il decoro) un campo pieno di smalti liquidi o di graniglie a secco. L’effetto estetico è analogo a quello di marmi o graniti levigati, in quanto si ottengono elevate brillantezze (gloss > 90).
Con materiale greificato o “grès” si intende una ceramica a struttura compatta con una bassa porosità residua, ovvero caratterizzata dalla formazione in cottura di fasi vetrose (vetrificazione) che densificano il materiale ceramico aumentandone la resistenza meccanica. Ovviamente questo processo di sinterizzazione comporta un ritiro elevato (5 ÷ 8%) con la possibile generazione di calibri diversi sul prodotto finito in funzione del grado di omogeneità della formatura e del processo di cottura.
Con materiale micronizzato si intende un singolo minerale o un impasto ceramico a granulometria estremamente fine (100% < 63 µm) ottenuto per macinazione a secco dello stesso materiale a granulometria più grossolana.
Con porcellana si identifica un particolare materiale ceramico costituito da un impasto a base di caolino, feldspato e quarzo, sottoposto a cottura ad alta temperatura (1300 ÷ 1400°C). Le principali proprietà della porcellana sono la bassissima porosità (alta percentuale di fase vetrosa), la resistenza meccanica (conferita dai cristalli di mullite) e l’elevato grado di bianco (determinata dalla purezza dei componenti).
Con sciroppo si intende una barbottina concentrata di impasto ad elevato contenuto in pigmenti o opacizzanti da utilizzare per colorare o sbiancare una composizione di impasto standard.
Con materiale superbianco si intende un supporto ceramico di elevata bianchezza (L = 80 ÷ 88), ottenuta attraverso l’aggiunta nella formulazione dell’impasto di composti opacizzati, quali silicato di zirconio e allumina.
A partire dai primi anni 2000, parallelamente al continuo sviluppo di impianti produttivi aventi sempre crescenti capacità, sono apparse sul mercato ceramico le lastre in grès porcellanato di grande formato [1]. La loro introduzione è stata in un certo senso una logica conseguenza del costante aumento dei formati che da sempre ha contraddistinto l’industria ceramica delle piastrelle. Come illustrato in Fig. 11.1 ogni decade è stata caratterizzata da un formato “principale” di dimensioni via via maggiori; di pari passo, i macchinari (la pressa in primis) si sono dovuti adeguare a questo aumento di dimensioni e quindi di produttività.





Fig. 11.1 Evoluzione della forza di pressatura e dei formati di riferimento per piastrelle e lastre ceramiche
Pur mancando tuttora una definizione generalmente accettata (e tanto meno una normativa internazionale specifica), con il temine “lastra di grande formato” si identifica una piastrella ceramica avente il lato inferiore di 1 m (o più) ed una superficie di almeno 2 m2. Ovviamente il materiale utilizzato per la realizzazione delle lastre è unicamente il grès porcellanato (si veda il precedente Capitolo 10) in conseguenza delle superiori prestazioni tecniche.
Un ulteriore impulso alla produzione di formati sempre più grandi è stato possibile grazie alla introduzione di tecnologie di formatura innovative rispetto alle presse tradizionali e tra queste, in particolare, la formatura Continua a rulli [2] [3].
Attualmente la pressatura continua consente infatti di realizzare lastre con una larghezza fino a 180 cm (dimensioni del cotto) e con una lunghezza, teoricamente illimitata, determinata con un opportuno sistema di taglio sulla base del formato desiderato (ad es. 180×360 cm).
L’operazione di taglio consente inoltre di ricavare immediatamente a valle della formatura della lastra anche eventuali sotto-formati, garantendo in tal modo una totale flessibilità produttiva e la saturazione della linea in funzione delle tipologie di prodotto richieste.

Fig. 11.2 Vista complessiva di una linea Continua+
Anche riguardo agli spessori, la formatura continua è decisamente più versatile rispetto alle tecnologie tradizionali in quanto può realizzare indifferentemente lastre e sotto-formati a spessori sottili (tipicamente 3 ÷ 6 mm), a spessori standard (8 ÷ 12 mm) e ad alti spessori (14 ÷ 20 mm) [4].
La necessità di spessori molto diversi è funzione della destinazione d’uso del prodotto, così come l’estetica e la tipologia possono variare notevolmente. In generale, comunque, si tratta di prodotti ad alto valore aggiunto, che riguardano specifici segmenti del mercato tuttora con bassi volumi ma costantemente in espansione [5].
Il mercato delle grandi lastre in grès porcellanato è relativamente recente e quindi non sono ancora disponibili raccolte di dati statistici. Inoltre, essendo la lastra un oggetto dai molteplici utilizzi e quindi destinazioni finali, la stessa tipologia di prodotto potrebbe interessare mercati diversi, con reti distributive diverse (ed anche con valori economici diversi!).
Anche la stima delle quantità prodotte risulta difficile, in quanto spesso le lastre di grande formato vengono poi ridotte di dimensioni con successive operazioni di taglio e squadratura. Una stima indicativa, verosimilmente prudente, ipotizza una produzione mondiale di lastre per circa 800 milioni m2 (anno 2024), pari a circa il 5% dell’intera produzione mondiale.
I principali utilizzi di questi materiali sono i rivestimenti di grande formato indoor e outdoor, come indicato dalla Tab. 11.1 che mostra, in modo puramente indicativo, le attuali segmentazioni del mercato delle piastrelle e lastre ceramiche.
Destinazione d’uso
Rivestimenti smaltati
INDOOR RESIDENZIALE
INDOOR COMMERCIALE
OUTDOOR
Pavimenti smaltati
Pavimenti tecnici
Rivestimenti in grande formato
Pavimenti heavy duty
Rivestimenti di facciate
GRÈS
GRÈS
GRÈS PORCELLANATO 90×90 120×120 30×180 10 ÷ 12 mm Matt
GRÈS PORCELLANATO 80÷120×240 160×320 5 ÷ 8 mm Lucidi
GRÈS
PORCELLANATO 60×60 80×80 60×120 12 ÷ 30 mm
GRÈS
PORCELLANATO 60×120 120×240 160×320 6 ÷ 10 mm
Matt (tecnico)
Matt
Tab. 11.1 Suddivisione merceologica dei principali utilizzi delle piastrelle ceramiche (tipologie, formati, spessori, finiture)
Un ulteriore settore particolarmente promettente dal punto di vista del valore merceologico è rappresentato dalle superfici di lavoro per l’arredamento (countertops); a riguardo la Fig. 11.3 riporta la distribuzione dei diversi materiali utilizzati oggi presenti sul mercato. Come si può osservare, lo scenario è molto variegato, con diversi materiali in concorrenza tra loro; predominano le tipologie solid surface ed engineered stone costituite da polveri minerali (idrato di alluminio e quarzo rispettivamente) agglomerate con leganti sintetici, seguiti
dalle vere pietre naturali (in particolare i graniti) che rappresentano una quota consistente, così come pure i laminati. Il volume totale di questo mercato (dati 2022) si attesta attorno a 600 milioni m2 di cui le lastre ceramiche sono solamente il 2%, pari a circa 12 milioni m2
Solid surface
Natural stone
Laminate
Engineered stone
Porcelain slab
Other materials
Fig. 11.3 Distribuzione della produzione mondiale di countertops (dati 2022)
La proiezione per il 2027 è di una crescita molto sostenuta per la ceramica, con un raddoppio della penetrazione al 4%. Si consideri che il settore dei countertops è ad elevato valore aggiunto, con prezzi finali anche 10 volte quelli di una normale piastrella da pavimento o rivestimento.
11.03 IT
Come evidenziato all’inizio del capitolo, le lastre di grande formato sono necessariamente realizzate in grès porcellanato, dovendo sfruttarne le elevate caratteristiche meccaniche e di resistenza all’uso.
Secondo la norma internazionale ISO 13006 [6] che classifica le piastrelle ceramiche in vari gruppi a seconda del metodo di formatura e del valore di assorbimento d’acqua del prodotto finito (vedi Tab. 11.2), il grès porcellanato appartiene al gruppo Bla, relativo ai materiali totalmente greificati, caratterizzati cioè da valori di porosità (espressi come assorbimento d’acqua) inferiori allo 0,5%. In realtà, i prodotti realizzati e commercializzati presentano una porosità nettamente inferiore a quanto previsto dalla normativa, anche minore di 0,1%. Metodo di formatura
pressatura a secco
Tipologia di prodotto Grès porcellanato Monocottura vetrificata
Monocottura semivetrificata
Pavimento poroso Rivestimento poroso
Tab. 11.2 Classificazione ISO 13006 delle piastrelle pressate a secco; in evidenza il gruppo di appartenenza del grès porcellanato [6]
Come descritto nel precedente Capitolo 10, il grès porcellanato è, fra i materiali ceramici per pavimentazione, quello che meglio si presta a sopportare usure di qualsiasi tipo, in virtù della sua elevata durezza superficiale. Inoltre, possiede ottime proprietà di antigelività, di resistenza meccanica a flessione e compressione, di resistenza all’attacco chimico e alle macchie, ecc.
I metodi di prova delle caratteristiche dimensionali, fisiche e chimiche delle piastrelle ceramiche sono definiti dalla norma ISO 10545 [7].
La Tab. 11.3 presenta un quadro esplicativo delle principali caratteristiche, mettendo a confronto i valori minimi prescritti dalle norme ed i valori reali rilevabili sui prodotti in commercio.
Caratteristiche
Assorbimento d’acqua
ISO 10545-3 ≤ 0,5% < 0,1%
Carico di rottura MOR ISO 10545-4 > 35 N/mm2 > 50 N/mm2
Sforzo di rottura ISO 10545-4 > 1300 N (spess. ≥7.5 mm) > 700 N (spess. <7.5 mm) > 2000 N (spess. ≥7.5 mm) > 1500 N (spess. <7.5 mm)
Resistenza abrasione profonda (UGL)
Resistenza abrasione superficiale (GL)
ISO 10545-6 < 175 mm3 < 150 mm3
ISO 10545-7 dichiarata dal produttore dichiarata dal produttore
Coefficiente dilatazione ISO 10545-8 test disponibile 7,0 × 10-6
Resistenza sbalzi termici ISO 10545-9 test disponibile nessuna alterazione
Espansione all’umidità
Resistenza cavillo
ISO 10545-10 test disponibile test disponibile
ISO 10545-11 richiesta conforme
Resistenza al gelo ISO 10545-12 richiesta senza difetti visibili
Resistenza ai prodotti chimici
Resistenza agli acidi e alle basi
Resistenza macchie
Estrazione Pb e Cd dai vetri
ISO 10545-13 classe GB min classe GB min
ISO 10545-13 test disponibile dichiarata dal produttore
ISO 10545-14 classe 3 min classe 3 min
ISO 10545-15 test disponibile dichiarata dal produttore
Differenza di colore ISO 10545-16 test disponibile dichiarata dal produttore
Tab. 11.3 Caratteristiche tecniche del grès porcellanato secondo la normativaISO 13006 [6] rispetto ai valori di mercato
Il grès porcellanato è di fatto il materiale ceramico che presenta le migliori caratteristiche tecniche, in quanto è un prodotto fondamentalmente costituito di fasi cristalline ad elevata durezza, sinterizzato ad alta temperatura.
Confrontando le caratteristiche delle lastre in grès porcellanato con i materiali “concorrenti” utilizzati per i countertops (Fig. 11.3), si possono sintetizzare in forma qualitativa le valutazioni di merito come riportato in Tab. 11.4.
Tab. 11.4 Comparazione delle caratteristiche tecniche dei materiali per countertops
Dalla Tab. 11.4 risulta evidente come la lastra ceramica in grès porcellanato presenti eccezionali prestazioni tecniche, superiori ad ogni altro materiale concorrente; solo per quanto riguarda la resistenza meccanica (a causa soprattutto della minor resistenza agli urti) la ceramica risulta meno performante rispetto ai materiali agglomerati con resine.
Formati
Le linee di produzione per lastre in grès porcellanato (in particolare le linee di formatura Continua) permettono una elevata flessibilità in termini di formato, sia nel senso trasversale (quello del fronte di avanzamento) quanto, soprattutto, in senso longitudinale. Il formato viene quindi scelto in base a criteri diversi: tendenze di mercato, strategie di differenziazione rispetto ai concorrenti, capitolati di specifici progetti e proposte architettoniche innovative, ecc. [8] La flessibilità della linea Continua consente di regolare la larghezza di caricamento ad un valore minimo di 1000 mm e fino ad un massimo di 1800 mm (valori del prodotto finito, dopo cottura e squadratura). Riferendosi alle dimensioni delle lastre cotte, è pertanto semplicissimo passare da un modulo 800 mm (fronte 1600 mm), ad un modulo 750 o 500 mm (fronte 1500 mm) o ad un modulo 600 mm (fronte 1200 mm), minimizzando in ogni condizione la percentuale di rifilo.
I formati realizzati con tecnologia Continua oggi maggiormente presenti sul mercato sono i seguenti:
1600 × 3200 mm
1600 × 1600 mm 800 × 2400 mm 800 × 800 mm
1500 × 3000 mm 1500 × 1500 mm 750 × 1500 mm 750 × 750 mm
1200 × 2400 mm 1200 × 1200 mm 600 × 1200 mm 600 × 600 mm
Ovviamente ulteriori sotto formati, in particolare i listoni, possono convenientemente essere ottenuti mediante taglio in cotto della lastra stessa (ad es. 300x1800 mm).
Questa duplice modalità di lavorazione dei formati consente anche una più moderna e razionale gestione dei magazzini in quanto risulta più conveniente ridurre il numero di linee rispetto al passato, sfruttando la loro maggiore produttività e specializzandole sui prodotti e formati a più alto indice di rotazione.
Una caratteristica estetica peculiare per le lastre di grande formato, destinate al settore dell’arredamento (countertops, tavoli da arredo ecc.), è la possibilità di imitare la pietra naturale con le tecniche di decorazione a vena passante. Le lastre con decoro sincronizzato ed effetti nella massa a vena passante (si veda come esempio il prodotto di Fig. 11.4), permettono di imitare fedelmente i materiali naturali e le loro prerogative, come la possibilità di successive lavorazioni funzionali alla posa in opera (tagli, forature, arrotondamenti del bordo, ecc.). In questi casi, grazie alla peculiare tecnica di produzione (descritta nel Volume II al Paragrafo 3.3), il decoro superficiale è sempre sincronizzato all’effetto colorato in massa (ottenuto con polveri atomizzate colorate distribuite secondo la grafica desiderata), garantendo un risultato finale spesso indistinguibile dal campione naturale di riferimento.

Fig. 11.4 Esempio di lastra con effetto a vena passante
Viceversa, l’effetto vena passante non ha significato per i normali prodotti da pavimento e rivestimento, dove si espone alla vista unicamente la superficie della lastra e non è necessario decorare lo spessore (terza dimensione).
I prodotti a vena passante sincronizzata rappresentano pertanto la massima espressione della decorazione di un manufatto ceramico. Tuttavia, la complessità di un decoro a tutto spessore e l’elevato costo di produzione limitano l’interesse per questa tipologia di materiali ai soli produttori di fascia alta, in grado di competere nei settori tipici dei marmi naturali e delle pietre ingegnerizzate.
Le materie prime usate per la composizione di impasti per le grandi lastre sono del tutto analoghe a quelle normalmente utilizzate per i formati standard in grès porcellanato. Come tali possono essere ricondotte ad alcuni gruppi di minerali specifici, ciascuno dei quali esercita una propria funzione caratteristica: le materie prime argillose conferiscono plasticità all’impasto, mentre quelle complementari, non plastiche, comprendono i minerali fondenti e quelli a funzione prevalentemente smagrante e strutturale.
Alla prima famiglia appartengono i minerali argillosi di natura illitico-caolinitica o montmorillonitica, a caratteristiche plastiche più o meno marcate in relazione alla propria struttura mineralogica e alla granulometria delle particelle. I minerali fondenti sono rappresentati da feldspati e feldspatoidi, talco, euriti, pegmatiti, quelli più refrattari, a funzione strutturale sono il quarzo e le quarziti in genere.
Per tutti i componenti è richiesta una bassa concentrazione di ossidi coloranti quali Fe2O3 e TiO2, onde evitare inquinamenti cromatici del colore naturale dell’impasto.
I rapporti quantitativi fra i componenti dipendono dalla natura mineralogica delle argille, dalla granulometria delle particelle argillose e, in ultima analisi, dalla loro reattività nei confronti dei minerali fondenti. Le Tab. 11.5 e Tab. 11.6 forniscono una sintesi delle caratteristiche chimiche e fisiche delle materie prime per lastre in grès porcellanato. Oltre al feldspato Na, è opportuno utilizzare anche il feldspato K in quanto l’introduzione della componente feldspatica potassica garantisce una maggior stabilità nella fase di greificazione, aspetto fondamentale nel caso dei grandi formati. Materia prima
Argilla caolinitica (A) 48÷50 35÷38 0,1÷0,4 0,2÷0,4 0,1÷0,2 0,1÷0,2 0,5÷1,5 0,1÷0,2 12÷13
Argilla plastica (B) 62÷66 23÷27 0,2÷0,5 0,3÷0,5 0,5÷0,8 0,3÷0,6 1,3÷1,6 0,5÷0,8 6÷7
Feldspato sodico 68÷72 17÷20 0,2÷0,4 0,2÷0,5 0,2÷1,0 0,1÷0,5 0,8÷1,5 6÷10 0,1÷0,9
Feldspato potassico 64÷68 16÷18 0,1÷0,5 0,1÷0,5 0÷1 0,1÷0,3 9÷14 0÷2 0÷0,5
Tab. 11.5 Composizione delle materie prime per grès porcellanato (lastre)
Caratteristiche
Argilla (A)
Argilla (B)
Carico di rottura (in crudo) N/mm2 0,4÷0,6 0,8÷1,0
Carico di rottura (in essiccato) N/mm2 1,0÷1,5 3,0÷4,0
Dopo cottura a 1100 °C
Porosità % 10÷12 3÷6
Ritiro % 5÷7 4÷6
Carico di rottura (in cotto) N/mm2 12÷15 25÷35
Tab. 11.6 Caratteristiche fisiche delle argille di Tab. 11.5
L’argilla plastica (B) ha il compito di fornire le caratteristiche plastiche in verde, quindi la lavorabilità nella fase di formatura e la resistenza meccanica dopo essiccamento.
È opportuno sottolineare l’importanza di una plasticità medio-alta nelle composizioni di impasto per grandi formati, soprattutto nel caso di spessori sottili (≤ 6 mm). Un buon grado di plasticità determina un maggior rapporto di compressione nella fase di compattazione (rapporto tra altezza della polvere soffice e spessore del pressato maggiore di 2) facilitando il conseguimento di elevate densità in verde, e quindi una più elevata resistenza meccanica in crudo e minori ritiri in cottura. Inoltre, un impasto più plastico presenta una minor espansione di pressatura che, nel caso della formatura di lastre, dovrebbe essere sempre <0,6% in considerazione della criticità di elevati movimenti di espansione in un materiale crudo di grandi dimensioni.
L’argilla caolinitica (A) è complementare alla precedente per quanto riguarda le caratteristiche in crudo; è fondamentale, invece, sotto il profilo chimico, per incrementare il contenuto di allumina.
I feldspati sodici e potassici sono i principali fondenti utilizzati negli usuali livelli termici di cottura (1190 ÷ 1220 °C). Il quarzo, quando partecipa alla fusione con i feldspati, è un componente equilibratore della viscosità e dei flussi vetrosi; quando invece non partecipa alle reazioni costituisce la matrice base della fase cristallina presente nel prodotto finito, insieme ad una modesta quantità di mullite, generatasi dalla decomposizione delle caoliniti. Nel caso di impasti per lastre è comunque opportuno limitare l’introduzione di quarzo per evitare elevati coefficienti di dilatazione termica che possono poi determinare rotture per shock termico (sfilo) nella fase di raffreddamento.
Preferibilmente una composizione di grès porcellanato per lastre dovrebbe presentare un coefficiente di dilatazione termica lineare α(30°÷400°) ≤ 72×10-7 °C-1 tra 30° e 400°C, mentre lo stesso coefficiente misurato tra 500° e 650°C (l’intervallo termico caratteristico del cambio di fase del quarzo) dovrebbe risultare α(500°÷650°) ≤ 105×10-7 °C-1
Il grès porcellanato costituisce l’evoluzione del materiale conosciuto originariamente con il nome di grès chimico, tradizionalmente prodotto nel passato in piccoli formati (5×5 cm, 10×10 cm) con la tecnologia di cottura in forni a tunnel. Successivamente, con l’introduzione dei forni a rulli e delle presse idrauliche di elevato tonnellaggio, sono state sviluppate nuove composizioni basate su un elevato contenuto di feldspato sodico in modo da renderle idonee alla cottura a cicli rapidi.
Nella recente produzione di grandi formati, è però opportuno porre una particolare attenzione alla maggior o minor tendenza dell’impasto alla deformazione piroplastica in cottura [9]. A livello composizionale, questo richiede un maggior contenuto in allumina e l’introduzione di feldspato potassico, con un aumento della stabilità nella fase di greificazione.
Le composizioni riportate in Fig. 11.5 sono indicative per le due principali tipologie di impasti per lastre, base e superbianchi; questi ultimi si ottengono con aggiunte di composti sbiancanti come silicato di zirconio, allumina e, in taluni casi, anche fritte calcio-zirconio.
Impasto base
Argilla caolinitica
Argilla plastica
10 ÷ 20%
25 ÷ 35%
Feldspato sodico 30 ÷ 40%
Feldspato potassico 15 ÷ 20%
Impasto superbianco
Argilla caolinitica
Argilla plastica
Feldspato sodico
Feldspato potassico
Opacizzanti
20 ÷ 30%
10 ÷ 20%
20 ÷ 30%
20 ÷ 30%
5 ÷ 10%
Fritta calcio-zirconio 0 ÷ 20%
Fig. 11.5 Intervalli composizionali di grès porcellanato base e superbianco per lastre
Come esempio specifico, la Tab. 11.7 riporta le analisi chimiche effettive di un impasto base e di un superbianco (L=85). Impasto
Tab. 11.7 Esempi di composizioni di impasti da grès porcellanato base e superbianco
In merito alla maggior o minor tendenza di un impasto per lastre alla deformazione piroplastica in cottura, è doveroso un approfondimento. Questa proprietà risulta infatti particolarmente critica nelle lastre di grande formato, soprattutto nel caso di spessori sottili (≤ 8 mm) in relazione anche a diametro/passo dei rulli e della larghezza del forno utilizzato per la cottura.
La deformazione piroplastica di una composizione di impasto può essere convenientemente misurata, nelle reali condizioni di cottura, attraverso la seguente metodologia.
Da una piastrella cruda vengono ricavate alcune barrette rettangolari di dimensione 15×3 cm (minimo 2 campioni per ogni temperatura di cottura). Le barrette vengono posizionate centralmente su due supporti refrattari distanti tra loro 10 cm, in modo da restare sospese durante il processo di cottura su piastra refrattaria. Dopo cottura si procede a misurare la deformazione utilizzando come punto “zero” il punto di appoggio tra barretta e supporto (si faccia riferimento alla Fig. 11.6). L = 100 mm
150 mm
Fig. 11.6 Test di deformazione piroplastica col metodo delle barrette
L’indice di piroplasticità [10] può essere determinato a partire dal valore della deformazione dopo cottura attraverso la seguente formula:
PI = 4 �������� ℎ2 3 ��������4
Fig. 11.06 IT
dove:
PI indice di piroplasticità [cm–1]
h spessore della barretta in crudo [cm]
L distanza tra i punti di appoggio [cm] (normalmente L=10 cm)
S distanza verticale tra i punti di appoggio e il punto di massima deformazione [cm]
Il valore così ottenuto esprime la tendenza alla deformazione piroplastica dell’impasto e può essere valutato secondo criteri di Tab. 11.8.
Nel caso di una eccessiva deformazione piroplastica è opportuno correggere la composizione dell’impasto, aumentando la percentuale di materie prime che apportano allumina e riducendo il contenuto di eventuali fondenti energici come talco, dolomite, nefelina, feldspato sodico o fritte eccessivamente vetrose (introdotte ad esempio nelle formulazioni di impasti iperbianchi translucidi).
Indice di piroplasticità
[×10–5 cm–1]
< 2,2
2,3 ÷ 2,6
2,7 ÷ 3,0
3,1 ÷ 3,4
MEDIA
SUFFICIENTE > 3,4
INSUFFICIENTE
Tab. 11.8 Criteri di valutazione dell’indice di piroplasticità (spessore del provino ≈ 10 mm)
Impasti compositi
Negli ultimi anni si stanno diffondendo sempre di più le tecniche di decorazione a secco direttamente nella fase di formatura, sia con applicazioni superficiali che in tutto lo spessore. Lo scopo è un arricchimento estetico del grès porcellanato tecnico tramite l’utilizzo di semilavorati, quali: pellettizzati, grani e scaglie di smalti o di impasti colorati (vedi Fig. 11.7).

Fig. 11.7 Esempi di grès porcellanato tecnico con scaglie
Questa tendenza decorativa si sta ulteriormente affermando (anche nelle lastre) grazie al continuo sviluppo di dispositivi sempre più sofisticati per il caricamento delle polveri atomizzate colorate e di loro miscele. Dal punto di vista tecnologico è fondamentale verificare la compatibilità tra i diversi semilavorati e gli impasti in cui vengono introdotti, con particolare riferimento a comportamento dilatometrico e fusibilità alle temperature di cottura richieste. Inoltre, per evitare problematiche in fase di formatura è opportuno limitare la quantità dei semilavorati a basse percentuali (<10%).
Le diverse tipologie di smalti impiegati nella produzione di lastre ceramiche di grès porcellanato sono di natura simile a quelli utilizzati per i formati medio-piccoli, ma necessariamente devono rispettare valori di coefficiente di dilatazione termica più ristretti.
Le tipologie di smalti si suddividono principalmente nelle seguenti classi:
- smaltobbio tradizionale o digitale
- inchiostri, riservanti e colle digitali
- graniglie con elevata trasparenza per prodotti lappati
- sbobba con micrograniglie per lappati con effetto lucido, satinato o matt
- smalti protettivi trasparenti matt o satinati.
Gli smaltobbi tradizionali per applicazioni con airless/vela e quelli digitali (più trasparenti rispetto alla versione tradizionale a causa del minor peso applicabile), possono essere più o meno opacizzati a seconda dell’effetto estetico specifico richiesto per le lastre; in ogni caso devono consentire un buon sviluppo cromatico degli inchiostri della decorazione digitale. I materiali per le applicazioni digitali come inchiostri, colle, ecc., sono gli stessi utilizzati nei formati più piccoli ed essendo impiegati con basse grammature non esercitano una significativa influenza sulla planarità delle lastre. I riservanti sono particolari sostanze, applicate in digitale, con un effetto impermeabilizzante tale da permettere alla successiva applicazione di smaltobbio o di smalto di asciugarsi e fare spessore solo ai bordi delle zone col riservante, realizzando così marcate strutture ad alta risoluzione.
Dopo le decorazioni con inchiostri digitali vengono applicati smalti trasparenti a funzione “protettiva” con diversi effetti superficiali matt o satinato. La trasparenza degli smalti ad effetto matt si ottiene con composizioni di fritte del sistema Ba-Al-Si che, nel corso della cottura delle lastre, portano alla formazione di cristalli di celsiana (feldspato di bario); questo minerale non altera l’indice di rifrazione rispetto alla fase vetrosa non cristallizzata, garantendo così una ottima trasparenza.
Gli smalti protettivi, la sbobba e le graniglie, che presentano un aspetto lucido/brillante dopo cottura, sono invece realizzati da miscele di fritte e materie prime che costituiscono un omogeneo vetro trasparente con assenza di fasi cristalline.
Le graniglie trasparenti per i prodotti lappati sono costituite da miscele di fritte ad alta trasparenza, con granulometria medio-fine per consentire la realizzazione di miscele con diverse proprietà di fusibilità e di coefficiente di dilatazione; vengono applicate a secco sulla superficie coperta da colle che aiutano l’adesione ed il fissaggio dei grani fino alla fase di rammollimento in cottura.
L’effetto estetico di maggior pregio nei prodotti sia matt-satinati che full-lappato si ottiene con alte quantità di vetri trasparenti applicati, in particolare di graniglie a secco, che però esasperano le problematiche di accordo dilatometrico tra impasto e smalto.
Una differenza di coefficiente di dilatazione termica tra l’impasto e gli smalti utilizzati può causare durante il raffreddamento delle lastre di medio-basso spessore (≤ 12 mm) problemi di planarità concava o convessa dei pezzi, rendendo difficoltose (se non impossibili) le successive lavorazioni di lappatura o rettifica. Per le lastre con spessori maggiori (> 12 mm) un coefficiente dilatometrico elevato dello smalto può ridurre la resistenza allo shock termico e portare alla rottura/fessurazione dello smalto (craquelé), mentre un coefficiente di dilatazione basso dello smalto può causare il distacco di scaglie durante le fasi di lavorazione del cotto come il taglio e la rettifica.
Oltre che dalla natura chimica e mineralogica delle materie prime costituenti l’impasto, le caratteristiche finali di lastre in grès porcellanato dipendono fortemente dai parametri tecnologici adottati nel corso del processo di lavorazione.
Grado di macinazione: per favorire le reazioni di vetrificazione e densificazione in cottura, il valore del residuo a 230 mesh (63 µm) della barbottina dopo macinazione deve essere molto contenuto, compreso tra 0,5 e 1%. A questi valori di residuo corrispondono normalmente diametri medi delle particelle compresi tra 15 e 20 µm. Questo grado di finezza contribuisce ad aumentare la superficie specifica delle particelle componenti la massa ceramica e quindi la loro reattività in cottura. Nel caso di macinazione continua modulare, più efficiente della macinazione discontinua, il diametro medio 15 ÷ 20 µm si raggiunge con un residuo leggermente più alto, intorno al 2% sul setaccio di controllo da 63 µm. Comunque può essere consentito anche un residuo di macinazione superiore (3 ÷ 4%), ad esclusione dei prodotti di fascia più alta come i countertops
Polveri atomizzate: possono avere una granulometria standard anche se, nella pressatura di grandi formati, è preferibile una distribuzione più spostata verso dimensioni maggiori che migliorano la disaerazione durante la fase di formatura grazie appunto all’assenza delle frazioni più fini (vedi Fig. 11.8). Di fondamentale importanza risulta altresì la costanza di umidità, da mantenere possibilmente in un intervallo di ± 0,3 rispetto al valore medio, in modo da garantire una miglior uniformità del pressato ed evitare difetti di bordo.
Fig. 11.8 Granulometria di un impasto di grès porcellanato standard e per lastre
Densità in crudo: l’obiettivo resta quello di raggiungere, in fase di pressatura, il massimo grado di compattazione in crudo delle polveri atomizzate compatibilmente con il controllo della degasazione nella fase di preriscaldo in cottura, in modo da evitare la possibile formazione di cuore nero in particolare con spessori elevati. La pressione di formatura normalmente utilizzata di 300 ÷ 400 daN/cm2 (1 daN/cm2 equivale a circa 1 kgforza/cm2) consente
Fig. 11.08 IT NEW
di ottenere valori di densità del pezzo pressato di 1,95 ÷ 2,00 g/cm3 o anche maggiori, dipendentemente dalla plasticità dell’impasto. Nella produzione di lastre ad elevato spessore (20 mm), in presenza di impasti ad alto contenuto di sostanze organiche, si possono ridurre i valori di densità fino a 1,90 ÷ 1,95 g/cm3 per limitare le problematiche di cottura. In ogni caso, trattandosi di grandi formati, la costanza della densità è un requisito fondamentale per evitare deformazioni in cottura dovuta a ritiri differenziati.
Ciclo e temperatura di cottura: è la fase che porta a compimento il processo, nella quale si manifesta il risultato della macinazione e quello della pressatura; temperatura e tempo sono i parametri fondamentali che devono essere attentamente valutati per raggiungere l’obiettivo prefissato, ovvero l’ottenimento di lastre sinterizzate a bassissima porosità. Mediamente la cottura rapida del grès porcellanato in grande formato richiede temperature di cottura dell’ordine di 1190 ÷ 1220 °C e cicli di 70 ÷ 90 min per spessori standard, 45 min per spessori sottili e fino a 180 min per spessori maggiorati.
Nel caso di impasti colorati e di loro miscele è molto importante considerare la compatibilità delle rispettive curve di greificazione, in quanto le diverse aggiunte di pigmenti possono modificare l’attitudine dell’impasto alla vetrificazione e spostare il punto ottimale di cottura, rendendo a volte necessario l’aggiustamento della formulazione.
Il grafico in Fig. 11.9 raffigura invece l’andamento di resistenza a flessione, ritiro in cottura ed assorbimento d’acqua in funzione della temperatura di cottura, analogamente a quanto già descritto per il grès porcellanato.
di cottura (°C)
cottura [°C]
Fig. 11.9 Variazione di carico di rottura (1), porosità (2) e contrazione (3) in funzione della temperatura di cottura
Le proprietà del prodotto finito (assorbimento d’acqua quasi nullo, valori molto elevati di resistenza alla flessione ed alla abrasione profonda, scarsa macchiabilità) sono ovviamente
Fig. 11.9 IT NEW
dipendenti dalla scelta delle materie prime, dalla composizione dell’impasto e dalle condizioni di processo (macinazione, pressatura, essiccamento e cottura).
Un ultimo aspetto da considerare nel processo di cottura dei grandi formati è la necessità di un raffreddamento ottimale al fine di evitare tensioni residue nel materiale cotto che possono ad esempio limitare la lavorabilità meccanica delle lastre cotte e addirittura provocare rotture a distanza di tempo nel pezzo in opera.
Come si può osservare nel grafico di Fig. 11.10, il gradiente medio di raffreddamento accettabile da una lastra ceramica sottoposta a cottura rapida è principalmente funzione del suo spessore. Ovviamente una lastra sottile si può raffreddare molto più rapidamente rispetto alle lastre di grosso spessore per le quali è necessario adottare cicli più lenti e lunghezze di forno adeguate, sfruttando per quanto possibile tutte le zone utilizzabili per la fase di raffreddamento.
Spessore [mm]
Fig. 11.10 Gradiente massimo di raffreddamento in funzione dello spessore della lastra
La realizzazione di lastre in grès porcellanato, oltre a prevedere l’utilizzo di linee opportunamente dimensionate per la decorazione di prodotti smaltati, richiede anche soluzioni impiantistiche dedicate alla produzione di tipologie affini al grès porcellanato tecnico, ovvero:
- la colorazione delle barbottine e/o delle polveri atomizzate base;
- il dosaggio e la miscelazione delle polveri colorate richieste per la realizzazione di prodotti decorati nella massa;
- la miscelazione di granuli e scaglie con le diverse polveri atomizzate colorate;
- la distribuzione delle polveri colorate in fase di formatura nella produzione di prodotti decorati in massa o nello strato superficiale;
Nella Fig. 11.11 si riporta in un diagramma a blocchi il flusso del processo produttivo completo per la fabbricazione di lastre in grès porcellanato smaltato e tecnico.
Materie prime
Dosaggio
Macinazione
Atomizzazione
Stoccaggio polveri atomizzate
Pressatura
Essiccamento
Smaltatura e decorazione
Stoccaggio crudo
Cottura
Stoccaggio cotto
Taglio / Squadratura
Scelta e imballo
Magazzino
Colorazione a umido
Colorazione a secco
Preparazione smalti
Levigatura / Lappatura
Fig. 11.11 Diagramma a blocchi del processo per la produzione di lastre in grès porcellanato
Fig. 11-11 IT NEW
Tecnologia Ceramica Piastrelle
La Fig. 11.12 riporta invece uno schema concettuale delle tipologie di impasti (base, superbianco, iperbianco e colorati) in relazione alle tipologie di prodotti finiti richiesti.
IMPASTO BASE
SUPER BIANCO
IPER BIANCO
IMPASTI COLORATI
Smaltati decorati
Smaltati decorati + graniglia + lappatura
Tinte unite
Miscelati tutta massa con grani e scaglie
Venati a tutta massa smaltati decorati
Fig. 11.12 Diagramma a blocchi della produzione delle varie tipologie di lastre in grès porcellanato
Fig. 11.12 IT
Il caso più semplice dal punto di vista impiantistico (vedi Fig. 11.12), è rappresentato dalla produzione di grès porcellanato con impasto base o bianco per realizzare prodotti smaltati decorati. Più complessa risulta invece l’ulteriore applicazione di graniglie a spessore con conseguente fase di lappatura per ottenere superfici lucide a specchio di particolare pregio estetico.
Ancor più critica risulta la produzione di prodotti in grès tecnico monocolori a causa dei possibili inquinamenti nella movimentazione su nastri trasportatori, nella setacciatura di controllo e nei dispositivi di dosaggio delle diverse polveri atomizzate.
Infine, la realizzazione di prodotti a tutta massa, sia costituiti da miscele di atomizzati con grani e scaglie, sia soprattutto decorati con vene passanti, risulta particolarmente complicata per la necessità di una attenta messa a punto estetica dei semilavorati. Nel caso specifico di un decoro a vena passante è ad esempio richiesta la preparazione di una grafica da riprodurre a tutto spessore con le polveri colorate, il controllo della stessa con opportuni sistemi di visione e la sincronizzazione con la grafica superficiale ad alta definizione delle stampanti inkjet.
La Fig. 11.13 riporta un esempio di lay-out produttivo di un impianto versatile, idoneo per la produzione di lastre in grès porcellanato smaltate e decorate. A destra si vede la zona di dosaggio materie prime e preparazione impasto (macinazione e atomizzazione); quindi, dal basso, la linea di formatura Continua+, direttamente collegata all’essiccatoio multipiano orizzontale. Segue poi, sempre in collegamento diretto (senza stoccaggio) la linea di smaltatura e decorazione digitale, che dopo un rinvio a 180° immette direttamente alla fase di cottura nel lungo forno a rulli monostrato. All’uscita dal forno le lastre cotte vengono immagazzinate nell’area di stoccaggio, asservita da veicoli a guida autonoma, e da questa alimentate verso la zona delle lavorazioni meccaniche in cotto (lappatura, taglio e squadratura). Infine, le lastre sono inviate alla linea di scelta e confezionamento (in casse o cavalletti).
A seguire si riporta una sintetica descrizione delle varie fasi di lavorazione per la produzione di lastre in grès porcellanato.
Dosaggio materie prime
Nel reparto di preparazione degli impasti per lastre sono generalmente previsti sistemi di pesatura e dosaggio continui gestiti da un sistema automatico di controllo, con successivo stoccaggio intermedio della formulazione in un apposito silo che alimenta in continuo il mulino. In questa fase il deflocculante liquido viene dosato e miscelato con la sospensione acquosa contenente i residui di setacciatura normalmente sottoposti a ricircolo.
In taluni casi, si può prevedere una scioglitura preventiva anche di una parte dell’argilla (generalmente la frazione più fine e a maggior plasticità) insieme allo scarto crudo dell’impasto base, con produzione di una sospensione argillosa che può essere inviata al mulino oppure miscelata a valle con la barbottina prodotta dal mulino stesso.
Macinazione
Per la produzione di lastre in grès porcellanato, l’utilizzo del mulino continuo per produrre la barbottina dell’impasto base è prioritario.
Allo scarico dei mulini sono collocati diversi setacci, il primo dei quali ha il compito di eliminare eventuali corpi solidi grossolani (es. residui dei corpi macinanti), mentre i successivi hanno la precisa funzione tecnologica di selezionare granulometricamente la barbottina da atomizzare. Il residuo di setacciatura viene trasportato dall’acqua predosata destinata al mulino e quindi riciclato.
Colorazione degli impasti
La colorazione dell’impasto base viene preferibilmente effettuata per addizione discontinua di sciroppi concentrati. Con il termine “sciroppo” si intende una barbottina colorata con una elevata concentrazione di pigmenti ottenuta per macinazione in mulini specifici discontinui o, se sufficientemente fini, mediante semplice scioglitura in apposite vasche agitate. Il dosaggio e la miscelazione continua degli sciroppi si ottiene per mezzo di dispositivi di misura e regolazione della portata volumetrica o massica.
Il sistema di dosaggio volumetrico degli sciroppi concentrati provenienti da apposite vasche di stoccaggio (ciascuna delle quali dedicata ad un colore), permette di effettuare la regolazione della portata volumetrica per differenti livelli di concentrazione. I sistemi massici, più precisi, effettuano il dosaggio tenendo conto direttamente della densità degli sciroppi. In ogni caso le unità di dosaggio sono dotate di un quadro di controllo predisposto all’impostazione ed alla gestione dei parametri del processo di miscelazione.
lappatura e squadratura
dosaggio e preparazione impasto essiccamento
scelta e confezionamento
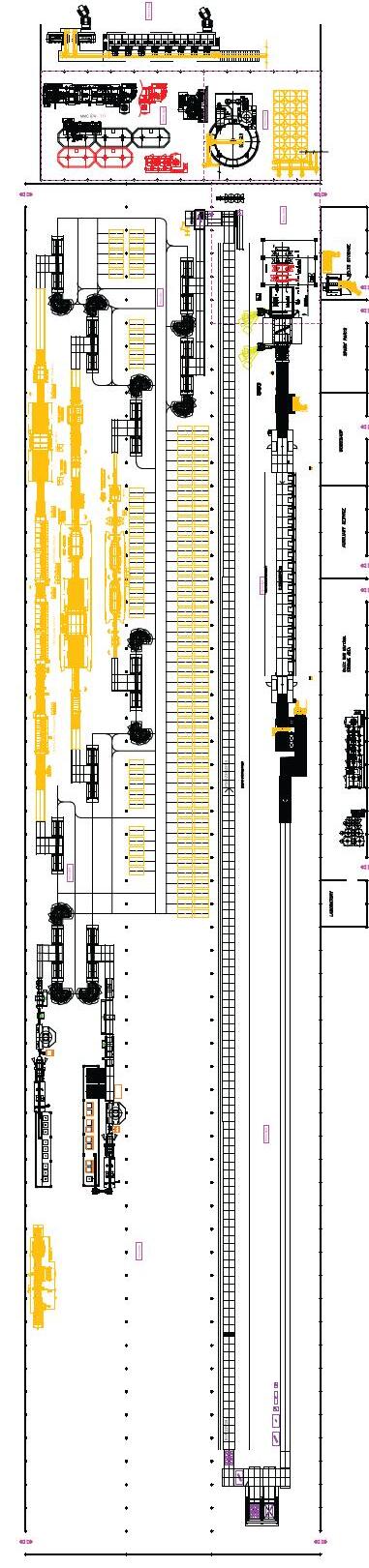
stoccaggio (prodotto cotto)
formatura Continua+
Fig. 11.13 IT
smaltatura e decorazione
cottura (forno monostrato)
Fig. 11.13 Esempio di layout di impianto per la produzione di lastre in grès porcellanato
Atomizzazione
La specifica tipologia di prodotto che si intende realizzare ed il numero di colori delle polveri atomizzate richieste costituiscono fattori determinanti nel dimensionamento del reparto di preparazione delle polveri. Infatti, nonostante l’operazione di atomizzazione sia concettualmente identica per tutti i prodotti ceramici, la complessità dei mix di polveri da produrre ed immagazzinare, determinata dalle esigenze estetiche e commerciali, richiede un adeguato numero di vasche e di sili specifici di stoccaggio, oltre che di atomizzatori con le potenzialità necessarie.
Miscelazione dei semilavorati
Qualora sia richiesta la miscelazione di due o più colori atomizzati, si utilizza solitamente un mescolatore continuo, inserito a valle di un sistema di dosaggio con nastri pesatori. Questo processo può risultare utile anche per effettuare la colorazione a secco, con idonei pigmenti, dell’impasto base o del bianco.
Pressatura
Il processo di compattazione, normalmente con tecnologia Continua+, al quale viene sottoposto il materiale deve garantire un elevato addensamento della massa di impasto, raggiungendo una densità di 1,95 ÷ 2,00 g/cm3 necessaria per una buona greificazione in fase di cottura (con contenimento del ritiro a valori di porosità prossimi a zero). Solamente nel caso di lastre a grosso spessore, il grado di densificazione in fase di pressatura può essere ridotto (1,85 ÷ 1,90 g/cm3) per facilitare l’ossidazione delle sostanze organiche e l’allontanamento dei gas generati nella fase di preriscaldo/cottura, riducendo il rischio di cuore nero nello spessore interno.
In generale, è comunque sempre necessario un perfetto caricamento delle polveri in modo da ottenere una densità omogenea e costante in tutta la sezione. A riguardo, la compattazione continua su nastro risulta ottimale rispetto ai tradizionali sistemi di caricamento discontinui.
Essiccamento
L’essiccamento del grès porcellanato non presenta particolari difficoltà nelle normali condizioni di lavoro di un essiccatoio, considerando ovviamente che il ciclo dipende dallo spessore dei pezzi e dal contenuto di umidità in ingresso.
Nel caso dei grandi formati, vengono ovviamente impiegati essiccatoi orizzontali multipiano in modo da poter gestire le lastre con elevate produttività. I cicli di essiccamento variano dai soli 15 min per lastre a spessore 3 mm, a 60 min con spessore 10 mm, fino a 160 min nel caso di spessore 20 mm, considerando una umidità in ingresso massima del 7%.
Smaltatura e decorazione
Le linee di decorazione per grandi formati sono così costituite:
- trasporti a nastro o con adeguato numero di cinghie in base a spessore/larghezza lastre
- decoratrice digitale per applicazioni di riservanti o smaltobbi
- cabina airless (o vela) per smaltobbio
- decoratrice digitale ad alta definizione per colori ed effetti
- decoratrice digitale a secco per colla e graniglie
- granigliatore a secco
- cabina airless per protettivi o sbobba.
Grande attenzione deve essere posta alla movimentazione delle lastre che subiscono maggiormente i dislivelli nei trasporti tra nastri-cinghie-rulli e sbalzi non supportati, possibile causa di crepe o incrinature che possono manifestarsi anche solo a cottura ultimata. L’utilizzo negli impasti di tenacizzanti per evitare la rottura o le crepe nelle grandi lastre non è sempre in grado ovviare alle problematiche meccaniche dei trasporti. Questo perché le lastre con spessori maggiori (15 ÷ 20 mm) non sono flessibili, mentre le lastre con bassi spessori (3 ÷ 6 mm) non sopportano sbalzi specialmente quando rimangono umide e deboli dopo la smaltatura. Nella produzione di lastre a basso spessore (≤ 6 mm) si limita la quantità di smalti applicati in forma liquida ed a volte si utilizzano lampade o essiccatoi per ridurre la quantità di acqua assorbita dai pezzi.
La velocità delle linee di smaltatura per lastre di grandi dimensioni con applicazioni ad airless mobili è di 10 ÷ 20 m/min. Alte produttività con velocità elevate richiedono una adeguata lunghezza delle linee per consentire l’asciugamento degli smalti e degli inchiostri tra le varie applicazioni.
Gli obiettivi da perseguire nel processo di cottura sono la vetrificazione della massa (valori di assorbimento d’acqua prossimi a 0%) e la stabilità dimensionale nell’intervallo di sinterizzazione.
Il raggiungimento di questi obiettivi è legato a molti fattori, tra i quali i più importanti sono: - la reattività tra i componenti dell’impasto - il grado di macinazione della barbottina
- la pressione di formatura
- la temperatura e il ciclo di cottura.
Il grado di sinterizzazione ottimale per lastre completamente greificate è funzione della reattività dei componenti argillosi, dell’azione fondente esercitata dai feldspati e della natura della fase vetrosa che si va gradualmente formando.
Anche altri fattori, come il grado di macinazione dell’impasto e la densità del pressato influenzano la cinetica della reazione, ovvero determinano la velocità alla quale essa avviene.
L’energia termica fornita ai componenti allo stato solido è esemplificata dalla curva di cottura (Fig. 11.14), disegnata in modo tale che le reazioni avvengano in modo graduale in tutte le fasi (preriscaldo, cottura e raffreddamento).
Fig. 11.14 Esempio di curva di cottura per lastre in grès porcellanato
I cicli di cottura delle lastre in grès porcellanato variano in funzione di spessore e formato, passando ad esempio da 40 min richiesti per la cottura di lastre a spessore 3 mm, 80 min necessari per uno spessore standard (10 mm), fino a 180 min per la cottura di prodotti spessorati (20 mm).
Fig. 11.14 IT
Le temperature massime di cottura variano solitamente tra 1180 e 1220 °C in funzione delle caratteristiche composizionali dell’impasto, del suo grado di macinazione, della densità delle polveri pressate, ecc.
Risulta di fondamentale importanza il mantenimento di condizioni stabili di cottura, con costanza delle temperature nella sezione del forno.
La gestione computerizzata permette di controllare in tempo reale e memorizzare i dati produttivi oltre che le specifiche condizioni di cottura, grazie ai numerosi sensori e dispositivi presenti nei nuovi forni.
Scelta
La fase di selezione delle lastre secondo i criteri stabiliti da ciascuna realtà aziendale è ovviamente fondamentale. La disponibilità di opportuni sistemi di visione che valutano la qualità della grafica, la presenza di difetti, i toni, le dimensioni e la planarità delle lastre consente oggi una totale automazione del processo.
Levigatura e lappatura
L’elevata compattezza del grès porcellanato tecnico permette di ottenere, tramite il processo di levigatura superfici lucidate a specchio, conferendo in tal modo un valore estetico di prestigio al prodotto.
Allo stesso modo una più blanda operazione di lappatura viene realizzata su lastre smaltate a spessore.
A seguire una descrizione dei due processi di finitura superficiale.
Levigatura
La levigatura di lastre in grès porcellanato si effettua con processo ad umido sui soli prodotti tecnici (non smaltati). Le macchine levigatrici sono composte da diverse stazioni in serie, dotate di teste operatrici rotanti che possono essere fornite di organi oscillanti con mole di materiale abrasivo. La levigatura consta di alcune fasi distinte:
- calibratura della superficie: si effettua con l’utilizzo di utensili diamantati. Questa operazione serve ad eliminare piccoli scostamenti dalla planarità e ad uniformare gli spessori delle piastrelle;
- spianatura della superficie: si effettua con abrasivi a base di carburo di silicio a grana grossa; anche questa operazione serve per rendere il più planare possibile le lastre ed eliminare le rigature;
- levigatura: rappresenta il primo stadio della lucidatura e viene realizzata riducendo progressivamente la rugosità superficiale mediante una successione di abrasivi a grana decrescente;
- lucidatura: è l’ultima operazione apportata sulla superficie per mezzo di abrasivi sempre più fini per ottenere una superficie a specchio.
Al termine di questi interventi effettuati sulla superficie, si deve operare con macchine squadratrici-bisellatrici per rifinire il perimetro delle lastre. La velocità teorica di avanzamento sulla linea di levigatura può arrivare a 8 m/min, ma nel caso di lastre in grande formato è solitamente inferiore.
Lappatura
Nel caso di lastre in grès porcellanato smaltato viene più convenientemente realizzata la sola lappatura, un processo più blando rispetto alla levigatura del grès tecnico.
Infatti, i prodotti smaltati mediante l’utilizzo di cristalline, di “sbobbe” o di graniglie applicate a secco, possono essere facilmente sottoposti ad un processo di lappatura e lucidatura che conferiscono alla superficie un aspetto più o meno brillante, permettendo di passare da una finitura satinata ad una lucida a specchio (gloss > 90).
Il processo di lappatura può essere realizzato:
- a campo pieno con finitura omogenea
- a campo pieno con stonalizzazioni lucido/matt
- in cresta, ovvero sulle sole superfici in rilievo realizzate con smalti e graniglie strutturanti.
Taglio e squadratura
Qualora il mix produttivo richieda anche la realizzazione di formati sottomultipli o listoni, le lastre sono preventivamente sottoposte ad una operazione di taglio in cotto che può essere effettuata con differenti tecnologie:
- incisione e spacco: le lastre sono incise con una rotella di metallo duro (o diamante) e poi inflesse fino a rottura controllata della linea di demarcazione;
- dischi di taglio diamantati: è la tecnica tradizionale, a umido, che permette di evitare la successiva fase di squadratura dei lati;
- taglio ad idrogetto: utilizzato in particolare nel caso di forme anche non rettangolari.
La squadratura si ottiene con apposite macchine rettificatrici funzionanti soprattutto a secco, dotate di mandrini squadratori e bisellatori che utilizzano utensili diamantati. Nel caso delle lastre, lo spessore di materiale asportato può andare da 10 a 25 mm per lato, in funzione delle dimensioni.
La produzione di lastre in grès porcellanato è divenuta una realtà consolidata, frutto soprattutto delle capacità di innovazione dell’industria ceramica italiana.
L’affermazione di questa nuova tipologia produttiva nelle sue varie declinazioni merceologiche (rivestimento, pavimento, piani cucina, ecc.) è stata possibile anche grazie alle recenti evoluzioni impiantistiche, con particolare riferimento ai sistemi di formatura ed alla decorazione digitale.
Dal punto di vista tecnico, infatti, la produzione di lastre può essere realizzata su un ampio intervallo di spessori, formati e sottoformati, rappresentando una valida soluzione per tutte le esigenze del mercato.
Nondimeno il fattore determinante per un prodotto a così alto valore aggiunto è il livello estetico raggiungibile, con la possibilità di realizzare prodotti decorati a tutto spessore, grafiche digitali ad alta risoluzione, finiture superficiali con strutture ad effetto pietra o lappature lucide e matt superiori ai marmi naturali.
[1] A. Bresciani, M. Dardi, M. Federici e C. Ricci, «Porcelain tile large-size ceramic slabs» in Qualicer, 2000.
[2] A. Bresciani e C. Ricci, «Innovative process for ceramic tile manufacture by double pressing with continous precompaction» in Qualicer, 2004.
[3] A. Bresciani, C. Ricci e B. Spinelli, «The evolution of ceramic tile pressing: from the origins to Continua+», Ceramic World Review, n. 118, pp. 170-181, 2016.
[4] G. Pederzini e A. Bresciani, «Slabs and sub-sizes: high flexibility and a productivity of 21,500 sqm/day», Ceramic World Review, n. 140, pp. 58-60, 2021.
[5] A. Bresciani e R. Pelliconi, «Large-size ceramic panels and slabs: the evolution continues», Ceramic World Review, n. 125, pp. 88-89, 2018.
[6] ISO, International Organization for Standardization, «ISO 13006 Ceramic tiles –Definitions, classification, characteristics and marking», 2018.
[7] ISO, International Organization for Standardization, «ISO 10545 Ceramic tiles – Part 1-20», 2014-2022.
[8] D. Biserni, «Form and function: the new age of digital ceramics», Ceramic World Review, n. 160, pp. 72-75, 2025.
[9] A. Bresciani e B. Spinelli, «Porcelain tile pyroplastic deformation during firing and post-firing variations in planarity», Ceramic Forum Int.l 89, 2012.
[10] Adcock, D.S. et al., «Pyroplastic Index and Firing Deformation of Ceramic Bodies», Journal of the American Ceramic Society, vol. 42, n. 11, p. 525, 1959.
Traliccio in legno o metallo, su cui è possibile appoggiare le lastre quasi in verticale, con un angolo prossimo a 90° rispetto al piano orizzontale. Queste sono generalmente disposte col lato lungo orizzontale, in modo da gravare minimamente col proprio peso sulle lastre sottostanti. È un sistema molto valido per resistere alle sollecitazioni durante il trasporto, anche se non permette grandi densità di carico.
Piani di lavoro ed in particolare piani cucina solitamente in pietra naturale, quarzo ricomposto o in grès porcellanato, materiali caratterizzati da ottime caratteristiche estetiche e tecniche (elevata durezza, resistenza all’abrasione, alla macchiabilità ed all’attacco chimico).
Trattamento della superficie di lastre in grès porcellanato smaltato, realizzato con macchine lucidatrici dotate di utensili abrasivi. Lo strato di smalto applicato in superficie (solitamente trasparente) deve avere uno spessore sufficiente per permettere la lucidatura completa dell’intera lastra (full), ed è ottenuto applicando (sopra il decoro) un campo pieno di smalti liquidi o di graniglie a secco. L’effetto estetico è analogo a quello di marmi o graniti levigati, in quanto si ottengono elevate brillantezze (gloss > 90).
Con materiale iperbianco si intende un supporto ceramico di estrema bianchezza (L > 90) ottenuto attraverso l’aggiunta di composti opacizzanti, anche sotto forma di fritte vetroceramiche, e l’eliminazione pressoché totale delle materie prime cromofore (in particolare delle argille plastiche).
Piastrella rettangolare stretta e lunga (ad esempio di formato 20×120, 30×180 cm ecc.), ottenuta per taglio di lastre di grandi dimensioni, solitamente utilizzata per pavimentazioni in grès porcellanato ad effetto legno ma applicabile anche nel rivestimento di pareti.
MATERIALE GREIFICATO
Con materiale greificato o "grès" si intende una ceramica con una bassa porosità residua, ovvero caratterizzata dalla formazione in cottura di fasi vetrose (vetrificazione) che densificano il materiale ceramico aumentandone la resistenza meccanica. Ovviamente questo processo di sinterizzazione comporta un ritiro elevato (5 ÷ 8%) con la conseguente generazione di calibri diversi sul prodotto finito in funzione del grado di omogeneità della formatura e del processo di cottura, che possono richiedere una squadratura finale.
Deformazione di un materiale ceramico dovuta alla formazione di fasi vetrose a bassa viscosità durante il processo di sinterizzazione alle alte temperature di cottura.
La sbobba ceramica è una sospensione acquosa con un alto contenuto di materia solida. Il termine viene utilizzato per smalti liquidi, applicabili su piastrelle o lastre già decorate in linea di smaltatura, costituiti prevalentemente da una o più miscele di micro-graniglie trasparenti di fritte ceramiche in una base acquosa addizionata con leganti e sospensivanti sia organici che ceramici. Queste miscele risultano idonee all’applicazione a spruzzo airless oppure con cortina (vela).
Con materiale superbianco si intende un supporto ceramico di elevata bianchezza (L = 80 ÷ 88), ottenuta attraverso l’aggiunta nella formulazione dell’impasto di composti opacizzati, quali silicato di zirconio e allumina.
12.1 Tavola periodica degli elementi
12.2 Pesi atomici degli elementi 12-3
12.3 Composti inorganici di interesse ceramico 12-4
12.4 Diagrammi di fase di composti ceramici 12-12
12.5 Composti opacificanti per smalti 12-26
12.6 Tensione superficiale di ossidi per fritte e smalti 12-27
12.7 Pigmenti artificiali e sintetici
12.8 Scale di durezza dei minerali
12.9 Densità dei materiali solidi
12.10 Caratteristiche fisiche di materiali ceramici incoerenti 12-31
12.11 Tabella comparativa dei setacci 12-32
12.12
12.13
12.15 Coefficiente di dilatazione termica di materiali 12-36
12.16 Coefficiente di dilatazione termica dei principali ossidi 12-37
12.17 Temperatura di fusione di composti inorganici e minerali 12-38
12.18 Entalpie di trasformazione di alcuni composti ceramici 12-39
12.19 Calore per la cottura di materie prime e impasti 12-40
12.20 Tabelle di conversione delle unità di misura 12-41
Fonte: https://commons.wikimedia.org
(see boehmite)
Anhydrite (see calcium sulphate)
Baddeleyite (see zirconium oxide)
Calcium borate, hydrate (see colemanite)
Calcium
(see calcite)
Calcium feldspar (see anorthite)
Calcium orthophosphate (see apatite)
Calcium silicate (see wollastonite)
Calcium
anhydrous
Calcium sulphate, hydrated (see gypsum, selenite)
Cassiterite (see tin oxide)
Celestine (see strontium sulphate)
Chalcopyrite
Enstatite (see magnesium silicate) Eucryptite
Fluorite (see calcium fluoride)
Forsterite (see magnesium silicate)
Galena (see lead sulphide)
Gibbsite (see aluminium hydroxide)
(see iron
(II) disulphide (see pyrite)
Iron (II,III) oxide (magnetite)
Iron (III) oxide hydroxide (see goethite)
(II) sulphate (copperas)
Magnesite (see magnesium carbonate)
Magnesium aluminate (see spinel)
Magnesium silicate, hydrated (see talc)
Inorganic compound
Magnetite (see iron oxide)
Manganese (II) oxide, green (pyrolusite)
(IV) oxide, black
Manganese (II) sulphate, anhydrous
Manganese (II) sulphate, hydrated
Microcline
Minium (see lead oxide)
Periclase (see magnesium oxide)
Potassium feldspar (see orthoclase)
Pyrolusite (see manganese oxide)
(see
Siderite (see iron carbonate)
Sodium feldspar (see albite)
Sodium
Zinc aluminate (see gahnite)
I diagrammi riportati in questo paragrafo sono tratti da: National Bureau of Standards, “Phase diagrams for ceramists”, vol. 1, The American Ceramic Society, 1964.
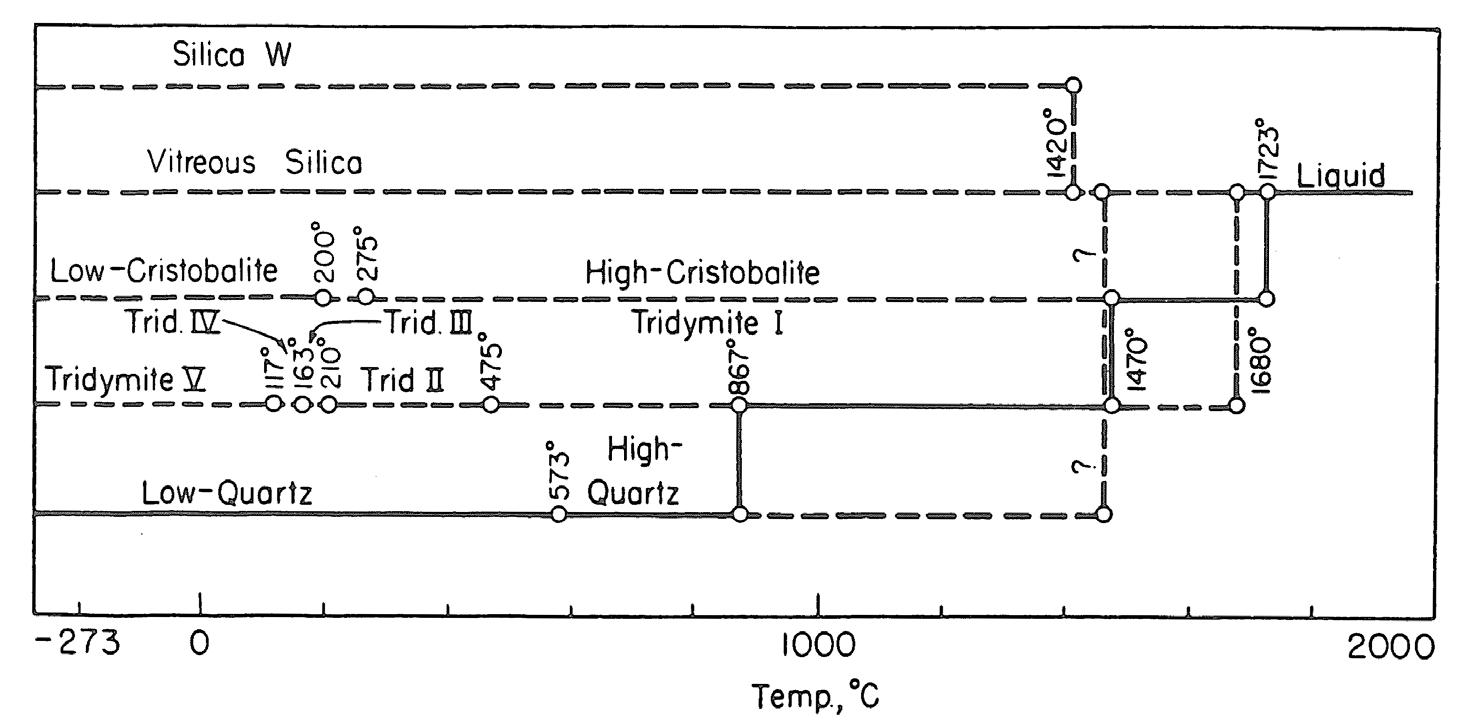
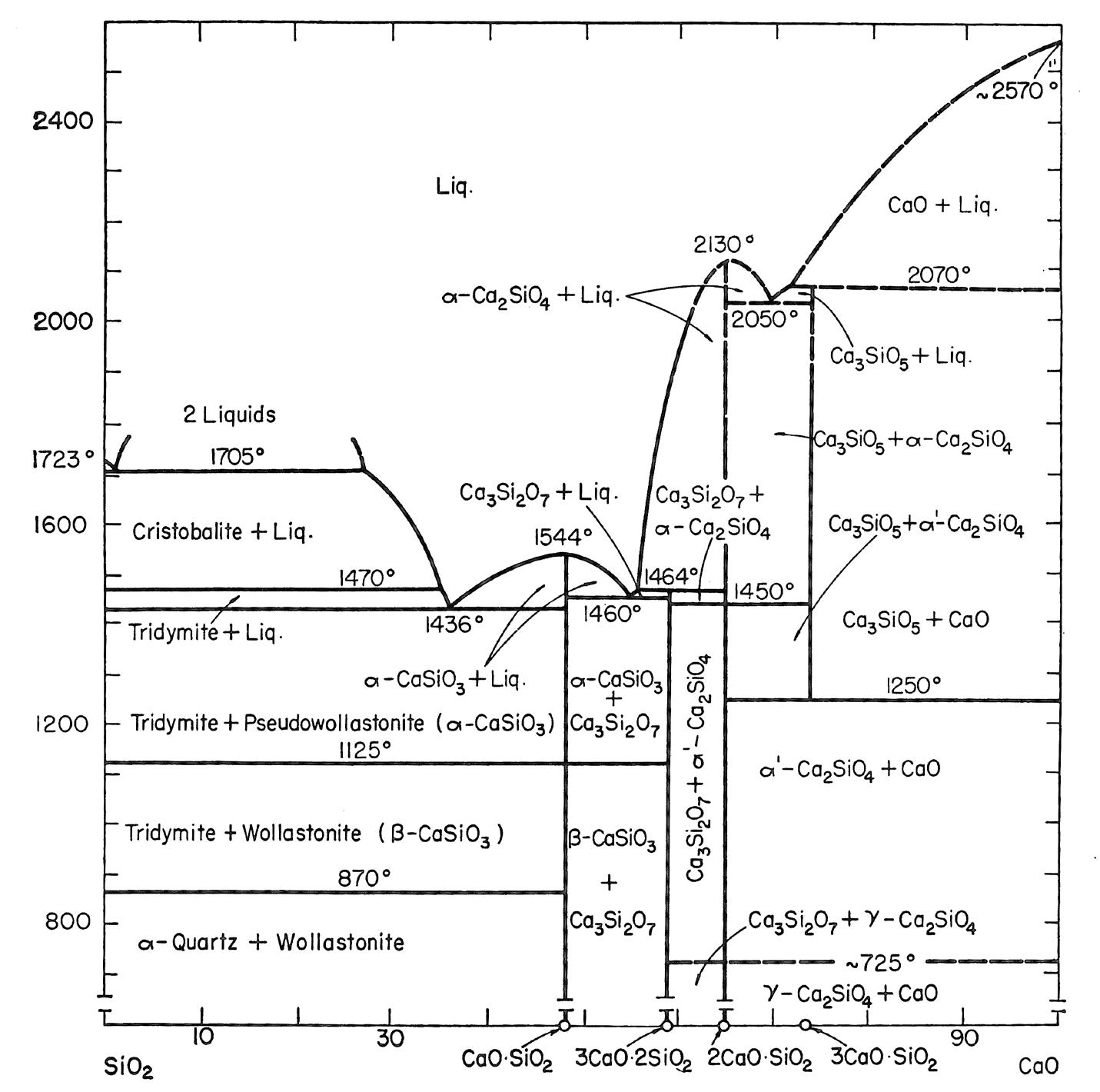
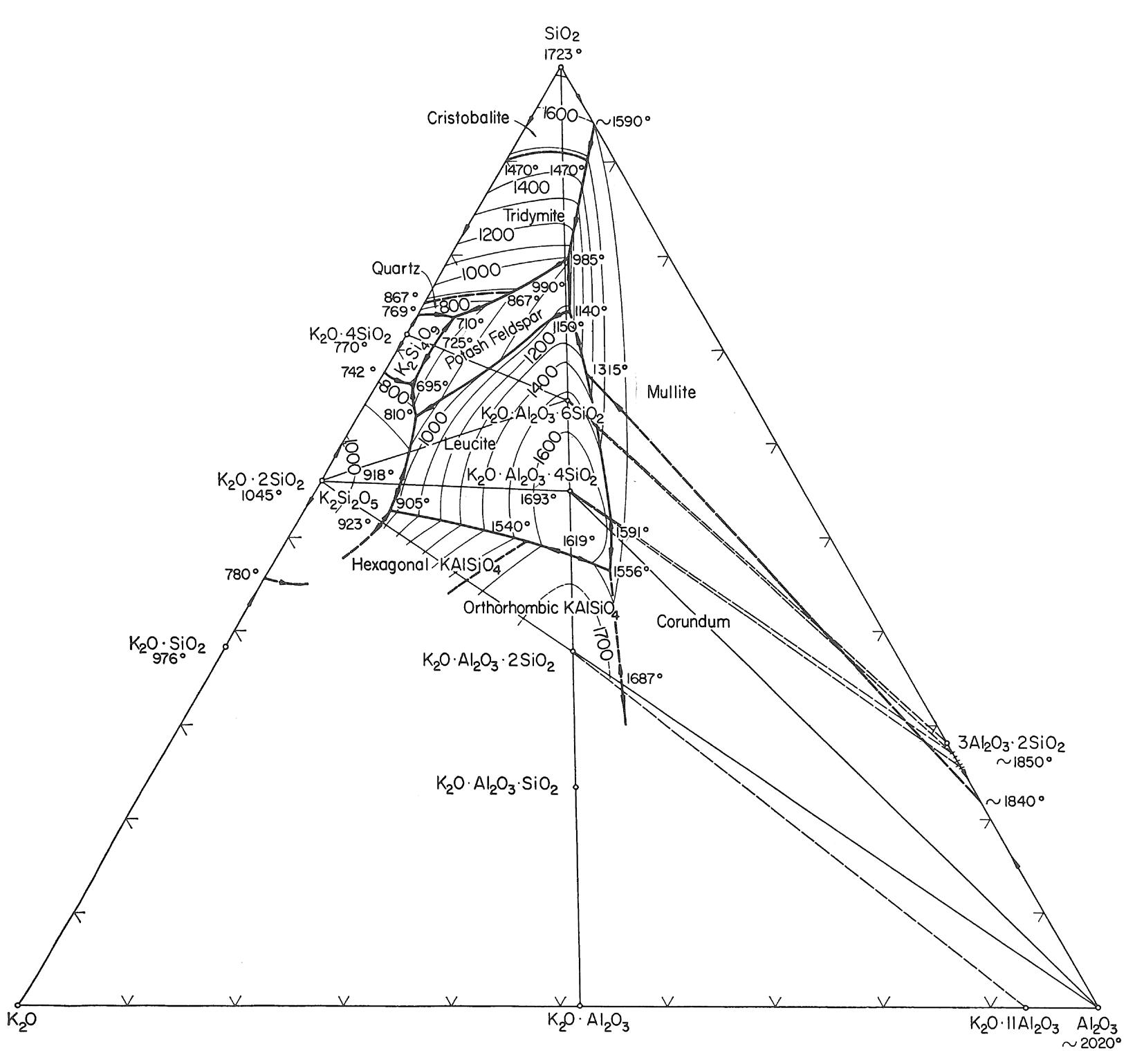
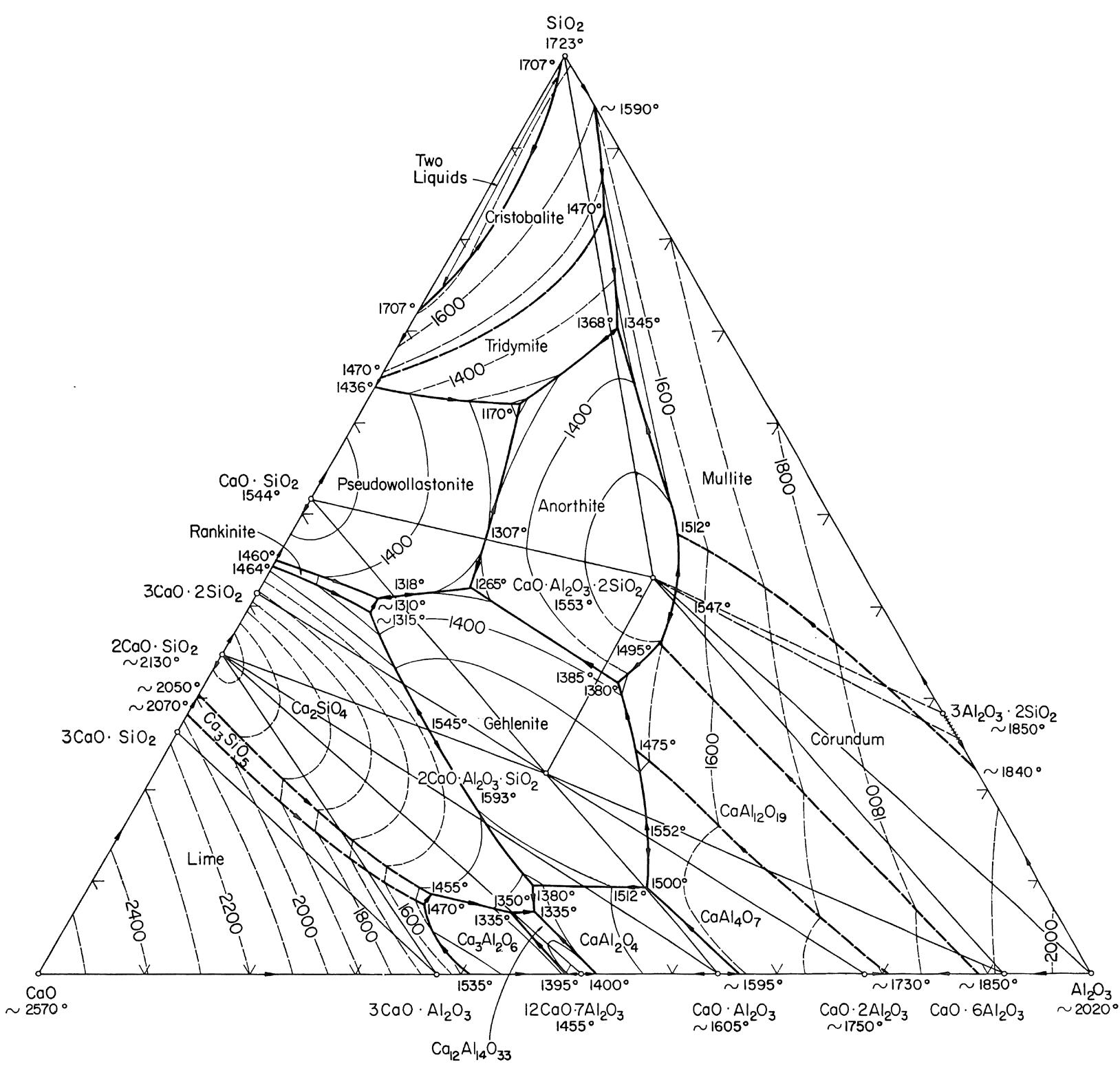
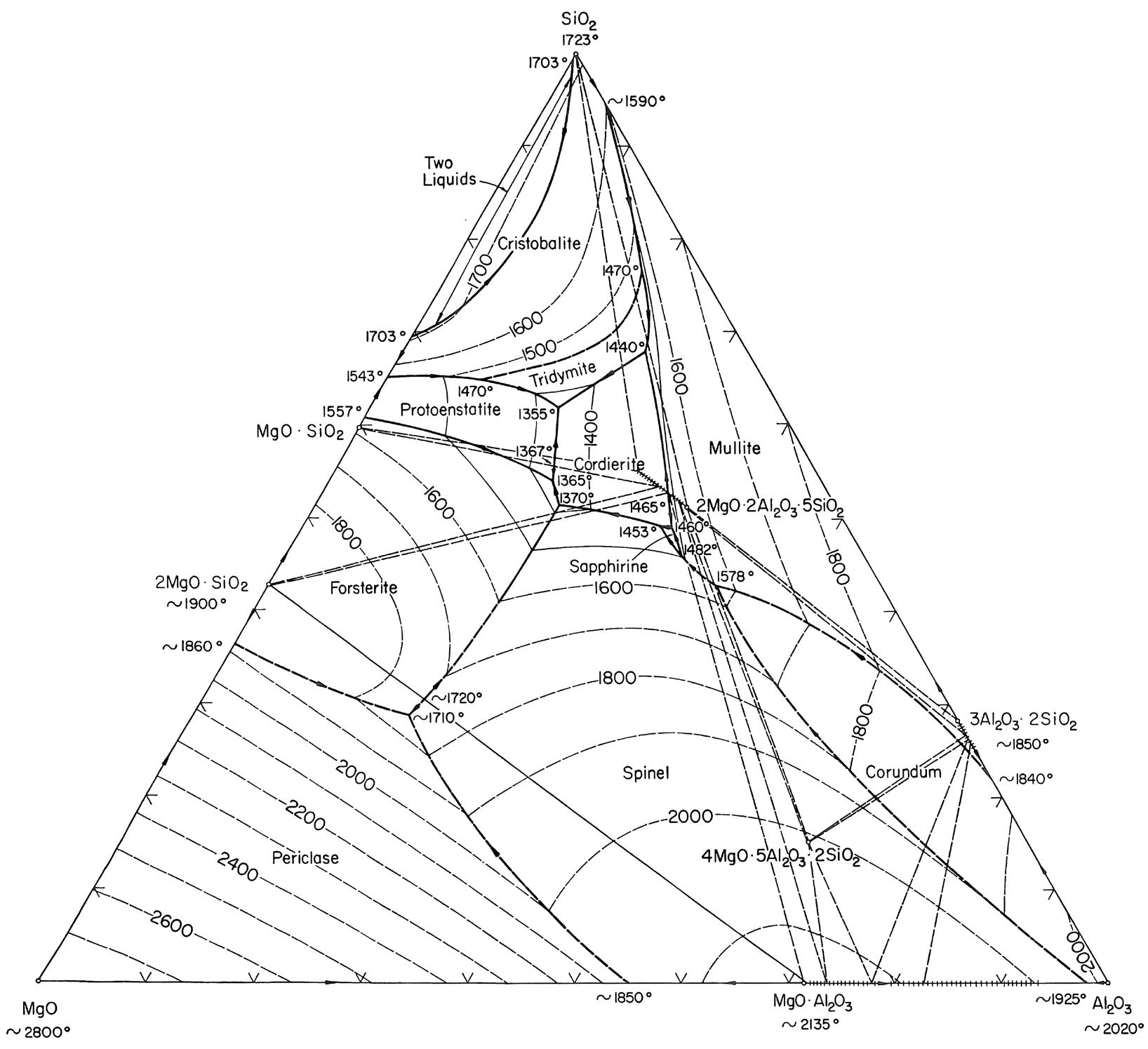
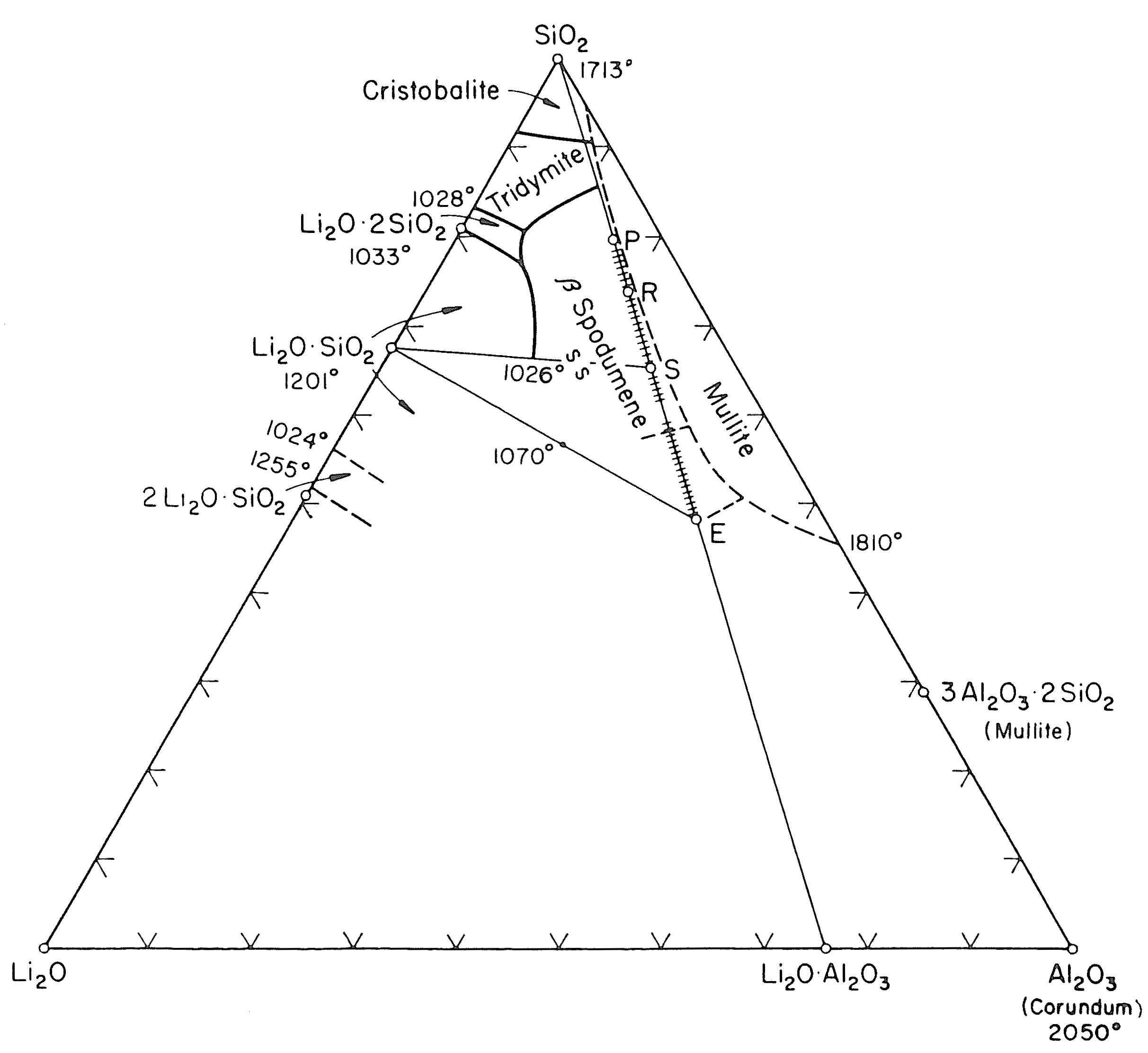
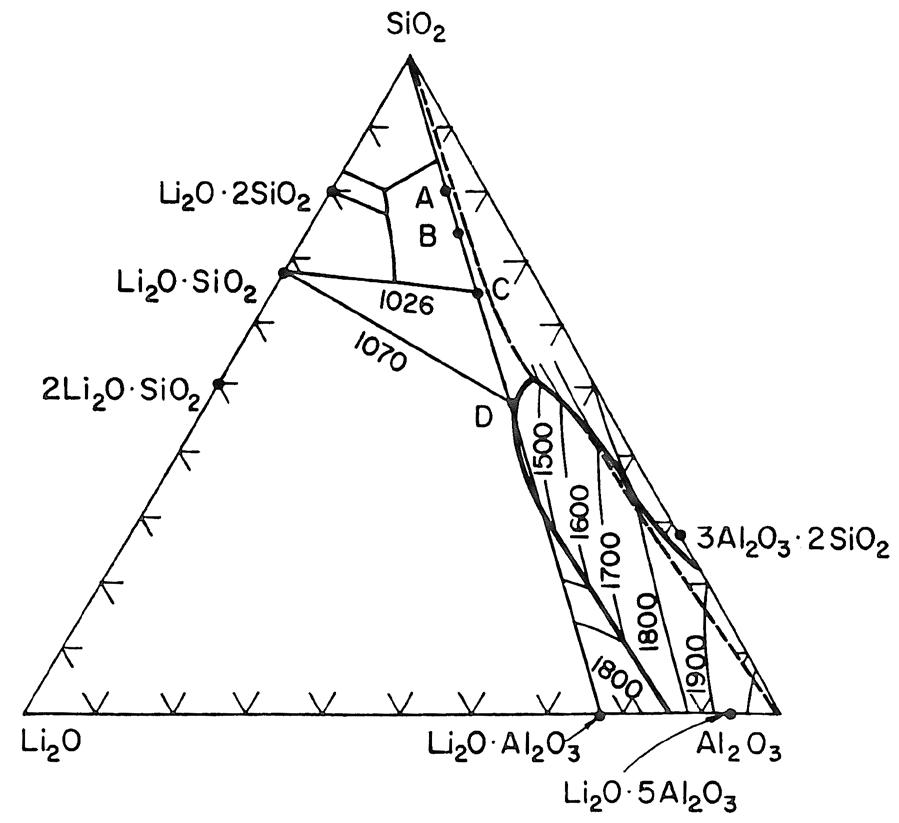
A = Li2O·Al2O3·8SiO2 Petalite
B = Li2O·Al2O3·6SiO2 Lithium feldspar
C = Li2O·Al2O3·4SiO2 Spodumene
D = Li2O·Al2O3·2SiO2 Eucryptite
Fig. 12.13 Diagramma ternario SiO2 – Al2O3 – Li2O
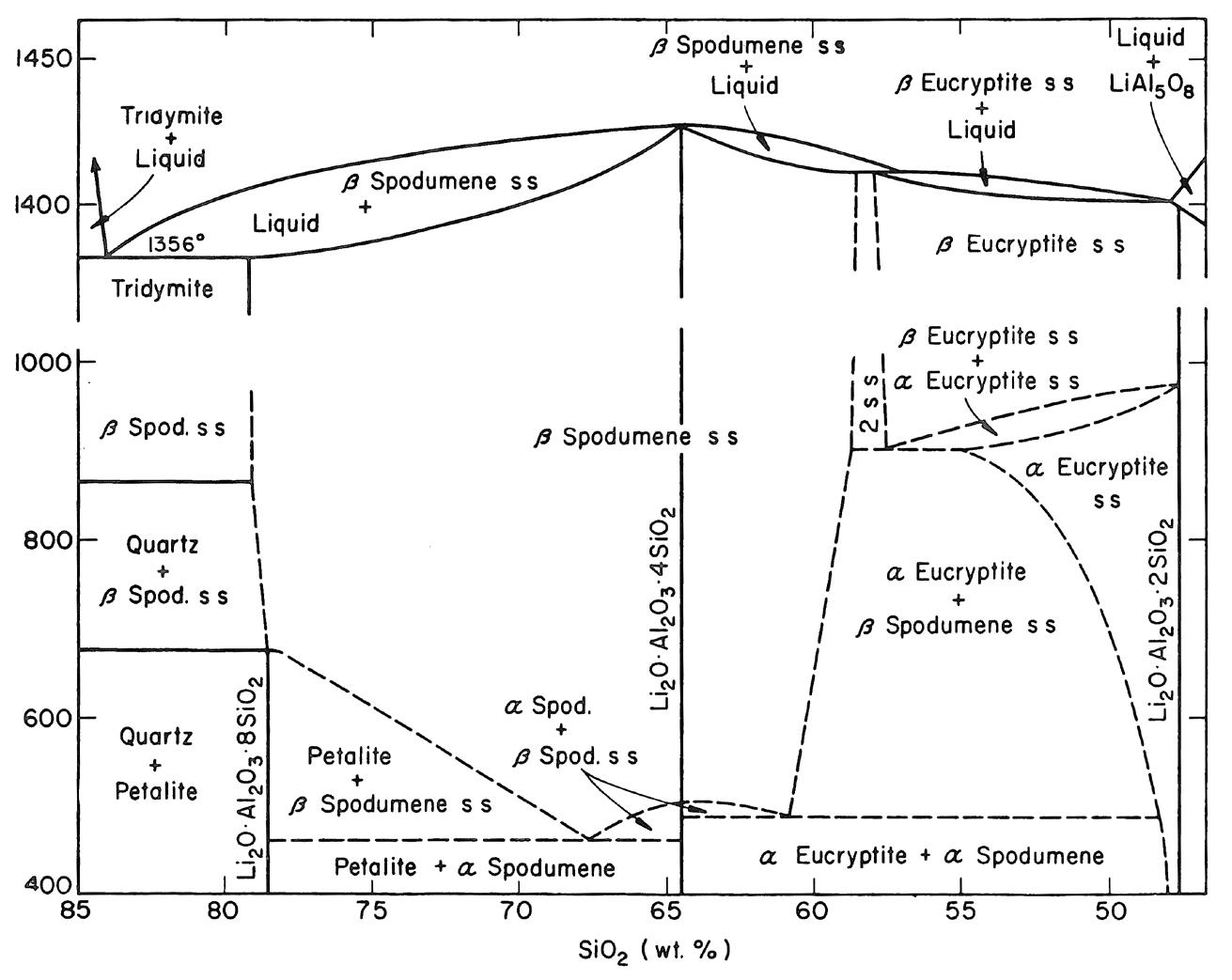
Fig. 12.14 Diagramma di stato Li2O·Al2O3·2SiO2 (Eucryptite) – SiO2
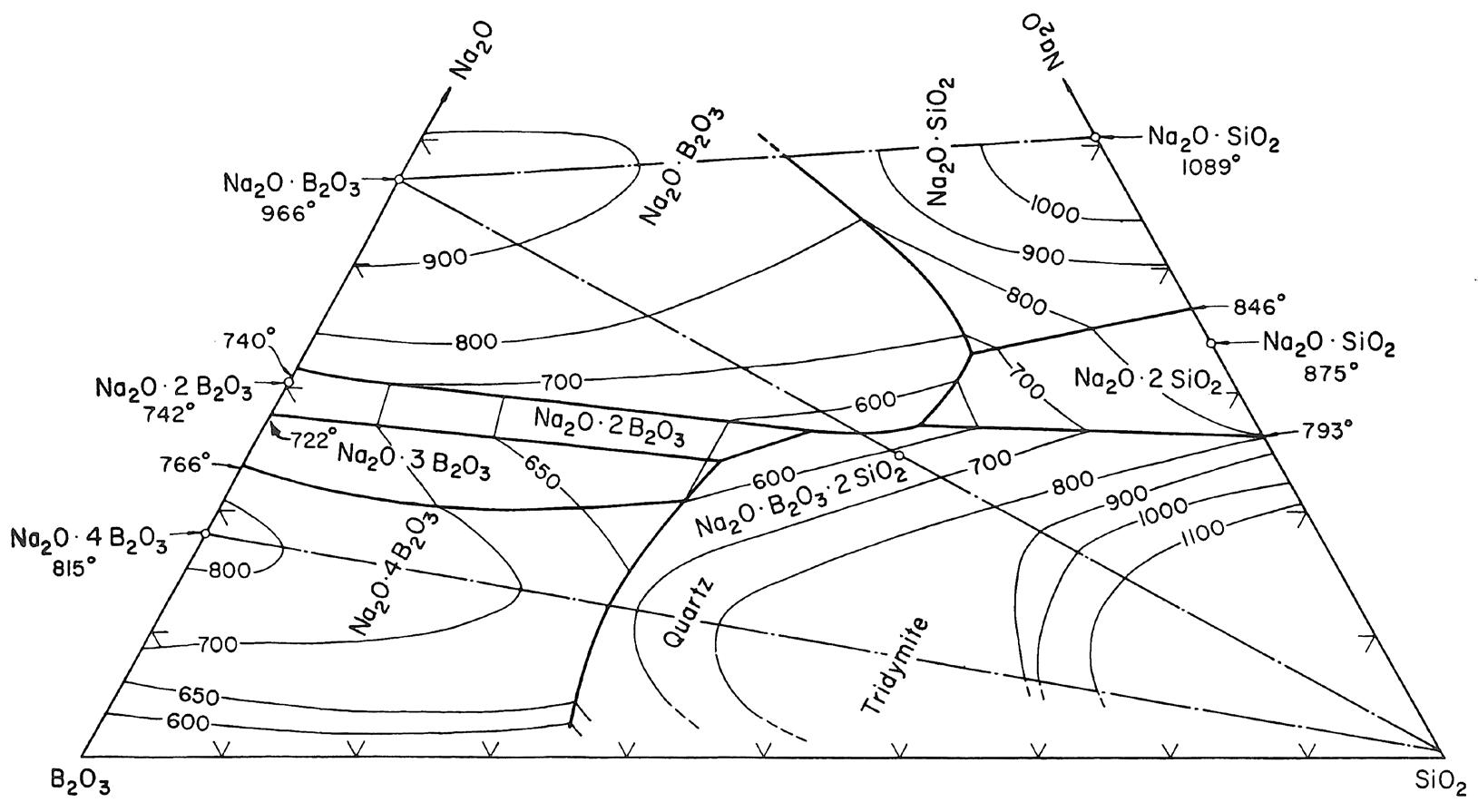
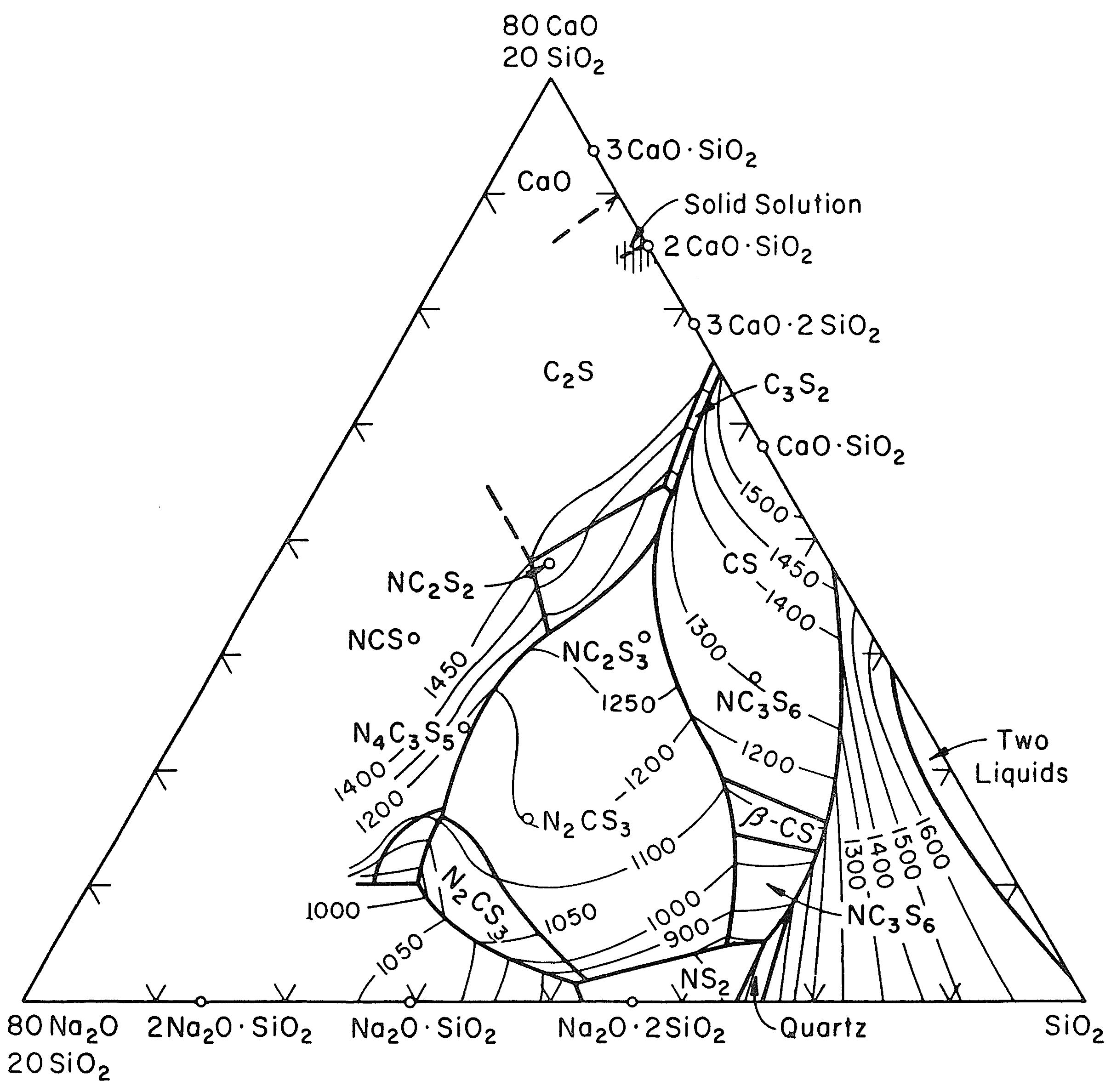
Composto % Tipo di bianco Uso
SnO2 2 ÷ 3 Caldo, intenso Non in frittaggio, per effetti particolari
Contro indicazioni Note
Non adatto per smalti alcalini Costoso
ZrO2 2 ÷ 3 Freddo, intenso Non in frittaggio, per effetti particolari Costoso
Silicato di zirconio (ZrSiO4) 10 ÷ 12 Freddo, intenso
In frittaggio o in macinazione per una opacificazione a tutte le temperature Varie granulometrie in commercio
TiO2 2 ÷ 6 Caldo, intenso A tutte le temperature e in varie composizioni
Ce2O3 3 ÷ 10 Molto intenso, reattivo Per effetti reattivi
Con minime tracce di Fe vira al giallo Solo per effetti speciali reattivi
Con minime tracce di Fe vira al giallo Molto costoso. Si preferisce ZrO2
Al2O3 7 ÷ 15 Lattiginoso Per alta temperatura Resta non fuso
Ossidi dei metalli alcalino terrosi 12 ÷ 20 Lattiginoso, favorisce la devetrificazione, con effetto matt Per tutte le temperature
ZnO 10 ÷ 20 Effetto matt
In frittaggio aumenta l’opalescenza alle basse temperature
Ostacola la formazione di alcuni colori (verde e rosa)
Contributo specifico (percentuale) degli ossidi alla tensione superficiale γ di uno smalto secondo il modello di Dietzel (valori in mN/m). I numeri tabellati, moltiplicati per la percentuale dell’ossido relativo, danno una stima della tensione superficiale (espressa in mN/m).
Es. per uno smalto composto da 20% Na2O, 10% CaO e 70% SiO2 risulta una tensione superficiale γ stimata pari a:
γ = 20·1,5 + 10·4,8 + 70·3,4 = 316 mN/m
Type Base Used range [%]
Yellow Sn-V Low viscous 1 ÷ 5 Lemon yellow 1300 good very bad
Yellow Zr-V Highly viscous 2 ÷ 8 Straw yellow 1350 good good
Yellow Pr-Si-Zr Any 1 ÷ 5 Lemon yellow 1350 good good
Yellow Ti-Sb-Ni Titanium 1 ÷ 5 Lemon yellow 1300 good very bad
Yellow Ti-Sb-Cr Titanium 1 ÷ 4 Ochreorange 1300 good very bad
Green Si-Ca-Cr Calcium (no Zn) 5 ÷ 10 Grass green 1200 good very bad
Green Cr-Al-Si Any 1 ÷ 4 Deep green 1350 good good
Green Cr-Co Any 1 ÷ 5 Blue green 1300 good good
Green Si-Zr-V Any 2 ÷ 5 Light green 1300 good good
Blue Al-Co Aluminium 1 ÷ 3 Light blue 1350 good good
Blue Si-Co Any 1 ÷ 3 Sevres blue 1400 good good
Blue Si-Zr-V Any 2 ÷ 5 Turquoise blue 1300 good good
Pink AI-Zn-Cr Zinc and aluminium 3 ÷ 8 Light pink 1400 good good
Pink AI-P-Mn Aluminium 2 ÷ 5 Light pink 1400 good good
Red Si-Fe Any 1 ÷ 10 Brick red 1300 good good
Pink Sn-Ca-Cr Calcium 5 ÷ 15 Pinkcarmine red 1300 good very bad
Red CdS-Cd-Se Special 2 ÷ 5 Red 1000 good good
Red Si-Zr-Fe Any 2 ÷ 5 Salmon red 1350 good good
Brown Cr-Zn
Cr-Fe-Zn
Cr-Fe-Zn-AI
Cr-Mn Any (presence of Zn determinant) 2 ÷ 5 Beige, brown reddish 1400 good good
Grey
Zr-Co-Ni
Sn-Co-Ni
Sn-Sb Any 1 ÷ 5 Grey - dark grey late 1400 good good
Black Cr-Ni-Fe Any 1 ÷ 5 Black 1350 good very bad
Densità barbottina Percent. acqua Percent. solido (*) Contenuto acqua Contenuto solido (*)
1,450 50,0 50,0 726 724
1,460 49,3 50,7 720 740
1,470 48,5 51,5 713 757
1,480 47,8 52,2 707 773
1,490 47,1 52,9 701 789
1,500 46,3 53,7 695 805
1,510 45,6 54,4 689 821
1,520 44,9 55,1 683 837
1,530 44,2 55,8 677 853
1,540 43,6 56,4 671 869
1,550 42,9 57,1 665 885
1,560 42,2 57,8 659 901
1,570 41,6 58,4 652 918
1,580 40,9 59,1 646 934 1,590 40,3 59,7 640 950 1,600 39,6 60,4 634 966
1,610 39,0 61,0 628 982
1,620 38,4 61,6 622 998
1,630 37,8 62,2 616 1014 1,640 37,2 62,8 610 1030
1,650 36,6 63,4 604 1046 1,660 36,0 64,0 598 1062
1,670 35,4 64,6 591 1079 1,680 34,8 65,2 585 1095 1,690 34,3 65,7 579 1111
1,700 33,7 66,3 573 1127
Formule di calcolo della densità di una barbottina: 1 Db = x Ds + y Da
dove:
Db densità barbottina (g/cm3)
D s densità solido (g/cm3)
D a densità acqua (g/cm3) x frazione in peso di solido y frazione in peso di acqua (1–x)
1,710 33,2 66,8 567 1143 1,720 32,6 67,4 561 1159 1,730 32,1 67,9 555 1175 1,740 31,5 68,5 549 1191
da cui (assumendo Da = 1 g/cm3): Db = Ds (x + y⋅Ds ) Db(x)= 1 1 x+x/Ds Db(y)= Ds 1 y+y Ds Ds = x⋅Db 1 − y Db x = 1 1/Db 1 1/Ds y = Ds Db Db (Ds 1) [g/cm3] [%]
1,750 31,0 69,0 543 1207 1,760 30,5 69,5 537 1223 1,770 30,0 70,0 530 1240 1,780 29,5 70,5 524 1256 1,790 29,0 71,0 518 1272
1,800 28,5 71,5 512 1288 1,810 28,0 72,0 506 1304
1,820 27,5 72,5 500 1320 1,830 27,0 73,0 494 1336
1,840 26,5 73,5 488 1352
1,850 26,0 74,0 482 1368 (*) valori calcolati con densità del solido = 2,64 g/cm3
DEFINIZIONI
1 grado Francese = 10 mg/L CaCO3 (contenuto equivalente di Ca e Mg)
1 grado Tedesco = 10 mg/L CaO (equivalente a 17,85 mg/L CaCO3)
1 grado Inglese = 1 grain/imperial gallon CaCO3 (equivalente a 14,25 mg/L CaCO3)
NOTA: 1 mg/L (acqua) corrisponde a 1 ppm (parte per milione = 10−6)
Si
Si riportano i coefficienti di Mayer e Havas per il calcolo della dilatazione termica volumica (cubica) per le miscele di ossidi. I numeri tabellati, moltiplicati per la percentuale dell’ossido relativo, danno una stima del coefficiente 3α (×10−7 °C−1).
Es. per una miscela di 5% Na2O, 25% Al2O3 e 70% SiO2 risulta: 3α = 5·10 + 25·5 + 70·0,8 = 231 ×10−7 °C−1
NOTA
Per ricavare il coefficiente di dilatazione termica lineare α è sufficiente dividere per 3 il valore di dilatazione volumica 3α sopra calcolato.
Material
Temp. [°C]
Alumina 2050
Barium carbonate 811 (D)
Barium sulphate 1580 (D)
Bauxite 2035
Calcium oxide 2613
Chromium oxide 1900
Cobalt nitrate 55
Cobalt oxide 895 (D)
Copper oxide (Cu2O) 1210
Copper oxide (CuO) 1064
Corundum 2044
Dolomite 750 (D)
Ferric oxide 1565
Ferrous oxide 1377
Fluorite 1330
Gibbsite 165 (D)
Halloysite 1775
Kaolinite 1770
Kyanite 1820
Lime 2613
Material
Temp. [°C]
Litharge 880
Magnesium oxide 2852
Magnetite 1538
Manganese dioxide 1058
Minium 830
Mullite 1840
Nickel oxide 1957
Potassic feldspar 1250
Potassium carbonate 891 (D)
Quartz 1713
Silicon carbide 2200
Sillimanite 1850
Sodium carbonate 851 (D)
Sodium chloride 792
Sodium nitrate 307
Sodium sulphate 884 (D)
Tin oxide 1727
Titanium oxide 1640
Zircon 2550
Zirconia 2715
(D) = decomposing temperature
Unità di forza
Unità di energia
Unità di potenza
Unità di pressione
Nella tabella seguente sono riportati i fattori di conversione tra alcune unità di uso comune del Sistema metrico con le corrispondenti unità dei Sistemi imperiale (GB) e statunitense (US).
Es.
