
12 minute read
ATION


Aberrant Ca2+ Signaling by IP3Rs in Adipocytes Links Inflammation to Metabolic Dysregulation in Obesity
Signaling, December 2021 (DOI: 10.1126/scisignal.abf2059)
Advertisement

The role of inflammation in obesity and associated metabolic diseases is well established. This study demonstrates a key missing link between inflammation and metabolic dysregulation in adipocytes that have remained enigmatic for many decades. During chronic overnutrition or obesity, adipocytes, the cells that store lipids, become increasingly enlarged and dysfunctional and cannot effectively buffer nutrients. This, and potential additional stress signals, results in metaflammation, which is the chronic metabolic inflammation originating from target cells such as adipocytes, in response to excess nutrients. This response involves intracellular stress, recruitment of immune cells, and abnormal production and secretion of inflammatory molecules. Over time, this unfavorable situation ultimately results in systemic metabolic failure, including insulin resistance, dyslipidemia, and metabolic disease. Despite playing important roles in obesity, insulin resistance, and diabetes, the initiation and propagation mechanisms of metaflammation are not fully understood and established, thereby limiting the ability to effectively intervene with this critical pathological mechanism. This study clarifies these mechanisms by demonstrating that proper regulation of the calcium (Ca2+) channel inositol triphosphate receptor (IP3R) is a central process for the maintenance of adipocyte metabolic health. Moreover, the study shows that the alteration of this fine balance is a key driver of adipocyte metaflammation and dysfunction in the context of obesity.
Inflammation, either induced by cytokine exposure in cells or by obesity in animals, leads to increased expression and activity of IP3Rs and phosphorylation of a stress-transmitting enzyme called Ca2+/calmodulindependent protein kinase II in adipocytes. This mechanism is dependent on the c-Jun N-terminal kinase (JNK), a molecule we have previously discovered to be a key mediator of obesity-induced insulin resistance. Studies in cells demonstrated that the JNK-PI3R axis plays a critical role in tumor necrosis factor-alpha’s inflammatory and metabolic activity, a pro-inflammatory cytokine elevated in obese mice and humans. Obese mice, either those fed a high-fat diet or those with a genetic defect causing obesity, have altered Ca2+ homeostasis in adipocytes at least in part due to higher IP3R activity. Moreover, mice lacking IP3R isoforms in adipocytes show protection against adipose tissue inflammation and insulin resistance, despite substantial diet-induced weight gain. This development of increased adiposity uncoupled from metabolic complications, possibly represents a model of metabolically healthy obesity. Interestingly, genetic evidence both in fruit flies and humans, also supports that loss of function of IP3R in body fat also causes increased adiposity. Most intriguingly, human genome-wide association studies have shown that an ITPR genetic polymorphism is associated with increased body-mass index, fat mass, and waist-to-hip ratio, but not metabolic complications. Thus, the regulation of IP3R-mediated Ca2+ homeostasis in adipocytes is a key signal, linking metabolic stress to inflammation and metabolic deterioration in obesity, a mechanism conserved across species.
This study lays the foundation for understanding the impact of intracellular Ca2+ homeostasis on adipocyte metabolism and metaflammation. Also, approaches to prevent aberrant IP3R activity in adipocytes could be a powerful strategy to rescue systemic glucose metabolism and inflammatory stress in obesity. Pharmacological research in this direction is emerging, such as the use of SERCA agonists or Azoramide, although it remains to be elucidated whether targeting this mechanism can be safely and effectively implemented in humans.
Regulation of Liver Subcellular Architecture Controls Metabolic Homeostasis
Nature, March 2022 (DOI: 10.1038/s41586-022-04488-5)

This study reports the most detailed characterization of the subcellular molecular architecture in the liver tissue native environment, in both healthy and obese contexts, shedding critical light on the role of regulation of molecular architectural structure for metabolic programming and homeostasis in health and disease.
Organs and tissues exhibit distinct features of structural organization at various scales to meet functional demands and adapt to challenges for maintaining homeostasis and viability. Within cells, the vast diversity and highly dynamic nature of tasks demand subcellular structural complexity and flexibility. For example, cells that secrete large amounts of protein, such as pancreatic acinar or plasma cells, are filled with rough endoplasmic reticulum (ER) in the form of stacked sheets, whereas Leydig cells, which secrete steroids or lipid hormones, have a vast network of smooth tubular ER. Unfortunately, subcellular structures have been difficult to capture in detail in the native tissue setting, especially for organelles with complex architecture such as the ER, limiting the ability to determine their precise topology and functional profiles of alterations. This study visualized hepatocytes in their native liver tissue environment using enhanced focused ion beam scanning electron microscopy imaging, which made it possible to image large intact volumes of the liver. To identify, segment, and quantify each subcellular structure within the cell, we employed artificial intelligence and machine learning-based approaches, along with convolutional neural networks to automatically segment organelles. Finally, threedimensional reconstruction of the images through multiple analytical platforms resolved the organization of organelles in unprecedented detail in the liver tissue.

This study also examined structure-function relationships in the context of metabolic homeostasis in health and disease, using obesity as a model. A comparative analysis of subcellular structures in liver tissue of lean and obese mice revealed massive structural reorganization of the ER, characterized by marked disorganization of stacks of ER sheets and predominance of ER tubules. Notably, experimental recovery of the ER’s structural organization through multiple genetic and synthetic interventions resulted in improved ER function, as well as systemic glucose and lipid metabolism. These data demonstrate that the hepatic ER’s molecular architecture is highly dynamic, integrated with the metabolic state, and critical for adaptive homeostasis and tissue health.
Together, our findings indicate that cellular structures can be a prerequisite for metabolic programming, and such regulatory circuits can open new avenues for understanding endocrine and metabolic homeostasis. The fundamental regulatory mechanism identified could be applied to evaluate the susceptibility—or resistance—of individuals to a disease state like obesity, and determine what steps, such as diet, nutrients, or fasting, will reduce, eliminate, or exacerbate these states. Finally, the workflow delineated in this study could be exploited in future studies to further understand the structure-function relationship in native tissue context, and how this may be linked to metabolic function in healthy and diseased tissue.
A Hormone Complex of FABP4 and Nucleoside Kinases Regulates Islet Function
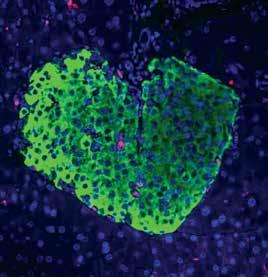
Nature, December 2021 (DOI: 10.1038/s41586-021-04137-3)

This study reports a new factor—the Fabkin complex—that integrates energy status with the function of metabolic organs, representing a promising target for preventing and treating metabolic disease.
Lipolysis, a process in which adipocytes release energy, is necessary for survival. However, uncontrolled or chronic lipolysis in the context of insulin resistance or insulin insufficiency, can cause disruptions in metabolic homeostasis. We have recently identified a hormone, fatty acid-binding protein 4 (FABP4), as the only hormone known to be released during lipolysis. Our previous work has also shown that mice lacking FABP4 have significantly higher levels of beta cells, which play a central role in the control of lipolysis and the development of diabetes.
FABP4 levels are known to be elevated in type 2 diabetes and correlate with body mass index, yet the role of FABP4 in type 1 diabetes has remained unclear. In this study, we detected significantly higher serum levels of FABP4 in type 1 diabetes patients compared with normoglycemic individuals, indicating that FABP4 may indeed play a role in type 1 diabetes. We added upon these findings by examining non-obese mice, and confirmed that circulating FABP4 serum levels are significantly elevated just before and immediately after impairment of glycemic control, further confirming a potential role of FABP4 in the pathogenesis of both type 1 and type 2 diabetes.
Although circulating FABP4 levels are strongly associated with cardiometabolic diseases in preclinical models and humans, the mechanism of action of FABP4 has not yet been determined. Moreover, although FABP4 influences beta-cell mass and function in vivo, previous studies have not shown a consistent effect of recombinant FABP4 in vitro. These conflicting findings suggest that FABP4 may combine with other proteins to elicit its biological function. Based on pulldown and mass spectroscopy of serum samples from mice with diabetesinduced obesity, this study revealed that adenosine kinase (ADK) and nucleoside diphosphate kinase (NDPK) bind FABP4 to form a complex (termed Fabkin), that regulates extracellular adenosine triphosphate (ATP) and adenosine diphosphate (ADP) levels. The findings suggest that when FABP4, NDPK, and ADK are combined in a complex, the ADP:ATP ratio decreases, inhibiting purinergic receptors on beta cells and thus, reduces glucose-stimulated insulin secretion. This study also demonstrated that the Fabkin complex alters calcium homeostasis in the endoplasmic reticulum (ER), resulting in ER dysfunction and beta-cell death in vitro, which are critical mechanisms in both.
Finally, we demonstrated in this study using a prototype monoclonal antibody, that this complex is critical in both type 1 and type 2 diabetes pathogenesis. After mapping the affinities of each components of the Fabkin complex and antibody interactions, we administered into multiple genetic models of disease to test therapeutic potential. These studies showed that therapautic targeting of the Fabkin complex improves metabolic outcomes, enhances beta-cell function, and preserves beta-cell integrity to prevent both type 1 and type 2 diabetes.
These findings elucidate the mechanism of action of FABP4, demonstrating that FABP4 couples energy fluxes with the metabolic response, and establish an endocrine axis of metabolic regulation. The Fabkin complex likely targets other tissues to influence immunometabolic pathologies such as inflammation, insulin resistance, and cardiac pathophysiologies. Importantly, this discovery also provides a straightforward translational path for applications to human disease.
Endothelial FABP4 Constitutes the Majority of Basal Circulating Hormone Levels and Regulates Lipolysis-Driven Insulin Secretion
In this publication, we report the novel discovery that the adipocyte hormone, fatty acid binding protein 4 (FABP4), is also secreted from endothelial cells—the cells which line the blood vessels. Importantly, and unexpectedly, we found that endothelial FABP4 is the major source of basal hormonal FABP4, contributing to ~75% of basal circulating FABP4 levels in lean mice. Moreover, we showed that mice lacking endothelial FABP4 showed altered insulin responses to lipolysis, indicating that endothelial FABP4 is critical for lipolysis-induced insulin secretion.
Our lab previously discovered that FABP4 is a lipid chaperone secreted from adipocytes upon stimulation of lipolysis. Mice lacking FABP4 are protected against obesityassociated metabolic diseases, and circulating FABP4 levels correlate positively with body mass index and indicators of metabolic syndrome in humans, and are increased in mice with dietary or genetic obesity. Taken together, all of these raise the possibility that excessive adipose tissue release of FABP4 may, in fact, be the strong contributor to metabolic dysregulation seen in mice and humans. While adipocytes have always been presumed to be the major source of hormonal FABP4, this question has not been experimentally shown definitively in vivo. To address this question, we generated a new genetic model of mice where FABP4 deletion can be achieved selectively in the desired cells known to express the gene—adipocytes, endothelial cells, myeloid cells, and the whole body—to examine the contribution of these cell types to basal and stimulated plasma FABP4 levels. Unexpectedly, we found that mice with adipocyte deletion of FABP4 only showed ~25% reduction in basal circulating FABP4 levels, whereas mice with deletion of FABP4 from endothelial cells showed ~75% decreases in basal FABP4, compared to wild-type controls. Our data showed, for the first time, that the majority of baseline circulating FABP4 in lean mice is derived from vascular endothelial cells, rather than adipocytes.

FABP4 secretion by cultured adipocytes and in vivo is inducible, stimulated by prolonged fasting and lipolytic signals. Long ago, it was shown that acute β-adrenergic stimulation of lipolysis in vivo is associated with a rapid, strong, and transient induction of insulin secretion in general, this is seen as a pathological response. Interestingly, our previous studies in FABP4-deficient animals demonstrated that the presence of FABP4 is an important requirement for this lipolysis-driven insulin secretion, as insulin responses to lipolysis are markedly blunted in the absence of FABP4 in the whole body. In this study, we found that mice with adipocyte deletion of FABP4 exhibited ~62% reduction in FABP4 responses to lipolysis, while there was only a minimal reduction in mice with endothelial FABP4 deletion, indicating that adipocytes are indeed the main FABP4 source during lipolysis. Surprisingly, though mice with adipocyte deletion had blunted FABP4 responses to lipolysis, insulin secretory responses remained largely intact. In contrast, mice with endothelial FABP4 deletion had nearly intact FABP4 responses but showed blunted lipolysis-induced insulin secretion, identical to whole-body FABP4 deletion mice. These data demonstrated, for the first time, that endothelial FABP4 is a key requirement for lipolysis-induced insulin secretion.
We conclude that the endothelium is the major source of baseline hormonal FABP4, and is required for the insulin response to lipolysis. Endothelial dysfunction is a key aspect of many metabolic diseases, and thus, our discovery of the contribution of the endothelium to circulating FABP4 and its regulatory role in insulin secretion identifies a new role of the endothelium in metabolic regulation, and a potential source for FABP4 in metabolic disease.
ATGL is Differentially Required for Adipocyte FABP4 Secretion In Vivo and Ex Vivo
10.1101/2022.10.14.512277)
For the vast majority of human history, mankind has faced challenges to survival including famine and disease. As such, various biological pathways have evolved to combat conditions of deprivation, orchestrating incremental responses to promote survival, the central function of the adipose tissue is as a storage depot for excess nutrients, accumulated in periods of nutrient excess to ensure survival in conditions of famine. Liberation of these excess nutrients is not only coupled to nutrient scarcity, but also to conditions of sudden high energy demand, the socalled “fight-or-flight” response mediated by the central nervous system. Whereas ancient man faced a plethora of environmental threats, modern humans overwhelmingly exist in a world of nutrient excess, lack of predation, and low physical demand. Thus, our modern world is at odds with our biology, and metabolic programs that evolved to ensure our survival are now aberrantly promoting metabolic disease.
In recent decades, our understanding of adipose tissue has evolved significantly, with a greater appreciation for the role of adipose tissue as a central signaling organ in the regulation of metabolism. Numerous adipokines have now been identified as playing critical roles in controlling appetite and satiety, glucose and lipid metabolism, as well as inflammation. However, only one recently identified adipokine, Fatty Acid Binding Protein 4 (FABP4), is known to be secreted from the adipose tissue in response to fasting and the breakdown of energy stores in adipocytes. As a lipid-chaperone protein, it was hypothesized that the free fatty acid products of lipolysis may bind FABP4 to potentiate its release. This process of lipolysis involves the breakdown of triglycerides into fatty acids and glycerol, which are subsequently released into circulation to fuel distant tissues, such as the liver and muscle. While essential for survival when starving, this process is paradoxically inappropriately and chronically engaged in obesity, leading to the chronic release of lipolysis products. Concomitantly, circulating FABP4 levels are increased in obesity, and strongly associated with numerous metabolic disorders including diabetes, atherosclerosis, and various cancers in humans. Therefore, it is pertinent to understand the sources and regulation of this protein.
In this study, we investigated the regulation of FABP4 secretion from the adipose tissue in response to lipolysis. To study the contribution of the process of lipolysis to FABP4 secretion in physiology, we generated new genetic models with or without FABP4, and specifically lacking the first enzyme of the lipolysis pathway (ATGL) in their adipose tissue (ATGLAdpKO). Unexpectedly, when a stimulus is given to induce lipolysis in these mice, FABP4 secretion was even greater than the wildtype controls, despite the lack of triglyceride breakdown. However, the adipose tissue, when removed from these mice and stimulated to undergo lipolysis outside the body, was completely deficient in its ability to secrete FABP4. This paradoxical finding raised several possibilities; 1) FABP4 secretion in ATGLAdpKO mice is derived from a non-adipocyte cell type; or 2) a factor present in ATGLAdpKO mice, but not in cell culture conditions, stimulates FABP4 release.
To address the first question, we generated mice specifically lacking both ATGL and FABP4 in the adipose tissue (double knockout, DKO). Upon stimulation of lipolysis, DKO mice exhibited no induction of FABP4 secretion, indicating that the potentiated FABP4 release in the ATGLAdpKO mice was indeed sourced from the adipose tissue. By removing the adipose tissue from the mice, cells are disconnected from the central nervous system. It has been reported previously, that obese mice and humans exhibit hyperactivation of the sympathetic nervous system, which may also be the case in the ATGLdeficient models. Thus, we investigated whether sympathetic nervous system signaling in the mice, which is absent from tissue explants, was associated with enhanced FABP4 secretion. Our experiments using synthetic inhibitors demonstrated that the sympathetic system is, indeed, critical for FABP4 secretion. Therefore, the activity of a key enzymatic step of lipolysis mediated by ATGL, per se, is not required for stimulated in vivo FABP4 secretion from adipocytes, which can be induced through sympathetic signaling. These studies provide us with a novel system wherein we can dissect the biology of FABP4 and the products of lipolysis in generating the function of FABP4 and controlling its secretion.
Differential IL18 Signaling via IL18 Receptor and Na-Cl Co-transporter Discriminating Thermogenesis and Glucose Metabolism Regulation

Xian Zhang, Songyuan Luo, Minjie Wang, Qiongqiong Cao, Zhixin Zhang, Qin Huang, Jie Li, Zhiyong Deng, Tianxiao Liu, Cong-Lin Liu, Mathilde Meppen, Amelie Vromman, Richard A. Flavell, Gökhan S. Hotamışlıgil, Jian Liu, Peter Libby, Zhangsuo Liu, Guo-Ping Shi
Nature Communication, December 2022 (DOI: 10.1038/s41467-022-35256-8)
White adipose tissue (WAT) plays a role in storing energy, while brown adipose tissue (BAT) is instrumental in the re-distribution of stored energy when dietary sources are unavailable. Interleukin-18 (IL18) is a cytokine playing a role in T-cell polarization, but also for regulating energy homeostasis via the dimeric IL18 receptor (IL18r) and Na-Cl co-transporter (NCC) on adipocytes. Here we show that IL18 signaling in metabolism is regulated at the level of receptor utilization, with a preferential role for NCC in brown adipose tissue (BAT) and dominantly via IL18r in WAT. In Il18r-/Ncc-/- mice, high-fat diet (HFD) causes more prominent body weight gain and insulin resistance than in wild-type mice. The WAT insulin resistance phenotype of the doubleknockout mice is recapitulated in HFD-fed Il18r-/- mice, whereas decreased thermogenesis in BAT upon HFD is dependent on NCC deletion. BAT-selective depletion of either NCC or IL18 reduces thermogenesis and increases BAT and WAT inflammation. IL18r deletion in WAT reduces insulin signaling and increases WAT inflammation. In summary, our study contributes to the mechanistic understanding of IL18 regulation of energy metabolism and shows clearly discernible roles for its two receptors in brown and white adipose tissues.







