

¿QUÉ

¿QUÉ
La espectrometría de masas aplicada a la proteómica es una técnica analítica avanzada que mide la relación masa-carga (m/z) de proteínas y péptidos previamente ionizados en fase gaseosa para determinar su peso molecular exacto y deducir su estructura química. A través de este proceso, la técnica genera información detallada sobre la identidad de las proteínas (mediante la lectura de su secuencia de aminoácidos), su cantidad relativa o absoluta en una muestra y el mapeo de sus modificaciones post-traduccionales (como fosforilaciones o glicosilaciones)
Esto la convierte en la herramienta clave de la proteómica porque es la única metodología con la sensibilidad y el rendimiento necesarios para identificar y cuantificar simultáneamente miles de proteínas distintas en mezclas biológicas complejas, permitiendo mapear el dinamismo celular real que la genómica no puede predecir.
Moléculas a las que se aplica
Proteinas y peptidos (principal aplicacion en proteomica)
Acidos nucleicos (ADN y ARN)
Metabolitos
Lípidos
Fármacos y compuestos químicos pequeños

A principios del siglo XX, J J Thomson desarrolló los primeros experimentos que separaban iones según su relación masa/carga.
Francis William Aston perfeccionó el espectrógrafo de masas, permitiendo descubrir isótopos en 1922.
Durante varias décadas, la técnica se utilizó principalmente para moléculas pequeñas debido a la dificultad de ionizar compuestos grandes sin destruirlos.
AVANCES
Desarrollo de técnicas de ionización “suaves” (finales de XX):
Ionización por electrospray (ESI)
John B. Fenn, permite convertir proteínas en iones sin fragmentarlas significativamente.
• Desorción/ionización láser asistida por matriz (MALDI)
Koichi Tanaka, facilita el análisis de biomoléculas grandes mediante láser.
Técnica de ionizacion
suave para peptidoa y proteínas
Ionizacion por Electrospray (ESI)
Se considera técnica de ionización suave por qué…
Aplicaciones
Permite ionizar peptidos y proteínas sin fragmentarlos significativamente pues no requiere tanta energía, manteniendo los enlaces químicos de la molecula intactos
Técnicas de separación en fase líquida como la cromatografía líquida de alta resolución (HPLC) y estudio de interacciones no covalentes (enzima-sustrato) entre otras
Desorción/ionización láser asistida por matriz (MALDI)
Genera iones de biomoléculas grandes casi intactas, ya que la matriz orgánica con la que se mezcla la muestra absorbe la radiacion laser, evitando que la energía impacte directamente en la biomolécula.
Identificación rápida de bacterias y otros MO en diagnóstico de infecciones, mapeo de distribucion de farmacos, metabolitos y proteínas en tejidos y control de calidad de peptidos sintéticos, entre otros.
Ionización Química (CI)
Se ioniza un gas reactivo a alta presión, formando iones reactivos que reaccionan con la muestra transfiriendo protones mediante reacciones ión-molécula, resultando en una baja fragmentación
Análisis de aliento y biomarcadores, Detección de contaminantes ambientales halogenados y Identificacion de peso molecular en compuestos orgánicos.
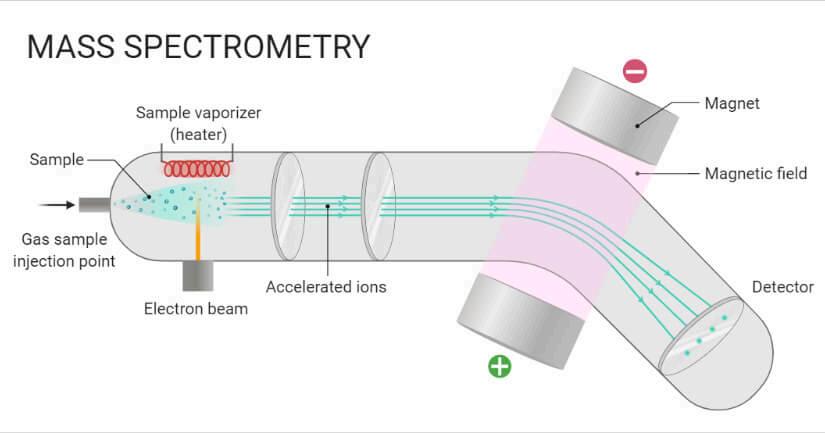
1 SISTEMA DE INTRODUCCIÓN DE MUESTRA (INLET SYSTEM)
INTRODUCE LA MUESTRA BIOLÓGICA (USUALMENTE UNA MEZCLA COMPLEJA DE PÉPTIDOS DIGERIDOS) EN EL INSTRUMENTO GENERALMENTE ESTÁ ACOPLADO A UN SISTEMA DE CROMATOGRAFÍA LÍQUIDA (LC)
AL SEPARAR LOS PÉPTIDOS POR SU HIDROFOBICIDAD ANTES DE ENTRAR AL MS, REDUCE LA COMPLEJIDAD DE LA MUESTRA ESTO EVITA QUE LOS PÉPTIDOS MÁS ABUNDANTES "TAPEN" A LOS MENOS ABUNDANTES, PERMITIENDO IDENTIFICAR PROTEÍNAS DE BAJA EXPRESIÓN
EL ÁREA BAJO EL PICO CROMATOGRÁFICO DE UN PÉPTIDO A LO LARGO DEL TIEMPO ES DIRECTAMENTE PROPORCIONAL A SU ABUNDANCIA, LO QUE PERMITE LA CUANTIFICACIÓN RELATIVA
4 DETECTOR
CUENTA EL NÚMERO DE IONES QUE LLEGAN A ÉL O REGISTRA
LA CORRIENTE INDUCIDA POR SU MOVIMIENTO
INFLUENCIA EN IDENTIFICACIÓN: GENERA LA SEÑAL QUE PRODUCE EL ESPECTRO DE MASAS (LOS PICOS EN LA GRÁFICA).
LA INTENSIDAD DEL PICO (LA ALTURA O EL ÁREA) EN EL DETECTOR ESTÁ DIRECTAMENTE RELACIONADA CON LA CANTIDAD DE IONES QUE IMPACTARON O PASARON POR ÉL. A MAYOR CORRIENTE DETECTADA, MAYOR ES LA CANTIDAD DE ESE PÉPTIDO ESPECÍFICO EN LA MUESTRA ORIGINAL
2 FUENTE DE IONIZACIÓN
CONVIERTE LAS MOLÉCULAS NEUTRAS EN IONES EN FASE GASEOSA EN PROTEÓMICA, LOS MÉTODOS MÁS COMUNES SON ESI (ELECTROSPRAY) Y MALDI (DESORCIÓN/IONIZACIÓN LÁSER ASISTIDA POR MATRIZ)
DETERMINA QUÉ TAN "ENTERA" LLEGA LA MOLÉCULA AL ANALIZADOR ESI Y MALDI SON IONIZACIONES "SUAVES", LO QUE SIGNIFICA QUE NO DESTRUYEN EL PÉPTIDO ANTES DE MEDIRLO ESI, ADEMÁS, PRODUCE IONES CON MÚLTIPLES CARGAS (+2, +3, ETC ), FACILITANDO LA LECTURA DE PROTEÍNAS GRANDES EN ANALIZADORES ESTÁNDAR
LA EFICIENCIA DE LA IONIZACIÓN DEBE SER CONSTANTE SI UN PÉPTIDO NO SE IONIZA BIEN (EFECTO MATRIZ), NO SE DETECTARÁ NI SE PODRÁ CUANTIFICAR CORRECTAMENTE
3 ANALIZADOR DE MASA
SEPARA LOS IONES SEGÚN SU RELACIÓN MASA/CARGA (M/Z) LOS MÁS COMUNES SON EL ORBITAL (ORBITRAP), EL TIEMPO DE VUELO (TOF) Y EL CUADRUPOLO.
LA RESOLUCIÓN Y LA EXACTITUD DE MASA DEL ANALIZADOR SON CRÍTICAS AQUÍ UN ANALIZADOR DE ALTA RESOLUCIÓN PUEDE DISTINGUIR ENTRE DOS PÉPTIDOS QUE DIFIEREN SOLO POR MILÉSIMAS DE DALTON, ASEGURANDO QUE LA IDENTIFICACIÓN DE LA PROTEÍNA SEA INEQUÍVOCA
EN EXPERIMENTOS DIRIGIDOS (COMO SRM/MRM USANDO CUADRUPOLOS), EL ANALIZADOR ACTÚA COMO UN FILTRO ULTRAESPECÍFICO QUE SOLO DEJA PASAR MASAS EXACTAS, LOGRANDO CUANTIFICACIONES EXTREMADAMENTE REPRODUCIBLES Y SENSIBLES
Una vez que el espectrómetro de masas ha procesado la muestra, el resultado primario obtenido no es una lista de proteínas, sino un conjunto masivo de datos crudos (archivos .raw) que contienen miles de espectros con picos de intensidades y relaciones masa-carga (m/z). Para transformar estos datos físicos en información biológica coherente y determinar qué proteínas y modificaciones postraducionales (PTMs) están presentes, se recurre a la bioinformática mediante las siguientes vías de análisis:
Estrategias de Identificación
Dependiendo del tipo de datos colectados por el instrumento y los objetivos del estudio, se emplean principalmente dos métodos algorítmicos:
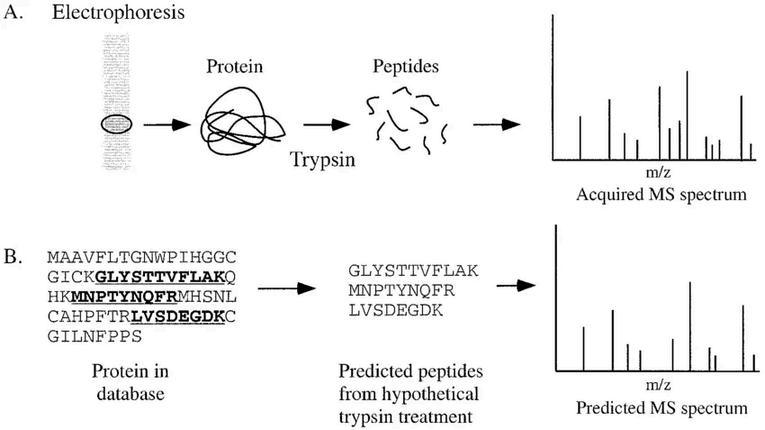
(Peptide Mass Fingerprinting - PMF): Se utiliza generalmente cuando los datos provienen de un espectrómetro MS simple (como MALDI-TOF) tras separar las proteínas por electroforesis en gel (2D-PAGE) El software toma las masas experimentales de los péptidos intactos y las compara con una digestión teórica de todas las proteínas conocidas en una base de datos Si un número estadísticamente significativo de masas coincide, se identifica la proteína.
Espectrometría de Masas en Tándem (MS/MS): Es el método estándar de la proteómica moderna (Shotgun proteomics) Aquí no solo se mide la masa del péptido intacto (MS1), sino que este se fragmenta deliberadamente dentro del equipo para obtener un segundo espectro (MS/MS) que revela el peso de los aminoácidos individuales. Esto permite leer la secuencia primaria del péptido de manera inequívoca.
Debido a la complejidad matemática de cruzar miles de espectros, el análisis manual es imposible Se utilizan potentes motores de búsqueda y plataformas de procesamiento como:
MaxQuant: Una de las plataformas gratuitas más utilizadas a nivel mundial para el análisis de datos de proteómica de alta resolución
Mascot (Matrix Science): El motor de búsqueda clásico por excelencia para PMF y MS/MS.
Proteome Discoverer: Software comercial de Thermo Scientific muy robusto para la identificación y cuantificación.
Para que los programas anteriores funcionen, necesitan un diccionario contra el cual comparar los espectros obtenidos. Las bases de datos biológicas más rigurosas y utilizadas son:
UniProt (específicamente UniProtKB/Swiss-Prot): Es la base de datos preferida porque está curada manualmente por expertos Contiene secuencias de proteínas verificadas y con anotaciones biológicas confiables, reduciendo drásticamente los falsos positivos.
NCBI (GenBank / RefSeq): Utilizada frecuentemente cuando se trabaja con organismos no modelo que aún no están del todo mapeados en UniProt
Una vez obtenidos los espectros de fragmentación (MS/MS), el análisis de modificaciones postraduccionales (PTMs) representa uno de los mayores retos bioinformáticos A diferencia de una proteína nativa, una proteína modificada cambia su masa molecular de forma precisa (por ejemplo, una fosforilación añade +79.96 Da y una acetilación +42.01 Da). Si no se le indica al software que busque estos cambios, el espectro simplemente no se emparejará con la base de datos y la proteína pasará desapercibida Para solucionar esto, en la proteómica moderna se utilizan principalmente los siguientes 2 enfoques
Búsquedas de tipo "cerrado" o dirigido
En programas como MaxQuant (Algoritmo Andromeda) y Mascot el usuario debe predefinir en una lista qué modificaciones biológicas específicas sospecha que están en la muestra (por ejemplo, buscar solo fosforilaciones en los aminoácidos Serina, Treonina y Tirosina).
MaxQuant es el estándar para la cuantificación y cuenta con un módulo de localización probabilística que calcula con precisión matemática en qué aminoácido exacto de la cadena ocurrió la modificación. Mascot se utiliza por su robustez estadística al procesar muestras donde las modificaciones esperadas ya están bien caracterizadas.
Búsqueda Abierta" (Open Search)
Los programas MSFragger y MetaMorpheus En lugar de buscar una modificación en específico, comparan el espectro real con la base de datos sin restricciones, identificando cualquier diferencia de masa (picos desplazados) de manera masiva
Se emplean para el descubrimiento "ciego" de PTMs MSFragger es sumamente utilizado debido a su velocidad sin precedentes; puede procesar en minutos búsquedas masivas de modificaciones que a otros programas les tomaría días. MetaMorpheus se prefiere cuando el objetivo es el análisis simultáneo de múltiples modificaciones sobre la misma muestra biológica.

La espectrometría de masas (MS) es una técnica analítica y cuantitativa fundamental que permite obtener la relación masacarga (m/z) de los analitos para determinar la masa molecular de péptidos y proteínas. A partir de los espectros obtenidos, se genera información crítica que, al ser contrastada con bases de datos, permite la identificación precisa de proteínas específicas en muestras complejas. Asimismo, la intensidad de las señales en el espectro facilita la cuantificación de la abundancia proteica, funcionando como una herramienta predictiva para seleccionar tratamientos eficaces o detectar biomarcadores clave
En conclusión, estos resultados permiten desarrollar aplicaciones clínicas avanzadas, como "biopsias líquidas" de alta sensibilidad para el diagnóstico temprano de enfermedades.
DIAGNÓSTICO DE CÁNCER DE PÁNCREAS
Objetivo: Identificar biomarcadores en vesículas extracelulares (exosomas) para detectar el cáncer de páncreas en etapas donde aún es operable
Resultados: Se identificó una firma de 3 proteínas de membrana exclusivas de exosomas tumorales mediante LC-MS/MS de alta resolución. Conclusión: La proteómica permite una "biopsia líquida" mucho más sensible que las pruebas de imagen tradicionales.
Objetivo: Entender por qué solo el 20% de los pacientes con melanoma responden al tratamiento
Resultados: El perfil proteómico reveló que los "respondedores" tienen una alta expresión de proteínas de transporte de antígenos antes de iniciar la terapia
Conclusión: La MS funciona como una herramienta predictiva para seleccionar el tratamiento más eficaz para cada paciente
Karp, G., Iwasa, J., y Marshall, W. (2020). Biología celular y molecular: Conceptos y experimentos (8.ª ed.). McGraw-Hill.
Hoffmann, E., & Stroobant, V. (2013). Mass Spectrometry: Principles and Applications (3rd ed.). Wiley-Interscience.
Espectrometría de masas de proteínas. (n.d.). Merckmillipore.com. Retrieved March 29, 2026, from https://www.merckmillipore.com/HN/es/applications/protein-biology/proteinmass-spectrometry?
Dasgupta. (2019). Critical issues in alcohol and drugs of abuse testing (A. Dasgupta, Ed.; 2nd ed.). Academic Press.
National Center for Biotechnology Information. (2023). Mass spectrometry in proteomics. https://pmc.ncbi.nlm.nih.gov/articles/PMC7216093/pdf/ijms-21-02873.pdf
Seemann, A. S. T. (n.d.). Protein Identification - ABRPI-Training. https://sepsisomics.github.io/tutorials/modules/xtandem/