8 gerais
quinta-feira, 19 de novembro de 2020
Covid-19
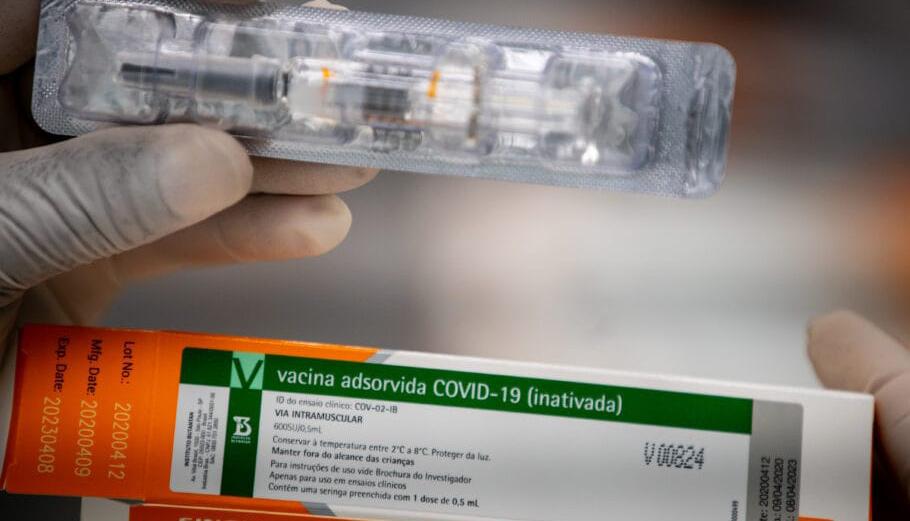
120mildosesdevacinachegam ao Brasil prontas para uso >> Distribuição terá início assim que for registrada e autorizada pelo Anvisa Mike Segar/Reuters/ABr
Vacina CoronaVac, do laboratório chinês Sinovac, está pronta para ser usada Por Época
A
s primeiras 120 mil doses da vacina Coronavac, produzidas pelo laboratório chinês Sinovac, desembarcam na manhã desta quinta-feira, 19, no Aeroporto Internacional de Guarulhos, um dia antes da data prevista pelo governo de São Paulo. Segundo o Instituto Butantan, a carga especial saiu da China na segunda-feira (16) e deve chegar a São Paulo nesta quinta-feira por volta das 9h. Após desembaraço aduaneiro, o material será transportado até o Instituto em um caminhão que receberá escolta especial. As 120 mil doses já estão prontas para uso assim que o imunizante for registrado e autorizado pela Agência Nacional de Vigilância Sanitária (Anvisa). O lote faz parte da carga de 6 milhões de doses que o governo de São Paulo comprou do laboratório chinês e ficará armazenado em local não informado por questões de segurança. Na próxima semana, outra remessa da China é aguardada: com 600 litros de insumos da vacina que serão
!
Até o momento, cerca de 10 mil voluntários já receberam ao menos uma das doses da vacina ou do placebo CIANO MAGNTA AMARELO PRETO
formulados e envazados no Butantan. Tanto as vacinas já prontas quanto a matéria-prima para a formulação das novas doses precisam ser transportadas refrigeradas entre 2 e 8 graus – a mesma temperatura em que é mantida a vacina da gripe, por exemplo. O insumo é líquido e vem armazenado em bolsas de 200 litros, dentro de contêineres nas aeronaves. Em coletiva no início da semana, o governador João Dória afirmou que até o dia 30 de dezembro o Butantan deverá receber todas as doses prontas (6 milhões) e toda a matéria prima necessária para a produção de mais 40 milhões de doses da vacina contra o coronavírus no país. Segundo o Butantan, todo material viajará em três voos de carga comerciais e seis voos fretados exclusivamente para trazer esse carregamento especial. Os voos com as doses e com os insumos da vacina não acontecerão todos no mesmo dia e sim ao longo das semanas. “Essa é uma carga muito preciosa e todos os cuidados com a segurança desse transporte estão sendo tomados”, afirmou Dimas Covas, em entrevista anterior à ÉPOCA. O Butantan possui duas fábricas para formulação e envase de vacinas com capacidade de finalizar 2 milhões de doses de vacina por dia. Segundo Covas, uma unidade inteira será destinada exclusivamente para a produção das 40 milhões de doses da Coronavac. Essas linhas de formulação são as mesmas usadas para a produção da vacina contra gripe Influenza. De acordo com o Butantan, as linhas já passaram por manutenção e estão prontas para início dos trabalhos. Se for preciso, as duas
linhas serão usadas, já que a produção da vacina tradicional da gripe deve começar apenas em meados de fevereiro. Eficácia A Coronavac está atualmente na terceira fase de testes clínicos para avaliação da eficácia – no Brasil os testes são conduzidos pelo Instituto Butantan. Até o momento, cerca de 10 mil voluntários já receberam ao menos uma das doses da vacina ou do placebo. Os dados sobre a eficácia da vacina ainda não foram divulgados, pois a pesquisa ainda não alcançou um número mínimo de voluntá-
rios contaminados pelo SARS-COV-2. Ao todo, segundo o instituto, 13 mil pessoas devem participar dessa fase da pesquisa. Nesta terça-feira (17), foram publicados na revista científica The Lancet os resultados dos estudos de fase 1 e 2, feitos com 743 pacientes, mostrando que a vacina é segura e induz resposta imune satisfatória. A fase 2 verifica justamente a segurança e a capacidade de gerar resposta do sistema de defesa do paciente. Segundo a pesquisa, as respostas de anticorpos foram induzidas no prazo de 28 dias após a primeira dose do imunizante.
Novo procedimento da Anvisa deve acelerar registro de vacina Andreia Verdélio
Repórter da Agência Brasil
A Agência Nacional de Vigilância Sanitária (Anvisa) definiu os procedimentos de submissão contínua de dados técnicos para o registro de vacinas contra a covid-19. A Instrução Normativa nº 77/2020 foi aprovada pela diretoria colegiada do órgão e publicada nesta quarta-feira, 18, no Diário Oficial da União. Segundo a Anvisa, os diretores também dispensaram a análise de impacto regulatório e consulta pública para o registro devido ao grau de urgência da vacina e gravidade da doença. “A medida possibilitará acelerar a disponibilização à população brasileira de vacinas contra o novo coronavírus, desde que demonstradas qualidade, segurança e eficácia conforme os requerimentos técnicos e regulatórios vigentes”, informou a agência, em comunicado. No procedimento de submissão contínua, os dados técnicos deverão ser encaminhados à Anvisa conforme forem gerados. Assim, as empresas interessadas no registro de vacinas não precisam ter em mãos todos os documentos reunidos para apresentá-los ao órgão regulador. Submissão contínua Esse procedimento será normatizado apenas para as vacinas contra co-
vid-19 a serem registradas no país. Segundo a Anvisa, outras autoridades regulatórias de referência, como a dos Estados Unidos, da Europa, da Suíça e da China, já utilizam a submissão contínua em situações específicas. A proposta da Anvisa prevê o atendimento a dois critérios para uso desse procedimento diferenciado. Um deles se refere à exigência de um dossiê de desenvolvimento clínico de medicamento referente à vacina proposta, protocolado na agência. Outro critério é que a pesquisa esteja em fase 3 de desenvolvimento clínico. As vacinas que tiverem a análise iniciada pelo procedimento de submissão contínua poderão ser submetidas ao pedido de registro formal após a conclusão da análise do último aditamento protocolado. Além disso, a empresa deve ter dados suficientes de qualidade, eficácia e segurança para o estabelecimento de uma relação positiva de benefício e risco da vacina, considerando a indicação terapêutica solicitada à Anvisa. A instrução normativa publicada hoje regulamenta artigo da Resolução nº 55/2010 da Anvisa, que diz que a empresa solicitante do registro poderá procurar a Coordenação de Produtos Biológicos para discutir aspectos relacionados ao desenvolvimento do produto, antes da submissão da documentação de registro.