
42 minute read
Calls for Increased Use of Off-Patent Medicines


The European Parliament has adopted a report calling for increased use of off-patent medicines in Europe to improve patient access, highlighting lessons learned during the Covid-19 pandemic and urging the European institutions to address them directly in future healthcare policy initiatives. The Parliament, in its own initiative report, stresses the central role of generic, biosimilar and valueadded medicines as the European Commission works to revise the legislative framework of the European pharmaceutical industry. Off-patent medicines already account for close to 70% of those used by patients in Europe. The call for increased use of off-patent medicines also implies a tailored regulatory pathway for value added medicines innovation, an area where the EU has been lagging behind the US for years.1 The report highlights that an efficient and optimised regulatory system for pharmaceuticals is also a priority. This includes digitalising regulatory processes and embracing environmentally friendly solutions like electronic patient information leaflets. The report calls on the European Commission to actively remove barriers to competition from loss of exclusivity. This includes reviewing the use of intellectual property incentives and assessing the framework for off-patent orphan and paediatric medicines. Adapting these frameworks could enable increased access for the rare disease population but a level playing field is needed to facilitate the development and manufacturing of these medicines. Commenting on the European Parliament vote, Medicines for Europe President ad-interim, Rebecca Guntern says, “It is highly encouraging to see the European Parliament take such clear directions to increase access to off-patent medicines. I am particularly pleased to see the European Parliament factor in the lessons learned from COVID-19. Generics, biosimilars and valueadded medicines are key pillars for the long-term resilience and sustainability of European healthcare systems. “The review of the EU pharmaceutical legislation is a one-time opportunity to make sure Europe has a fit-for-purpose, efficient, digital and simplified regulatory framework. We look forward to continuing our work with the European institutions and Member States in the coming months. We cannot afford to miss this opportunity.” 1 For more on this, see https:// www.cep.eu/fileadmin/ user_upload/cep.eu/Studien/ cepInput_Off_Patent_Innovation/ cepInput_Incentivising_ Pharmaceutical_OffPatent_ Innovation_in_the_EU.p
Breakthrough in Motor Neuron Disease
Researchers at Trinity College Dublin have made a major discovery in understanding motor neuron disease (MND). The research team has found that MND has 4 distinct patterns of changes in electrical signals that can be identified using EEG (electroencephalography). This breakthrough will be extremely valuable in identifying patients for clinical trials and will assist in finding new treatments for this devastating disease. MND is a devastating condition which causes progressive paralysis, changes in thinking, increasing physical disability and ultimately death within an average of two to three years. There are over 500 people in Ireland with MND, and one person is diagnosed every 3 days with the condition. There is currently no effective treatment. While trials of new drugs are being undertaken, MND is known to be very heterogeneous with different patterns of disability and life expectancy. Predicting in advance the pattern of disability and life expectancy is one of the major challenges in designing modern clinical trials.
The world class electrical signal analysis research developed within Trinity College has discovered different patterns of brain network disruption reflect the underlying disease process. The Trinity Professor Orla Hardiman, Professor of Neurology, Trinity College Dublin
researchers have now shown that these patterns of brain network disruptions in MND cluster into 4 distinct subtypes that are predictive of how the disease progresses. The team’s findings move the Trinity researchers one step closer to building better and more effective treatments for different sub-categories of the disease. The work was performed by Mr Stefan Dukic, a PhD student within the Academic Unit of Neurology at Trinity, under the supervision of Dr Bahman Nasseroleslami, Fr Tony Coote Assistant Professor in Neuroelectric Signal Analysis. Professor Orla Hardiman, Professor of Neurology and a world leader in MND research says, “This is a very important and exciting body of work. A major barrier to providing the right drug for the right patient in MND is the heterogeneity of the disease. This breakthrough research has shown that it is possible to use patterns of brain network dysfunction to identify subgroups of patients that cannot be distinguished by clinical examination. The implications of this work are enormous, as we will have new and reliable ways segregate patients based on what is really happening within the nervous system in MND.”
Access to Essential Medicines under Threat
Access to off-patent medicines is essential to increase competition, offer accessible and affordable treatments and for the budgetary sustainability of healthcare systems in Europe, as highlighted by a European Parliament report1 adopted recently. Medicines for Europe states however that ‘as EU governments struggle to cope with the economic and financial effects of the Covid-19 pandemic, several countries are proposing further cost containment measures for off-patent medicines, which cover 70% of prescription medicine supply across Europe at just 25% of the total cost.’
Many essential off-patent medicines have already been subject to strict price regulation (often referred to as reference pricing), successive price cutting measures, and budget adjustment measures (such as clawback) for many years. Says the representative body, “These measures have led to substantial price erosion, which is now compounded by mounting inflation across Europe and the recent increase in global production costs: raw material costs have tripled2, shipping rates are 6 times more expensive3 , airfreight costs have more than tripled4, energy costs rose by more than 230% last year5 with overall producer prices increasing by 10%.6 “Against this background further price cuts would be simply unsustainable for manufacturers. This creates serious risks for medicines supply and availability. The European Commission has collected a growing body of evidence showing that extreme cost containment policies applied to generic medicines are counterproductive, driving industrial consolidation and supply risks and potential medicine withdrawals from the
New Appointment for Paul
Paul Neill, Director, Generic Medicines Ireland with Teva Pharmaceuticals, has recently been appointed as Vice Chairperson of Medicines for Ireland. Paul says, “I'm really proud to have worked for almost 20 years in the Generic Medicines Industry in Ireland and to continue to play my part in developing sustainable and affordable healthcare for Irish Patients.” Best of luck in the new role Paul. market for important older, less expensive medicine. “Medicines for Europe calls on the Commission to react by legally mandating Member States, which use monopsony buying power, to integrate security of supply considerations into the EU Transparency Directive (which governs medicines pricing and reimbursement decisions) and the Public Procurement Directive (which governs medicine tendering).” Director General of Medicines for Europe, Adrian van den Hoven, says, “Just this week the European Parliament recognized that off-patent medicines provide the majority of accessible and affordable treatments and contribute greatly to the budgetary sustainability of healthcare systems, generating costs savings while underpinning the high quality of healthcare. Our industry is part of the solution, not part of the problem: it’s time to treat it as such. Security of supply needs to be factored into the pharmaceutical policies of Member States. We all need to remember that healthcare is an essential investment, not just a short-term cost.” 1 Report on a pharmaceutical strategy for Europe (2021/2013(INI)) 2 The Print – Pharma industry warns of Covid drug shortages as raw materials prices surge 200% (May 2021) 3 McKinsey & Company – What’s going on with shipping rates? (August 2021) 4 Air Cargo News - Airfreight rates expected to continue to rise (November 2021) 5 Euronews - Why Europe's energy prices are soaring and could get much worse (October 2021) 6 Eurostat - Producer prices in industry, total - monthly data (Jan 2020 vs September 2021)

HSE Excellence Awards 2021
The Maternal & Newborn Clinical Management System National (MN-CMS) Team was delighted to be chosen as Finalist in the recent HSE excellence Awards. The award recognised the hard work of the team and in particular the work of Gwen Malone in collaboration with the laboratories and back office staff of CUMH, UHK, The Rotunda and National Maternity Hospital. This work included the development and implementation of new electronic requests for laboratory testing of SARSCoV-2 (COVID-19) tests across all 4 hospitals, and is focused on the incorporation of real time electronic reporting of all SARS-CoV-2 test results from the individual hospital laboratories into MN-CMS. As part of this, Gwen has developed a customised electronic test request form to include fields for mandatory data required by the labs for statistical reporting to the HSE, CIDR, etc. She has also developed a COVID-19 order set of laboratory investigations, comprising a suite of diagnostic lab tests which is focused around disease management. This greatly speeds up and allows for safer processes both at the patient bedside and within the lab; and allows for instant access to test results from anywhere in the hospital, from outpatient clinics, and from remote-access devices.
Standardisation across all MN-CMS hospitals and laboratories of the requests and results of SARS-CoV-2 tests streamlines and optimises workflows for clinical staff across hospitals, and allows for the prompt collation of data on both the testing status and test results of patients across multiple hospitals. The rapidly changing nature of SARS-CoV-2 testing in response to scaling of laboratory capacity around the country, challenges with supplies and equipment, and expansion of the scope of referral testing proved challenging to reflect and maintain in an EHR setting but was achieved in a timely manner. The presentation was made by Dr. Colm Henry, CCO, to members of the team.

WITH ITS RAPID RESPONSES AND PROVEN MAINTENANCE OF EFFICACY SEE THE DIFFERENCE XELJANZ®• CAN MAKE FROM THE START

XELJANZ®• (tofacitinib) Prescribing Information:
Please refer to the Summary of Product Characteristics (SmPC) before prescribing XELJANZ 5 mg or 10 mg film-coated tablets, XELJANZ 11 mg prolonged release tablets or XELJANZ 1 mg/mL oral solution. Presentation: Film-coated tablet containing tofacitinib citrate, equivalent to 5 mg or 10 mg tofacitinib. Prolonged-release tablets containing tofacitinib citrate, equivalent to 11 mg tofacitinib. Oral solution containing tofacitinib citrate, equivalent to 1 mg/mL tofacitinib. Indications: Please note not all presentations are licensed for all indications, please see dosage section for details: In combination with methotrexate (MTX) for the treatment of moderate to severe active rheumatoid arthritis (RA) in adult patients who have responded inadequately to, or who are intolerant to one or more disease-modifying antirheumatic drugs. Can be given as monotherapy in case of intolerance to MTX or when treatment with MTX is inappropriate. In combination with MTX for the treatment of active psoriatic arthritis (PsA) in adult patients who have had an inadequate response or who have been intolerant to a prior disease modifying antirheumatic drug (DMARD) therapy. For the treatment of adult patients with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent. For the treatment of active polyarticular juvenile idiopathic arthritis (rheumatoid factor positive [RF+] or negative [RF-] polyarthritis, and extended oligoarthritis), and juvenile psoriatic arthritis (PsA) in patients 2 years of age and older, who have responded inadequately to previous therapy with DMARDs. Can be given in combination with methotrexate (MTX) or as monotherapy in case of intolerance to MTX or where continued treatment with MTX is inappropriate. Dosage: Treatment should be initiated and supervised by specialist physicians experienced in the diagnosis and treatment of the condition for which tofacitinib is indicated. Tofacitinib is given with or without food. RA and PsA: The recommended dose is 5 mg orally twice daily or 11 mg once daily which should not be exceeded. Treatment with tofacitinib 5 mg film coated tablets twice daily and tofacitinib 11 mg prolonged release tablet once daily may be switched between each other on the day following the last dose of either tablet. UC: The recommended dose is 10 mg given orally twice daily for induction for 8 weeks. For patients who do not achieve adequate therapeutic benefit by week 8, the induction dose of 10 mg twice daily can be extended for an additional 8 weeks (16 weeks total), followed by 5 mg twice daily for maintenance. Tofacitinib induction therapy should be discontinued in any patient who shows no evidence of therapeutic benefit by week 16. The recommended dose for maintenance treatment is tofacitinib 5 mg given orally twice daily. Tofacitinib 10 mg twice daily for maintenance treatment is not recommended in patients with UC who have known venous thromboembolism (VTE) risk factors, unless there is no suitable alternative treatment available. For patients with UC who are not at increased risk for VTE, tofacitinib 10 mg orally twice daily may be considered if the patient experiences a decrease in response on tofacitinib 5 mg twice daily and failed to respond to alternative treatment options for ulcerative colitis such as tumour necrosis factor inhibitor (TNF inhibitor) treatment. Tofacitinib 10 mg twice daily for maintenance treatment should be used for the shortest duration possible. The lowest effective dose needed to maintain response should be used. Polyarticular JIA and juvenile PsA: The recommended dose in patients 2 years of age and older: 10 kg - < 20 kg: 3.2 mg (3.2 mL of oral solution) twice daily, 20 kg - < 40 kg: 4 mg (4 mL of oral solution) twice daily, and ≥ 40 kg 5 mg (5 mL of oral solution or 5 mg film-coated tablet) twice daily. Patients ≥ 40 kg treated with tofacitinib 5 mL oral solution twice daily may be switched to tofacitinib 5 mg film-coated tablets twice daily. Patients < 40 kg cannot be switched from tofacitinib oral solution. Dose interruption in adults and paediatric patients: Tofacitinib treatment should be interrupted if a patient develops a serious infection until the infection is controlled. Interruption of dosing may be needed for management of dose-related laboratory abnormalities including lymphopenia, neutropenia, and anaemia. It is recommended not to initiate dosing in patients with an absolute lymphocyte count (ALC) less than 0.75 x 109/L, an absolute neutrophil count (ANC) less than 1x109 /L or with haemoglobin less than 9 g/dL. It is recommended not to initiate dosing in paediatric patients with an absolute neutrophil count (ANC) less than 1.2 x 109/L or with haemoglobin less than 10 g/dL. Renal impairment: No dose adjustment is required in patients with mild or moderate renal impairment. Patients with severe renal impairment the dose should be reduced to 5 mg once daily when the indicated dose in the presence of normal renal function is 5 mg twice daily or 11 mg prolonged-release tablet once daily. Dose should be reduced to 5 mg twice daily when the indicated dose in the presence of normal renal function is 10 mg twice daily. Patients with severe renal impairment should remain on a reduced dose even after haemodialysis. Hepatic impairment: No dose adjustment is required in patients with mild hepatic impairment. Patients with moderate hepatic impairment dose should be reduced to 5 mg once daily when the indicated dose in the presence of normal hepatic function is 5 mg twice daily or 11 mg prolonged-release tablet once daily. Dose should be reduced to 5 mg twice daily when the indicated dose in the presence of normal hepatic function is 10 mg twice daily. Tofacitinib should not be used in patients with severe hepatic impairment. Elderly: No dose adjustment is required in patients aged 65 years and older. Use with caution as increased risk and severity of adverse events. See also Warnings & Precautions for use in patients over 65 years of age. Interactions: Tofacitinib total daily dose should be reduced by half in adult and paediatric patients receiving potent inhibitors of cytochrome P450 (CYP) 3A4 (e.g. ketoconazole) and in patients receiving 1 or more concomitant medicinal products that result in both moderate inhibition of CYP3A4 as well as potent inhibition of CYP2C19 (e.g. fluconazole). Coadministration of tofacitinib with potent CYP inducers (e.g. rifampicin) may result in a loss of or reduced clinical response. Coadministration of potent inducers of CYP3A4 with tofacitinib is not recommended. Only in paediatric patients: available data suggest that clinical improvement is observed within 18 weeks of initiation of treatment with tofacitinib. Continued therapy should be carefully reconsidered in a patient exhibiting no clinical improvement within this timeframe. The safety and efficacy of tofacitinib in children less than 2 years of age with polyarticular JIA and juvenile PsA has not been established. The safety and efficacy of tofacitinib in children less than 18 years of age with other indications (e.g. ulcerative colitis) has not been established. The safety and efficacy of tofacitinib prolonged-release formulation in children aged less than 18 years have not been established. Contraindications: Hypersensitivity to any of the ingredients, active tuberculosis (TB), serious infections such as sepsis, or opportunistic infections, severe hepatic impairment, pregnancy and lactation. Warnings and Precautions: Patients treated with tofacitinib should be given a patient alert card. Use in patients over 65 years of age: Considering the increased risk of serious infections, myocardial infarction, and malignancies with tofacitinib in patients over 65 years of age, tofacitinib should only be used in patients over 65 years of age if no suitable treatment alternatives are available. Combination with other therapies: There was a higher incidence of adverse events for the combination of tofacitinib with MTX versus tofacitinib as monotherapy in RA clinical studies. Tofacitinib should be avoided in combination with biologics and potent immunosuppressants such as azathioprine, 6-mercaptopurine, ciclosporine and tacrolimus. Venous thromboembolism (VTE): Serious VTE events including pulmonary embolism (PE), some of which were fatal, and deep vein thrombosis (DVT), have been observed in patients taking tofacitinib. In a randomised post authorisation safety study in patients with rheumatoid arthritis who were 50 years of age or older with at least one additional cardiovascular risk factor,
aFrom the start of treatment with XELJANZ in patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent.3 XELJANZ 10 mg BID achieved higher rates of remission (primary endpoint) at week 8 vs placebo in OCTAVE Induction 1 (19% [88/476] vs 8% [10/122]; P=0.007) and OCTAVE Induction 2 (17% [71/429] vs 4% [4/112]; P<0.001)1 XELJANZ 10 mg BID achieved higher rates of clinical response at week 8 vs placebo in OCTAVE Induction 1 (60% [285/476] vs 33% [40/122]; P<0.001) and OCTAVE Induction 2 (55% [236/429] vs 29% [32/112]; P<0.001)1 In OCTAVE Sustain, for patients showing remission or clinical response following induction, XELJANZ 5 mg BID achieved higher rates of remission at week 52 (primary endpoint) vs placebo (34% [68/198] vs 11% [22/198]; P<0.001).1 Decreases in rectal bleeding and stool frequency Mayo subscores were observed as early as day 3 in patients treated with XELJANZ 10 mg BID2
RAPID RESPONSES AND PROVEN MAINTENANCE OF EFFICACY, CAN MAKE FROM THE START1,2,a
a dose dependent increased risk for VTE was observed with tofacitinib compared to TNF inhibitors. In a post hoc exploratory analysis within this study, in patients with known VTE risk factors, occurrences of subsequent VTEs were observed more frequently in tofacitinib-treated patients that, at 12 months treatment, had D-dimer level greater than or equal to twice the upper limit of normal (2× ULN) versus those with D-dimer level <2×ULN. For patients with RA with known risk factors for VTE, consider testing D-dimer levels after approximately 12 months of treatment. If D-dimer test result is ≥ 2× ULN, confirm that clinical benefits outweigh risks prior to a decision on treatment continuation with tofacitinib. Tofacitinib should be used with caution in patients with known risk factors for VTE, regardless of indication and dosage. Tofacitinib 10 mg twice daily for maintenance treatment is not recommended in patients with UC who have known VTE risk factors, unless there is no suitable alternative treatment available. Promptly evaluate patients with signs and symptoms of VTE and discontinue tofacitinib in patients with suspected VTE, regardless of dose or indication. Infections: Serious and sometimes fatal infections have been reported in patients administered tofacitinib. Rheumatoid arthritis patients taking corticosteroids may be predisposed to infection. Patients should be closely monitored for infections, with prompt diagnosis and treatment. Treatment should be interrupted if a serious infection develops. As there is a higher incidence of infections in the elderly and in the diabetic populations in general, caution should be used when treating the elderly and patients with diabetes. Tuberculosis: Patients should be evaluated for both active and latent TB prior to being treated with tofacitinib. Patients who test positive for latent TB should be treated with standard antimycobacterial therapy before administering tofacitinib. Viral Reactivation: In clinical studies viral reactivation and cases of herpes zoster have been observed. Screening for viral hepatitis should be performed in accordance with clinical guidelines prior to starting therapy with tofacitinib. The impact on chronic viral hepatitis is not known. Major adverse cardiovascular events (MACE): MACE have been observed in patients taking tofacitinib. In a randomised post authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, an increased incidence of myocardial infarctions was observed with tofacitinib compared to TNF inhibitors. In patients over 65 years of age, patients who are current or past smokers, and patients with other cardiovascular risk factors, tofacitinib should only be used if no suitable treatment alternatives are available. Vaccinations: Prior to initiating tofacitinib, it is recommended that all patients, particularly pJIA and jPsA patients, be brought up to date with all immunisations in agreement with current immunisation guidelines. Live vaccines should not be given concurrently with tofacitinib. Malignancy and lymphoproliferative disorder: Tofacitinib may affect host defences against malignancies. In a randomised post authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, an increased incidence of malignancies excluding non-melanoma skin cancer (NMSC), particularly lung cancer and lymphoma, was observed with tofacitinib compared to TNF inhibitors. Lung cancers and lymphoma in patients treated with tofacitinib have also been observed in other clinical studies and in the post marketing setting. Other malignancies in patients treated with tofacitinib were observed in clinical studies and the post marketing setting, including, but not limited to, breast cancer, melanoma, prostate cancer, and pancreatic cancer. In patients over 65 years of age, patients who are current or past smokers, and patients with other malignancy risk factors (e.g. current malignancy or history of malignancy other than a successfully treated non-melanoma skin cancer) tofacitinib should only be used if no suitable treatment alternatives are available. NMSCs have been reported in patients treated with tofacitinib; the risk of NMSC may be higher in patients treated with tofacitinib 10 mg twice daily than in patients treated with 5 mg twice daily. Periodic skin examination is recommended in patients at increased risk for skin cancer. Interstitial lung disease: Caution is recommended in patients with a history of chronic lung disease as they may be more prone to infection. Asian patients are known to be at higher risk of ILD, caution should be exercised with these patients. Gastrointestinal perforations: Tofacitinib should be used with caution in patients who may be at increased risk, e.g. history of diverticulitis or concomitant use of corticosteroids or NSAIDs. Hypersensitivity: Cases of drug hypersensitivity associated with tofacitinib administration have been reported. Allergic reactions included angioedema and urticaria; serious reactions have occurred. If any serious allergic or anaphylactic reaction occurs, tofacitinib should be discontinued immediately. Laboratory Parameters: Increased incidence of lymphopenia and neutropenia have been reported, and decreases in haemoglobin, which should be monitored in accordance with the SmPC. Monitor ANC and haemoglobin at baseline, 4-8 weeks and 3 monthly, ALC at baseline and 3 monthly. Tofacitinib has been associated with increases in lipid parameters, maximal effects were observed within 6 weeks. Monitoring should be performed 8 weeks after initiation and managed according to hyperlipidaemia guidelines. Increases in liver enzymes greater than 3x ULN were uncommonly reported; use caution when initiating with potential hepatotoxic medicinal products. Gastrointestinal obstruction with a non-deformable prolonged-release formulation: Caution should be used when administering tofacitinib 11 mg prolonged release tablets to patients with pre-existing severe gastrointestinal narrowing (pathologic or iatrogenic). Pregnancy & Lactation: Use of tofacitinib during pregnancy and breast-feeding is contraindicated. Side Effects: RA: The most common serious adverse reactions were serious infections; pneumonia, cellulitis, herpes zoster, UTIs, diverticulitis, appendicitis and opportunistic infections. The most commonly reported adverse reactions during the first 3 months in controlled clinical trials were headache, upper respiratory tract infections, nasopharyngitis, diarrhoea, nausea and hypertension. UC: The most commonly reported adverse reactions in patients receiving tofacitinib 10 mg twice daily in the induction studies were headache, nasopharyngitis, nausea, and arthralgia. Commonly reported adverse reactions (>1/100 to <1/10) across all indications were pneumonia, influenza, herpes zoster, urinary tract infection, sinusitis, bronchitis, viral upper respiratory tract infection, pharyngitis, anaemia, headache, hypertension, cough, abdominal pain, vomiting, diarrhoea, nausea, gastritis, dyspepsia, rash, arthralgia, pyrexia, oedema peripheral, fatigue, blood creatine phosphokinase increased. Refer to section 4.8 of the SmPC for further information on side effects, including description of selected adverse reactions. Legal Category: S1A. Marketing Authorisation Number: EU/1/17/1178/003 – 5 mg (56 film-coated tablets); EU/1/17/1178/007 – 10 mg (56 film-coated tablets); EU/1/17/1178/012 – 11 mg (28 prolonged-release tablets); EU/1/17/1178/015 1mg/mL oral solution. Marketing Authorisation Holder: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium. For further information on this medicine please contact: Pfizer Medical Information on 1800 633 363 or at medical.information@pfizer.com. For queries regarding product availability please contact: Pfizer Healthcare Ireland, 9 Riverwalk, National Digital Park, Citywest Business Campus, Dublin 24, + 353 1 467 6500. ˸ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information.
Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 of the SmPC for how to report adverse reactions. Last revised: 10/2021. Ref: XJ 13_0.
References:
1. Sandborn WJ, Su C, Sands BE, et al; for the OCTAVE Induction 1, OCTAVE Induction 2, and OCTAVE Sustain Investigators. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723-1736. 2. Hanauer S et al. Tofacitinib Induction Therapy Reduces Symptoms Within 3 Days for Patients With Ulcerative Colitis. Clin Gastroenterol Hepatol. 2019;17(1):139-147. 3. XELJANZ Summary of Product Characteristics.
Cancer in Ireland: NCR Annual Report
The National Cancer Registry Ireland (NCRI) has announced its updated statistics on cancer incidence, mortality and survival for patients diagnosed with cancer in Ireland 1994 – 2019.
Highlights this year include:
• Five-year net survival averaging 65% for patients diagnosed between 2014 and 2018, a substantial increase from twenty years previously when 42% was the average. • The number of cancer survivors living through or after cancer treatment in Ireland continuing to increase, year on year. At the end of 2019, there were nearly 200,000 patients living after a cancer diagnosis. • Indications of substantial inroads being made in the progress to control the four commonest cancers (prostate, breast, lung and colorectal), which comprise over half of all invasive tumours (other than non-melanoma skin cancers). • Mortality rates falling for these four major cancers (or stabilising for lung cancer in females), and incidence rates falling for both lung and colorectal cancers, in both sexes, though relatively recently for lung cancer in females. Professor Deirdre Murray, Director of the National Cancer Registry said, “The report also draws attention to other opportunities to improve the health of the nation. The incidence and mortality rates of melanoma skin cancer continue to increase in females, pointing to the importance of sun safety. “Incidence rates for oropharyngeal and liver cancers are increasing, as are mortality rates for liver cancer in both sexes. This report is a timely reminder that HPV vaccination and adopting a healthy lifestyle can reduce the risk of developing these cancers. “The incidence rate of cervical cancer continues to fall, reflecting the impact of the screening programme. It must be remembered from a service planning perspective, that despite the successes in reducing cancer incidence and mortality rates, the number of patients diagnosed with cancer every year is rising and will continue to rise in future decades as the Irish population continues to grow and the average age increases.” The improved cancer survival figures revealed by the report are being put at serious risk by pandemic-related disruption, according to the Irish Cancer Society. Commenting on the National Cancer Registry Ireland (NCRI) annual report, the Society’s CEO Averil Power said, “While it is heartening to hear that progress is being made for devastating cancers like breast, lung and prostate according to latest figures up to 2019, we are very worried that significantly less cancers were diagnosed last year. “This will present a major challenge for years to come, and is unfortunately no surprise as already struggling cancer services have been stretched to breaking point during the pandemic. Lengthy waiting lists and disruptions to vital diagnostic and screening services are now all too commonplace. “Patients are telling us that they are terrified of having their treatment delayed given the current spike in Covid case numbers and are very distressed about the worrying consequences to their health from catching the virus, and the further risk of treatment delays that this would bring. “The Irish Cancer Society is collaborating with researchers from the Royal College of Surgeons Ireland and others on a much-needed project to get a clearer picture of the impact of the pandemic on cancer services. In the meantime we need the Government to step up and put a massive effort into clearing waiting lists and securing cancer services for patients nearly two years into this crisis. “The NCRI report shows that as many as 1 in 8 cancers that were predicted to be diagnosed in 2020 were not. Although there has been an encouraging trend of people seeking medical help so far this year, waiting times for diagnostic tests remain too long and these must be addressed as a matter of urgency. “It is frightening to think that there are people in our community with cancer who don’t know it yet.” Earlier this year, another report released by the National Cancer Registry highlighted that at least a third of invasive cancers can be prevented through modifying some of our lifestyle behaviours. The report analysed skin cancer rates in Ireland using just two of the factors that contribute to our skin cancer risk: a single sunburn episode and using sunbeds. It found that these two factors alone contributed to: • More than 400 cases of melanoma, the most serious form of skin cancer. • Almost 3,000 diagnoses of nonmelanoma skin cancer (NMSC). Although melanoma is not the most frequently diagnosed skin cancer, it is associated with significant ill-health, is much more likely to spread to other parts of the body and can be fatal. While NMSC is rarely fatal, treatment may require disfiguring surgery and/or lifetime treatment and monitoring.
WITH THE FLEXIBILTY OF 3 WEEKLY OR 6 WEEKLY DOSING2

44% REDUCTION IN THE RISK OF DISEASE RECURRENCE OR DEATH VS PLACEBO3
Infusion 25mg/mL
3-YEAR RECURRENCE-FREE SURVIVAL (RFS) OF THE OVERALL POPULATION IN KEYNOTE-0543,a
100 90 80 70 60 50 40 30 20 10 0
0
75
%60
HRb=0.56 (95% CI, 0.47–0.68)
KEYTRUDA Placebo
6
Number at risk KEYTRUDA Placebo 514 505 412 360 12
374 298
68 %64
%47 %44
18
351 259 24 30
Time, months
333 226 314 215 36 42
189 126 29 28 48
0 0
IMMUNE-RELATED ADVERSE EVENTS (IRAES) IN KEYNOTE-0544,c
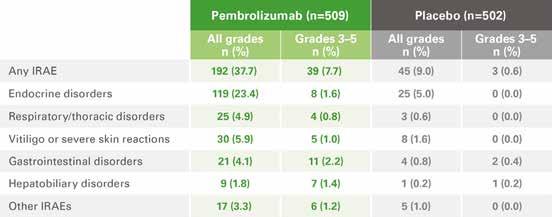
a RFS was estimated by the Kaplan-Meier method.3 b The HR and its confidence interval (CI) were estimated using the Cox model stratified by stage provided at randomization.3 c Regardless of investigator attribution.4
KEYTRUDA® (pembrolizumab) ABRIDGED PRODUCT INFORMATION Refer to Summary of Product Characteristics before prescribing. PRESENTATION KEYTRUDA 25 mg/mL: One vial of 4 mL of concentrate contains 100 mg of pembrolizumab. INDICATIONS KEYTRUDA as monotherapy is indicated for the treatment of advanced (unresectable or metastatic) melanoma in adults. KEYTRUDA as monotherapy is indicated for the adjuvant treatment of adults with Stage III melanoma and lymph node involvement who have undergone complete resection. KEYTRUDA as monotherapy is indicated for the first-line treatment of metastatic non-small cell lung carcinoma (NSCLC) in adults whose tumours express PD-L1 with a ≥50% tumour proportion score (TPS) with no EGFR or ALK positive tumour mutations. KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of metastatic non-squamous NSCLC in adults whose tumours have no EGFR or ALK positive mutations. KEYTRUDA, in combination with carboplatin and either paclitaxel or nab-paclitaxel, is indicated for the first-line treatment of metastatic squamous NSCLC in adults. KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic NSCLC in adults whose tumours express PD-L1 with a ≥1% TPS and who have received at least one prior chemotherapy regimen. Patients with EGFR or ALK positive tumour mutations should also have received targeted therapy before receiving KEYTRUDA. KEYTRUDA as monotherapy is indicated for the treatment of adult and paediatric patients aged 3 years and older with relapsed or refractory classical Hodgkin lymphoma (cHL) who have failed autologous stem cell transplant (ASCT) or following at least two prior therapies when ASCT is not a treatment option. KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic urothelial carcinoma in adults who have received prior platinum-containing chemotherapy. KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic urothelial carcinoma in adults who are not eligible for cisplatin-containing chemotherapy and whose tumours express PD L1 with a combined positive score (CPS) ≥ 10. KEYTRUDA as monotherapy or in combination with platinum and 5-fluorouracil (5-FU) chemotherapy, is indicated for the first-line treatment of metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC) in adults whose tumours express PD-L1 with a CPS ≥ 1. KEYTRUDA as monotherapy is indicated for the treatment of recurrent or metastatic HNSCC in adults whose tumours express PD-L1 with a ≥ 50% TPS and progressing on or after platinum-containing chemotherapy. KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of advanced renal cell carcinoma (RCC) in adults. KEYTRUDA as monotherapy is indicated for the first line treatment of metastatic microsatellite instability high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer in adults. KEYTRUDA, in combination with platinum and fluoropyrimidine based chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic carcinoma of the oesophagus or HER-2 negative gastroesophageal junction adenocarcinoma in adults whose tumours express PD-L1 with a CPS ≥ 10. DOSAGE AND ADMINISTRATION See SmPC for full details. Therapy must be initiated and supervised by specialist physicians experienced in the treatment of cancer. The recommended dose of KEYTRUDA in adults is either 200 mg every 3 weeks or 400 mg every 6 weeks administered as an intravenous infusion over 30 minutes. The recommended dose of KEYTRUDA as monotherapy in paediatric patients aged 3 years and older with cHL is 2 mg/kg bodyweight (up to a maximum of 200 mg), every 3 weeks administered as an intravenous infusion over 30 minutes. KEYTRUDA must not be administered as an intravenous push or bolus injection. When administering KEYTRUDA as part of a combination with intravenous chemotherapy, KEYTRUDA should be administered first. Treat patients until disease progression or unacceptable toxicity. Atypical responses (i.e., an initial transient increase in tumour size or small new lesions within the first few months followed by tumour shrinkage) have been observed. Recommended to continue treatment for clinically stable patients with initial evidence of disease progression until disease progression is confirmed. For the adjuvant treatment of melanoma, KEYTRUDA should be administered until disease recurrence, unacceptable toxicity, or for a duration of up to one year. KEYTRUDA, as monotherapy or as combination therapy, should be permanently discontinued (a) For Grade 4 toxicity except for: endocrinopathies that are controlled with replacement hormones; or haematological toxicity, only in patients with cHL in which KEYTRUDA should be withheld until adverse reactions recover to Grade 0-1; (b) If corticosteroid dosing cannot be reduced to ≤10 mg prednisone or equivalent per day within 12 weeks; (c) If a treatment-related toxicity does not resolve to Grade 0 1 within 12 weeks after last dose of KEYTRUDA; (d) If any event occurs a second time at Grade ≥ 3 severity. Patients must be given the Patient Alert Card and be informed about the risks of KEYTRUDA. Special populations Elderly: No dose adjustment necessary. Data from patients ≥ 65 years are too limited to draw conclusions on cHL population. Data are limited in patients ≥ 75 years for pembrolizumab monotherapy in patients with resected Stage III melanoma and MSI-H or dMMR CRC; for pembrolizumab in combination with axitinib in patients with advanced RCC; for chemotherapy combination in patients with metastatic NSCLC and oesophageal carcinoma; for pembrolizumab (with or without chemotherapy) in patients receiving first line treatment for metastatic or unresectable recurrent HNSCC. Renal impairment: No dose adjustment needed for mild or moderate renal impairment. No studies in severe renal impairment. Hepatic impairment: No dose adjustment needed for mild hepatic impairment. No studies in moderate or severe hepatic impairment. Paediatric population: Safety and efficacy in children below 18 years of age not established except in paediatric patients with cHL. CONTRAINDICATIONS Hypersensitivity to the active substance or to any excipients. PRECAUTIONS AND WARNINGS Assessment of PD-L1 status When assessing the PD-L1 status of the tumour, it is important that a well-validated and robust methodology is chosen to minimise false negative or false positive determinations. Immune-related adverse reactions Immune-related adverse reactions, including severe and fatal cases, have occurred in patients receiving pembrolizumab. Most immune related adverse reactions occurring during treatment with pembrolizumab were reversible and managed with interruptions of pembrolizumab, administration of corticosteroids and/or supportive care. Immune related adverse reactions have also occurred after the last dose of pembrolizumab. Immune-related adverse reactions affecting more than one body system can occur simultaneously. See SmPC for full details. Immune-related pneumonitis: Patients should be monitored for signs and symptoms of pneumonitis.. Suspected pneumonitis should be confirmed with radiographic imaging and other causes excluded. Refer to SmPC for information on management of immune-related pneumonitis. Immune-related colitis: Patients should be monitored for signs and symptoms of colitis, and other causes excluded. Consider the potential risk of gastrointestinal perforation. Refer to SmPC for information on management of immune-related colitis. Immune-related hepatitis: Patients should be monitored for changes in liver function (at the start of treatment, periodically during treatment and as indicated based on clinical evaluation) and symptoms of hepatitis, and other causes excluded. Refer to SmPC for information on management of Immune-related hepatitis. Immune-related nephritis: Patients should be monitored for changes in renal function, and other causes of renal dysfunction excluded. Refer to SmPC for information on management of immune-related nephritis. Immune-related endocrinopathies: Severe endocrinopathies, including adrenal insufficiency, hypophysitis, type 1 diabetes mellitus, diabetic ketoacidosis, hypothyroidism, and hyperthyroidism have been observed with pembrolizumab treatment and patients should be monitored for these endocrinopathies. Refer to SmPC for information on management of immune-related endocrinopathies. Immune-related skin adverse reactions: Patients should be monitored for suspected severe skin reactions and other causes should be excluded. Cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported in patients receiving pembrolizumab. If SJS or TEN is confirmed, pembrolizumab should be permanently discontinued. Other clinically significant immune-related adverse reactions: The following additional clinically significant, immune-related adverse reactions, have been reported in clinical studies or in post-marketing experience: uveitis, arthritis, myositis, myocarditis, pancreatitis, Guillain-Barré syndrome, myasthenic syndrome, haemolytic anaemia, sarcoidosis, encephalitis, myelitis, vasculitis, cholangitis sclerosing and gastritis. Refer to SmPC for information on management of significant immune-related adverse reactions. Complications of allogeneic Haematopoietic Stem Cell Transplant (HSCT): Allogeneic HSCT after treatment with pembrolizumab: Cases of graft-versus-host-disease (GVHD) and hepatic veno-occlusive disease (VOD) have been observed in patients with classical Hodgkin lymphoma undergoing allogeneic HSCT after previous exposure to pembrolizumab. Allogeneic HSCT prior to treatment with pembrolizumab: In patients with a history of allogeneic HSCT, acute GVHD, including fatal GVHD, has been reported after treatment with pembrolizumab. Infusion-related reactions: For Grades 3 or 4infusion reactions including hypersensitivity and anaphylaxis, stop infusion and permanently discontinue pembrolizumab. With Grades 1 or 2 infusion reactions, infusion may continue with close monitoring. Premedication with antipyretic and antihistamine may be considered. Overdose: There is no information on overdose with pembrolizumab. In case of overdose, monitor closely for signs or symptoms of adverse reactions and treat appropriately. INTERACTIONS No formal pharmacokinetic drug interaction studies have been conducted with pembrolizumab. No metabolic drug drug interactions are expected. The use of systemic corticosteroids or immunosuppressants before starting pembrolizumab should be avoided because of their potential interference with the pharmacodynamic activity and efficacy of pembrolizumab. Corticosteroids can be used as premedication, when pembrolizumab is used in combination with chemotherapy, as antiemetic prophylaxis and/or to alleviate chemotherapy-related adverse reactions. FERTILITY, PREGNANCY AND LACTATION Women of childbearing potential Women of childbearing potential should use effective contraception during treatment with pembrolizumab and for at least 4 months after the last dose of pembrolizumab. Pregnancy No data on use in pregnant women. Do not use during pregnancy unless the clinical condition of the woman requires treatment with pembrolizumab. Breast-feeding It is unknown whether pembrolizumab is secreted in human milk. A risk to newborns/ infants cannot be excluded. Fertility No clinical data available. SIDE EFFECTS Refer to SmPC for complete information on side effects. Pembrolizumab is most commonly associated with immune-related adverse reactions. Most of these reactions resolved with appropriate medical treatment or withdrawal of pembrolizumab. The most serious adverse reactions were immune-and infusion-related adverse reactions. Monotherapy: Very Common: anaemia, hypothyroidism, decreased appetite, headache, dyspnea, cough, abdominal pain, nausea, vomiting, constipation, musculoskeletal pain, arthralgia, asthenia, oedema, pyrexia, diarrhoea, rash, pruritus, fatigue. Common: pneumonia, thrombocytopenia, neutropenia, lymphopenia, hyponatraemia, hypokalaemia, hypocalcaemia, insomnia, neuropathy peripheral, lethargy, dry eye, cardiac arrhythmia (including atrial fibrillation), hypertension, hyperthyroidism, thyroiditis, insomnia, dizziness, dysgeusia, pneumonitis, colitis, dry mouth, severe skin reactions, vitiligo, dry skin, alopecia, eczema, dermatitis acneiform, erythema, dermatitis, myositis, pain in extremity, arthritis, influenza like illness, chills, AST and ALT increases, hypercalcaemia, increase in blood alkaline phosphatase, blood bilirubin increased, blood creatinine increased, infusion related reaction. Combination with chemotherapy: Very Common: pneumonia, anaemia, neutropenia, thrombocytopenia, hypothyroidism, hyponatraemia, hypokalaemia, decreased appetite, insomnia, dizziness, neuropathy peripheral, headache, dyspnoea, cough, alopecia, nausea, diarrhoea, vomiting, abdominal pain, constipation, rash, pruritus, musculoskeletal pain, arthralgia, pyrexia, fatigue, asthenia, oedema, blood creatinine increased. Common: febrile neutropenia, leukopenia, lymphopenia, infusion related reaction, hyperthyroidism, hypocalcaemia, dysgeusia, lethargy, dry eye, cardiac arrhythmia (including atrial fibrillation), vasculitis, hypertension, pneumonitis, colitis, dry mouth, gastritis, hepatitis, severe skin reactions, dry skin, erythema, dermatitis, myositis, pain in extremity, arthritis, acute kidney injury, influenza-like illness, chills, hypercalcaemia, ALT increase, AST increased, blood alkaline phosphatase increased, blood bilirubin increased. Combination with axitinib: Very Common: hyperthyroidism, hypothyroidism, decreased appetite, headache, dysgeusia, hypertension, dyspnoea, cough, dysphonia, diarrhoea, abdominal pain, nausea, vomiting, constipation, palmar-plantar erythrodysaesthesia syndrome, rash, pruritus, musculoskeletal pain, arthralgia, pain in extremity, fatigue, asthenia, pyrexia, alanine aminotransferase increased, aspartate aminotransferase increased, blood creatinine increased. Common: pneumonia, anaemia, neutropenia, leukopenia, thrombocytopenia, infusion related reaction, hypophysitis, thyroiditis, adrenal insufficiency, hypokalaemia, hyponatraemia, hypocalcaemia, insomnia, dizziness, lethargy, neuropathy peripheral, dry eye, cardiac arrhythmia (including atrial fibrillation), pneumonitis, colitis, dry mouth, gastritis, hepatitis, severe skin reactions, dermatitis acneiform, dermatitis, dry skin, alopecia, eczema, erythema, myositis, arthritis, tenosynovitis, acute kidney injury, nephritis, oedema, influenza like illness, chills, blood alkaline phosphatase increased, hypercalcaemia, blood bilirubin increased PACKAGE QUANTITIES KEYTRUDA 25 mg/mL: 4 mL of concentrate in a 10 mL Type I clear glass vial. Legal Category: POM. Marketing Authorisation numbers EU/1/15/1024/002 Marketing Authorisation holder Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands. Date of revision: June 2021. © Merck Sharp & Dohme B.V. 2021. All rights reserved. Further information is available on request from: MSD, Red Oak North, South County Business Park, Leopardstown, Dublin D18 X5K7 or from www.medicines.ie. PSUSA-202009-II-097
Adverse events should be reported. Reporting forms and information can be found at www.hpra.ie. Adverse events should also be reported to MSD (Tel: 01-2998700)
References:
1. https://www.hse.ie/eng/services/list/5/cancer/profinfo/chemoprotocols/melanoma/melanoma%20protocols.html 2. Keytruda Summary of Product characteristics, available from www.medicines.ie 3. Eggermont AMM, Blank CU, Mandala M, et al. Longer follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: updated results from the EORTC 1325-MG/KEYNOTE-054 trial. J Clin Oncol. 2020
Sep18;38:3925-3936. 4. Eggermont AMM, Blank CU, Mandala M, et al. Supplementary Appendix to: Longer follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: updated results from the EORTC 1325- MG/KEYNOTE-054 trial. (J Clin Oncol. Epub 2020.) doi:10.1200/JCO.20.02110.
Red Oak North, South County Business Park, Leopardstown, Dublin D18 X5K7, Ireland. IE-KEY-00374 Date of Preparation: October 2021
Vaccine Hesitancy Down
COVID-19 vaccine hesitancy fell by 45 points in 12 months, according to an analysis of surveys conducted by Ipsos MRBI for the Irish Pharmaceutical Healthcare Association (IPHA), the representative body for the research-based biopharmaceutical industry. In November 2020, 17% of people said they would refuse a COVID-19 vaccine. By last month, that figure had dropped to 5%. Between November 2020 and November 2021, the proportion of people undecided about taking a COVID-19 fell from 35% to 2%. That places the overall drop in hesitancy at 45 points. Among 18 to 34-year-olds, the proportion of people who would refuse a vaccine for COVID-19 has dropped from 19% in November 2020 to 6% in November 2021. The proportion of people in that age category undecided about taking a COVID-19 fell from 38% to 3% in the same period. That means hesitancy among people aged between 18 and 34 dropped by 48 points in 12 months. Around 1.2 million people are aged between 18 and 34 out of a population of about five million. Overall, 93% of people either intend to get vaccinated for COVID-19 or have already received a vaccine for the disease, according to the latest survey*. The research-based biopharmaceutical industry said COVID-19 vaccines are saving and protecting millions of lives around the world.
Bernard Mallee, Director of Communications and Advocacy at IPHA, said, “From childhood to later in life, the development of vaccines has protected us from serious, and sometimes deadly, diseases. In Ireland, smallpox, rubella, polio, tuberculosis, diphtheria, pneumonia and measles used to be part of life. Now, we don’t have to worry about them as much.
“Vaccines for COVID-19, developed in record time without compromising on safety and quality, are saving and protecting millions of lives around the world. That COVID-19 vaccine hesitancy is low in Ireland has helped to make us one of the most vaccinated countries in the world. Vaccination reduces serious illness and mortality, giving us a very effective weapon in the battle against COVID-19. “Vaccines, allied with public health measures, protect health and save lives. The emergence of the Omicron variant, and the increased transmissibility of the Delta variant, show that we need to keep searching for new scientific paths to combat the virus. We are fortunate to live at a time of unprecedented medical innovation. We should continue to trust science and to heed public health advice.”
With the exception of clean, safe drinking water, vaccination is among the most successful and cost-effective public health interventions ever. The World Health Organisation estimates that vaccines save up to three million lives every year. IPHA’s new life-course immunisation campaign, ‘Progress. Developed by Vaccines’, is live on Twitter, Facebook and Instagram.
Accord Healthcare Maintain Leading Position

The High-Tech medicines scheme was first introduced in Ireland in 1996. The scheme involves the supply & dispensing of High-Tech medicines through community pharmacies. Expenditure on this scheme has continually increased with spend rising by 173% between 2009 and 2019.2 Consistency of supply is extremely important in the context of High-Tech medicines as many of these medicines are prescribed for long term illnesses. Accord Healthcare Ireland’s High-Tech portfolio consists of sixteen medicines of which they are the leading generic supplier1 in fifteen of these. 2022 will see many new High-Tech launches. Demonstrating their commitment to these medicines Accord conducted market research with Behaviour and Attitudes in 2021. The research demonstrated that 63% of pharmacists who use the High-Tech hub had a positive experience with it.3 On average, pharmacists placed orders for 33 scripts through the hub per month.3 Another positive highlighted in the research is that
Padraic O’ Brien, Managing Director, Accord Healthcare Ireland 59% do not have concerns about other therapeutic areas joining the hub.3 Pharmacists also nominated Accord Healthcare Ireland as the supplier they would be most likely to recommend in this research. The reasons for this were; reliability, product availability and price to name a few.4 Padraic O’ Brien, Managing Director of Accord Healthcare Ireland said, “We at Accord Healthcare Ireland have established ourselves as a leader in the generic High-Tech market, we want Irish pharmacists to know that we are always here to support them and to ‘Think High-Tech’s, Think Accord’. We identified the opportunity in this market some time ago and our portfolio has increased by 45% since 2017 alone. We have an extensive range of generic hightech medicines demonstrating our commitment to ensuring more Irish patients have access to the medicines they need when they need them. The next 12 months will be an important time for the High-Tech space and Accord Healthcare Ireland will be playing its part as an industry leader promising a very strong pipeline for the year ahead.” As the generic High-Tech product is priced up to 60%5 below the branded product, when pharmacists dispense items from the Accord Healthcare Ireland High-Tech portfolio, they are not only helping to provide a quality treatment for Irish patients, they are also helping the state to make considerable savings. In turn, these savings are used to provide access to new medicines allowing more patients access to the treatments they require. Think High-Tech’s, Think Accord.
References
1. IQVIA Retail Data accessed
November 2021.
2. Figures from The Medical
Independent articles; ‘HSE spend on High-Tech Drugs
Scheme increases by almost 100%’ and ‘Figures reveal spiralling cost of HSE high-tech drugs’, accessed November 2021.
3. Data on file IE-01721, page 35. 4. Data on file IE-01721, page 11. 5. Framework Agreement between the Association of
Pharmaceutical Manufacturers of Ireland and the Department of Health and the Health
Service Executive on the
Supply Terms, Conditions and
Prices of Generic Medicines, section 5.2.





