Sara Tolaney, Sarah Blagden, and Jianjun Zhang discuss advancements in the field
Comparison of Breast Sensibility Following Breast Reconstruction with Two Different Techniques Editor's Pick:
04 Editorial Board
07 Welcome
09 Foreword
Congress Review
10 Review of the European Society for Medical Oncology (ESMO) Congress 2024, 13th–17th September 2024
Congress Features
18 Advances in Treatment for Oestrogen Receptor-Positive Breast Cancer
Katie Wright
23 Immunotherapy in Endometrial Cancer: What Should We Know?
Ada Enesco
Symposium Reviews
27 Treatment Strategies and Sequencing After Endocrine Therapy Plus CDK4/6 Inhibitors in Patients with ER+/HER2- Advanced/ Metastatic Breast Cancer
39 Evolving Patient-Centred Therapies for Metastatic NSCLC
49 Maximising the Synergy of Tumour Tissue and Liquid Biopsy Testing in Oncology Clinical Practice
57 Therapeutic Advances in Gastrointestinal Cancers: Immuno-oncology and Beyond
69 Platinum Resistance in Ovarian Cancer: Is This the End of the Line?
Abstract Review
80 Predisposition, Clinical Characteristics, and Management of Immune Checkpoint Inhibitor-Induced Nephritis: A Series of 190 Cases
Olsson-Brown et al.
82 Abstract Highlights
Congress Interviews
88 Jianjun Zhang
Interviews
91 From Podium into Practice: Working Together to Revolutionise Cancer Care in the Real World
96 Sara Tolaney
101 Sarah Blagden
Infographics
106 Cancer Vaccines
108 Unravelling HER2+ Breast Cancer
Feature
110 Novel Approaches to Treat Glioblastoma Multiforme
Malkin et al.
Articles
116 Editor's Pick: Comparison of Breast Sensibility Following Breast Reconstruction with Two Different Techniques: Deep Inferior Epigastric Perforator Flap and Implant
Soraya Tadimi Tazi
129 BRCA Mutation in Ovarian Cancer: Implications for Screening, Diagnosis, and Preventive Measures
Roy et al.
138 MammaPrint Genomic Assay Providing Prognostic Information in Early Breast Cancer: 10-Year Follow-Up From a Retrospective German Breast Cancer Registry Analysis
Jackisch et al.
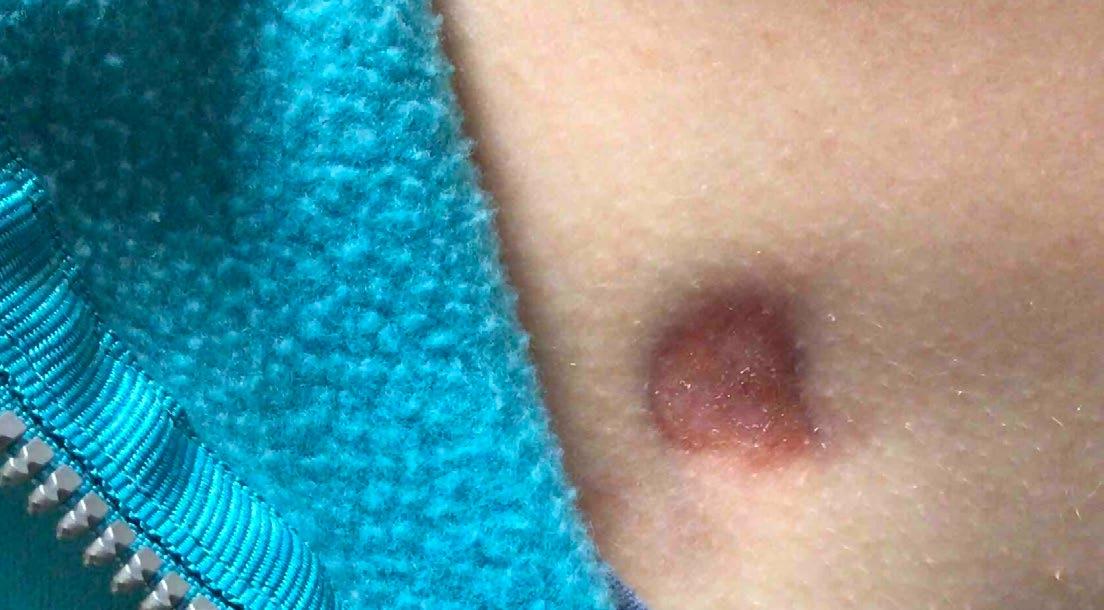
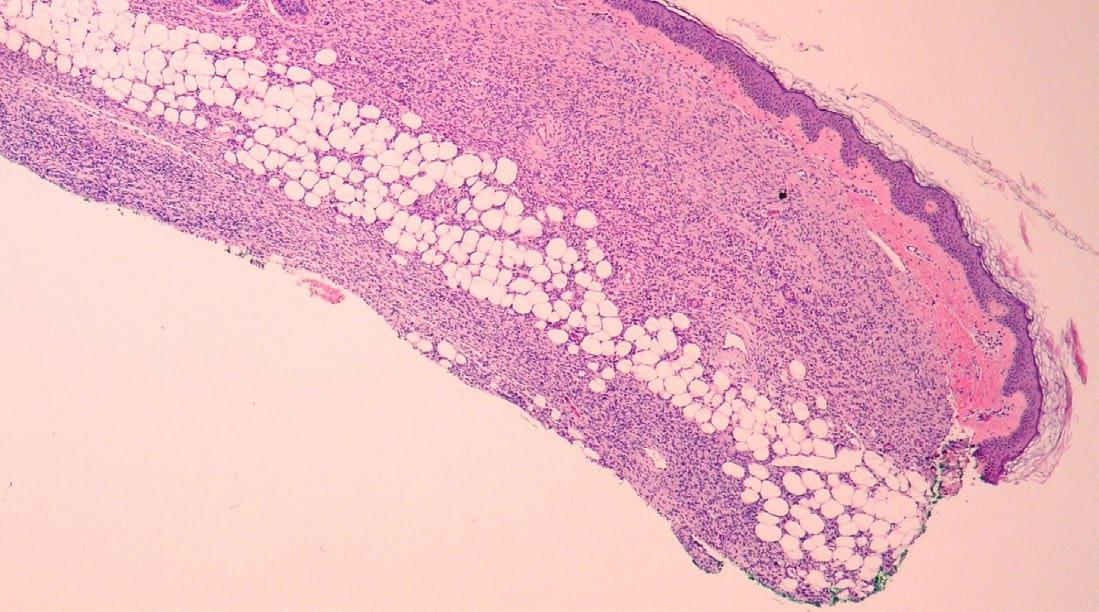
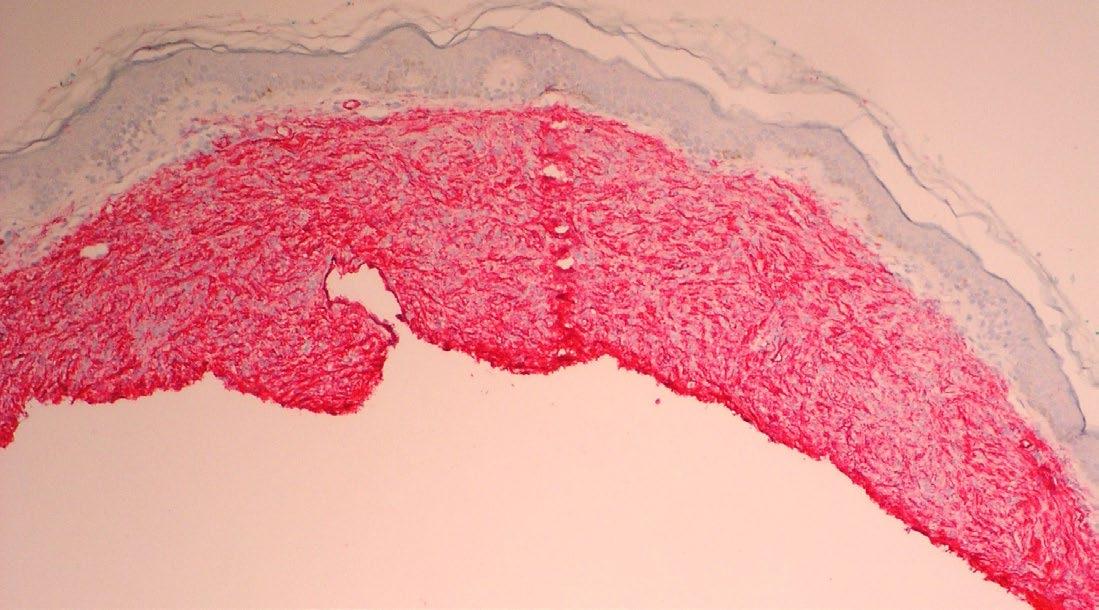
148 Congenital Dermatofibrosarcoma Presenting as an Atrophic Plaque
Jenna Koblinski and Katarina Lequeux Nalovic
153 Effectiveness of Vinorelbine in the Management of Pseudomyogenic Hemangioendothelioma: A Case Report
Toumi et al.
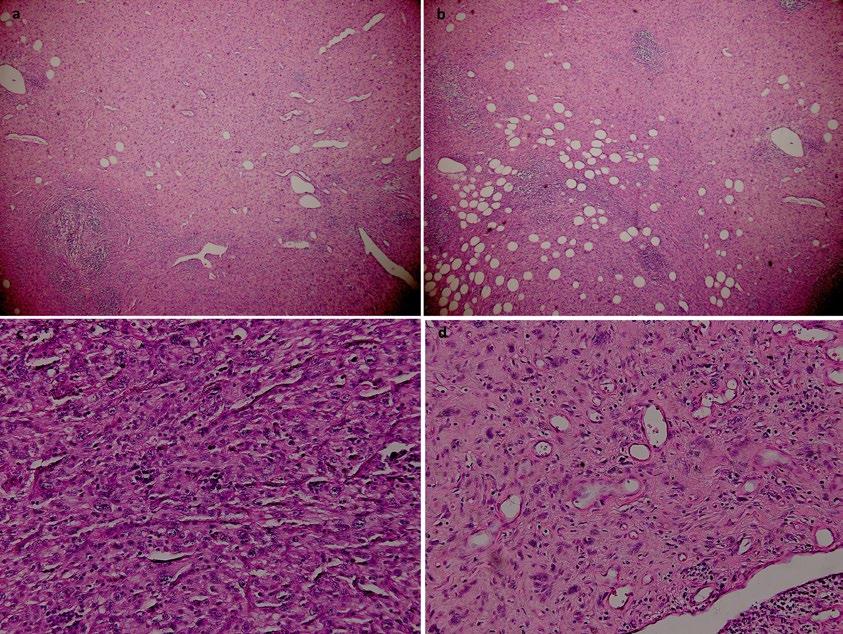
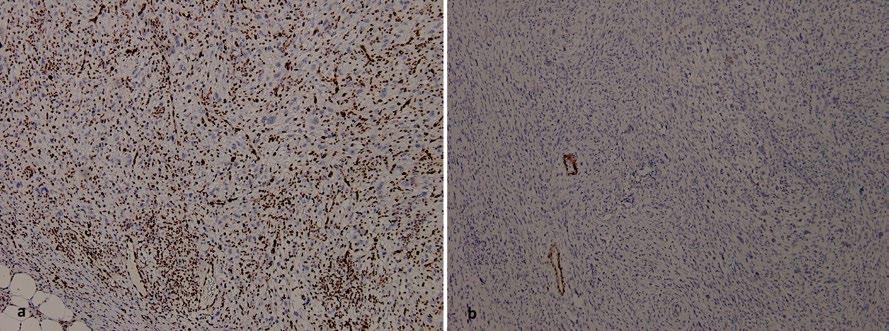
159 Ependymoma Alongside Hereditary ParagangliomaPhaeochromocytoma Syndrome Due to a SDHB Mutation: A Case Report
Ragguett et al.
Editorial Board
Editor-in-Chief
Prof Ahmad Awada
Université Libre de Bruxelles, Belgium
Head of the Oncology Department and Director of the Chirec Cancer Institute in Brussels, Belgium. With over 35 years experience in the field, he is a renowned and trusted expert
Dr Divyanshu Dua
Canberra Hospital, Australia
Dr Caroline Michie
Edinburgh Cancer Centre & University of Edinburgh, UK
Dr Aniket Mohite
Arogyam Multispeciality
Clinic and Kolhapur Cancer Centre, India
Dr Mohammad Akheel
Greater Kailash Hospitals, India
Dr Jyoti Dabholkar
King Edward Memorial Hospital and Seth
Gordhandas Sunderdas Medical College, India
Dr Jad Degheili
Ibn Sina Hospital, Kuwait
Dr Aniket Mohite
Arogyam Multispeciality Clinic and Kolhapur Cancer Centre, India
Prof Paul Dent
Virginia Commonwealth University, USA
Dr Abdulmajeed Hammadi
Alyermook Teaching Hospital, Iraq
Prof Antoine Italiano
Institut Bergonié, France
Dr Katarazyna Rygiel
Medical University of Silesia (SUM), Poland
Dr Francesco Sclafani
Institut Jules Bordet, Belgium
Dr Klaus Seiersen
Aarhus University Hospital, Denmark
Prof Yong Teng
Georgia Cancer Center, USA
Aims and Scope
EMJ Oncology is an open access, peer-reviewed ejournal committed to helping elevate the quality of practices in interventional cardiology globally by informing healthcare professionals on the latest research in the field.
The journal is published annually, six weeks after the European Society for Medical Oncology (ESMO) Annual Congress, and features highlights from this event, alongside interviews with experts in the field, reviews of abstracts presented at ESMO, as well as in-depth features on sessions from this event. The journal also covers advances within the clinical and pharmaceutical arenas by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ Oncology also publishes peer-reviewed research papers, review articles, and case reports in the field. In addition, the journal welcomes the submission of features and opinion pieces intended to create a discussion around key topics in the field and broaden readers’ professional interests. The journal is managed by a dedicated editorial team that adheres to a rigorous double-blind peer-review process, maintains high standards of copy editing, and ensures timely publication.
EMJ Oncology endeavours to increase knowledge, stimulate discussion, and contribute to a better understanding of practices in the field. Our focus is on research that is relevant to all healthcare professionals in this area. We do not publish veterinary science papers or laboratory studies not linked to patient outcomes. We have a particular interest in topical studies that advance knowledge and inform of coming trends affecting clinical practice in interventional cardiology.
Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
Peer Review
On submission, all articles are assessed by the editorial team to determine their suitability for the journal and appropriateness for peer review.
Editorial staff, following consultation with either a member of the Editorial Board or the author(s) if necessary, identify three appropriate reviewers, who are selected based on their specialist knowledge in the relevant area.
All peer review is double blind. Following review, papers are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected.
Editorial staff have final discretion over any
proposed amendments.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields across Europe.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®; selected articles are indexed in PubMed Central®
EMJ is available through the websites of our leading partners and collaborating societies. EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
This Publication Launch Date: 2013 Frequency: Yearly Online ISSN: 2054-619X
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. EMJ is completely independent of the review event (ESMO 2024) and the use of the organisations does not constitute endorsement or media partnership in any form whatsoever. The cover photo is of Barcelona, Spain the location of ESMO 2024.
~50% of your patients with endometrial carcinoma may be TP53 wild-type1
Learn about a clinical trial for eligible TP53 wild-type patients, a key molecular profile in endometrial carcinoma
Welcome to the 2024 issue of EMJ Oncology, highlighting key advancements from this year’s European Society for Medical Oncology (ESMO) Congress, which was hosted in Barcelona, Spain. Once again, this year’s research presentations are a testament to the increasing importance of precision oncology
We have secured a plethora of interviews and Congress content, including abstract highlights that showcase the use of AI in breast cancer screening, racial disparities in clinical trial enrollment, and germline variants associated with non-small cell lung cancer, amongst others. This research helps to emphasise the need for ensuring equitable representation in clinical trials, and the benefits of genetic testing in ensuring early cancer detection and prevention.
Be sure to not miss our two highly informative infographics: one explores the state-of-the-art in cancer vaccines, while the other explores HER2+ breast cancer, its current treatments, and ongoing promising clinical trials. An insightful article also provides a critical evaluation of the current landscape in glioblastoma multiforme treatment, discussing two recently completed clinical trials.
I would like to thank our reviewers, contributors, and Editorial Board for bringing this issue together. Until our next issue, stay updated on advancements in oncology through our newly launched 'Onc Now' podcast, available on the EMJ website!
accountsreceivable@emjreviews.com
Evgenia Koutsouki Editor
Reprints: info@emjreviews.com Media enquiries: marketing@emjreviews.com
ER+/HER2- metastatic breast cancer (mBC): Acquired mutations can be resistance mechanisms in metastatic breast cancer leading to worse prognosis.
mBC.
Foreword
Dear Colleagues,
Welcome to the latest issue of EMJ Oncology, where we spotlight the most exciting breakthroughs in cancer research and treatment. This edition features a diverse array of peer-reviewed articles, expert interviews, engaging infographics, and an in-depth recap of the much-anticipated 2024 European Society for Medical Oncology (ESMO) Congress.
The EMJ team and I were fortunate enough to attend the ESMO Congress 2024, held in Barcelona, Spain, in-person. You can read a curated, comprehensive review capturing the key insights and learnings from the meeting in this journal issue.
Alongside this, the edition features exclusive interviews with three renowned oncology experts: Sara Tolaney, Sarah Blagden, and Jianjun Zhang. Conversations centre around their career journeys, their thoughts on the latest innovations, and the challenges the oncology field faces today.
We are proud to feature six peer-reviewed articles in this issue. Among them is a rare case of posterior fossa ependymoma in a patient with a genetic predisposition, highlighting the complexities of managing hereditary cancers. Another article delves into the impact of BRCA mutations in
ovarian cancer, providing valuable insights into advancements in prevention and early detection strategies. Additionally, we present a comparative study on breast reconstruction techniques, examining their effects on patient outcomes, along with a long-term analysis of the role of genomic testing in early-stage breast cancer prognosis.
You can read a curated, comprehensive review capturing the key insights and learnings from the meeting in this journal issue
Finally, we bring you insightful infographics on HER2+ breast cancer and the growing role of cancer vaccines in disease prevention.
I would like to extend my heartfelt thanks to the EMJ team, Editorial Board, interviewees, authors, and peer reviewers for their continued efforts in delivering an exceptional issue.
Enjoy reading!
Prof Ahmad Awada Université Libre de Bruxelles, Belgium
ESMO 2024
The ESMO Congress 2024 opening ceremony set the stage for a landmark event in oncology
2,186 research abstracts
1,828 posters 208 programme sessions
Review of the European Society for Medical Oncology (ESMO) Congress 2024 Congress Review
KNOWN for its research excellence in the fields of oncology, bionanomedicine, and cardiovascular disease, Barcelona, Spain, provided the ideal setting for the European Society for Medical Oncology (ESMO) Congress 2024. The event was attended by 34,000 participants from across the globe, including almost 600 speakers from 149 different countries, and saw the presentation of 2,186 research abstracts, featured 1,828 posters, and offered 208 programme sessions.
The Congress formally opened with the welcome address, delivered by the current (2023–2024) President of ESMO, Andrés Cervantes, University of Valencia, Spain. During this speech, he highlighted several new initiatives to aid in the research and practice of clinical oncologists. The establishment of an ESMO fellowship on digital and computational pathology, and a webinar series on genomics-guided care, were among some of the initiatives mentioned.
The scientific address was delivered by Rebecca Dent, National Cancer Center, Singapore. She highlighted that over 2,000 abstracts, including 151 proffered papers and 207 mini oral sessions, covering a wide range of topics from precision oncology to AI-driven cancer care, were embedded into the 2024 programme. The programme also included exciting keynote lectures on leveraging DNA repair pathways, the transformative potential of AI in oncology, and the global epidemic of young-onset cancers.
Several prestigious awards were presented during the opening ceremony. The first of which was bestowed to Ann H. Partridge, Dana-Farber Cancer Institute, Boston, Massachusetts, USA. Partridge discussed the long-term impact of cancer care, particularly focusing on survivorship issues. Her presentation highlighted how the majority of cancer survivors face unintended consequences, such as quality of life deterioration, particularly in young adults diagnosed with breast cancer. Partridge emphasised the need for targeted support systems for survivors to address issues such as fertility, mental health, and adherence to post-treatment therapies. She showcased the success of programmes like 'Young and Strong', which provides a comprehensive support system for young patients with breast cancer.
Serena Nik-Zainal, University of Cambridge, UK, presented a thought-provoking lecture on how mutational signatures derived from cancer genomes can be leveraged for clinical applications. Her work has made
substantial contributions to understanding cancer genomics and how whole-genome analysis can inform treatment strategies. In recognition of this, she was awarded the ESMO Award for Translational Research. Nik-Zainal emphasised the use of mutational signatures to decode environmental and genetic influences on cancer, pushing the boundaries of precision medicine. The evolution of this field was traced from early studies to current AI-driven platforms that allow clinicians to analyse mutational signatures and apply this knowledge to personalise treatment.
The ESMO Women for Oncology Award for bridging gaps in lung cancer research and gender equity was awarded to MyungJu Ahn, Samsung Medical Center, Seoul, Republic of Korea. Ahn delivered an insightful talk touching on her career path as well as research advancements in the field of nonsmall cell lung cancer. Finally, John Haanen, Netherlands Cancer Institute, Amsterdam, the Netherlands, was awarded the ESMO Lifetime Achievement Award for his work in cancer immunotherapy. Haanen’s presentation shared insights into T cell therapies for cancer, particularly in the context of adoptive T cell transfer. Haanen highlighted the success of these treatments in metastatic melanoma, showcasing how adoptive T cell transfer can provide durable responses and effective tumour targeting.
The ESMO Congress 2024 opening ceremony set the stage for a landmark event in oncology, highlighting groundbreaking advances in personalised medicine, immunotherapy, and the transformative role of AI in cancer care. With over 2,000 abstracts presented and key sessions spanning global collaborative efforts, the congress emphasised the importance of multidisciplinary approaches in tackling cancer’s most pressing challenges.
Read on for more coverage from the 2024 ESMO Congress, and stay tuned for the ESMO Congress 2025, which will take place in Berlin, Germany, from 17th–21st October 2025!
Long-Term Outcomes of Early Switch from Targeted Therapy to Immunotherapy in Advanced BRAFV600-Positive Melanoma:
Results from the ImmunoCobiVem Trial
RESULTS of the ImmunoCobiVem trial were presented at the ESMO Congress 2024. The trial explored the optimal sequencing of targeted therapies (TT) and immune checkpoint inhibitors (ICI) in patients with advanced BRAFV600-positive melanoma, aiming to determine whether starting with TT and then switching early to ICI would improve patient outcomes compared to continuing TT until disease progression.
In this randomised Phase II study, one group (Arm A) received continuous treatment with the targeted drugs vemurafenib (960 mg twice daily) and cobimetinib (60 mg, daily d 21–28, then every 4 weeks) until disease progression, at which point they switched to the immunotherapy drug atezolizumab (1200 mg every 3 weeks). The other group (Arm B) switched early to atezolizumab after a 3-month period of TT, with the option to switch back to the targeted therapies if progression occurred. The primary endpoint was progressionfree survival (PFS) during the initial phase of treatment, while secondary outcomes included overall survival (OS), further progression-free survival (PFS2 and PFS3), overall response rates, and safety.
The final analysis, after a median followup of 57 months (interquartile range [IQR]: 22.7–63.0), showed that continuous TT (Arm A [69 patients]; hazard ratio [HR]: 0.61; 95% CI: 0.41–0.91; p=0.006) provided better initial tumour control, as seen in higher PFS during the initial phase. However, patients who switched early to immunotherapy (Arm B) had better long-term OS at the 4- and 5-year marks (3-, 4- and 5-year landmark OS were 55% [95% CI: 41–66], 42% [95% CI: 29–55], and 40% [95% CI: 27–53] for Arm A; 55% [95% CI: 41–67], 53% [95% CI: 38–65],
and 45% [95% CI: 31–58] for Arm B; and descriptive HR [A versus B] was 1.17; 95% CI: 0.71–1.91). In terms of response rates, both groups performed well, but Arm B had a slightly higher overall response rate than Arm A (89% versus 81%), with more patients achieving complete responses (29% versus 22%, respectively).
Importantly, when patients in Arm B who had switched to ICI experienced disease progression and were retreated with TT, and had better outcomes than those in Arm A who switched to ICI after failing on TT.
In conclusion, the early switch to immunotherapy after an initial period of targeted therapy showed a trend toward better long-term survival. However, the overall benefit remained modest, and there was no clear evidence that any specific subgroup gained a distinct clinical advantage from this approach.
The early switch to immunotherapy after an initial period of targeted therapy showed a trend toward better long-term survival
10-Year Study Confirms Long-Term Survival Benefit of Nivolumab in Advanced Melanoma
THE FINAL results of the landmark CheckMate 067 trial were presented at the ESMO Congress 2024. The presentation showed the results of a minimum 10-year follow-up and revealed the long-term survival benefits of nivolumab (NIVO), alone or in combination with ipilimumab (IPI), for patients with advanced melanoma.
The study was the longest Phase III trial of a PD-1-based therapy to date and aimed to investigate the transformative impact of these immune checkpoint inhibitors on melanoma prognosis.
The study involved 945 patients with untreated advanced melanoma, and the trial compared three treatment arms: NIVO combined with IPI, NIVO alone, and IPI alone. The results showed a difference in overall survival (OS) across these groups. Median OS reached 71.9 months for the NIVO + IPI group, compared to 36.9 months for the NIVO-only group, and 19.9 months for the IPI group. The combination therapy reduced the risk of death by 47% compared to IPI alone, demonstrating consistent benefits across all subgroups, including patients with varying PD-L1 expression levels and BRAF mutation statuses.
Furthermore, melanoma-specific survival (MSS) rates were notably high. For patients receiving the NIVO + IPI combination, the median MSS was not reached, indicating survival beyond 120 months. Patients treated with NIVO alone had a median MSS of 49.4 months, while those on IPI alone had a median MSS of 21.9 months. In patients who achieved progression-free survival for at least 3 years, the 10-year MSS rates were
96% for the NIVO + IPI group, 97% for the NIVO group, and 88% for the IPI group.
The study also showed the durability of response in patients who discontinued treatment due to adverse events during the induction phase. These patients demonstrated similar 10-year OS and MSS rates as the broader intention to treat population, showing that early discontinuation due to side effects did not compromise long-term survival benefits.
These final results from CheckMate 067 confirm the sustained efficacy of NIVObased therapies, offering hope for a potential cure in responsive patients and marking a significant milestone in the treatment of advanced melanoma.
In patients who achieved progression-free survival for at least 3 years, the 10-year MSS rates were 96% for the NIVO + IPI group, 97% for the NIVO group, and 88% for the IPI group.
Neoadjuvant and Adjuvant Pembrolizumab Improves Survival for High-Risk Early-Stage
Triple-Negative Breast Cancer
RESULTS of the Phase III KEYNOTE-522 study have shown that compared to neoadjuvant chemotherapy alone, neoadjuvant and adjuvant pembrolizumab with chemotherapy significantly improves overall survival in patients with high-risk early-stage triple-negative breast cancer (TNBC) (Stage T1c N1-2 or T2-4 N0-2 per the American Joint Committee on Cancer staging).
In the study, 1,174 patients with untreated, non-metastatic, centrally confirmed TNBC either received neoadjuvant pembrolizumab 200 mg every 3 weeks (n=784), or placebo (n=390), before four cycles of paclitaxel and carboplatin, followed by four cycles of doxorubicin or epirubicin, and cyclophosphamide. After definitive surgery, patients received adjuvant pembrolizumab or placebo for nine cycles, or until recurrence or unacceptable toxicity occurred.
At the prespecified data cutoff of 22 March 2024, the median follow-up was 75.1 months. At this point, 115 patients (14.7%) in the pembrolizumab group and 85 patients (21.8%) in the placebo group had died. The analysis revealed that the hazard ratio for mortality was 0.66 (95% CI: 0.50–0.87; p=0.0015), meaning that the treatment group had a 34% lower mortality risk than the placebo group. The data also showed that the 5-year overall survival rate was 86.6% (95 CI: 84.0–88.8) for pembrolizumab and 81.7% (95% CI: 77.5–85.2) for placebo. Improvements in overall survival were across different subgroups of patients, such as PDL1 expression level and nodal status.
The 5-year event-free survival rate for patients in the pembrolizumab group was 81.2% (95% CI: 78.3–83.8), compared to 72.2% (95% CI: 67.4–76.4) in the placebo group, with a hazard ratio of 0.65 (95% CI: 0.51–0.83). Meanwhile, the rate of Grade 3 or higher adverse events was 77.1% in the pembrolizumab group and 73.3% in the placebo group, and immune-related adverse events occurred in 35.0% of patients who received pembrolizumab, compared to 13.1% in the placebo group.
Overall, the results presented at the ESMO Congress 2024 revealed that patients treated with neoadjuvant and adjuvant pembrolizumab had a lower mortality risk, higher overall survival rate, and higher 5-year event-free survival rate. However, pembrolizumab was associated with a higher risk of Grade 3 or higher adverse events and immune-related adverse events. Nevertheless, the study demonstrates pembrolizumab's efficacy in improving overall and event-free survival, highlighting a beneficial treatment strategy for patients with high-risk early-stage TNBC.
The treatment group had a 34% lower mortality risk than the placebo group
Targeted Enzyme Inhibitor Shows Promise in Treating Deleted Gene Tumours
A NEW clinical trial presented at the ESMO Congress 2024 has detailed the safety, tolerability, and preliminary antitumour efficacy of AMG 193, an investigational oral inhibitor designed to selectively target protein arginine methyltransferase 5 (PRMT5) in tumours with methylthioadenosine phosphorylase (MTAP) deletions.
MTAP deletions are present in approximately 10–15% of solid tumours, including nonsmall cell lung cancer (NSCLC), pancreatic ductal adenocarcinoma (PDAC), and biliary tract cancer (BTC). By exploiting synthetic lethality, AMG 193 aims to destroy cancer cells while sparing healthy tissues.
In the trial, patients with NSCLC, PDAC, BTC, and other MTAP-deleted tumours received AMG 193 orally, either once daily (QD) or twice daily. All 143 patients included in the study had a history of a median of two prior therapies before starting AMG 193, 80 of whom were placed in a dose escalation (dES) phase treatment group, receiving doses ranging from 40–1,600 mg. The remaining 63 patients were in the dose expansion (dEX) phase, all at a dose of 1200 mg. The trial evaluated safety, anti-tumour activity, pharmacokinetics, and pharmacodynamics.
The most common treatment-related adverse events were nausea (50%), fatigue (30%), vomiting (29%), and decreased appetite (19%). Dose-limiting toxicities, specifically Grade 3 vomiting and Grade 3 hypokalaemia, were observed in two patients in the 1200 mg QD cohort, which was determined to be the maximum tolerated dose. Notably, no dose-limiting cytopaenias were reported, distinguishing AMG 193 from earlier PRMT5 inhibitors.
Efficacy results showed that patients receiving active doses (800 mg and 1200 mg) demonstrated promising responses. Two out of 11 patients with NSCLC, two out of 16 patients with PDAC, and two out of 11 patients with BTC achieved objective responses, with the median duration of response lasting 8.3 months. Tumour biopsies revealed that AMG 193 disrupts key pathways including RNA splicing, cell cycle regulation, and DNA damage response.
The most common treatmentrelated adverse events were...
Non-operative Management Shows Promise for Patients with Rectal Cancer in NO-CUT Trial
EARLY results of the NO-CUT trial presented at the ESMO Congress 2024 suggest that non-operative management (NOM) following total neoadjuvant treatment (TNT) could be a viable alternative to surgery for patients with proficient mismatch repair locally advanced rectal cancer (pMMR LARC).
The Phase II trial involved 180 patients across four cancer centres over 6 years, and aimed to examine the effectiveness of TNT followed by either surgery or NOM for patients who achieved a clinical complete response (cCR).
Patients in the NOM group demonstrated a DRFS rate of 96.9% at 30 months, compared to 74% for those who underwent surgery
In the trial, patients received a combination of chemotherapy (4 cycles of CAPOX) followed by long-course chemoradiotherapy. Of the patients who completed treatment, 46 (25.5%) were assigned to the NOM cohort based on achieving a cCR after TNT. These patients were closely monitored with intensive follow-up instead of undergoing rectal surgery.
The primary goal of the trial was to assess whether NOM compromises distant relapsefree survival (DRFS). The results were promising, with patients in the NOM group demonstrating a DRFS rate of 96.9% at 30 months, compared to 74% for those who underwent surgery. Local regrowth was slightly higher in the NOM group (15%) than in the surgical group (9%), but overall survival outcomes remained strong, with 12 deaths reported among the entire cohort.
Additionally, early multiomic analyses, including circulating tumour DNA from liquid biopsies, showed correlations with cCR and DRFS, highlighting potential biomarkers for future treatment decisions.
The findings from the NO-CUT trial indicate that NOM, with rigorous follow-up, could be a safe and effective option for a subset of patients with rectal cancer, reducing the need for invasive surgery while maintaining high survival rates. Further analysis of biomarkers may help refine patient selection for this non-operative approach.
Advances in Treatment for Oestrogen Receptor-Positive Breast Cancer
THIS YEAR the European Society for Medical Oncology (ESMO) Congress was hosted in Barcelona, Spain from 13th–17th September. Among the many impactful sessions, a symposium titled 'Incorporating Novel Treatment Insights for Estrogen Receptor-Positive (ER+) Early Breast Cancer Patients' garnered particular attention. Featuring presentations by Stephen Johnston, Royal Marsden, London, UK; Nadia Harbeck, Ludwig Maximilian University of Munich, Germany; and Etienne Brain, Institut Curie in Paris & Saint-Cloud, France, the session provided a comprehensive overview of current advances in ER+ early breast cancer treatment. The speakers explored adjuvant endocrine therapies, prognostic and predictive factors for clinical decision-making, and the unique challenges of optimising treatment for older patients, underscoring the evolving landscape of personalised care in ER+ early breast cancer.
SPEAKER 1: STEPHEN JOHNSTON
Johnston began by describing the benefits of adjuvant endocrine therapies in postmenopausal ER+ early breast cancer, paying tribute to the late Virgil Craig Jordan widely known as the father of tamoxifen. Adjuvant endocrine therapy, particularly tamoxifen, has shown significant benefits, reducing the risk of recurrence by nearly 40%, while aromatase inhibitors offer a smaller additional gain.1 Cyclindependent kinase (CDK) 4/6 inhibitors, crucial in managing hormone-resistant cancer, have been described as gamechangers in advanced disease, but early breast cancer trials, especially with palbociclib, have yielded mixed results while managing toxicity remains a key challenge in treatment.
CDK 4/6 inhibitors, crucial in managing hormone-resistant cancer, have been described as gamechangers in advanced disease
Recent advancements in breast cancer treatment are highlighted by two pivotal clinical trials: monarchE2 and NATALEE.3 These studies focus on high-risk nodepositive populations and a broader group of Stage II and III breast cancer patients, respectively. Both trials have shown promising results, demonstrating the efficacy of newer therapies that could significantly reduce recurrence rates.
monarchE Trial
The monarchE trial encompasses two cohorts. The first includes patients with high clinical risk features, such as having four or more positive nodes or large tumour sizes, while the second, added at the request of regulatory agencies, examines smaller tumours with one to three nodes that possess a high Ki-67 proliferation index.
Five-year data presented by Harbeck at ESMO 2023, indicated that, despite 95% of high-risk patients undergoing chemotherapy, one in four relapses in the control arm.4 However, the addition of abemaciclib reduces this recurrence risk by
approximately 32%, significantly benefitting premenopausal women and patients with neoadjuvant therapy and large tumours. While distant relapse-free survival rates also reflect a positive trend, Johnston advised that it remains too early to draw meaningful conclusions from the overall survival data.
NATALEE Trial
NATALEE was designed as an open-label Phase III trial that includes a broader patient demographic, administering a 400 mg dose over 3 years. It also encompasses node-negative patients, particularly those classified as Stage IIa with either Grade III tumours or high-risk Grade II tumours based on Ki-67 or genomic features. The initial findings have indicated a 25% reduction in the risk of invasive disease-free survival and a 3.3% absolute difference at 27 months of follow-up.
Subgroup analyses reveal benefits across various categories, including node-negative patients, although the small sample size results in confidence intervals that cross one, indicating uncertainty in these findings.
Biomarker Research
Biomarker studies in monarchE have revealed that Ki-67 is a strong prognostic factor but does not predict treatment response. Furthermore, a more extensive exploratory biomarker analysis involving whole exome sequencing and RNA sequencing has identified intrinsic subtypes, Oncotype recurrence scores, and mutation profiles. Johnston noted that patients with a high Oncotype recurrence score showed no significant difference in benefit from abemaciclib treatment compared to those with a low score. This analysis underscores the complexity of treatment decisionmaking, as traditional pathology parameters may not reliably predict patient outcomes.
Additionally, circulating tumour DNA (ctDNA) detection has emerged as a promising prognostic marker, as patients with positive ctDNA after chemotherapy and before study enrolment have poorer outcomes, whereas those with negative ctDNA exhibit better prognosis. Monitoring ctDNA dynamics during treatment may further guide therapeutic decisions and identify patients who are not benefiting from their current regimen.
As Johnston concluded his presentation, he emphasised how vital it is to reflect on the significant developments in breast cancer treatment, particularly regarding the ongoing the ADAPTcycle trial.5 This trial is exploring a preoperative selection strategy that integrates dynamic Ki-67 response to therapy and baseline Oncotype recurrence scores. The trial stratifies patients into three intermediate risk groups based on biological markers, enabling a randomisation between CDK inhibitor combined with endocrine therapy versus chemotherapy in the adjuvant setting. It is anticipated that this will provide crucial insights into whether patients with intermediate risk can achieve comparable outcomes with CDK inhibitor-based therapy instead of traditional chemotherapy.
Furthermore, the GEICAM group is conducting the CARABELA trial, which compares neoadjuvant treatment with letrozole and abemaciclib against chemotherapy in high and intermediate risk patients is providing head-to-head comparisons in early breast cancer treatments, which echo similar efforts seen in metastatic settings.6
SPEAKER 2: NADIA HARBECK
Harbeck provided a comprehensive overview of treatment indications for ER+, HER2- early breast cancer, highlighting the significance of biomarkers, prognostic factors, and predictive factors, especially considering the recently published ESMO early breast cancer guidelines.7 Unlike HER2+and triple-negative breast cancers, which have more defined treatment pathways, hormone receptor-positive cases present diverse therapeutic options, and this variability necessitates careful consideration in determining treatment indications. Emphasising the critical role of validated biomarkers in guiding treatment decisions, Harbeck quoted former ASCO President Den Hayes, University of Michigan, USA when he stated: “A bad biomarker is as bad for a patient as a bad drug.” She underscored the risks associated with unvalidated biomarkers, which could either withhold effective treatments or lead
to unnecessary therapies. Understanding a patient’s risk profile, whether low-risk (luminal A-like) or high-risk (luminal B-like), is essential for informing therapy choices.
Assessing the Need for Chemotherapy
One of the most pressing questions from patients is whether chemotherapy is necessary in addition to endocrine therapy. For patients with 0–3 positive lymph nodes, gene expression assays can assist in making this determination. Harbeck noted that only two assays, TAILORx and the recently reported results, have been prospectively validated in clinical settings.8
The TAILORx trial found that patients with intermediate-risk recurrence scores (11–25) do not benefit from chemotherapy, while premenopausal patients show some uncertainty, with marginal benefits for recurrence scores of 16–20 and clearer benefits for scores of 21–25. This highlights the need for careful evaluation of cutoff scores when utilising genomic testing.
Insights From Recent Clinical Trials
Harbeck discussed findings from studies such as MINDACT9 and RxPONDER,10 which suggested that postmenopausal women with low genomic risk scores and high clinical risk do not require chemotherapy. However, the data for younger women remain ambiguous, with indications of a potential 5% benefit from chemotherapy. She expressed concern over the ASCO committee’s decision not to recommend gene expression assays for node-positive young women, arguing that many of these patients might not need chemotherapy.
Furthermore, she highlighted that recent data from the RxPONDER trial revealed the influence of ovarian function on determining chemotherapy necessity for younger women. Specifically, patients with low AMH levels showed no significant differences in outcomes between chemotherapy and endocrine therapy, whereas those with preserved ovarian function appeared to benefit from chemotherapy.
Harbeck reiterated the complexity of treatment decisions for ER+ and HER2-
early breast cancer and concluded her presentation by underscoring the importance of addressing the specific needs of younger patients and the necessity for further investigation into treatment strategies to ensure appropriate care without unnecessary interventions.
SPEAKER 3: ETIENNE BRAIN
The third, and final, speaker, Brain, addressed the challenges and considerations in optimising treatment for older patients diagnosed with ER+ early breast cancer. His discussion highlighted the unique needs of this patient demographic, emphasising the importance of tailoring treatment strategies to account for the complexities associated with ageing.
Future Directions: Combination Therapies and New Strategies
In light of these challenges, Brain expressed optimism regarding the potential of CDK4/6 inhibitors in treating older
populations. He referenced ongoing trials that, despite mixed results, indicate that the benefits observed with these agents do not significantly differ across age groups. However, he cautioned that older patients often experience higher rates of treatment discontinuation and dose adjustments, which could influence the overall effectiveness of these therapies. Furthermore, Brain pointed out that efforts are underway to explore chemofree regimens combining aromatase inhibitors with CDK4/6 inhibitors as a viable alternative for older patients. Such regimens aim to mitigate the toxicities associated with chemotherapy while maintaining treatment efficacy.
Finally, Brain addressed the emerging interest in neoadjuvant strategies as a way to better assess treatment efficacy and safety before surgical interventions, stressing that patient-centred care must take precedence and acknowledging the unique values older patients prioritise, such as safety, quality of life, and active participation in their treatment decisions.
Brain expressed optimism regarding the potential of CDK4/6 inhibitors in treating older populations
ER+ Breast Cancer in Older Women
Brain noted a critical misconception in cancer statistics: it is often stated that one in eight women will develop breast cancer in their lifetime. While there is a preconceived notion that this regards young womens' risk, he clarified that this statistic is based on a model where all women live to the age of 71 years. This statistic underscores the significance of older populations in breast cancer care, as the majority of cases diagnosed in women over 65 years are ER+.
Despite the prevalence of ER+ cases in older women, Brain pointed out a notable lack of specific data regarding this population in clinical trials. He lamented that while older patients often represent a significant portion of breast cancer cases, they are underrepresented in clinical research. For instance, only about 5% of participants in registration trials for CDK4/6 inhibitors were aged 75 years and older, indicating a clear gap in data that could inform treatment guidelines.
References
1. Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Adjuvant bisphosphonate treatment in early breast cancer: metaanalyses of individual patient data from randomised trials. Lancet. 2015;386(10001):1353-61. Corrected and republished from: Lancet. 2017;389(10088):2472.
2. Johnston SRD et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 2023;24(1):77-90.
3. Slamon D et al. Ribociclib plus endocrine therapy in early breast cancer. N Engl J Med. 2024;390(12):1080-91.
Brain emphasised the importance of understanding competing risks for older patients. Many individuals over the age of 70 years do not die from cancer but rather from other health conditions, highlighting the necessity for an integrated approach to treatment. He argued in favour of the integration of geriatric assessments in the treatment decision-making process, noting that such evaluations can significantly influence management strategies.
Brain stated that integrating geriatric assessments can lead to treatment modifications in up to 40% of cases, often resulting in de-escalation of treatment and his presentation as a whole underscored the need for a more nuanced understanding of treatment strategies for older patients with ER+ early breast cancer. He advocated for a tailored approach that takes into account the complexities of ageing, emphasising the importance of geriatric assessments and individualised treatment plans.
4. Rastogi P et al. Adjuvant abemaciclib plus endocrine therapy for hormone receptor-positive, human epidermal growth factor receptor 2-negative, high-risk early breast cancer: results from a preplanned monarche overall survival interim analysis, including 5-year efficacy outcomes. J Clin Oncol. 2024;42(9):987-93. Corrected and republished from: J Clin Oncol. 2024;42(17):2111.
5. Harbeck N et al. ADAPTcycle: adjuvant dynamic marker-adjusted personalized therapy (ADAPT) comparing endocrine therapy plus ribociclib versus chemotherapy in intermediate-risk HR+/HER2- early breast cancer (EBC) J. Clin. Oncol. 2020;38:TPS601.
6. Guerrero A et al. Evaluating Ki67 and oncotype DX breast recurrence score during neoadjuvant treatment with letrozole/abemaciclib or chemotherapy
in patients with highly proliferative HR+/HER2- breast cancer participating in the GEICAM CARABELA trial. J Clin Oncol. 2024;42(Suppl 16):576.
7. Loibl S et al. Early breast cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2024;35(2):159-82.
8. Sparano et al. Abstract GS1-05. SABCS, December 6-10, 2022.
9. Kalinsky K et al. Adjuvant trial randomized ER+ patients who had a recurrence score <25 and 1-3 positive nodes to endocrine therapy (ET) versus ET + chemotherapy. Abstract GS3‐01. San Antonio Breast Cancer Symposium (SABCS), December 8-11, 2020.
10. Kalinsky et al. Abstract GS3-00. American Society of Clinical Oncology, May 29-31, 2020.
Immunotherapy in Endometrial Cancer: What Should We Know?
Author: Ada Enesco, EMJ, London, UK
Citation: EMJ Oncol. 2024;12[1]:23-26.
https://doi.org/10.33590/emjoncol/ZJLA9296.
IMMUNOTHERAPY with chemotherapy is emerging as a new standard firstline treatment in advanced endometrial cancer (EC). In an insightful session presented at this year’s European Society for Medical Oncology (ESMO) Congress, held in Barcelona, Spain from the 13th–17th September, experts in the field discussed what we know, and what we should know, on immunotherapy and EC.
IMMUNOTHERAPY: NEW STANDARD OF CARE IN ADVANCED ENDOMETRIAL CANCER?
EC is the most common gynaecological malignancy globally, with over 400,000 new cases reported in 2020 and mortality rates increasing annually by 1.8% on average. Ana Oaknin, Vall d’Hebron Institute of Oncology, Barcelona, Spain, raised the current challenges in the treatment of EC. While early-stage EC has a favourable prognosis, she stressed that patients diagnosed at an advanced stage (FIGO Stage III/IV) face a much lower 5-year survival rate of around 17%, largely due to limited treatment options for advanced disease.
Traditionally, standard first-line therapy for advanced EC involved either carboplatin and paclitaxel chemotherapy, or hormonotherapy, depending on clinical and histological characteristics. These approaches, however, have had limited effectiveness, with median progression-free survival often under 1 year, particularly with hormonal therapies.
Oaknin stated that major progress has now been made through the Cancer Genome Atlas (TCGA) project, which classified endometrial cancer into four molecular subgroups: POLE ultramutated, microsatellite instability-high (MSI-high), copy-number low, and copy-number high.
This classification not only provides relevant prognostic information but can also predict responses to different therapies.
EC is the solid tumour with the greatest percentage of MSI-high cases (31%), which are associated with higher rates of mutation, higher neoantigen expression, increased tumour-infiltrating lymphocytes, and higher PD-(L)1 expression. Oaknin explained that this specific microenvironment makes mismatch repair-deficient (dMMR)/MSI-high EC an ideal candidate for immune checkpoint inhibitors (ICI). Recently, dostarlimab and pembrolizumab showed compelling results in patients with dMMR/MSI-high EC after platinum failure,1,2 leading to the regulatory approval of these two agents. The logical next step, continued Oaknin, is to try to incorporate ICIs into first-line therapy, either with chemotherapy only, or in combination with poly-ADP ribose polymerase (PARP) inhibitors to yield a potential synergistic anti-tumour effect.
“Would the addition of an anti-PD(L)1 antibody to first-line platinum-based chemotherapy sufficiently improve outcomes in advanced dMMR/MSI-high EC to become a new standard of care?” This is the question that currently needs to be addressed, explained Oaknin.
Oaknin highlighted results from four key trials for dMMR EC. RUBY, a Phase III
64
Atezolizumab (anti-PD-L1 antibody) + chemotherapy reduced the risk of progression or death by % compared to placebo in patients with advanced/ recurrent dMMR EC
randomised multicentre study, enrolled patients with advanced/recurrent EC who had not yet undergone therapy for advanced stages.3 Patients were randomised 1:1 to receive chemotherapy (paclitaxel and carboplatin) + placebo, or chemotherapy + dostarlimab (antiPD-1 antibody) for a duration of 3 years. Combining dostarlimab with chemotherapy led to a 72% lower risk of progression or death in patients with dMMR EC, and a significant increase in overall survival (OS).3
In another important Phase III trial, NRGGY018, pembrolizumab (anti-PD-1 antibody) + chemotherapy reduced the risk of progression or death by 70% versus placebo + chemotherapy in patients with advanced/ recurrent dMMR EC.4
In the AtTEnd study, atezolizumab (antiPD-L1 antibody) + chemotherapy reduced the risk of progression or death by 64% compared to placebo in patients with advanced/recurrent dMMR EC, with a dramatically higher OS also observed in the atezolizumab group.5
Finally, the Phase III DUO-E trial demonstrated that durvalumab + chemotherapy followed by maintenance durvalumab with or without PARP inhibitor, olaparib, resulted in significantly lower risk of disease progression or death compared with chemotherapy alone for patients with advanced/recurrent EC.6 Oaknin stated that all these data highlight the clinical benefit of integrating immunotherapy into first-line chemotherapy.
While dMMR is a known predictor of how certain cancers respond to immunotherapy, there is variability within the dMMR patient population. Oaknin explained that two key mechanisms can lead to MMR deficiency: epigenetic promoter methylation or germline/somatic mutations in mismatch repair genes. These differences could influence how tumours respond to ICIs, and some preliminary findings seem to support this hypothesis. However, results from the NRG-GY018 trial7 suggested that pembrolizumab provided benefits in both methylated and non-methylated dMMR groups, indicating that dMMR status alone
might not fully predict response. Similarly, RUBY trial3 results showed significant benefit from dostarlimab irrespective of the specific dMMR mechanism.
ICIs are transforming treatment for advanced/ recurrent EC
Beyond dMMR, other biomarkers such as PD-L1 and tumour mutational burden (TMB) are also investigated for their role in predicting responses to immunotherapy. However, PD-L1 expression remains an ambiguous predictor. Analysis from the NRG-GY018 trial showed that in dMMR subgroups, progression-free survival was similar regardless of whether patients were PD-L1-positive or -negative, indicating that PD-L1 expression alone is not a reliable marker for determining outcomes.7 In contrast, the DUO-E trial, which used an assay called tandem affinity purification (TAP), found a significant benefit for patients with TAP values ≥1% when given experimental treatments compared to the control arm.8 However, this effect was not observed in PD-L1-negative patients. This raises the question of how PD-L1 status intersects with other markers like TMB and dMMR in predicting outcomes.
The GARNET trial further explored this interaction by combining multiple biomarkers to predict overall response rates. For dMMR patients who were also PD-L1-positive or had a high TMB, the response rate was significantly higher (60%) compared to dMMR patients without these additional markers.9 Ongoing trials like KEYNOTE-C93 and DOMENICA now aim to refine these findings to identify which patients may benefit most from immunotherapy, potentially allowing for treatment de-escalation and more personalised approaches.
Oaknin concluded that ICIs are transforming treatment for advanced/recurrent EC. Patients with dMMR/MSI-high tumours obtain a clinically meaningful benefit by combining these ICIs with paclitaxel/ carboplatin, and this regimen must be considered a new standard of care, she
stressed. However, work is still needed to identify which patients with dMMR EC might not benefit from these therapies.
OVERCOMING RESISTANCE TO IMMUNO-ONCOLOGY
Frederik Marmé, Heidelberg University, Germany, addressed the key topic of immuno-oncology (IO) resistance, focusing on three different scenarios: primary resistance, acquired resistance, and progression after IO treatment. This classification is crucial because many studies address IO resistance in various diseases, but the setting in which they are conducted must be specified. Furthermore, patterns of resistance might vary between these categories and could respond differently to next-generation immunotherapies. Currently, there is no standardised definition of IO resistance in EC, though definitions exist for other cancers.
For his talk, Marmé focused on ECs with dMMR, a subgroup expected to respond to immunotherapy. He proposed a clinical definition of IO resistance, adapted from that of non-small cell lung cancer. While
not intended for routine clinical use, it is important to establish stringent inclusion criteria for clinical trials to ensure data comparability. Marmé distinguished primary resistance, which is non-response to IO therapy from the outset, from acquired resistance, which he defined by three criteria: receiving PD-(L)1 blockade, achieving an objective response such as a complete or partial response, and experiencing disease progression within 6 months of the last PD-(L)1 inhibitor treatment. He added that stable disease does not fall under the definition of acquired resistance, as the focus is on patients who initially respond, but later experience progression.
Combining dostarlimab with chemotherapy led to a 72% lower risk of progression or death in patients with dMMR EC, and a significant increase in overall survival
“When does resistance occur in dMMR EC?” Marmé reviewed data from recent ICI monotherapy trials for dMMR EC, emphasising that, while approximately half
of patients achieve an initial response to IO therapy (50–55% primary resistance), this rate of response diminishes significantly in subsequent courses, with approximately 30% of acquired resistance. Marmé explained that mechanisms of resistance are complex and diverse, but key drivers include low neoantigen presentation, multiple immune checkpoints, neutrophil and T-regulatory cell immunosuppression, and inflammation and immunosuppression.
Because primary resistance is the most common form of IO resistance in dMMR EC, finding new strategies to overcome this initial resistance is crucial. Marmé suggested a role for IO combination, with promising preliminary data on the effectiveness of dual immune checkpoint blockade for advanced EC (anti-TIGIT and anti-PD-L1).10 However, combination of ICIs with a different class of inhibitors, PARP inhibitors, was not shown to overcome primary resistance.
References
1. O'Malley DM et al. Pembrolizumab in patients with microsatellite instabilityhigh advanced endometrial cancer: results from the KEYNOTE-158 study. J Clin Oncol. 2022;40(7):752-61.
2. Antill Y et al. Clinical activity of durvalumab for patients with advanced mismatch repair-deficient and repair-proficient endometrial cancer. A nonrandomized phase 2 clinical trial. J Immunother Cancer. 2021;9(6):e002255.
3. Tesaro, Inc. A study to evaluate dostarlimab plus carboplatin-paclitaxel versus placebo plus carboplatinpaclitaxel in participants with recurrent or primary advanced endometrial cancer (RUBY). NCT03981796. https:// clinicaltrials.gov/study/NCT03981796.
4. National Cancer Institute (NCI). Testing the addition of the immunotherapy drug pembrolizumab to the usual chemotherapy treatment (paclitaxel and carboplatin) in stage III-IV or recurrent endometrial cancer.
Finally, Marmé stressed that identifying immune predictors of response to ICIs will be crucial for advancing treatment of dMMR EC. He drew attention to a recent study that conducted an unsupervised hierarchical clustering based on immune markers to identify biomarkers associated with ICI response, such as PD-L1 and HLA-I.11
Currently, there are no data indicating the appropriate course of action in case of progression after PD-(L)1 in dMMR EC. However, Marmé pointed out that other IO-sensitive solid tumours, like non-small cell lung cancer, urothelial carcinoma, and melanoma, have been shown to regain some degree of sensitivity to PD-(L)1 blockade after a treatment-free interval of at least 6 months.
Marmé concluded that precise classification of resistance types, alongside novel IO combinations and biomarker discovery, will be pivotal in optimising treatment strategies for dMMR EC.
5. Mario Negri Institute for Pharmacological Research. Atezolizumab trial in endometrial cancer - AtTEnd (AtTEnd). NCT03603184. https://clinicaltrials. gov/study/NCT03603184.
6. AstraZeneca. Durvalumab with or without olaparib as maintenance therapy after first-line treatment of advanced and recurrent endometrial cancer (DUO-E). NCT04269200. https://clinicaltrials.gov/study/ NCT04269200.
7. Eskander A et al. Updated response data and analysis of progression free survival by mechanism of mismatch repair loss in endometrial cancer (EC) patients (pts) treated with pembrolizumab plus carboplatin/ paclitaxel (CP) as compared to CP plus placebo (PBO) in the NRG GY018 trial. LBA43. ESMO Congress 2023, October 20-24, 2023.
8. Westin SN et al. Durvalumab plus
carboplatin/paclitaxel followed by maintenance durvalumab with or without olaparib as first-line treatment for advanced endometrial cancer: the phase III DUO-E trial. J Clin Oncol. 2024;42(3):283-99.
9. Oaknin A et al. Clinical activity and safety of the anti-programmed death 1 monoclonal antibody dostarlimab for patients with recurrent or advanced mismatch repair-deficient endometrial cancer: a nonrandomized phase 1 clinical trial. JAMA Oncol. 2020;6(11):1766-72.
10. Rojas C et al. Vibostolimab coformulated with pembrolizumab (vibo/pembro) for previously treated advanced mismatch repair–deficient (dMMR) endometrial cancer: results from cohort B1 of the phase 2 KEYVIBE-005 study. J Clin Oncol. 2024;42(16):5502.
11. Grau Bejar JF et al. Immune predictors of response to immune checkpoint inhibitors in mismatch repair-deficient endometrial cancer. J Immunother Cancer. 2024;12(7):e009143.
Treatment Strategies and Sequencing After Endocrine Therapy Plus CDK4/6 Inhibitors
in Patients with ER+/HER2Advanced/Metastatic Breast Cancer
This industry symposium took place during the European Society for Medical Oncology (ESMO) Congress held in Barcelona, Spain, from 13th–17th September 2024.
Chairperson: Peter Schmid1,2
Speakers: Virginia Kaklamani,3 Frederik Marmé4
1. Barts Cancer Institute, Queen Mary University London, UK
2. St. Bartholemew Cancer Centre, Barts Hospital, London, UK
3. University of Texas Health Sciences Center, San Antonio, USA
4. University Hospital Mannheim, Germany
Disclosure: Schmid has received consulting fees from Eli Lilly, Gilead, and Menarini Stemline. Kaklamani has received consulting fees from AstraZeneca, Daiichi Sankyo, Eli Lilly, Genentech, Gilead, Menarini Stemline, Novartis, and TerSera; funding from Eisai; and is on the speakers’ bureau for AstraZeneca, Eli Lilly, and Gilead. Marmé has received financial support/ sponsorship for research, consultation, speaker fees, or travel grants from AGO Study Group, AstraZeneca, BioNTech, Boehringer-Ingelheim, Clovis (Pharma&), Daiichi Sankyo, Eisai, Eli Lilly, German Breast Group, Gilead, GSK, Immunogen (AbbVie), Immutep, Menarini Stemline, MSD, Myriad Genetics, Nerviano Medical Sciences, Novartis, Novocure, Pfizer, Roche, and Seagen (Pfizer).
Acknowledgements: Writing assistance was provided by Nicola Humphry, Nottingham, UK.
Support: The publication of this article was funded by Menarini Stemline. The views and opinions expressed are exclusively those of the speakers. PHARMA
Meeting Summary
This symposium took place on the first day of the 2024 European Society for Medical Oncology (ESMO) Congress in Barcelona, Spain. The goal was to present recommendations for treatment strategies and sequencing for patients with oestrogenreceptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-), advanced/metastatic breast cancer after first-line (1L) therapy with endocrine therapy (ET) plus inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6i).
An expert panel of clinicians explained that most patients will eventually develop resistance to ET regimens during the advanced/metastatic setting, and they discussed the current ESMO recommendations for second- or later-line (2L+) treatment, which are driven by endocrine sensitivity status and biomarkers. Trial data that support the therapeutic recommendations in this patient population were presented, and the benefits and risks associated with different treatment options were summarised.
The panel emphasised the importance of testing for emergent ESR1 mutations at each progression during the advanced/metastatic treatment course, ideally by analysing circulating DNA from a liquid biopsy, in order to identify patients for whom elacestrant will be particularly beneficial.
The Treatment Landscape for ER+/HER2- Advanced/Metastatic Breast Cancer
Over 70% of breast cancers are ER+/HER2-, for which the backbone of treatment is ET.1-3 Advanced breast cancer (aBC) can be considered to include both inoperable, locally advanced breast cancer and metastatic breast cancer (mBC). While aBC/mBC remains largely incurable, important advances over the past 20 years have improved overall survival in patients with ER+/HER2- disease.
Virginia Kaklamani, Professor of Medicine in the Division of Hematology/Oncology at the University of Texas Health Sciences Center, San Antonio, USA, and leader of the breast cancer programme at the Mays Cancer Center, San Antonio, USA, explained that treatment choices for patients with ER+/ HER2- mBC are affected by the complexity and heterogeneity of the disease, the characteristics of the individual patient (e.g., performance status, imminent organ failure, menopausal status, and prior lines of therapy), and the genomic landscape in terms of endocrine sensitivity/resistance and biomarkers (Figure 1).2-5
Treatment Choices at First-Line
The 1L standard of care (SoC) in ER+/HER2mBC is ET plus CDK4/6i.2,6,7 ETs used for this indication include aromatase inhibitors (AI), such as anastrozole, letrozole, and exemestane; and selective oestrogen receptor degraders (SERD), such as fulvestrant.8 The addition of a CDK4/6i such
as palbociclib, ribociclib, or abemaciclib to ET provides significant benefits both in terms of progression-free survival (PFS) and overall survival (OS) through the suppression of cell proliferation.9-14 The median duration of treatment with SoC at 1L is approximately 15–22 months (based on pivotal trials).12,15,16
Median PFS (mPFS) in the PALOMA-2 (palbociclib plus letrozole), MONALEESA-2 (ribociclib plus letrozole), MONALEESA-7 (ribociclib plus ET), and MONARCH-3 (abemaciclib plus non-steroidal AI) trials was 24.8 months (95% CI: 22.1 to not estimable), 25.3 months (95% CI: 23.0–30.3), 23.8 months (95% CI: 19.2 not reached [NR]), and 28.2 months (95% CI: not reported), respectively,9-12 with a median overall survival (mOS) of 53.8 months (95% CI: 49.8–59.2), 63.9 months (95% CI: 52.4–71.0), 58.7 months (95% CI: not reported), and 63.7 months (95% CI: not reported), respectively.13-15,17
As these data imply, most patients will eventually develop resistance to ET.2,4 Kaklamani explained that in ER+/HER2aBC/mBC, resistance to ET can be classified by clinical and molecular variables. In clinical terms, ET resistance in the aBC/mBC setting can be considered primary (disease progression within the first 6 months of 1L ET-based therapy) or secondary (disease progression after more than 6 months of 1L ET-based therapy, or after any duration of 2L+ ET therapy).4,18 In molecular terms, ET resistance can be considered to be intrinsic (e.g., alterations of the PI3K/AKT/mTOR, RAS-MAPK pathway or fibroblast growth factor receptor 1 pathway, or mutations
Figure 1: Treatment choices are driven by endocrine sensitivity status and biomarkers.
Patients with ER+/HER2- mBC1
imminent organ failure
organ failure or short PFS on ET
If PIK3CAm+: Alpelisib + fulvestrant
If ESR1m+: Elacestrant
T-DXd Everolimus + exemestane or Everolimus + fulvestrant or Switch ET ± CDK4/6i or Fulvestrant monotherapy If germline
/PALB2m+: PARP inhibitor
If PIK3CAm/AKT/PTENalteration: Capivasertib + fulvestrant Chemotherapy or sacituzumab govitecan if not used before
Adapted from Gennari A et al.2 2021, and ESMO Metastatic Breast Cancer Living Guidelines 2023.3
1L: first line; 2L+: second and later lines; mBC: metastatic breast cancer; BRCA: breast cancer gene; CDK4/6i: cyclin-dependent kinase 4/6 inhibitor; ER: oestrogen receptor; ESR1: oestrogen receptor 1; ET: endocrine therapy; HER2: human epidermal growth factor receptor 2; m: mutation; PALB2: partner and localiser of BRCA2; PARP: poly(ADP-ribose) polymerase; PD: progressive disease; PFS: progression-free survival; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; T-DXd: trastuzumab deruxtecan.
in BRCA1/2, RB1, or TP53) or acquired mechanisms of resistance (e.g., ESR1 mutations, occurring after prior ET in aBC/mBC).4,19-21
Kaklamani emphasised that different treatment mechanisms are effective for different mechanisms of resistance (Kaklamani, personal communication).
Treatment Choices at Secondor Later-Line are Driven by Endocrine Sensitivity, Biomarker Status, and Toxicity
In patients with ER+/HER2- mBC who do not have imminent organ failure and who experience disease progression after a long PFS on prior ET plus CDK4/6i (suggesting continued ET sensitivity), guidelines recommend exhausting ET options.2,3,6 Sequential ET in combination with a CDK4/6i, mTOR inhibitor (everolimus), PIK3 inhibitor (alpelisib), AKT inhibitor
(capivasertib), or ET monotherapy are therefore used at 2L+ in this population.2,3,6 However, Kaklamani stressed that there remains a considerable margin for therapeutic improvement.
For example, ET monotherapies only provide an mPFS of around 2–4 months.22-25 In addition, combination therapies such as ET plus CDK4/6i or ET plus PI3K/ AKT/mTOR inhibitors can be associated with toxicity. For example, CDK4/6i are associated with adverse events (AE) such as neutropenia, leukopenia, and anaemia, and sometimes with diarrhoea,16,26,27 with discontinuation due to AEs in up to 19% of patients,16,26,28 and PI3K/AKT/mTOR inhibitors are associated with AEs such as diarrhoea, rash, hyperglycaemia, and stomatitis,29-31 with discontinuation rates due to AEs in up to 24% of patients.32-34 As an intramuscular injection, monotherapy or combination therapy with fulvestrant can also be associated with injection site pain, as well
as musculoskeletal pain, back pain, and peripheral neuropathy.35
Mutations in key genes are used as therapeutically relevant biomarkers to guide treatment choices. For example, mutations in the genes PIK3CA, ESR1, or BRCA/PALB2. 2,3
Treatment choices at secondor later-line for patients without specific biomarkers
In the absence of specific mutation biomarkers, ESMO guidelines for 2L+ treatment of ER+/HER2- mBC (without imminent organ failure and with a long PFS on prior ET) include switching ET and/or CDK4/6i, combining everolimus with either fulvestrant or exemestane, or fulvestrant monotherapy.2,3
Unfortunately, rechallenge with a CDK4/6i has been associated with mixed results in clinical trials. Positive findings were reported in the MAINTAIN trial for ribociclib plus fulvestrant/exemestane versus fulvestrant/ exemestane monotherapy (mPFS: 5.3 months versus 2.8 months, respectively).36
A statistically significant, but not clinically meaningful, efficacy improvement was also found with abemaciclib plus fulvestrant versus fulvestrant monotherapy in the postMONARCH study (mPFS: 6.0 months versus 5.3 months, respectively), though benefits were not observed in patients with prior ribociclib therapy in the latter study.37,38 However, no significant improvements were reported from palbociclib plus fulvestrant versus fulvestrant monotherapy in the PACE trial (mPFS: 4.6 months versus 4.8 months, respectively),39 or from palbociclib plus fulvestrant/letrozole versus fulvestrant/ letrozole monotherapy in the PALMIRA study (mPFS: 4.2 months versus 3.6 months, respectively).40
Though the combination of an mTOR inhibitor plus fulvestrant/exemestane has shown positive results in the overall ER+/ HER2- aBC/mBC population at 2L+, patients with an ESR1 mutation appear to receive less benefit.41-45 For example, the BOLERO-2 trial of everolimus plus exemestane versus exemestane monotherapy, conducted in a population not previously exposed to
CDK4/6 inhibitors, was associated with an mPFS of 7.8 months versus 3.2 months across all patients, yet an mPFS of 5.4 months versus 2.8 months among patients with an ESR1 mutation.44,45
Treatment choices at secondor later-line for patients with AKT/PIK3CA/PTEN alterations ESMO guidelines recommend 2L+ treatment with fulvestrant plus alpelisib for patients with ER+/HER2- mBC (without imminent organ failure and with a long PFS on prior ET) who are positive for a pathogenic mutation in PIK3CA (PIK3CAmut) and who have prior exposure to an AI.2,3
Fulvestrant plus alpelisib is approved for use in patients with ER+/HER2- PIK3CAmut aBC/mBC after disease progression following endocrine monotherapy.29 This is based on results from the SOLAR-1 study, which enrolled patients with prior AI therapy (only 6% had received prior CDK4/6i therapy) into two cohorts based on tumourtissue PIK3CA mutation status.46 In the PIK3CAmut cohort, 169 patients received alpelisib plus fulvestrant, and 172 patients received placebo plus fulvestrant. Over the course of the trial, the mPFS was 11 months in the alpelisib-fulvestrant group versus 5.7 months in the placebo-fulvestrant group, with a hazard ratio (HR) of 0.65 (95% CI: 0.50–0.85; p<0.001).46
However, Kaklamani pointed out that analysis of the subsequent BYLieve study, in which all patients had prior CDK4/6i therapy, alpelisib-fulvestrant tended to be less effective (overall mPFS: 8.0 months).47 The mPFS with alpelisib-fulvestrant was just 5.6 months in the ESR1mut group (n=27), compared with 8.3 months in the wild-type ESR1 group (n=75).47
The AKT kinase inhibitor, capivasertib, has also been approved for use in combination with fulvestrant, in ER+/HER2- aBC/mBC with one or more PIK3CA, AKT1, or PTEN mutations following recurrence/progression on ET.31 In the CAPItello-291 study, patients with aBC/mBC and mutations in PIK3CA, AKT1, or PTEN had an mPFS of 7.3 months with capivasertib-fulvestrant (n=155) versus 3.1 months with placebo-fulvestrant
(n=134), with an adjusted HR of 0.5 (95% CI: 0.38–0.65; p<0.001).25
Kaklamani noted that subgroup analyses showed that mPFS was shorter for both capivasertib-fulvestrant and placebofulvestrant in patients with prior CDK4/6i exposure (5.5 months versus 2.6 months), and shorter still with prior chemotherapy for aBC/mBC (3.8 months versus 2.1 months), or with liver metastases at baseline (3.8 months versus 1.9 months).48 Data on the efficacy of capivasertib-fulvestrant in patients with ESR1mut is not available.
Treatment choices at secondor later-line for patients with BRCA/PALB2 mutation
ESMO guidelines recommend the consideration of 2L+ treatment with poly ADP ribose polymerase inhibitor (PARPi) monotherapy (olaparib or talazoparib) for patients with ER+/HER2- mBC (without imminent organ failure and with a long PFS on prior ET) with a pathogenic germline mutation in BRCA1/2 (BRCAmut) or PALB2 (PALB2mut).2,3
In the OlympiAD study, patients with HER2- mBC and germline BRCA1/2mut, and up to two prior chemotherapy regimens for metastatic disease, received olaparib (n=205) or the physician’s choice of chemotherapy (n=97).49 The mPFS was 7.0 months in the olaparib group versus 4.2 months in the chemotherapy group, with an adjusted HR of 0.58 (95% CI: 0.43–0.80; p<0.001).49
A similar benefit of PARPi over chemotherapy was reported in the EMBRACA study, with an mPFS of 8.6 months in patients treated with talazoparib (n=287) and 5.6 months in patients treated with chemotherapy (n=144), with an adjusted HR of 0.54 (95% CI: 0.41–0.71; p<0.001).50
PALB2, like BRCA1/2, is involved in DNA repair, and some limited data have confirmed that PARPi are likely to have a benefit in patients with PALB2mut.51,52 However, because of the low frequency of the PALB2 mutation, dedicated studies may not be possible.53
Summary of treatment options at second- or later-line Kaklamani emphasised that the use of fulvestrant monotherapy or ET combination therapy appears to be associated with a consistently lower PFS duration in patients with prior CDK4/6i therapy than in those without, and PFS duration appears to be lower still in those patients who also harbour an ESR1 mutation.15,22,25,29,30,36-38,42,47,48,54-61
Treatment choices at second- or laterline for patients with ESR1 mutation ESMO guidelines recommend elacestrant at 2L for patients with ER+/HER2- mBC (without imminent organ failure and with a long PFS on prior ET) who harbour a mutation in ESR1 (ESR1mut) and experience disease progression after at least 1 line of ET.2,3
This recommendation is based on results from the EMERALD study, which enrolled 477 patients with ER+/HER2- aBC/mBC who had progressed/relapsed after 1–2 lines of ET for aBC/mBC, one of which had to be combined with a CDK4i.22 Patients were stratified by ESR1mut status and were randomised 1:1 to treatment with elacestrant (n=239) or the investigator’s choice of SOC (an AI or fulvestrant; n=238) until disease progression. Kaklamani stressed that all patients in the trial had received prior CDK4/6i therapy, and that across the elacestrant group and the SOC group, 68% and 71% of patients had visceral metastases, respectively, and 48% and 47% were ESR1mut, respectively.22
Among patients with ESR1mut, elacestrant (n=115) versus SOC (n=113) was associated with a 45% reduction in the risk of progression or death (HR: 0.55; 95% CI: 0.39–0.77; p=0.0005). The 6-month PFS in the elacestrant group versus the SOC group was 40.8% versus 19.1%, respectively, and the 12-month PFS was 26.8% versus 8.2%, respectively.22
As EMERALD patients population included primary endocrine resistance, an exploratory analysis showed that duration of prior ET plus CDK4/6i therapy may be positively associated with mPFS in patients with ESR1mut. Among patients with ≥6
months of prior ET plus CDK4/6i, mPFS with elacestrant (n=103) was 4.1 months, compared with 1.9 with SOC (n=102). However, among patients with ≥12 months of prior ET plus CDK4/6i, mPFS reached 8.6 months (n=78) versus 1.9 months (n=81), respectively; and in those with ≥18 months of prior ET plus CDK4/6i, mPFS reached 8.6 months (n=55) versus 2.1 months (n=56), respectively.38 The clinically meaningful improvement in PFS versus SOC in patients with longer prior exposure to prior ET plus CDK4/6i has been demonstrated regardless of the metastatic site location or number; coexistence of PIK3CAmut, TP53mut, or HER2-low expression; or ESR1mut variant (Table 1).38 Kaklamani explained
Patients with longer prior ET + CDK4/6i (≥12 months)
that, because the benefit observed with elacestrant versus SOC was not impacted by other commonly coexisting mutations or molecular expressions, it is highly likely that ESR1 mutations were the main driver of disease in this population.
To explain the significance of these findings, Kaklamani stressed that if a patient has received ≥12 months of prior ET plus CDK4/6i before experiencing disease progression, their tumour is likely to be endocrine sensitive, whereas the tumour of a patient with disease progression after, for example, 4 months of prior ET plus CDK4/6i therapy is likely to be endocrine resistant. Ultimately, clinicians need to
Table 1: PFS in subgroups of patients with ESR1-mutated tumours and longer prior ET+CDK4/6i.
select tumours that are endocrine sensitive to have confidence in further ET, and longer PFS with prior exposure to ET+CDK4/6i (>6 months)18 is a good guideline to use (Kaklamani, personal communication).
In the overall EMERALD population, the majority of adverse events that occurred were Grade 1 or 2;22 no Grade 4 treatmentrelated AEs were reported.62 In the elacestrant and SOC arms of the study, 3.4% and 0.9% of patients discontinued treatment due to treatment-related AEs.22 Nausea was responsible for elacestrant discontinuation in 1.3% of patients, though Kaklamani pointed out that the use of antiemetics in the elacestrant group was actually less than in the SOC (AI) group.38 No haematologic safety signal was observed, and none of the patients in either treatment arm had sinus bradycardia.38
Kaklamani stressed that elacestrant is not the only endocrine-based therapy being developed for patients with ER+/HER2mBC, and that data for other drugs are expected in the next future (Kaklamani, personal communication).
Treatment choices at second- or laterline for patients with imminent organ failure or primary endocrine resistance For those patients with ER+/HER2- mBC who have imminent organ failure or who had a short PFS (<6 months) on ET at 1L (indicative of primary endocrine resistance), ESMO guidelines recommend 2L+ treatment with chemotherapy-based regimens at 2L+.2,3,18
In the recent DESTINY-Breast06 study, T-DXd was evaluated in patients with HER2low or -ultralow after disease progression on ET (≥2 prior lines of ET or 1 line of ET and primary endocrine resistance) but with no prior chemotherapy for mBC.63 Among patients with HER2-low, the mPFS in the T-DXd group (n=359) was 13.2 months, while the mPFS in the physician’s choice of chemotherapy group (n=354) was 8.1 months, indicating that T-DXd significantly improved PFS versus treatment with physician’s choice of chemotherapy (HR: 0.62; 95% CI: 0.51–0.74; p<0.0001).63
Sacituzumab govitecan, an antibody-drug conjugate consisting of a trop-2-directed antibody and a topoisomerase inhibitor, should be considered for patients in this population with HER2-0 and ≥2 prior lines of chemotherapy.3,64 In patients with HER2low and ≥1 prior line of chemotherapy, trastuzumab deruxtecan (T-DXd), an antibody drug conjugate consisting of an HER2-directed antibody and a topoisomerase inhibitor, should be considered.2,3,65
These data demonstrate that ADCs should be considered for patients with ER+/HER2- mBC who have imminent organ failure or who had a short PFS (<6 months ) in a prior line of endocrine therapy, or are no longer eligible for endocrine therapybased regimens.
Biomarkers of Acquired Resistance in Breast Cancer
Frederik Marmé, Professor of Experimental and Translational Gynecologic Oncology at University Hospital Mannheim, Germany, and co-chair of the AGO Study Group, explained that breast cancer is a dynamic disease in which mutations may emerge over the course of 1L mBC treatment.2-5
One of the key mechanisms of resistance to ET is the emergence of mutations in the ESR1 gene.21 Marmé stressed that because ESR1 mutations are acquired during 1L mBC treatment, they are sub-clonal, which means that the molecular profile can vary between and within tumour sites.4,5
Mutations in ESR1 that alter the ligandbinding domain of the oestrogen receptor result in constitutive activation of the oestrogen receptor, which confers ligand independence.20,66 Constitutive oestrogen receptor signalling leads to increased proliferation, differentiation, and survival in the affected cancer cells.67,68 ESR1 mutations have been associated with endocrine resistance, visceral metastases, and poorer outcomes. 21,67-70
ETs exert their anti-tumour activity by binding to the ligand-binding pocket of the oestrogen receptor and inhibiting the activation of downstream targets.21 By altering the ligand-binding domain, ESR1 mutations can induce resistance to ETs.21
Marmé explained that the longer mBC is exposed to ET, the greater the risk of developing ESR1 mutations during treatment, which eventually emerge in up to 40% of patients (Figure 2).21,22,71-77
As described above, ESR1 mutations drive treatment decisions because the biomarker profile of ER+/HER2- mBC influences the choice of therapy in 2L+.2 In light of this, ESMO, NCCN, and ASCO recommend testing for ESR1 mutations at each progression if not detected previously.79-83
Unlike BRCA and PIK3CA mutations, ESR1 mutations are typically undetectable in the primary tumour because they are sub-clonal, and archival tissue from the primary tumour should not be used to
identify ESR1mut.79,84 For this reason, testing of ESR1mut is best performed in liquid biopsy (ctDNA).79,83 Blood-based ctDNA is also preferred for ESR1mut testing because it is more sensitive for these mutations compared with tissue sampling. For example, the ESR1mut prevalence rate in liquid biopsy is higher than in tissue, especially when ctDNA tumour fraction is ≥1.85 Kaklamani described the interpretation of ctDNA results as relatively straightforward, especially when the laboratory performing the test provides support.
What About Patients with a Tumour That is Positive for More Than One Biomarker?
Marmé explained that, in a tumour with ESR1mut that is also positive for other biomarkers, thinking about ctDNA allele frequency is not particularly useful when making treatment decisions. He emphasised that intrinsic mutations such as PIK3CA or AKT would likely have a higher allelic
Tissue biopsy to confirm breast cancer and testing for intrinsic mutations - PIK3CA/AKT/PTEN - BRCA1/2, PALB2
Liquid biopsy
Testing for acquired mutations - ESR1
As ESR1 mutations occur almost exclusively after ET in the mBC setting,73 testing for ESR1-mut should occur at each progression if not
Figure 2: Longer exposure to ET in mBC increases the chance of developing ESR1mut during treatment.
Figure courtesy of Menarini Stemline.
frequency than an acquired ESR1 mutation, which will probably have occurred in a subclone of the original tumour. However, he stressed that elacestrant will also work in those tumour cells that express wildtype ESR1, and it is important to use a treatment that will effectively treat the ESR1mut cells (Marmé, personal communication).
Kaklamani added that, if there are more than one treatment option available for a patient with co-mutations, she would consider the toxicity of the available treatments, and in general, single-agent ET tends to be better tolerated than combination therapies (Kaklamani, personal communication).
Closing Remarks
Peter Schmid, Chair of Cancer Medicine at Barts Cancer Institute, Queen Mary University, and Clinical Director of the Breast Cancer Centre at the St. Bartholomew Cancer Centre, Barts Hospital,
References
1. Zhou FH et al. CDK4/6 inhibitor resistance in estrogen receptor positive breast cancer, a 2023 perspective. Front Cell Dev Biol. 2023;11:1148792.
2. Gennari A et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann Oncol. 2021;32(12):1475-95.
3. ESMO. ESMO metastatic breast cancer living guideline: er-positive her2-negative breast cancer. Available at: https://www.esmo.org/livingguidelines/esmo-metastatic-breastcancer-living-guideline/er-positiveher2-negative-breast-cancer. Last accessed: 4 Septemebr 2024.
4. Hartkopf AD et al. Endocrineresistant breast cancer: mechanisms and treatment. Breast Care (Basel). 2020;15(4):347-54.
5. Bennett C et al. Breast cancer genomics: primary and most common metastases. Cancers. 2022;14(13):3046.
6. Burstein HJ et al. Endocrine treatment and targeted therapy for hormone receptor-positive, human epidermal
London, UK, concluded the symposium by emphasising the key takeaways.
Essentially, a biomarker-driven treatment algorithm is needed to ensure optimal treatment selection for patients with ER+/ HER2- mBC.6,12,22,25,47 Biomarkers include intrinsic mutations in BRCA or PIK3CA and acquired ESR1 mutations that emerge over time in up to 40% of patients after ET.21,22,76,77,86 Longer PFS on prior ET + CDK4/6i can be used as a surrogate for endocrine sensitivity in ESR1mut tumours,22,38,87 and endocrine-sensitive tumours with coexisting PIK3CA and ESR1 mutations may benefit from elacestrant monotherapy before PI3K/AKT inhibitors, as data suggest the endocrine receptor pathway may drive disease progression in these patients.38
Schmid explained that fundamentally, at 1L progression, patients with ER+/HER2aBC/mBC should be tested for genomic alterations in liquid biopsy to define the optimal treatment.2,3
7. Cardoso F et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. 2020;31(12):1623-49.
8. Belachew EB et al. Molecular mechanisms of endocrine resistance in estrogen-positive breast cancer. Front Endocrinol (Lausanne). 2021;12:599586.
9. Finn RS et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925-36.
10. Johnston S et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. 2019;5:5.
11. Tripathy D et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptorpositive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018;19(7):90415.
12. Hortobagyi GN et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2negative advanced breast cancer. Ann Oncol. 2018;29(7):1541-7.
13. Hortobagyi GN et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Engl J Med. 2022;386(10):942-50.
14. Lu YS et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2- advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res. 2022;28(5):851-9.
15. Slamon DJ et al. Overall survival with palbociclib plus letrozole in advanced breast cancer. JCO. 2024;42(9):9941000.
16. Pfizer. IBRANCE (palbociclib). EMA summary of product characteristics. Available at: https://www.ema. europa.eu/en/documents/productinformation/ibrance-epar-productinformation_en.pdf. Last accessed: 4 September 2024.
17. Goetz MP et al. Abemaciclib plus a nonsteroidal aromatase inhibitor
as initial therapy for HR+, HER2advanced breast cancer: final overall survival results of MONARCH 3. Ann Oncol. 2024;35(8):718-27.
18. Cardoso F et al. 6th and 7th International consensus guidelines for the management of advanced breast cancer (ABC guidelines 6 and 7). Breast. 2024;76:103756.
19. Rani A et al. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol (Lausanne). 2019;10:245.
20. Xu XQ et al. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol Sin. 2021;42(2):171-8.
21. Brett JO et al. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 2021;23(1):85.
22. Bidard FC et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from the randomized phase III EMERALD trial. J Clin Oncol. 2022;40(28):3246-56.
23. Lindeman GJ et al. VERONICA: Randomized phase II study of fulvestrant and venetoclax in ERpositive metastatic breast cancer post-CDK4/6 inhibitors - efficacy, safety, and biomarker results. Clin Cancer Res. 2022;28(15):3256-67.
24. Oliveira M et al. Abstract GS3-02: GS3-02 Camizestrant, a nextgeneration oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2negative breast cancer: Results of the randomized, multi-dose Phase 2 SERENA-2 trial. Cancer Research. 2023;83(Suppl 5):GS3-02.
25. Turner NC et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. 2023;388(22):2058-70.
26. Novartis Pharma. KISQALI (ribociclib). EMA summary of product characteristics. Available at: https:// www.ema.europa.eu/en/documents/ product-information/kisqali-eparproduct-information_en.pdf. Last accessed: 4 September 2024.
27. Eli Lilly. VERZENIOS (abemaciclib). EMA summary of product characteristics. Available at: https:// www.ema.europa.eu/en/documents/ product-information/verzenios-eparproduct-information_en.pdf. Last accessed: 4 September 2024.
28. Lilly USA. VERZENIO (abemaciclib). FDA prescribing information.
Available at: https://www.accessdata. fda.gov/drugsatfda_docs/ label/2023/208716s010s011lbl.pdf. Last accessed: 4 September 2024.
29. Novartis. PIQRAY (alpelisib). EMA summary of product characteristics. Available at: https://www.ema. europa.eu/en/documents/productinformation/piqray-epar-productinformation_en.pdf. Last accessed: 4 September 2024.
30. Novartis. AFINITOR (everolimus). EMA summary of product characteristics. Available at: https://www.ema. europa.eu/en/documents/productinformation/afinitor-epar-productinformation_en.pdf. Last accessed: 4 September 2024.
31. AstraZeneca. TRUQAP (capivasertib). EMA summary of product characteristics. Available at: https:// www.ema.europa.eu/en/documents/ product-information/truqap-eparproduct-information_en.pdf. Last accessed: 4 September 2024.
32. AstraZeneca. TRUQAP (capivasertib). FDA prescribing information. Available at: https://www.accessdata. fda.gov/drugsatfda_docs/ label/2023/218197s000lbl.pdf. Last accessed: 4 September 2024.
33. Novartis. AFINITOR (everolimus). FDA prescribing information. Available at: https://www.novartis.com/us-en/ sites/novartis_us/files/afinitor.pdf. Last accessed: 4 September 2024.
34. Novartis PIQRAY (alpelisib). FDA prescribing information. Available at: https://www.accessdata. fda.gov/drugsatfda_docs/ label/2024/212526s009lbl.pdf. Last accessed: 17 September 2024.
35. AstraZeneca. FASLODEX (fulvestrant). EMA summary of product characteristics. Available at: https:// www.ema.europa.eu/en/documents/ product-information/faslodex-eparproduct-information_en.pdf. Last accessed: 4 September 2024.
36. Kalinsky K et al. Randomized phase II trial of endocrine therapy with or without ribociclib after progression on cyclin-dependent kinase 4/6 inhibition in hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: MAINTAIN Trial. J Clin Oncol. 2023;41(24):4004-13.
37. Kalinsky K et al. Abemaciclib plus fulvestrant vs fulvestrant alone for HR+, HER2- advanced breast cancer following progression on a prior CDK4/6 inhibitor plus endocrine therapy: primary outcome of the phase 3 postMONARCH trial. JCO. 2024;42(Suppl 17):LBA1001-LBA1001.
38. Bardia A et al. Elacestrant in ER+, HER2- MBC with ESR1-mutated
tumors: subgroup analyses from the phase III EMERALD trial by prior duration of endocrine therapy plus CDK4/6 inhibitor and in clinical subgroups. Clin Cancer Res. 2024;DOI:10.1158/1078-0432.CCR-241073.
39. Mayer EL et al. PACE: a randomized phase II study of fulvestrant, palbociclib, and avelumab after progression on cyclin-dependent kinase 4/6 inhibitor and aromatase inhibitor for hormone receptorpositive/human epidermal growth factor receptor-negative metastatic breast cancer. J Clin Oncol. 2024;42(17):2050-60.
40. Llombart-Cussac A et al. Second-line endocrine therapy (ET) with or without palbociclib (P) maintenance in patients (pts) with hormone receptor-positive (HR[+])/human epidermal growth factor receptor 2-negative (HER2[]) advanced breast cancer (ABC): PALMIRA trial. JCO. 2023;41(Suppl 16):1001.
41. Vasseur A et al. Fulvestrant and everolimus efficacy after CDK4/6 inhibitor: a prospective study with circulating tumor DNA analysis. Oncogene. 2024;43(16):1214-22.
42. Bardia A et al. Phase I/II trial of exemestane, ribociclib, and everolimus in women with HR+/HER2- advanced breast cancer after progression on CDK4/6 inhibitors (TRINITI-1). Clin Cancer Res. 2021;27(15):4177-85.
43. Cook MM et al. Everolimus plus exemestane treatment in patients with metastatic hormone receptor-positive breast cancer previously treated with CDK4/6 inhibitor therapy. Oncologist. 2021;26(2):101-6.
44. Chandarlapaty S et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2(10):1310-5.
45. Yardley DA et al. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progressionfree survival analysis. Adv Ther. 2013;30(10):870-84.
46. André F et al. Alpelisib for PIK3CAmutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929-40.
47. Turner N et al. Abstract PD1501: Impact of ESR1 mutations on endocrine therapy (ET) plus alpelisib benefit in patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-), PIK3CA-mutated, advanced breast cancer (ABC) who progressed on or after prior cyclindependent kinase inhibitor (CDK4/6i)
therapy in the BYLieve trial. Cancer Res. 2022;82(Suppl 4):PD15-01.
48. Oliveira M et al. 187O Capivasertib and fulvestrant for patients (pts) with aromatase inhibitor (AI)-resistant HR+/ HER2– advanced breast cancer (ABC): Subgroup analyses from the phase III CAPItello-291 trial. ESMO Open. 2023; 8(1):101376.
49. Robson M et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523-33.
50. Litton JK et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753-63.
51. Tung NM et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38(36):4274-82.
52. Gruber JJ et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat Cancer. 2022;3(10):1181-91.
53. ASCO Daily News. What is the potential role of PARP Inhibitors for metastatic breast cancer with a germline PALB2 pathogenic variant?. Available at: https:// dailynews.ascopubs.org/doi/10.1200/ ADN.23.201519. Last accessed: 4 September 2024.
54. Chia S et al. Alpelisib + endocrine therapy in patients with PIK3CA -mutated, hormone receptor–positive, human epidermal growth factor receptor 2-negative, advanced breast cancer: analysis of all 3 cohorts of the BYLieve study. JCO. 2023;41(Suppl 16):1078.
55. Stemline Therapeutics. ORSERDU (elacestrant). EMA summary of product characteristics. Available at: https://www.ema.europa.eu/en/ documents/product-information/ orserdu-epar-product-information_ en.pdf. Last accessed: 4 September 2024.
56. Johnston SR et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentre, phase 3 randomised trial. Lancet Oncol. 2013;14(10):989-98.
57. Rozenblit M et al. Patterns of treatment with everolimus exemestane in hormone receptor-positive HER2negative metastatic breast cancer in the era of targeted therapy. Breast
Cancer Res. 2021;23(1):14.
58. Baselga J et al. Everolimus in postmenopausal hormone-receptorpositive advanced breast cancer. N Engl J Med.. 2012;366(6):520-9.
59. Rugo HS et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, noncomparative study. Lancet Oncol. 2021;22(4):489-98.
60. Fillbrunn M et al. PIK3CA mutation status, progression and survival in advanced HR + /HER2- breast cancer: a meta-analysis of published clinical trials. BMC Cancer. 2022;22(1):1002.
61. André F et al. Alpelisib for PIK3CAmutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929-40.
62. Bardia A. EMERALD phase 3 trial of elacestrant versus standard of care endocrine therapy in patients with ER+/HER2- metastatic breast cancer: updated results by duration of prior CDK4/6i in metastatic setting. Presentation GS3-01. SABCS22, 6-10 December, 2022.
63. Curigliano G et al. Trastuzumab deruxtecan (T-DXd) vs physician’s choice of chemotherapy (TPC) in patients (pts) with hormone receptorpositive (HR+), human epidermal growth factor receptor 2 (HER2)-low or HER2-ultralow metastatic breast cancer (mBC) with prior endocrine therapy (ET): primary results from DESTINY-Breast06 (DB-06). JCO. 2024;42(Suppl 17):LBA1000.
64. Bardia A et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU132-01 basket trial. Ann Oncol. 2021;32(6):746-56.
65. Martín M et al. Trastuzumab deruxtecan in breast cancer. Crit Rev Oncol Hematol. 2024;198:104355.
66. Lei JT et al. Endocrine therapy resistance: new insights. Breast. 2019;48(Suppl 1):S26-30.
67. Williams MM et al. Steroid hormone receptor and infiltrating immune cell status reveals therapeutic vulnerabilities of ESR1-mutant breast cancer. Cancer Res. 2021;81(3):73246.
68. Jeselsohn R et al. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12(10):573-83.
69. Bardia A et al. Case 35-2018: A 68-year-old woman with back pain
and a remote history of breast cancer. N Engl J Med. 2018;379(20):1946-53.
70. Jeselsohn R et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. 2018;33(2):173-186.e5.
71. Jeselsohn R et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757-67.
72. Allouchery V et al. Circulating ESR1 mutations at the end of aromatase inhibitor adjuvant treatment and after relapse in breast cancer patients. Breast Cancer Res. 2018;20(1):40.
73. Schiavon G et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7(313):313ra182.
74. Toy W et al. ESR1 ligand-binding domain mutations in hormoneresistant breast cancer. Nat Genet. 2013;45(12):1439-45.
75. Jhaveri K et al. 383MO Imlunestrant with or without everolimus or alpelisib, in ER+, HER2- advanced breast cancer (aBC): results from the phase Ia/b EMBER study. Annals of Oncology. 2023;34:S338-9.
76. Lin NU et al. 382MO Updated results from the phase I/II study of OP-1250, an oral complete estrogen receptor (ER) antagonist (CERAN) and selective ER degrader (SERD) in patients (pts) with advanced or metastatic ERpositive, HER2-negative breast cancer. Annals of Oncology. 2023;34:S338.
77. Bhave M et al. Abstract PO2-16-05: ESR1 mutations (ESR1mut) in HR(+) HER2(-)patients with metastatic breast cancer (MBC): prevalence along treatment course and predictive value for endocrine therapy (ET) resistance in real-world practice. Cancer Research. 2024;84(Suppl 9):PO2-1605.
78. Lee N et al. Currently applied molecular assays for identifying ESR1 mutations in patients with advanced breast cancer. International Journal of Molecular Sciences. 2020;21(22):8807.
79. Burstein HJ et al. Biomarker testing and endocrine and targeted therapy in metastatic breast cancer expert panels. testing for ESR1 mutations to guide therapy for hormone receptorpositive, human epidermal growth factor receptor 2-negative metastatic breast cancer: ASCO guideline rapid recommendation update. J Clin Oncol. 2023;41(18):3423-5.
80. Mosele MF et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced
cancer in 2024: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2024;35(7):588606.
81. Pascual J et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2022;33(8):750-68.
82. National Comprehensive Cancer Network (NCCN). Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Breast Cancer. Available at: https://www.nccn.org/ professionals/physician_gls/pdf/breast.
pdf. Last accessed: 4 September 2024.
83. Gradishar WJ et al. NCCN guidelines® insights: breast cancer, version 4.2023. J Natl Compr Canc Netw. 2023;21(6):594-608.
84. Turner NC et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020;21(10):1296-308.
85. Lone SN et al. Liquid biopsy: a step closer to transform diagnosis, prognosis and future of cancer treatments. Mol Cancer. 2022;21(1):79.
86. Santiago Novello RG et al. 220P Oral selective estrogen receptor degraders for metastatic hormone receptorpositive, HER2 negative breast cancer according to ESR1 mutation: a systematic review and meta-analysis of randomized control trials. ESMO Open. 2023;8(1):101409.
87. Bardia A et al. Abstract GS3-01: GS3-01 EMERALD phase 3 trial of elacestrant versus standard of care endocrine therapy in patients with ER+/HER2- metastatic breast cancer: updated results by duration of prior CDK4/6i in metastatic setting. Cancer Res. 2023;83(Suppl 5):GS3-01.
Evolving Patient-Centred Therapies for Metastatic NSCLC
This symposium took place on 14th September 2024, as part of the European Society for Medical Oncology (ESMO) Annual Congress, held in Barcelona, Spain.
Chairperson: Jarushka Naidoo1
Speakers: Terri Conneran,2 Luis Paz-Ares,3 Alexander Drilon4
1. Beaumont RCSI Cancer Centre, Dublin, Ireland
2. KRAS Kickers, Charlotte, North Carolina, USA
3. Hospital Universitario 12 de Octubre, Madrid, Spain
4. Memorial Sloan Kettering Cancer Center, New York, USA
Disclosure: Naidoo has received advisory board/lecture fees from AbbVie, Amgen, Arcus Biosciences, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Elevation Oncology, Kaleido Biosciences, NGM Biopharmaceuticals, Pfizer, Regeneron, Roche/Genentech, and Takeda; research funding from Amgen, Arcus Biosciences, AstraZeneca, Bristol Myers Squibb, Novartis, Pfizer, Roche/Genentech, and Takeda. Conneran has received advisory board/lecture fees from 23andMe, Agilent, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Diaceutics, Frontier Medicines, Jaguar Health, Janssen, Labcorp, Loxo@ Lilly, Merck, Novartis, QIAGEN, Revolution Medicines, Roche, and Sanofi. Paz-Ares has received advisory board/lecture fees from AbbVie, Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Gilead, GSK, Janssen, Lilly, Merck, Merck KGaA (Darmstadt, Germany), Novartis, Roche, Pfizer, PharmaMar, Regeneron, Sanofi, and Takeda; research funding from AstraZeneca, Bristol Myers Squibb, Merck, and Pfizer. Drilon has received advisory board/lecture fees from 14ner Oncology/Elevation Oncology, AbbVie, Amgen, AnHeart, ArcherDX, AstraZeneca, BeiGene, BerGenBio, Blueprint Medicines, Bristol Myers Squibb, Chugai, EcoR1 Capital, EMD Serono, Entos, Exelixis, Helsinn, Hengrui, Ignyta/Genentech/Roche, Janssen, Loxo/Bayer/Lilly, Merus, Monopteros Therapeutics, Monte Rosa Therapeutics, Novartis, Nuvalent, Pfizer, Prelude Therapeutics, Regeneron, Repare Therapeutics, Springer Healthcare, Takeda/ARIAD Pharmaceuticals/Millennium Pharmaceuticals, Treeline Biosciences, Turning Point Therapeutics, Tyra Biosciences, and Verastem Oncology; royalties from Wolters Kluwer; CME honoraria from Answers in CME, Applied Pharmaceutical Science, AXIS Pharmaceuticals, Clinical Care Options, Doc Congress, EPG Health, Harborside, i3 Health, Imedex, Liberum IME, Medendi, Medscape, Med Learning Group, MEDtalks, MJH Life Sciences, MORE Health, Ology Medical Education, OncLive, Paradigm Biopharmaceuticals, PeerView, PeerVoice, Physicians’ Education Resource, Projects In Knowledge, Resources, Remedica, Research To Practice, RV More, Targeted Oncology, touchIME, WebMD; other provisions (food/beverage) from Merck, Puma, Merus, and Boehringer Ingelheim; and research funding from Foundation Medicine, GSK, Teva Pharmaceuticals, Taiho, and PharmaMar. The speakers have declared no conflict of interest.
Acknowledgements: Medical writing assistance provided by OPEN Health Scientific Communications, London, UK.
Disclaimer The opinions expressed in this article belong solely to the named speakers.
Support: The publication of this article was supported by Bristol Myers Squibb.
Meeting Summary
The metastatic non-small cell lung cancer (mNSCLC) treatment landscape has vastly expanded over the past two decades as a result of advancements in biomarker testing. However, unmet needs remain both in terms of treatment options for some patient groups, and patient support throughout the treatment journey. In this symposium, Jarushka Naidoo, Consultant Medical Oncologist, Beaumont RCSI Cancer Centre, Dublin, Ireland; Terri Conneran, KRAS Kickers, Charlotte, North Carolina, USA; Luis Paz-Ares, Chair of the Medical Oncology Department, Hospital Universitario 12 de Octubre, Madrid, Spain; and Alexander Drilon, Chief of Early Drug Development and Thoracic Oncology, Memorial Sloan Kettering Cancer Center, New York, USA, focused on patient-centric approaches to mNSCLC treatment, starting with a patient and patient advocacy group perspective on what patients want from their care team during their treatment journey. The panel also discussed both immuno-oncology (I-O) monotherapy and combination therapy, including dual I-O therapies for patients with programmed death-ligand 1 (PD-L1) tumour expression <1%, as well as the treatment landscape for KRASG12C-mutated mNSCLC, and ongoing trials of KRAS-targeted agents. In addition, the latest data on tyrosine kinase inhibitors (TKI) for patients with alterations in ROS1 and NTRK genes were discussed, focusing on next-generation TKIs. Finally, the panel discussed patient cases, taking into account specific considerations and how to best approach treatment decisions.
Introduction
Naidoo provided an introductory overview of the mNSCLC treatment landscape and its vast expansion over the past two decades.1-4 In addition, she highlighted the multitude of targetable biomarkers in mNSCLC while underscoring the importance of treating patients with the appropriate therapy. However, while testing can help change patient outcomes, the important role of other members of the multidisciplinary team and patient advocacy groups should not be overlooked.5
The Importance of Patient Empowerment During Their Journey
Conneran introduced herself as a lung cancer survivor of 7.5 years and gave an overview of her personal cancer journey, which included five separate recurrences. She highlighted that it was only after she experienced two recurrences and received additional opinions that she was diagnosed with a KRAS-mutated tumour. Upon researching KRAS mutations, she recalled feeling scared after finding out that her KRASG12C cancer was described as ‘undruggable’. This motivated her to seek knowledge, and connect with other individuals with KRAS-mutated cancers.
PHARMA PARTNERSHIP
KRAS Kickers
Conneran explained that patients tend to connect to information that is available to them and that without it, they feel very vulnerable, especially in terms of deciding next steps. As a result, Conneran founded KRAS Kickers in January 2020, initially as a Facebook group, to connect with other patients experiencing the same issues worldwide. In particular, she used this opportunity to learn more about KRAS mutations, what they mean for patients, and why patients should care. Patients within the group were seeking similar information regarding their cancer, including Knowledge, Research, Advocacy, and Survivorship (KRAS). In doing so, KRAS Kickers sought to use the term ‘KRAS’ as a call to action for patients and not just a driver of their cancer. They also introduced the term ‘KRAS HOLEs’; gaps in, or barriers to, optimal Healthcare Outcomes Literacy and Equity (HOLE), which includes full biomarker analyses and result sharing with patients.
Patient Empowerment
Patients need to be involved in treatment decisions and feel empowered. Patient empowerment involves sharing new information from a rapidly evolving field with patients directly to help identify what they need to know and enable them to take the next steps. It is crucial for patients to be able to make decisions strategically so that they can make the right decisions for themselves in the future, such as choosing to take part in a clinical trial or taking their own personal next steps. Physicians need to share information with patients, such as what their cancer is and what is driving it, to help eliminate the anxiety that patients feel. Conneran reminded the audience that patients trust physicians with their lives at a time when they are potentially the most scared and vulnerable.5
Conneran concluded the session with a summary of how KRAS Kickers is empowering over 10,000 patients in 117 countries, and highlighted the importance of connecting with patient groups, providing examples of other biomarker-specific patient groups (Figure 1).
Paz-Ares, Hospital Universitario 12 de Octubre, Madrid, Spain, set the scene with an overview of the increasing number of
first-line (1L) treatment options available for patients with mNSCLC and no targetable mutations, including I-O monotherapy and combination therapy with chemotherapy, describing I-O as the cornerstone of treatment for these patients. He highlighted that, while I-O monotherapy is an option for patients with high tumour PD-L1 expression, a combination of I-O therapy
Figure 1. Biomarker patient advocacy groups.
Patient biomarker advocacy groups
and chemotherapy may be the most suitable option for other patients, including those tumors without PD-L1 expression.3,4
Outcomes for Patients with Tumour PD-L1 <1%
There remains an unmet need for treatments with long-lasting efficacy in patients with tumour PD-L1 expression <1%.6 In the KEYNOTE-189 trial, an overall survival (OS) benefit with pembrolizumab + chemotherapy was observed across all levels of tumour PD-L1 expression versus placebo + chemotherapy. At year 5, OS rates were 19% and 10%, for pembrolizumab + chemotherapy, in the ITT population and those with tumour PD-L1 expression <1%, respectively. However, within 3 years, most patients with tumour PD-L1 expression <1% had experienced disease progression, with a median progression-free survival (PFS) of 6.2 months for pembrolizumab + chemotherapy versus 5.1 months for placebo + chemotherapy (hazard ratio [HR]: 0.67; 95% CI: 0.49–0.92).7 This was followed by 5-year data from a pooled analysis of results from KEYNOTE-189 and KEYNOTE-407, in which limited clinical benefit was shown for 1L pembrolizumab + chemotherapy treatment for patients with PD-L1 tumour expression <1% versus placebo + chemotherapy. At year 5, OS rates were 13% and 9%, for pembrolizumab + chemotherapy, and placebo + chemotherapy, respectively. Median OS was 18.3 months for pembrolizumab + chemotherapy, versus 11.4 months for placebo + chemotherapy (HR 0.64; 95% CI: 0.51–0.79).8 Similarly, real-world data demonstrated that patients with tumour PD-L1 expression <1% had poorer longterm outcomes compared with those with tumour PD-L1 expression ≥50% receiving 1L I-O therapy + chemotherapy; at year 4, OS rates were 12% and 23% , respectively, in patients with sqamous mNSCLC, and 15% and 29% in those with non-squamous mNSCLC, respectively6
Dual Immuno-oncology Therapies for Patients with Tumour PD-L1 Expression <1%
A possible treatment option for patients with mNSCLC is dual I-O therapy such as an anti-CTLA-4 combined with anti-PD(L)1 therapy, with or without chemotherapy. CTLA-4 signalling is important in the priming of the immune response and emergence of memory T cells, while inhibiting PD-1 signalling is particularly relevant for restoring the cytotoxic response to tumour cells.9-11 Dual I-O therapy ± chemotherapy has elicited survival benefits in patients with mNSCLC and tumour PD-L1 expression <1% in the CheckMate 9LA, CheckMate 227 Part 1b, and POSEIDON trials.10,12-14
In CheckMate 9LA, the 5-year OS rate was 22% for nivolumab + ipilimumab ± chemotherapy versus 8% for chemotherapy alone.13 Similarly, in CheckMate 227 Part 1b, the 5-year OS rate was 19% for nivolumab + ipilimumab compared with 10% for nivolumab + chemotherapy and 7% for chemotherapy. Furthermore, 6-year OS rates were 16%, 10%, and 5% for nivolumab + ipilimumab, nivolumab + chemotherapy, and chemotherapy, respectively.12
Conversely, in the POSEIDON trial, the 5-year OS rate was 6.1% for durvalumab + tremelimumab + chemotherapy compared with 6.5% for durvalumab + chemotherapy, and 4.0% for chemotherapy.15
In the CheckMate 227 and CheckMate 9LA trials, not only were responses to dual I-O therapy in patients with tumour PD-L1 expression <1% better than those to chemotherapy, but the quality of these responses were also higher, with longer median durations of response (DOR) versus chemotherapy. Paz-Ares highlighted the 5-year DOR rates, which demonstrate the number of patients who still respond to treatment after 5 years and show that they can achieve long-term survival with dual I-O therapy.12-14
In CheckMate 9LA, the 5-year DOR rate was 25% for nivolumab + ipilimumab + chemotherapy compared with 0% for chemotherapy.13
Similarly, in CheckMate 227 Part 1b, the 5-year DOR rate was 25% for nivolumab + ipilimumab versus 3% for chemotherapy.12
Long-term efficacy benefits were also seen in other difficult-to-treat populations treated with dual I-O therapy-based regimens, such as those with squamous histology,13,15,16 baseline brain metastases,15,17 or STK11 mutations.15,17 Paz-Ares ended the session with a reminder that there is an increased risk of treatment-related adverse events (AE) with all I-O therapies, with approximately double the rate of treatment discontinuation with dual I-O combination therapy versus chemotherapy.6,12,15,18 However, he highlighted that dual I-O combination therapies such as nivolumab + ipilimumab ± chemotherapy and durvalumab + tremelimumab + chemotherapy are viable treatment options for those with low or no tumour PD-L1 expression.5
Kicking Off Targeted Medicine for Patients with Metastatic KRASG12C NSCLC
Naidoo built on Conneran’s perspective on KRAS-mutated NSCLC to present complementary scientific data on treatment options for this previously ‘undruggable’ cancer. She introduced KRAS mutations, detailing that approximately 44% of patients with KRAS-mutated NSCLC have KRASG12C mutations,19 and that KRAS proteins have historically been undruggable due to the lack of available binding sites for small molecule inhibitors.20 Discovery of the switch II binding pocket led to the development of KRASG12C-selective inhibitors that trigger tumour cell death.21
KRASG12C Inhibitors
There have now been two Phase III trials in patients with previously treated advanced or metastatic KRASG12C NSCLC that have led to the accelerated or conditional approval of KRASG12C inhibitors adagrasib and sotorasib, in the USA, UK, and EU. Adagrasib was investigated in the randomised KRYSTAL-12 trial, while sotorasib was investigated in the CodeBreaK 200 trial. In both trials, the
comparator arm was docetaxel and the primary endpoint PFS.22,23
In KRYSTAL-12, adagrasib elicited a significant improvement in median PFS: 5.5 months versus 3.8 months for docetaxel (HR: 0.58; 95% CI: 0.45–0.76; p<0.0001) and significant improvements in overall response rate (ORR; a key secondary endpoint). Improvements in median DOR, disease control rate (DCR), and intracranial ORR were also observed with adagrasib compared with docetaxel. In terms of safety, TRAEs occurred in 94% of patients receiving adagrasib compared with 86% of patients receiving docetaxel, with 48% of patients experiencing TRAEs leading to dose reduction and 59% experiencing TRAEs leading to dose interruption in the adagrasib arm. The majority of TRAEs observed with adagrasib were of a low grade in severity, and permanent discontinuation was relatively low at 8% compared with 14% for docetaxel.22
Naidoo commented that there is a learning curve in terms of how KRASG12C inhibitors are administered in clinical practice.5 In CodeBreaK 200, treatment with sotorasib also led to a significant improvement in mPFS: 5.6 months versus 4.5 months for docetaxel (HR: 0.66; 95% CI: 0.51–0.86; p=0.002), as well as numerical improvements in ORR, median DOR, DCR, and intracranial ORR. TRAEs occurred in 70% of patients receiving sotorasib compared with 86% in those receiving docetaxel,23,24 and while there have been some regulatory concerns from the US Food and Drug Administration (FDA) regarding sotorasib, both adagrasib and sotorasib are broadly available in the USA.25
There has been an increase in the number of KRASG12C inhibitors being investigated in Phase I and II clinical trials as monotherapy in the second-line setting, including olomorasib, gasorasib, glecirasib, fulzerasib, and divarasib.26
KRASG12C Inhibitors in Combination with Immuno-oncology Therapy
In the 1L setting, early-phase data on KRASG12C inhibitor + I-O therapy are
available. In the Phase II part of the KRYSTAL-7 trial, adagrasib + pembrolizumab treatment was associated with a 63% ORR and a DCR of 84% in patients with tumour PD-L1 expression ≥50%. Based on these data, the Phase III part of this trial was initiated.27,28 In the Phase I CodeBreaK 101 trial, concurrent or lead-in treatment with sotorasib + atezolizumab or pembrolizumab was associated with an ORR of 29% and an mOS of 15.7 months, which, alongside other data concerning hepatotoxicity, led to the conclusion that sotorasib + I-O therapy was not a viable combination.29,30 While olomorasib + pembrolizumab is now under investigation in the Phase III SUNRAY-01 trial, in the initial Phase I LOXO-RAS-20001 trial, treatment with the combination therapy was associated with an unconfirmed ORR of 77% and DCR of 88%.31 In the Phase I MK1084-001 trial, treatment with MK-1084 + pembrolizumab was associated with an ORR of 71% and DCR of 86% in patients with tumour PD-L1 expression ≥1%.32 It is now under investigation in a Phase III study for patients with tumour PD-L1 expression ≥50%.
In the Phase II KRYSTAL-17 trial, which is currently ongoing, adagrasib + pembrolizumab + platinum-doublet chemotherapy and maintenance adagrasib + pembrolizumab in patients who have previously received four cycles of pembrolizumab + platinum-doublet chemotherapy are being investigated.33 Other treatment combinations have also been investigated: sotorasib + chemotherapy in the Phase I CodeBreaK 101 and Phase II SCARLET trials,34,35 and fulzerasib + cetuximab in the Phase II KROCUS trial.36 Selected ongoing Phase III trials of KRASG12C inhibitors in NSCLC and the potential future directions of KRAS-directed therapy are summarised in Figure 2
Next-Generation
Tyrosine Kinase Inhibitors: The Evolving Treatment Landscape for Cancers with ROS1 Or NTRK Fusions
Drilon introduced ROS1 fusions, which are found in up to 2% of NSCLC cases in
the form of numerous fusion partners. Drilon emphasised that it is important to keep in mind that ROS1 and TRK TKIs are generational.5
ROS1 Tyrosine Kinase Inhibitors
As early-generation ROS1 TKIs, crizotinib treatment was associated with a median PFS of 19.3 months and an ORR of 72% in the PROFILE 1001 trial, while entrectinib treatment was associated with a median PFS of 15.7 months and ORR of 67.4% in the pooled ALKA-372-001, STARTRK-1, and STARTRK-2 trials.41-44 Drilon proceeded to talk about next-generation TKIs, which he defined as treatments designed for use after the failure of early-generation drugs.5
Next-generation TKIs include repotrectinib, which was associated with a median PFS of 35.7 months in the TRIDENT-1 trial (TKI-naïve patients).45 In the TRUST-I and TRUST-II trials, median PFS was not reached for taletrectinib. Drilon also highlighted the notable intracranial activity of next-generation ROS1 TKIs.46-48
Resistance to ROS1 Tyrosine Kinase Inhibitors
The ROS1 G2032R mutation was highlighted as the most relevant and commonly occurring mechanism of resistance to early generation ROS1 TKIs.49 As such, next-generation ROS1 TKIs, such as repotrectinib, taletrectinib, and zidesamtinib, have been developed to overcome resistance. In the TRIDENT-1 trial cohort consisting of patients with NSCLC who previously received a ROS1 TKI, 59% of patients with ROS1 G2032R mutations responded to treatment with repotrectinib.50 Similarly, in patients with NSCLC who previously received crizotinib in TRUST-I and TRUST-II, 67% of patients with ROS1 G2032R mutations responded to treatment with taletrectinib.46 With zidesamtinib, responses were reported in 78% of patients with ROS1 G2032R mutations who were previously treated with TKIs for NSCLC.51 Responses were also described in patients without ROS1 G2032R mutations for all three agents.46,50,51
Figure 2. Summary of ongoing Phase III trials of KRASG12C inhibitors in KRASG12C NSCLC and future directions for KRAS-directed therapy.
Drilon then presented on TRK TKIs, which are tumour agnostic (Figure 3).
The first TRK TKI to receive approval was larotrectinib, which elicited a median DOR of 43.3 months in patients with NTRK fusion-positive solid tumours in the pooled NAVIGATE, LOVO-TKR-14001, and SCOUT trials.52 Entrectinib was also discussed as a TRK TKI for NTRK fusion-positive solid
tumours, providing a median DOR of 20.0 months.53 Next-generation TRK TKIs, such as repotrectinib, were designed to overcome mechanisms of resistance to early-generation TRK TKIs, such as G595R mutations.50 Data of repotrectinib in TKI-naive and TKI-pretreated tumours were presented, showing a 12-month DOR of 86% and 39%, respectively.54 Drilon closed this session by providing an overview of the safety
profiles of ROS1 and TRK TKIs. Treatment with entrectinib and next-generation TKIs such as repotrectinib and taletrectinib may be associated with a risk of experiencing neurological adverse events.42,46,48,55-59 Drilon emphasised the importance of discussing the possible side effects of these treatments with patients in clinical practice.
Panel Discussion: What Would Your Treatment Decision Be?
In an interactive session, the speakers came together to discuss three patient cases, and asked the audience how they would approach treatment in a range of different scenarios. The first case, presented by Paz-Ares, focused on I-O treatment options, and the audience was asked to select which I-O therapy they would choose for various scenarios, such as a patient with Stage IV non-squamous NSCLC with tumour PD-L1 <1% and with or without brain metastases.
The second case, presented by Naidoo, focused on treatment options for patients with KRASG12C mutations and tumour PD-L1 expression <1%. Questions for the audience included whether they would choose standard-of-care I-O + chemotherapy or enrolment in a clinical trial of the treatment options discussed in Naidoo’s presentation earlier for a patient that was treatmentnaïve. The final patient case was presented by Drilon and the audience was asked to select treatment options for patients with tumour PD-L1 expression >50% and genomic alterations such as ROS1, as discussed in his presentation.
Before the end of the symposium, the panel answered questions submitted by the audience, across a range of topics including the importance of networking to learn about rare gene-related cancers, treatment options for patients with tumour PD-L1 expression <1%, and how patient support can vary between countries, with a particular focus on supporting those living in areas with low literacy levels.
Figure 3: Overview of ROS1 and NTRK TKIs that are approved and recommended or in development.
References
1. National Cancer Institute (NIH). Drugs Approved for Lung Cancer. Available at: https://www.cancer.gov/about-cancer/treatment/drugs/lung. Last accessed: October 2024.
2. European Medicines Agency. Medicines authorised for non-small-cell lung cancer. Available at: https://www. ema.europa.eu/en/medicines/download-medicine-data. Last accessed: October 2024.
4. Hendriks LE et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(4): 358-76.
5. Hardavella G et al. Multidisciplinary care models for patients with lung cancer. Breathe (Sheff). 2020;16(4):200076.
6. Waterhouse D et al. Real-world longterm survival outcomes of first-line immunotherapy-based regimens in advanced non-small cell lung cancer. Abstract 3819. AACR 2024, 5-10 April, 2024.
7. Garassino MC et al. Pembrolizumab plus pemetrexed and platinum in nonsquamous non-small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 study. J Clin Oncol. 2023;41(11):1992-8.
8. Gadgeel SM et al. Pembrolizumab plus chemotherapy for metastatic NSCLC with programmed cell death ligand 1 tumor proportion score less than 1%: pooled analysis of outcomes after five years of follow-up. J Thorac Oncol. 2024;19(8):1228-41.
9. Das R et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194(3):950-9.
10. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-64.
11. Wang C et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846-56.
12. Ramalingam SS et al. Six-year survival and HRQoL outcomes with 1L nivolumab + ipilimumab in patients with metastatic NSCLC from CheckMate227. Abstract OA14.03. IASLC WCLC, 9-12 September, 2023.
13. Reck M et al. Five-year outcomes with first-line nivolumab plus ipilimumab with 2 cycles of chemotherapy versus 4 cycles of chemotherapy alone in patients with metastatic non-small cell lung cancer in the randomized CheckMate 9LA trial. Eur J Cancer. 2024;211:114296.
14. Garon EB et al. A brief report of durvalumab with or without tremelimumab in combination with chemotherapy as first-line therapy for metastatic nonsmall-cell lung cancer: outcomes by tumor PD-L1 expression in the phase 3 POSEIDON study. Clin Lung Cancer. 2024;25(3):266-73.e5;
15. Peters S et al. Durvalumab (D) ± tremelimumab (T) + chemotherapy (CT) in first-line metastatic (m) NSCLC: 5-year overall survival (OS) update from the POSEIDON study. Abstract LBA3. ESMO Immuno-Oncology Congress, 6-8 December, 2023.
16. Brahmer JR et al. Five-year survival outcomes with nivolumab plus ipilimumab versus chemotherapy as first-line treatment for metastatic nonsmall-cell lung cancer in CheckMate 227. J Clin Oncol. 2023;41(6):1200-12.
17. Reck M et al. Systemic and intracranial outcomes with first-line nivolumab plus ipilimumab in patients with metastatic NSCLC and baseline brain metastases from CheckMate 227 Part 1. J Thorac Oncol. 2023;18(8):1055-69.
18. Novello S et al. Pembrolizumab plus chemotherapy in squamous nonsmall-cell lung cancer: 5-year update of the Phase III KEYNOTE-407 study. J Clin Oncol. 2023;41(11):1999-2006.
19. Veluswamy R et al. KRAS G12C-mutant non-small cell lung cancer: biology, developmental therapeutics, and molecular testing. J Mol Diagn. 2021;23(5):507-20.
20. Ghimessy A et al. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020;39(4):1159-77.
21. Ostrem JM et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548-51.
22. Mok TSK et al. KRYSTAL-12: Phase 3 study of adagrasib versus docetaxel in patients with previously treat-
ed advanced/metastatic non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Abstract LBA8509. ASCO, 31 May-4 June, 2024.
23. de Langen AJ et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRASG12C mutation: a randomised, open-label, phase 3 trial. Lancet. 2023;401(10378):733-46.
24. Dingemans AC et al. Intracranial efficacy of sotorasib versus docetaxel in pretreated KRAS G12C-mutated advanced non-small cell lung cancer (NSCLC): practice-informing data from a global, phase 3, randomized, controlled trial (RCT). Abstract LBA9016. ASCO, 2-6 June, 2023.
25. Food and Drug Administration (FDA) Center for Drug Evaluation and Research. Final summary minutes of the Oncologic Drugs Advisory Committee meeting October 5, 2023. Available at: https://www.fda.gov/media/174559/ download. Last accessed: 27 September 2024.
26. O’Sullivan É, et al. Treatment strategies for KRAS-mutated non-smallcell lung cancer. Cancers (Basel). 2023;15(6):1635.
27. Garrassino MC et al. Durvalumab after sequential chemoradiotherapy in patients with unresectable stage III NSCLC: final analysis from PACIFIC-6. Abstract LBA65. ESMO, 20-24 October, 2023.
28. Garrassino MC et al. KRYSTAL-7: A phase III study of first-line adagrasib plus pembrolizumab versus pembrolizumab alone in patients with advanced NSCLC with KRASG12C mutation. Abstract 1394TiP. ESMO, 13-17 September, 2024.
29. Li BT et al. CodeBreaK 100/101: First report of safety/efficacy of sotorasib in combination with pembrolizumab or atezolizumab in advanced KRAS p.G12C NSCLC. Abstract OA03.06. IASLC WCLC, 6-9 August, 2022.
30. Desai A, Dimou A. Toxicity from sotorasib after immune checkpoint inhibitors: A note of caution and reflections of future advancements in the field. J Thorac Oncol. 2023;18(10):1265-67.
31. Burns TF et al. Efficacy and safety of olomorasib (LY3537982), a second-generation KRAS G12C inhibitor (G12Ci), in combination with pembrolizumab in patients with KRAS G12C-mutant advanced NSCLC. Abstract 8510. ASCO, 31 May-4 June, 2024.
32. Rojas C et al. Safety and preliminary efficacy of the KRAS G12C inhibitor MK-1084 in solid tumors and in combination with pembrolizumab in NSCLC. Abstract 663P. ESMO 20–24 October, 2023
33. Mirati Therapeutics Inc. Combination therapies with adagrasib in patients with advanced NSCLC with KRAS G12C mutation. NCT05609578. https://clinicaltrials.gov/study/NCT05609578.
34. Li BT et al. Sotorasib plus carboplatin and pemetrexed in KRAS G12C advanced NSCLC: updated analysis from the international CodeBreaK 101 trial. Abstract 8512. ASCO, 31 May-4 June, 2024.
35. Yoshioka H et al. Final analysis of SCARLET study: a single-arm, phase II study of sotorasib plus carboplatin–pemetrexed in patients with advanced non-squamous, non-small cell lung cancer KRAS G12C mutation. Abstract 8616. ASCO, 31 May-4 June, 2024.
36. Gregorc V et al. KROCUS: A phase II study investigating the efficacy and safety of fulzerasib (GFH925) in combination with cetuximab in patients with previously untreated advanced KRAS G12C-mutated NSCLC. Abstract LBA8511. ASCO, 31 May-4 June, 2024.
37. Hoffmann-La Roche. A study evaluating the efficacy and safety of divarasib versus sotorasib or adagrasib in participants with previously treated KRAS G12C-positive advanced or metastatic non-small cell lung cancer (Krascendo 1). NCT06497556. https://www.clinicaltrials.gov/study/NCT06497556?term=NCT06497556&rank=1.
38. Miyashita H et al. KRAS G12C inhibitor combination therapies: current evidence and challenge. Front Oncol. 2024;14:1380584.
39. Verastem, Inc. Phase 1/2 study of avutometinib (VS-6766) + sotorasib with or without defactinib in KRAS G12C NSCLC patients (RAMP203). NCT05074810. https://clinicaltrials.gov/ study/NCT05074810.
40. Merck Sharp & Dohme LLC. A study of MK-1084 plus pembrolizumab (MK3475) in participants with KRAS G12C mutant, metastatic non-small cell lung cancer (NSCLC) with programmed cell death ligand 1 (PD-L1) tumor proportion score (TPS) ≥50% (MK-1084-004). NCT06345729. https://clinicaltrials.gov/ study/NCT06345729.
41. Negrao MV et al. SUNRAY-01, a pivotal, global study of olomorasib (LY3537982) in combination with pembrolizumab with or without chemotherapy for 1L treatment in KRAS G12C-mutant advanced NSCLC. Abstract TPS8649. ASCO, 31 May-4 June, 2024.
42. Shaw AT et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol. 2019;30(7):1121-26.
43. Shaw AT et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann Oncol.2019;30(7):1121–26. (supplementary appendix)
44. Drilon A et al. Long-term efficacy and safety of entrectinib in ROS1 fusion-positive NSCLC. JTO Clin Res Rep. 2022;3(6):100332.
45. Drilon A et al. Repotrectinib in tyrosine kinase inhibitor (TKI)-naïve patients (pts) with advanced ROS1 fusion-positive (ROS1+) NSCLC in the phase 1/2 TRIDENT-1 trial: Clinical update, treatment beyond progression and subsequent therapies. Abstract 386. ASCO, 31 May-4 June, 2024.
46. Li W et al. Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non–small cell lung cancer: the phase II TRUST-I study. J Clin Oncol. 2024;42(22):2660-70.
47. Pérol M et al. Efficacy and safety of taletrectinib in patients with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of global TRUST-II study. Abstract 1373P. ESMO, 20-24 October, 2023.
48. Liu G et al. Efficacy and safety of taletrectinib in patients with ROS1+ non-small cell lung cancer: the global TRUST-II study. Abstract MA06.03. IASLC WCLC, 7-10 September, 2024.
49. Tangpeerachaikul A et al. Mutagenesis screens support potential best-in-class profile for selective, brain-penetrant, and TRK-sparing ROS1 inhibitor zidesamtinib (NVL-520). Abstract LB182. AACR, 5-10 April, 2024.
50. Drilon A et al. Repotrectinib in ROS1 fusion-positive non-small-cell lung cancer. N Engl J Med. 2024;390(2):118-31.
51. Drilon A et al. Safety and preliminary clinical activity of NVL-520, a highly
selective ROS1 inhibitor, in patients with advanced ROS1 fusion-positive solid tumours. Abstract 8. EORTC–NCI–AACR, 26-28 October, 2022.
52. Drilon A et al. Efficacy and safety of larotrectinib in a pooled analysis of patients with tropomyosin receptor kinase fusion cancer. Abstract 668P. ESMO, 20-24 October, 2023.
53. Demetri GD et al. Updated integrated analysis of the efficacy and safety of entrectinib in patients with NTRK fusion-positive solid tumors. Clin Cancer Res. 2022;28(7):1302-12.
54. Solomon B et al. Repotrectinib in patients with NTRK fusion-positive advanced solid tumors, including NSCLC: update from the phase I/II TRIDENT-1 trial. Abstract 1372P. ESMO, 20-24 October, 2023.
55. Pfizer. XALKORI (crizotinib) prescribing information. Available at: https://www. accessdata.fda.gov/drugsatfda_docs/ label/2017/202570s021lbl.pdf. Last accessed: 14 September 2024
56. Genentech (Roche). ROZLYTREK (entrectinib) prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/212725s011lbl.pdf. Last accessed: 14 September 2024
57. Pfizer. LORBRENA (lorlatinib) prescribing information. Available at: https:// www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf. Last accessed: 14 September 2024
58. Bristol-Myers Squibb Company. AUGTYRO prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/218213s001lbl.pdf. Last accessed: 14 September 2024
59. Bayer HealthCare Pharmaceuticals Inc. VITRAKVI (Larotrectinib) prescribing information. Available at: https://www. accessdata.fda.gov/drugsatfda_docs/ label/2022/210861s008lbl.pdf. Last accessed: 14 September 2024
60. Li W et al. Updated efficacy and safety of taletrectinib in patients with ROS1+ non-small cell lung cancer. Abstract 14MO. ELCC, 29 March-1 April, 2023.
Maximising the Synergy of Tumour Tissue and Liquid Biopsy Testing in Oncology Clinical Practice
This industry symposium took place during the European Society for Medical Oncology (ESMO) Congress held in Barcelona, Spain, 13th–17th September 2024.
Chairperson: Christian Rolfo1
Speakers: Guilhem Roubaud2, Federico Cappuzzo3, Bence Sipos4, Sara Pilotto5
1. Ohio State University, Columbus, Ohio, USA
2. Institut Bergonié, Bordeaux, France
3. Istituto Nazionale Tumori IRCCS Regina Elena, Rome, Italy
4. BAG for Pathology and Molecular Pathology, Stuttgart, Germany
5. University Hospital, Verona, Italy
Disclosure:
Acknowledgements:
Keywords:
Rolfo has received advisory board fees from AstraZeneca, Daiichi Sankyo, Regeneron, Novocure, Bristol-Myers Squibb, Novartis, Invitae, Guardant Health, COR2ED, Bayer, Boehringer Ingelheim, Abbvie, Invitae, Janssen, and EMD Serono; and research grant from AstraZeneca, Thermo Fisher, Oncohost, Lung Cancer Research Foundation, National Foundation for Cancer Research, and U54 (National Institute of Health); and research collaboration with Guardant Health, Foundation Medicine, Roche Diagnostics, and EMD Serono; and is on the scientific advisory board of Imagene; and has leadership roles in the International Society of Liquid Biopsy, The European School of Oncology, the International Association for Study of Lung Cancer, and the Oncology Latin American Association; and has an editorial roles as editor in chief of CROH, honorary editor at Journal of Liquid Biopsy Elsevier, Editorial board lung cancer, and ILCN. Roubaud has received honoraria/grants/advisory fees from AAA, Astellas, AstraZeneca, Bayer, Janssen / Johnson & Johnson, Ipsen, Novartis, Pfizer, and Orion. Cappuzzo has received fees for membership of an advisory board or lectures from F. Hoffmann-La Roche Ltd, AstraZeneca, Bristol-Myers Squibb Company, Pfizer Ltd, Takeda Pharmaceutical Company Ltd, Eli Lilly and Company, Bayer Plc, Amgen Inc, Sanofi, PharmaMar, Novocure GmbH, Mirati Therapeutics Inc, Galecto Biotech, OSE Immunotherapeutics SA, Illumina Inc, BeiGene Ltd, and Merck Sharp & Dohme Ltd. Sipos has received advisory board fees and speakers honoraria Novartis, Ipsen, AstraZeneca, Daiichi Sankyo, Roche Diagnostics, Bristol-Myers Squibb, and Thermo Fisher; and is the practice owner/ managing director of Molecular Pathology Baden-Wurttembert, Practice of Pathology, Stuttgart. Pilotto has received research funding from AstraZeneca, Roche; speakers’ bureau from AstraZeneca, MSD, BMS, Roche, Amgen, Novartis, Eli-Lilly, Sanofi, Takeda, Regeneron, advisory board fees from AstraZeneca, Amgen, Novartis, Roche, MSD, Sanofi, Takeda, Daiichi Sankyo, Pfizer, and consultant from AstraZeneca, MSD, BMS, Roche, Amgen, Novartis, Eli-Lilly, Sanofi, and Takeda.
Writing support for this article was provided by the medical and scientific affairs team of Thermo Fisher Scientific and the medical affairs team of Johnson & Johnson. EM-167676 October 2024.
This industry symposium was funded and organised by Johnson & Johnson in collaboration with Thermo Fisher Scientific in EMEA and is intended for healthcare professionals only. PHARMA
PARTNERSHIP
Meeting Summary
The symposium took place during the 2024 European Society for Medical Oncology (ESMO) Congress in Barcelona, Spain, with the goal of highlighting the synergy between tissue and liquid biopsy testing in the diagnosis and treatment of solid tumours.
Christian Rolfo, Director of the Division of Medical Oncology at The Ohio State University Comprehensive Caner Centre, Columbus, USA, set the stage with a discussion on the state-of-the-art liquid biopsy for solid tumour testing. He was followed by Guilhem Roubaud from the Bergonié Institute in Bordeaux, France, and Federico Cappuzzo from Regina Elena Institute, Rome, Italy, who delved into molecular testing challenges and applications in genitourinary and non-small cell lung cancer (NSCLC), respectively. Practical insights on next-generation sequencing (NGS) implementation was provided by Bence Sipos, TBAG of Molecular Pathology Baden-Württemberg, Germany, with real-world case studies presented by Sara Pilotto, University Hospital, Verona, Italy, and Roubaud.
The faculty emphasised the importance of integrating liquid and tissue biopsies to perform tumour molecular profiling, which is crucial for accurate diagnosis and personalised treatment strategies. They also highlighted the critical role of rapid NGS in detecting a wide range of genetic alterations to enhance the precision of diagnosis and access to precision therapies.
Introduction
While tissue biopsy remains the gold standard method for obtaining pathological information, it has limitations such as difficulty in acquiring adequate and sufficient tissue and capturing tumour heterogeneity.1 Liquid biopsy, particularly circulating tumour DNA (ctDNA), provides advantages such as the ability to sample over time, high concordance with tissue analysis, and insights into tumour evolution.1
As precision medicine continues to evolve, the integration of cutting-edge techniques such as liquid biopsy and NGS is revolutionising the way cancer is being diagnosed and treated.1 This symposium provided a platform for renowned oncologists, pathologists, and researchers to share their insights and discuss the applications of these technologies in clinical practice.
Layering Diagnostic Steps May be Able to Shorten the Diagnostic Odyssey
Christian Rolfo
Multiple organisations, including the International Association for the Study of Lung Cancer (IASLC) and ESMO, have recommended the use of tumour NGS for the molecular analyses of patients with advanced cancers, including NSCLC, in routine practice.2 Rolfo outlined three strategies for the use of liquid biopsy in treatment-naïve advanced NSCLC setting.1
1. Sequential approach: utilising liquid biopsy when tissue samples are inadequate or unavailable.
2. Complementary approach: employing both tissue and liquid biopsy simultaneously.
3. Plasma-first approach: prioritising liquid biopsy, progressing to tissue if negative.
Rolfo emphasised that the complementary approach is often the most beneficial, providing a holistic view of the patient’s molecular profile from the outset.
Additionally, incorporating liquid biopsy early in the diagnostic process can shorten the time required to achieve a definitive diagnosis and therefore to initiate appropriate therapy.3 Rolfo presented data (Figure 1) showing that liquid biopsy, when used at baseline or during clinical suspicion, can provide quick identification of actionable
A) Of 56 patients with liquid biopsy testing ordered before confirmed diagnosis, 24 patients (43%) harboured oncogene drivers. Among driver-negative patients, an elevated cfDNA tumour fraction ≥1% was observed in 18 patients (true-negative cases), while the others (n=14) had no elevated ctDNA tumour fraction (<1%) and were potentially false-negative cases. B) Among the 56 cases, 42 had documented the line of therapy received, and 10 driver-positive patients received matched therapy with real-world response in 71% of the cases.
Figure 1: Genomic results and treatment outcomes in pre-diagnosed liquid biopsy cohort.
genetic alterations, thereby allowing for faster initiation of targeted therapies, ultimately reducing the diagnostic odyssey often faced by patients with cancer.3
Earlier Testing is Better, Particularly for BRCA-Altered Patients
Guihem Roubaud
Roubaud provided an in-depth analysis of molecular testing in genitourinary oncology. Molecular testing in prostate cancer and bladder cancer is crucial for identifying genetic alterations that inform prognosis, guide treatment decisions, and provide opportunities for personalised therapy.4,5 The timing and method of testing are essential to maximise the benefits of these advanced diagnostic tools.4
Prostate Cancer
In advanced prostate cancer, Roubaud highlighted the critical role of identifying homologous recombination repair (HRR) gene alterations, such as BRCA1/2 mutations.4 These genetic alterations are associated with poor prognosis and can predict responsiveness to poly ADP ribose polymerase (PARP) inhibitors, which have been shown to improve radiographic progression-free survival and overall survival in patients with BRCA1/2 alterations.6-12 However, important factors must be considered.13
1. Recent biopsies preferred: recent tissue biopsies are preferable to older samples for molecular testing as these are more likely to provide accurate and relevant genetic information.13
2. Extra-bone versus bone biopsies: biopsies from extra-bone localisations are generally more effective than bone biopsies. This is significant because a substantial number of patients may present with bone-only metastases, thus, special care must be taken during the decalcification process of bone biopsies to avoid DNA denaturation.13
3. Germline versus somatic mutations: if a somatic mutation is not detected in the tissue biopsy, germline testing can be performed. However, Roubaud highlighted that relying solely on germline testing may miss somaticonly mutations, which can be crucial for treatment decisions.13
4. Liquid biopsy testing: cell free deoxyribonucleic acid (ctDNA) analysis has shown good concordance with tissue and should be performed in the case of disease progression. Liquid biopsy testing should be performed before the initiation of androgen deprivation therapy, as ctDNA concentration rapidly decreases during effective treatment.13
Bladder Cancer
In bladder cancer, the precise molecular characterisation through tissue biopsies is becoming increasingly important to optimise therapy selection. This is essential for identifying genetic alterations such as FGFR3 mutations and fusions, which can guide the use of targeted therapies like erdafitinib (an FGFR tyrosine kinase inhibitor for treatment of unresectable or metastatic urothelial cancer in those with an FGFR3 alteration who have worsened on prior immunotherapy)14 in the metastatic urothelial carcinoma (mUC) setting.15
There are various techniques for molecular testing of FGFR3 alterations, including polymerase chain reaction (PCR) and NGS. However, obtaining high-quality tissue samples, particularly in the advanced stages of the disease remains a challenge.16
While emphasising the importance of tissue biopsies, Roubaud also highlighted the role of liquid biopsy as a complementary tool. Liquid biopsy can be particularly useful when tissue samples are not available or are insufficient for molecular analysis. As Roubaud summarised, liquid biopsy can help monitor tumour dynamics and detect resistance mechanisms, although it may present challenges for the accurate detection of fusions and should ideally be confirmed with tissue biopsy results when possible.
The Importance of a Complete Molecular Profile for All Patients with Non-Small Cell Lung Cancer, Regardless of the Stage
Federico Cappuzzo
Cappuzzo provided a thorough examination of the evolving landscape of NSCLC treatment, emphasising the critical role of molecular testing and the integration of liquid biopsy in clinical practice.
Neoadjuvant Chemoimmunotherapy
The transition from surgery alone to neoadjuvant chemoimmunotherapy for resectable NSCLC shows promise as a treatment strategy; however, the benefit of this treatment approach is not universal, particularly for patients with oncogeneaddicted tumours such as EGFR mutations, who do not benefit from the addition of immunotherapy.17,18
Adjuvant Immunotherapy
For patients who have already undergone surgery, Cappuzzo highlighted the role of adjuvant immunotherapy. He stressed the importance of identifying patients who are most likely to benefit from adjuvant immunotherapy, particularly those with high levels of PD-L1 expression. Conversely, oncogene-addicted patients, such as those with EGFR mutations, benefit more from targeted therapies in the adjuvant setting.19,20
Unresectable Non-Small Cell Lung Cancer
For unresectable NSCLC, the current standard of care is concomitant chemoradiation followed by durvalumab, based on the PACIFIC trial results.21 Cappuzzo noted that the benefit of adding durvalumab is not seen in patients with oncogene-addicted tumours, highlighting the need for molecular profiling to guide treatment decisions.
Figure 2: Applications of circulating tumour DNA analysis.
The Role of Liquid Biopsy
Liquid biopsies present several applications such as screening or identifying patients who have achieved a complete pathological response and may not require further treatment, thereby reducing the risk of overtreatment and associated toxicities. Additionally, there is potential in liquid biopsy to identify minimal residual disease and monitor treatment response, given that patients with detectable ctDNA after surgery are at a higher risk of relapse and may benefit from intensified adjuvant therapy (Figure 2).22
Liquid biopsy, when used alongside tissue biopsy, can provide a more comprehensive molecular profile of the tumour. Cappuzzo’s case studies illustrated the successful application of molecular testing and liquid biopsy for timely and accurate molecular diagnostics in personalising treatment and achieving better patient outcomes.
A Quiet Revolution has Been Unfolding in the Field of NextGeneration Sequencing
Bence Sipos
Sipos provided a practical overview of the implementation of NGS in clinical practice. From a pathologist’s perspective, the key clinical needs include:23
Molecular Characterisation
Pathologists are tasked with providing molecular profiling of malignant tumours, which is essential for identifying actionable genetic alterations. However, factors to consider include the molecular biomarkers specific to each tumour type and the reimbursement for testing in specific countries or regions.
Timely and Accurate Diagnosis
Rapid turnaround time (TAT) is crucial for timely clinical decision-making. Studies have demonstrated that upfront
characterisation of lung cancer is associated with survival benefits.24,25 Pathologists must ensure that NGS results are delivered within a few days to meet the needs of multidisciplinary tumour boards (MTB) and enable prompt treatment initiation.
High Reliability and Low Failure Rates
Sipos emphasised that ensuring the reliability of NGS results is paramount. He noted that pathologists strive to achieve low failure rates in molecular testing to provide clinicians with accurate and actionable data. In his presentation, Sipos highlighted that his laboratory achieved a failure rate of less than 1%, demonstrating the importance of robust and reliable testing protocols.
Pathologists also must efficiently utilise available tissue samples, especially when dealing with small biopsies or suboptimally processed samples.
Navigating Molecular Diagnostics
Sipos highlighted the diverse choices available to pathologists in molecular diagnostics.23 PCR is suitable for detecting specific, predefined genetic alterations and is quick and cost-effective; however, it is limited to a small number of targets, so it may be more suited for follow-up testing of known variants.
Within NGS, the target enrichment method has implications for clinical testing.26 Hybrid-capture NGS is highly flexible and capable of detecting an unlimited number of targets; however, it requires a relatively high amount of input DNA of 40–80 ng and involves a longer TAT due to a more complex workflow.
Amplicon-based NGS, which requires less input DNA, is particularly valuable for routine clinical diagnostics. While there is limited flexibility in target selection, less input DNA (as low as 5–20 ng) makes it suitable for small biopsies and degraded samples, and faster TAT is possible, with some methods delivering results in as little as 2 days.
Arrival of sample: DNA/RNA extraction
27 December @ 8:41
Library preparation, sequencing, and analysis
Ultra-deep NGS targeting 46 genes
Sequencing depth: 53,750x
Limit of detection: <0.3%
Turnaround time: 54 hours
Evaluation, reporting, and transmission of report
29 December @ 14:41
The process includes the arrival of the sample, DNA/RNA extraction, ultra-deep NGS targeting 46 genes with a sequencing depth of 53,750x and an LOD of <0.3%, followed by data evaluation, report generation, and transmission of the final report in 54 hours.
LOD: limit of detection; NGS: next generation sequencing.
Adapted from Sipos’ presentation at the symposium.
By selecting the appropriate methods, optimising sample handling, ensuring rapid and reliable results, and collaborating effectively with clinical teams, pathologists can significantly contribute to the success of personalised cancer treatment. Using an amplicon-based NGS panel, Sipos stated that with his team they can analyse both tissue and liquid biopsy samples, achieving a TAT of up to 3 days in 82% of cases and a failure rate of <1% (Figure 3). In his lab, the reliability, failure rates, and TATs of amplicon-based NGS are equivalent to routine methods in pathology, such as immunohistochemistry, resulting in optimal decision-making and patient care.
Case Studies Illustrate the Importance of Adequate Molecular Profiling
Three case studies were presented by Roubaud and Pilotto. These highlighted the
importance of comprehensive molecular profiling using both liquid and tissue biopsies at diagnosis to avoid delays in identifying actionable mutations and making appropriate treatment decisions in prostate, bladder, and lung cancer.
Conclusion
The symposium concluded with a panel discussion featuring all the presenters, where Pilotto emphasised the complementarity of liquid and tissue biopsy and their high concordance and ability to reduce time to treatment in her clinical practice. The symposium concluded with closing remarks from Rolfo, who emphasised the importance of continued collaboration and innovation in the field to improve patient outcomes.
Figure 3: Timeline of liquid biopsy testing from Sipos’ lab.
References
1. Rolfo C et al. Liquid biopsy for advanced NSCLC: a consensus statement from the international association for the study of lung cancer. J Thorac Oncol. 2021;16(10):1647-62.
2. Mosele MF et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced cancer in 2024: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2024;37(7):588606.
3. Russo A et al. Liquid biopsy of lung cancer before pathological diagnosis is associated with shorter time to treatment. JCO Precis Oncol. 2024:e2300535.
4. Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-28.
5. Powles T et al. Bladder cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(3):244-58.
6. Hussain M et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383(24):2345-57.
7. Fizazi K et al. Rucaparib or physician’s choice in metastatic prostate cancer. N Engl J Med. 2023;388(8):719-32.
8. Saad F et al. Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(10):1094-108.
9. Agarwal N et al. Talazoparib plus enzalutamide in men with first-line metastatic castration-resistant prostate cancer (TALAPRO-2): a randomised, placebo-
10. Fizazi K et al. First-line talazoparib with enzalutamide in HRR-deficient metastatic castration-resistant prostate cancer: the phase 3 TALAPRO-2 trial. Nat Med. 2024;30(1):257-64.
11. Chi KN et al. Niraparib plus abiraterone acetate with prednisone in patients with metastatic castration-resistant prostate cancer and homologous recombination repair gene alterations: second interim analysis of the randomized phase III MAGNITUDE trial. Ann Oncol. 2023;34(9):772-82.
12. Chi KN et al. Niraparib and abiraterone acetate for metastatic castrationresistant prostate cancer. J Clin Oncol. 2023;41(18):3339-51.
13. Teyssonneau D et al. PARP inhibitors as monotherapy in daily practice for advanced prostate cancers. J Clin Med. 2022;11(6):1734.
14. Krook M et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer. 2021;24(5):880-92.
15. European Medicines Agency. Balversa, Erdafitinib. Available at: https://www. ema.europa.eu/en/medicines/human/ EPAR/balversa. Last accessed: 14 October 2024.
16. De Luca A et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21(18):6856.
17. He J et al. OA12. 06 Neoadjuvant Durvalumab+ Chemotherapy Followed by Adjuvant Durvalumab in Resectable EGFR-mutated NSCLC (AEGEAN). J Thorac Oncol. 2023;18(11):S72-3.
18. Heymach JV et al. Perioperative durvalumab for resectable non–small-cell lung cancer. N Engl J Med.
2023;389(18):1672-84.
19. Felip E et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-smallcell lung cancer (IMpower010): a randomised, multicentre, openlabel, phase 3 trial. The Lancet. 2021;398(10308):1344-57.
20. Wu YL et al. Osimertinib in resected EGFR-mutated non–smallcell lung cancer. N Engl J Med. 2020:383(18):1711-23.
21. Naidoo J et al. Brief report: durvalumab after chemoradiotherapy in unresectable stage III EGFRmutant NSCLC: a post hoc subgroup analysis from PACIFIC. J Thorac Oncol. 2023;18(5):657-63.
22. Wan J et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223-38.
23. Sarhadi et al. Molecular biomarkers in cancer. Biomolecules. 2022;12(8):1021.
24. Scott JA et al. Compromised outcomes in stage IV non–small-cell lung cancer with actionable mutations initially treated without tyrosine kinase inhibitors: a retrospective analysis of real-world data. JCO Oncol Pract. 2024;20(1):145-53.
25. Aggarwal C et al. Association between availability of molecular genotyping results and overall survival in patients with advanced nonsquamous non–small-cell lung cancer. JCO Pract Oncol. 2023;7:e2300191.
26. Thermo Fisher Scientific. Targeted Sequencing Approaches for NGS. Available at: https://www.thermofisher. com/us/en/home/life-science/ sequencing/sequencing-learningcenter/next-generation-sequencinginformation/ngs-basics/targetedsequencing-approaches.html. Last accessed: 2 October 2024.
EM-167676 October 2024
Therapeutic Advances in Gastrointestinal Cancers: Immuno-oncology and Beyond
This Industry Satellite Symposium was presented at the European Society for Medical Oncology (ESMO) Congress 2024, held in Barcelona, Spain, from 13th–17th September.
Chairperson: Sara Lonardi1
Speakers: Tania Fleitas Kanonnikoff,2 Thomas Decaens3
1. Veneto Institute of Oncology, IOV-IRCCS, Padua, Italy
2. INCLIVA, Hospital Clínico Universitario de Valencia, Spain
3. Hepato-Gastroenterology and Digestive Oncology Department, University Grenoble-Alpes, France
Disclosure: Lonardi is affiliated with, has a financial interest in, or has received grants or research support from Amgen, Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Hutchinson, Incyte, Merck Serono, Mirati, MSD, Pfizer, Roche, and Servier; has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, Incyte, GSK, Lilly, Merck Serono, MSD, Pierre-Fabre, Roche, and Servier; and has served in an advisory role or expert testimony for Amgen, Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, GSK, Incyte, Lilly, Merck Serono, MSD, Servier, Takeda, Rottapharm, and BeiGene. Fleitas Kanonnikoff is affiliated with, has a financial interest in, or has received grants or research support from Gilead, BeiGene, Bristol Myers Squibb, Daiichi Sankyo, Adaptimmune, Roche, AstraZeneca, and Servier; has received honoraria from Bristol Myers Squibb, MSD, Amgen, Bayer, Roche, and Servier; and has served in an advisory role or expert testimony for Bristol Myers Squibb, MSD, AstraZeneca, and BeiGene. Decaens is affiliated with, has a financial interest in, or has received grants or research support from Genoscience Pharma, ENYO Pharma, Guerbet, NETRIS Pharma, and Transgene; has received honoraria from Bristol Myers Squibb, Ipsen, Becton Dickinson, AstraZeneca, Bayer, Roche, Eisai, SIRTeX, BTG, AbbVie, Gilead, Merck, and Guerbet; and has served in an advisory role or expert testimony for Bristol Myers Squibb, Becton Dickinson, AstraZeneca, and Roche.
Acknowledgements: Writing assistance was provided by Saba Jalali, PharmD, Amsterdam, the Netherlands.
Disclaimer The opinions expressed in this article belong solely to the named presenters.
Keywords: Colorectal cancer (CRC), first-line treatment, gastro-oesophageal cancers, hepatocellular carcinoma (HCC), human epidermal growth factor receptor 2 (HER2)-positive, immunotherapy, high microsatellite instability (MSI-H), nivolumab, PD-L1 expression, pembrolizumab.
Support: The publication of this article was fully funded by Bristol Myers Squibb.
Meeting Summary
At the European Society for Medical Oncology (ESMO) Congress 2024 in Barcelona, Spain, the latest advancements in immunotherapy for colorectal cancer (CRC), gastro-oesophageal cancers, and hepatocellular carcinoma (HCC) were presented. Sara Lonardi from the Veneto Institute of Oncology, Italy, discussed the role of neoadjuvant immunotherapy in patients with high microsatellite instability (MSI-H) CRC, highlighting promising data from the CheckMate 8HW and NICHE-2 trials. Tania Fleitas Kanonnikoff from INCLIVA, Hospital Clínico Universitario de Valencia, Spain, provided insight into the use of immunotherapy-based regimens for gastrooesophageal cancers, including treatment considerations based on key biomarkers and emerging treatment options. Thomas Decaens from the University of Grenoble-Alpes, France, presented results from several trials, including IMbrave150, HIMALAYA, and CheckMate 9DW, supporting the increasing role of immunotherapy combinations in first-line (1L) HCC treatment, which has been shown to improve overall survival in this challenging disease.
Colorectal Cancer: Novel Combination Therapies
Lonardi discussed recent advancements in immunotherapy for CRC, focusing on the impact of MSI-H status on treatment response. Historically, CRC was not considered responsive to immunotherapy until the identification of MSI-H, a marker of DNA mismatch repair associated with high mutation rates and neoantigen exposure.1-3 These insights have transformed the treatment landscape for MSI-H tumours, enabling the development of immune checkpoint inhibitors. Despite these advancements, MSI-H CRCs continue to exhibit complex immune evasion mechanisms, including overexpression of immune checkpoints such as programmed death-1 and cytotoxic T-lymphocyteassociated protein 4 (CTLA-4), underscoring the ongoing need to explore combination therapies.4
Development and Results of Combination Regimens
The CheckMate 142 trial, a Phase II multicohort study, evaluated the combination of nivolumab (NIVO) and low-dose ipilimumab (IPI) in patients with MSI-H metastatic (m) CRC who had progressed after prior treatments. In a cohort of 119 patients treated with NIVO + IPI, the combination
demonstrated a median progressionfree survival (PFS) that had not yet been reached, with 52% of patients remaining progression-free at 5 years. Grade 3 or 4 treatment-related adverse events (TRAE) occurred in 32% of patients.5
Pembrolizumab was subsequently investigated in the 1L setting through the KEYNOTE-177 trial, comparing it with standard chemotherapy in patients with MSI-H/mismatch repair deficient (dMMR) mCRC. The trial enrolled 307 patients, with 153 receiving pembrolizumab and 154 receiving chemotherapy. The median PFS was 16.5 months (95% CI: 5.4–32.4) in the pembrolizumab group and 8.2 months (95% CI: 6.2–10.2) in the chemotherapy group (hazard ratio [HR]: 0.60; 95% CI: 0.45–0.79). Grade 3 or higher TRAEs were reported in 21.6% of patients in the pembrolizumab group compared to 66% in the chemotherapy group.6
Ongoing Trials and Future Directions
The randomised Phase III trial 8HW is currently comparing nivolumab monotherapy, nivolumab + ipilimumab, and standard chemotherapy in the 1L setting for MSI-H patients. This study features dual primary endpoints: PFS between NIVO + IPI and chemotherapy and a direct comparison between
nivolumab monotherapy and nivolumab + ipilimumab. In the NIVO + IPI arm, which included 171 patients, the median PFS was not reached, with a CI of 38.4 months to not evaluable. This compared to a median PFS of 5.9 months (95% CI: 4.4–7.8) in the chemotherapy group of 84 patients (HR: 0.21; 97.91% CI: 0.13–0.35; p<0.0001). The study demonstrated that 79% of patients in the NIVO + IPI arm were progression-free at 12 months, and 72% remained progression-free at 24 months, compared to 21% and 14% in the chemotherapy group, respectively. The PFS benefit with NIVO + IPI was consistent across various analyses, including PFS by blinded independent central review. Data comparing nivolumab monotherapy versus nivolumab + ipilimumab are still awaited, and ongoing analysis aims to provide further insights. Additionally, NIVO + IPI had a different safety profile compared with chemotherapy, with fewer Grade 3/4 TRAEs, while improving health-related quality of life and reduced symptoms versus chemotherapy. Figure 1 demonstrates the data from CheckMate 8HW.7-9
Lonardi discussed the potential for further improving outcomes in MSI-H CRC through various dual immuno-oncology (IO) combinations. While multiple IO-based combinations are under investigation, she emphasised that using IO earlier in locally advanced resectable disease for MSI-H patients could deliver the most substantial improvement in outcomes.
Lonardi highlighted the NICHE-2 trial, which investigated neoadjuvant immunotherapy in patients with previously untreated, resectable clinical Stage II/III dMMR colon adenocarcinoma. In this trial, patients were treated with NIVO and low-dose IPI for one dose, followed 2 weeks later by a single dose of nivolumab monotherapy before surgery. A pathological complete response (pCR), defined as no residual viable tumour, was achieved in 67% of patients. Additionally, 95% of patients achieved a major pathological response, which includes both pCR and patients with ≤10% residual tumour. The trial also demonstrated promising long-term outcomes. With a median follow-up of 26.2 months, no disease recurrences were observed The safety profile of this
CheckMate 8HW
1L NIVO + low-dose IPI for MSI-H/dMMR mCRC (median follow-up: 31.5 months)
PFS per investigator
1L: first line; dMMR: deficient DNA mistmatch repair; HR: hazard ratio; IPI: ipilimumab; mCRC: metastatic colorectal cancer; MSI-H: microsatellite instability high; NIVO: nivolumab; NR: not reached; PFS: progression-free survival.
Figure 1: CheckMate 8HW study results.
neoadjuvant IO approach was notable, with only 4% of patients experiencing Grade 3 or 4 immune-related AEs.10,11
Data presented at ESMO 2024 of 3-year disease-free survival from NICHE-2 showed unprecedented 3-year DFS of 100% in patients with high-risk, locally advanced dMMR colon cancer with only two cycles of neoadjuvant immunotherapy. All patients were circular tumour (ct)DNA negative at minimal residual disease time point, in line with 0% recurrences. Association of (early) clearance with pCR: ctDNA may aid in organ preservation.12
KRAS Mutations and Other Colorectal Cancer Subtypes
Lonardi closed the session by noting that only about 5% of CRC patients are MSI-H,13 highlighting the need to develop new strategies for the remaining 95% of patients with CRC. One promising approach is targeting Kirsten rat sarcoma virus (KRAS) mutations, specifically KRASG12C, which is present in approximately 3% of patients with CRC.14 Preclinical data suggest that activation of epidermal growth factor receptor signalling can overcome KRASG12C inhibition in CRC.15,16
The CodeBreaK 300 trial evaluated sotorasib + panitumumab versus standard of care (SOC) in pre-treated KRASG12Cmutant mCRC. The median PFS was 5.62 months in the higher-dose group (sotorasib 960 mg + panitumumab) and 3.91 months in the lower-dose group, compared to 2.20 months with SOC. The objective response rate (ORR) was 30% in the higher-dose group, 8% in the lower-dose group, and 2% with SOC. Grade 3 or higher TRAEs occurred in 36% of the higher-dose group, 30% of the lower-dose group, and 43% of the SOC group.17
The KRYSTAL-1 trial evaluated the efficacy of adagrasib combined with cetuximab in pre-treated KRASG12C-mutant mCRC. In this trial involving 94 patients, the median PFS was 6.9 months (95% CI: 5.7–7.4), and the median OS was 15.9 months (95% CI: 11.8–18.8). The ORR was 34%, with a median duration of response of 5.8 months. Grade 3 or 4 TRAEs were reported in
28% of patients. These findings highlight the potential of targeted therapies like adagrasib combined with cetuximab in managing KRASG12C-mutant mCRC.18
Lonardi concluded that MSI is the more robust biomarker in CRC and recommended testing all patients from the outset. Pembrolizumab and NIVO + IPI are the SOC in the 1L setting for MSI-H patients,10 and NIVO + IPI in the neoadjuvant setting has shown high efficacy in resectable MSI-H colon cancer, with a 68% pCR rate. Data presented at ESMO 2024 of 3-year disease-free survival from NICHE-2 showed unprecedented 3-year DFS of 100% in patients with high-risk, locally advanced dMMR colon cancer with only two cycles of neoadjuvant immunotherapy.11,12 Lastly, targeting KRASG12C and epidermal growth factor receptor is an effective therapeutic approach for advanced, pre-treated mCRC with a KRASG12C mutation. The continued exploration of these strategies will be essential in refining treatment options and optimising outcomes for patients with CRC.15,16
Gastro-oesophageal Cancers: The Role of Immunotherapy
Tania Fleitas Kanonnikoff
Overview of Gastro-oesophageal Cancers
Kanonnikoff provided an overview of oesophageal and gastric cancers, which rank as the 7th and 5th most common causes of cancer-related death worldwide.19 The main types of oesophagal cancer are oesophageal squamous cell carcinoma (ESCC) and oesophageal adenocarcinoma (EAC). Gastro-oesophageal adenocarcinoma (gastric, gastro-oesophageal junction, or oesophageal adenocarcinoma) is treated in a similar way. Gastric cancer is known to exhibit significant heterogeneity. Biomarkers such as human epidermal growth factor receptor 2 (HER2), MSI-H, programmed death ligand-1 combined positive score (PD-L1 CPS), and recently Claudine 18.2 are essential in guiding treatment.
ESMO Guidelines for 1L Treatment in Oesophageal Squamous Cell Carcinoma
The ESMO guidelines recommend platinum plus fluoropyrimidine chemotherapy in the 1L treatment for PD-L1-negative patients. For PD-L1-positive patients with a tumour proportion score (TPS) of ≥1%, NIVO + chemo or NIVO + IPI is advised. For patients with a CPS score of ≥10, pembrolizumab plus chemotherapy is recommended.20 As of July 2024, the Committee for Medicinal Products for Human Use (CHMP) recommended the approval of toripalimab + chemotherapy for the 1L treatment of advanced ESCC.21
CheckMate 648: NIVO + Chemotherapy Versus Chemotherapy in Oesophageal Squamous
Cell Carcinoma
CheckMate 648 is a pivotal trial that evaluated NIVO + chemotherapy versus chemotherapy alone as a 1L treatment in ESCC. The trial included patients with varying levels of PD-L1 expression. The median OS for the entire population was 13.2 months (95% CI: 11.1–15.7) in the NIVO + chemotherapy group, compared to 10.7 months (95% CI: 9.4–12.1) in the chemotherapy-only group (HR: 0.77; 95% CI: 0.65–0.92). In patients with PD-L1 tumour cell expression ≥1%, also known as PD-L1 TPS, the median OS was 15.0 months (95% CI: 11.9–18.7) for the NIVO + chemotherapy arm versus 9.1 months (95% CI: 7.7–10.0) for chemotherapy alone (HR: 0.60; 95% CI: 0.47–0.77). In the subgroup of patients with tumour cell PD-L1 expression <1%, the HR for OS was 0.97, indicating that enriched benefit was observed in patients with higher PD-L1 expression.22
The long-term follow-up provided valuable insights into the survival benefit of immunotherapy combined with chemotherapy. Grade 3 or 4 TRAEs were reported in 49% of patients receiving NIVO + chemotherapy and 37% in the chemotherapy-only group.22
CheckMate 648: NIVO + IPI Versus Chemotherapy in Oesophageal Squamous Cell Carcinoma
An additional arm of CheckMate 648 assessed NIVO + IPI versus chemotherapy alone as a 1L treatment for ESCC. The trial reported a median OS of 12.7 months (95% CI: 11.3–15.5) for the NIVO + IPI group compared to 10.7 months (95% CI: 9.4–12.1) for the chemotherapy-only group (HR: 0.78; 95% CI: 0.65–0.92). In patients with tumour cell PD-L1 expression ≥1%, the median OS was 13.1 months (95% CI: 11.2–17.4) for NIVO + IPI versus 9.1 months (95% CI: 7.7–10.0) for chemotherapy (HR: 0.63; 95% CI: 0.49–0.81). For patients with tumour cell PD-L1 expression <1%, the HR for OS was 0.94, indicating that the benefit of NIVO + IPI was more pronounced in patients with higher PD-L1 expression. Grade 3 or 4 TRAEs were reported in 33% of the NIVO + IPI group and 37% in the chemotherapy-only group.22
KEYNOTE-590:
PEMBRO + Chemotherapy Versus Chemotherapy in Oesophageal Squamous Cell Carcinoma
The KEYNOTE-590 trial evaluated PEMBRO + chemotherapy versus chemotherapy alone in ESCC. In the overall ESCC population, the median OS was 12.6 months (95% CI: 10.2–14.2) for PEMBRO + chemotherapy versus 9.8 months (95% CI: 8.6–11.1) for chemotherapy alone (HR: 0.71; 95% CI: 0.60–0.85).23
For ESCC patients with PD-L1 CPS ≥10, the median OS was 13.9 months (95% CI: 11.1–16.0) in the PEMBRO + chemotherapy group compared to 8.8 months (95% CI: 7.8–10.5) in the chemotherapy group (HR: 0.59; 95% CI: 0.45–0.76). The 5-year OS rate was 13.8% for PEMBRO + chemotherapy and 3.7% for chemotherapy alone, highlighting the long-term benefit of immunotherapy in this subgroup.24
Additionally, in patients with PD-L1 CPS <10, the OS HR was 0.84, indicating a less pronounced benefit in this lower expression group.24 Grade 3 or higher TRAEs were reported in 71.9% of patients receiving PEMBRO + chemotherapy and 67.6% in the chemotherapy-only group.
Fleitas discussed the duration of 1L therapies in her clinical practice. Current guidelines recommend chemotherapy combined with either PEMBRO or NIVO.19 In her practice, she typically discontinues oxaliplatin after eight cycles and continues with immunotherapy and fluoropyrimidine if there is a clinical benefit for the patient.
KEYNOTE-811 in HER2Positive Gastric/Gastro-oesophageal Junction Cancer
For HER2-positive advanced gastric, gastro-oesophageal junction cancer, and EAC, the addition of PEMBRO to trastuzumab (TRAS) and chemotherapy is now a recommended treatment option for PD-L1 CPS ≥1, supported by results from the KEYNOTE-811 trial.24,25 The trial demonstrated significant improvements in PFS and OS when PEMBRO was added to the standard TRAS + chemotherapy regimen for the PD-L1 positive population.26,27
For patients with PD-L1 CPS ≥1, the median PFS was 10.9 months (95% CI: 8.5–12.5) in the PEMBRO group compared to 7.3 months (95% CI: 6.8–8.5) in the placebo group (HR: 0.71; 95% CI: 0.59–0.86). The median OS was 20.5 months in the PEMBRO group versus 15.6 months in the placebo group (HR: 0.79; 95% CI: 0.64–0.98). The ORR was 73.2% in patients with PD-L1 CPS ≥1 receiving PEMBRO + TRAS + chemotherapy, compared to 58.4% in the placebo group.26,27
Grade 3 or 4 TRAEs occurred in 58% of patients receiving PEMBRO and 50% of patients in the placebo group, reflecting the manageable safety profile of the combination therapy. This combination has become an established standard of care for HER2-positive patients with PD-L1 expression. Fleitas mentioned that in her practice she continues both pembrolizumab and trastuzumab until the disease progresses as a maintenance strategy.26,27
CheckMate 649 and KEYNOTE-859 in HER2-Negative Gastric/Gastrooesophageal Junction Cancer/ Oesophageal Adenocarcinoma
The CheckMate 649 and KEYNOTE-859 trials investigated the addition of immunotherapy to chemotherapy in advanced gastric, gastro-oesophageal junction cancer, and EAC, demonstrating that the addition of IO to chemotherapy showed superior benefit to chemotherapy alone. It also demonstrated that higher PD-L1 expression levels are associated with better treatment outcomes. EAC patients were only enrolled in CheckMate 649 study.
CheckMate 649 trial in HER2-negative gastric/gastro-oesophageal junction cancer/oesophageal adenocarcinoma28
• NIVO + chemotherapy was compared to chemotherapy alone.
• For PD-L1 CPS ≥5:
• Median OS: 14.4 months (95% CI: 13.1–16.2) with NIVO + chemotherapy.
• 11.1 months (95% CI: 10.1–12.1) in the chemotherapy group (HR: 0.70; 95% CI: 0.61–0.81).
• For PD-L1 CPS ≥1:
• Median OS: 13.8 months (95% CI: 12.4–14.8) with NIVO + chemotherapy.
• 11.4 months (95% CI: 10.7–12.3) in the chemotherapy group (HR: 0.75; 95% CI: 0.67–0.85).
• In all-randomised population:
• Median OS: 13.7 months (95% CI: 12.4–14.5) with NIVO + chemotherapy.
• 11.6 months (95% CI: 10.9–12.5) in the chemotherapy group (HR: 0.79; 95% CI: 0.71–0.88).
• For lower PD-L1 expression (CPS <1 and CPS <5), OS HRs were 0.98 and 0.95, respectively.
• Grade 3 or 4 TRAEs occurred in 60% of patients receiving NIVO + chemotherapy.
• 45% of patients experienced Grade 3 or 4 TRAEs in the chemotherapyonly group.
KEYNOTE-859 trial of pembrolizumab + chemotherapy versus chemotherapy in HER2-negative gastric/gastrooesophageal junction cancer29,30
• PEMBRO + chemotherapy was compared to chemotherapy alone.
• For PD-L1 CPS ≥10:
• Median OS: 15.8 months (95% CI: 14.0–19.3) with PEMBRO + chemotherapy.
• 11.8 months (95% CI: 10.3–12.7) in the placebo group (HR: 0.64; 95% CI: 0.53–0.78).
• For PD-L1 CPS ≥1:
• Median OS: 13.0 months (95% CI: 11.6–14.2) with PEMBRO + chemotherapy.
• 11.4 months (95% CI: 10.5–12.0) in the placebo group (HR: 0.75; 95% CI: 0.66–0.85).
• Grade 3 or higher TRAEs were reported in 59.4% of patients in the PEMBRO + chemotherapy group.
• 51.3% of patients experienced Grade 3 or higher TRAEs in the placebo group.
Both trials highlight the critical role of PD-L1 expression as a predictive biomarker, with higher expression correlating with improved survival outcomes when immunotherapy is added to chemotherapy.
Fleitas emphasised the critical role of biomarker testing in the management of gastro-oesophageal cancers. Ongoing research and future trials will continue to refine these treatment strategies, with a focus on expanding therapeutic options and improving outcomes across different patient subgroups.
Hepatocellular Carcinoma: 1L Treatment Options in the Metastatic Setting
Thomas Decaens
Rationale for Immunotherapy in Hepatocellular Carcinoma
Decaens discussed immunotherapy’s role in HCC. HCC often arises from chronic inflammatory liver conditions such as non-alcoholic steatohepatitis, alcohol-related liver disease, and viral aetiologies, like hepatitis B and C infection.31-33 These chronic conditions create an immunosuppressive environment characterised by a low tumour mutation burden and fewer tumour-associated antigens, contributing to the resistance of HCC to immune-modulating therapies.34-36 The inherent immunotolerance of the liver further complicates effective immune engagement.37 In most cases in the West, cirrhosis is present, leading to modifications in liver endothelial cells that hinder tumour-associated antigen presentation.38 Additionally, the fibrotic liver alters cell trafficking, affecting the entry of immune cells from the blood.39
Mechanisms of Combination Therapy
Due to these challenges, combination therapy has become necessary in HCC treatment, as trials investigating single agents have often failed. In the current landscape, anti-PD-L1 agents, which enhance pre-existing T cell responses and cytokine production, are combined either with anti-vascular endothelial growth factor therapies to normalise tumour vasculature and reduce immunosuppression or anti-CTLA-4 therapies to increase T cell priming.40-42 This combination shifts a cold tumour to a hot tumour. Anti-CTLA-4 therapies, in addition to increasing T cell priming in the lymph node, enhance tumour antigen presentation and activate T cells, providing a broader immune response against the tumour.43,44
Clinical Trials and Efficacy of 1L Treatments
Decaens highlighted pivotal Phase III clinical trials comparing immunotherapy combinations to standard tyrosine kinase inhibitors (TKI), sorafenib (SOR), or lenvatinib in the 1L setting:
1. IMbrave 150 Trial: This trial compared atezolizumab (ATEZO, PD-L1i) + bevacizumab (BEV, VGEFi) against SOR (TKI) in previously untreated patients with HCC. ATEZO + BEV demonstrated a median OS of 19.2 months compared to 13.4 months with SOR (HR: 0.66; 95% CI: 0.52–0.85; p<0.001). Grade 3 or 4 TRAEs were reported in 43% of patients receiving ATEZO + BEV and 46% of those receiving SOR. The rate of survival at 18 months was 52% in the immunotherapy group.45
2. HIMALAYA Trial: This trial compared the combination of tremelimumab (TREME, CTLA-4) + durvalumab (DURVA, PD-L1I) versus SOR in a 1L setting. At a predetermined interim analysis, the study was positive, and the median OS was 16.4 months for the TREME + DURVA group compared to 13.8 months for SOR (HR: 0.78; 95% CI: 0.67–0.92; p=0.0037). The median follow-up of this trial was 48 months, and 25% of the patients survived at the end of the follow-up. The TREME + DURVA combination was associated with a lower incidence of serious TRAEs (17.5%) compared to the current SOC (ATEZO + BEV). The SOR group registered a cumulative serious TRAE rate of 9.4%.46 Serious AEs, regardless of attribution, occurred in 41.2%, 31.7%, and 29.7% of participants receiving STRIDE (tremelimumab + durvalumab), durvalumab, and sorafenib, respectively. Serious treatment-related AEs occurred in 17.5% of participants treated with STRIDE, 8.5% of participants treated with durvalumab, and 9.6% of participants treated with sorafenib. No new serious treatmentrelated AEs occurred after the primary analysis for STRIDE. The updated 5-year OS for STRIDE (tremelimumab + durvalumab) demonstrated a sustained
OS benefit versus sorafenib, with OS rates of 19.6% versus 9.4% at 5 years and the OS rate ratios for STRIDE versus sorafenib increasing over time. There were no new serious treatmentrelated AEs after the primary analysis for STRIDE. This was presented at ESMO 2024.47
3. CheckMate 9DW Trial: This recent trial evaluated NIVO (PD1i) + IPI (CTLA-4) versus lenvatinib (LENVA) or SOR. In 85% of the patients, the investigators chose lenvatinib, the strongest TKI in the market. This is a huge distinguishing factor compared to other studies in the 1L space, as competitors used SOR only. SOR is a first-generation TKI with a narrower efficacy profile than LENVA. NIVO + IPI showed a median OS of 23.7 months compared to 20.6 months for LENVA/ SOR (HR: 0.79; 95% CI: 0.65–0.96; p=0.018). Grade 3 or 4 TRAEs were reported in 41% of patients in the NIVO + IPI group and 42% in the LENVA/SOR arm. The median follow-up was 35.2 months, which was relatively longer than the other trials, and in the NIVO + IPI arm, 38% of the patients were alive at the end of the follow-up.48
4. CARES-310 Trial: The trial compared camrelizumab (CAMRE, PDL1-i)) + rivoceranib (RIVO, VGEFR-2i) versus SOR in the 1L setting for HCC. The combination of CAMRE + RIVO showed a median OS of 23.8 months compared to 15.2 months with SOR (HR: 0.64; 95% CI: 0.52–0.79; p<0.0001). The survival rates at 12 months were 76.6% for the CAMRE + RIVO group and 60.9% for the SOR group, highlighting the significant benefit of the combination therapy. Grade 3 or 4 TRAEs were reported in 81% of patients receiving CAMRE + RIVO and 52% in the SOR group.49,50 Table 151,52 refers to the first-line systemic therapy recommendations for first-line HCC. The results of the trial are summarised in Table 2 45-50
Regimen(s)
ATEZO + BEVc
TREME + DURVA
SOR, LENVA
DURVA, TISLE, PEMBRO
Repotrectinib
EASL Clinical Practice Guidelines 201851
ESMO Clinical Practice Guidelines 202152
Standard
Recommended
Additional options
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) v2.2024*
Preferred regimens
Other recommended regimens
Useful in certain circumstances (for NTRK+ solid tumors)
ATEZO: atezolizumab; BEV: bevacizumab; DURVA: durvalumab; EASL: European Association for the Study of the Liver; ESMO: European Society for Medical Oncology; HCC: hepatocellular carcinoma; LENVA: lenvatinib; NCCN: National Comprehensive Cancer Network; PEMBRO: pembrolizumab; SOR: sorafenib; TISLE: tislelizumab; TREME: tremelimumab.
Clinical Considerations and Future Directions
Decaens discussed important clinical considerations, including the potential between increased exposure to CTLA-4 inhibitors and its assumed prolonged clinical benefit.50,53,54 Another consideration is that any patient with HCC is at risk of gastrointestinal bleeding due to the existing liver disease and the possibility of portal hypertension. The bleeding risk increases with advanced stages and antiangiogenic agents in combination regimens, as they could exacerbate portal hypertension.55-57
Looking ahead, Decaens highlighted the evolving landscape of HCC treatment. He noted that NIVO + IPI shows promise as a new SOC in 1L treatment. Researchers continue to explore new IO agents and combination therapies, with ongoing trials potentially defining a more optimised approach to managing advanced HCC. Decaens concluded that the recent advancements in 1L immunotherapy have significantly improved survival outcomes in HCC, offering a brighter outlook for patients with this challenging disease.
Table 2: Summary of immuno-oncology based 1L hepatocellular carcinoma treatments.
IMbrave15045,*
HIMALAYA46,47,†
CheckMate 9DW48,‡ CARES-31049,§
Cross-trial comparisons should not be made due to differences in study design, patient populations, treatment interventions, and duration of follow-up, among others. Data are presented side-by-side for ease of viewing. Unless otherwise specified, all data are reported as mo.
* Median follow-up 15.6 mo.
† Median follow-up for median OS and 48-mo OS rate: –48 mo; median follow-up for other outcomes: –32 mo.
1. Dienstmann R et al. Molecular subtypes and the evolution of treatment decisions in metastatic colorectal cancer. Am Soc Clin Oncol Educ Book. 2018;38:231-8.
2. Wu Wet al. Intratumor heterogeneity: the hidden barrier to immunotherapy against MSI tumors from the perspective of IFN-γ signaling and tumor-infiltrating lymphocytes. J Hematol Oncol. 2021;14(1):160.
3. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015;5(1):16-8.
4. Vesely MD et al. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-71.
5. Overman MJ et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/ microsatellite instability-high metastatic colorectal cancer. Abstract 3510. ASCO Annual Meeting, 3-7 June, 2022.
6. Shiu KK et al. Pembrolizumab versus chemotherapy in microsatellite instability-high (MSI-H)/mismatch repair-deficient (dMMR) metastatic colorectal cancer (mCRC): 5-year follow-up of the randomized phase III KEYNOTE-177 study. Abstract LBA32. ESMO Annual Meeting, 20-24 October, 2023.
7. Lenz HJ et al. Nivolumab (NIVO) plus ipilimumab (IPI) vs chemotherapy (chemo) as first-line (1L) treatment for microsatellite instability-high/ mismatch repair-deficient (MSI-H/ dMMR) metastatic colorectal cancer (mCRC): expanded efficacy analysis from CheckMate 8HW. ASCO Annual Meeting, 31 May-4 June, 2024.
8. Bristol-Myers Squibb. A study of nivolumab, nivolumab plus ipilimumab, or investigator's choice chemotherapy for the treatment of participants with deficient mismatch repair (dMMR)/ microsatellite instability high (MSI-H) metastatic colorectal cancer (mCRC) (CheckMate 8HW). NCT04008030.
https://clinicaltrials.gov/study/ NCT04008030.
9. Andre T et al. Nivolumab (NIVO) plus ipilimumab (IPI) vs chemotherapy (chemo) as first-line (1L) treatment for microsatellite instability-high/ mismatch repair-deficient (MSI-H/ dMMR) metastatic colorectal cancer (mCRC): first results of the CheckMate 8HW study. Abstract LBA768. ASCOGI Annual Meeting, 18-20 January, 2024.
10. Chalabi M et al. Neoadjuvant immunotherapy in locally advanced mismatch repair-deficient colon cancer. N Engl J Med. 2024;390(21):1949-58.
11. Chalabi M et al. Neoadjuvant immune checkpoint inhibition in locally advanced MMR-deficient colon cancer: the NICHE-2 study. Abstract LBA7. ESMO Congress, 9-13 September, 2022.
12. Chalabi M et al. Neoadjuvant immunotherapy in locally advanced MMR-deficient colon cancer: 3-year disease-free survival from NICHE-2. Abstract LBA24. ESMO Congress, 1317 September, 2024.
13. Garcia-Carbonero R et al. Real-world study on microsatellite instability and mismatch repair deficiency testing patterns among patients with metastatic colorectal cancer in Spain. Clin Transl Oncol. 2024;26(4):864-71.
14. Strickler JH et al. Prevalence of KRAS G12C mutation and co-mutations and associated clinical outcomes in patients with colorectal cancer: a systematic literature review. Oncologist. 2023;28(11):e981-94.
15. Ryan MB et al. KRASG12Cindependent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep. 2022;39(12):110993.
16. Amodio V et al. EGFR blockade reverts resistance to KRASG12C inhibition in colorectal cancer. Cancer Discov. 2020;10(8):1129-39.
17. Fakih MG et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. 2023;389(23):2125-39.
18. Yaeger R et al. Efficacy and safety of adagrasib plus cetuximab in patients with KRASG12C-mutated metastatic colorectal cancer. Cancer Discov. 2024;14(6):982-93.
19. Bray F et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229-63.
20. Obermannová R et al. Oesophageal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and
follow-up. Ann Oncol. 2022;33:9921004.
21. European Medicines Agency. CHMP summary of opinion: Loqtorzi .Available at: https://www.ema.europa. eu/en/documents/smop-initial/chmpsummary-positive-opinion-loqtorzi_ en.pdf. Last accessed: 10 October 2024.
22. Chau I et al. Nivolumab (NIVO) plus chemotherapy (chemo) or ipilimumab (IPI) vs chemo as first-line (1L) treatment for advanced esophageal squamous cell carcinoma (ESCC): 45-month (mo) follow-up from CheckMate 648. Abstract 4034. ASCO Annual Meeting, 1-4 June, 2024.
23. Shah MA et al. First-line pembrolizumab plus chemotherapy for advanced esophageal cancer: 5-year outcomes from the phase 3 KEYNOTE-590 study. Abstract 250. ASCO-GI Annual Meeting, 18-20 January, 2024.
24. Metges JP et al. First-line pembrolizumab plus chemotherapy versus chemotherapy in advanced esophageal cancer: longer-term efficacy, safety, and qualityof-life results from the phase 3 KEYNOTE-590 study. Abstract 241. ASCO-GI Annual Meeting, 18-20 January, 2024.
25. Lordick F et al. Gastric cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(10):1005-20.
26. Janjigian YY et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet. 2023;402(10418):2197-208.
27. Janjigian YY et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2+ metastatic gastric or gastroesophageal junction (mG/GEJ) adenocarcinoma: Survival results from the phase III, randomized, double-blind, placebo-controlled KEYNOTE-811 study. Abstract 15510. ESMO Congress, 20-24 October, 2023.
28. Shitara K et al. Nivolumab (NIVO) + chemotherapy (chemo) vs chemo as first-line (1L) treatment for advanced gastric cancer/gastroesophageal junction cancer/esophageal adenocarcinoma (GC/GEJC/EAC): 4 year (yr) follow-up of CheckMate 649. Abstract 306. ASCO-GI Annual Meeting, 18-20 January, 2024.
29. Rha SY et al. Pembrolizumab (pembro) + chemotherapy (chemo) for advanced HER2-negative gastric or gastroesophageal junction (G/GEJ) cancer: Updated results from the
30. Rha SY et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for HER2negative advanced gastric cancer (KEYNOTE-859): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(11): 1181-95.
31. Sanyal AJ et al. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15(Suppl 4):14-22.
32. Tacke F et al. EASL–EASD–EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024;81:492-542.
33. Kew MC. Aflatoxins as a cause of hepatocellular carcinoma. J Gastrointestin Liver Dis. 2013;22(3):305-10.
34. Vogelstein B et al. Cancer genome landscapes. Science. 2013;339(6127):1546-58.
35. Gabbia D, De Martin S. Tumor mutational burden for predicting prognosis and therapy outcome of hepatocellular carcinoma. Int J Mol Sci. 2023;24(4):3441.
36. Lu L et al. Targeting neoantigens in hepatocellular carcinoma for immunotherapy: a futile strategy?. Hepatology. 2021;73(1):414-21.
37. Roth GS, Decaens T. Liver immunotolerance and hepatocellular carcinoma: pathophysiological mechanisms and therapeutic perspectives. Eur J Cancer. 2017;87:101-12.
38. Eggert T, Greten TF. Tumor regulation of the tissue environment in the liver. Pharmacol Ther. 2017;173:47-57.
39. Ding DY et al. Collagen in hepatocellular carcinoma: a novel biomarker and therapeutic target. Hepatol Commun. 2024;8(7):e0489.
40. Llovet JM et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151-72.
41. Wang C et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2(9):846-56.
42. Hamanishi J et al. Programmed cell death 1 ligand 1 and tumorinfiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360-65.
43. Carlino MS et al. Immune checkpoint inhibitors in melanoma. Lancet. 2021;398(10304):1002-14.
44. Wei SC et al. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069-86.
45. Cheng AL et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76(4):862-73.
46. Sangro B et al. Four-year overall survival update from the phase III HIMALAYA study of tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. Ann Oncol. 2024;35(5):448-57.
47. Rimassa L et al. Five-year overall survival (OS) and OS by tumour response measures from the Phase 3 HIMALAYA study of tremelimumab plus durvalumab in unresectable hepatocellular carcinoma (uHCC). ESMO Congress, 13-17 September, 2024.
48. Galle PR et al. Nivolumab (NIVO) plus ipilimumab (IPI) vs lenvatinib (LEN) or sorafenib (SOR) as first-line treatment
for unresectable hepatocellular carcinoma (uHCC): first results from CheckMate 9DW. Abstract LBA4008. ASCO Annual Meeting, 31 May-4 June, 2024.
49. Vogel A et al. First-line treatment for hepatocellular carcinoma: CheckMate 9DW trial results. Abstract 4110. ASCO Annual Meeting, 31 May-4 June, 2024.
50. Qin S et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, openlabel, international phase 3 study. 2023;402(10408):1133-46.
51. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
52. Vogel A et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann Oncol. 2021;32(6):801-5.
53. Centanni M et al. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019;58:835-57.
54. Wang E et al. Population pharmacokinetic and pharmacodynamic analysis of tremelimumab in patients with metastatic melanoma. J Clin Pharmacol. 2014;54(10):1108-16.
55. Sangro B et al. Exposure-response analysis for nivolumab plus ipilimumab combination therapy in patients with advanced hepatocellular carcinoma (CheckMate 040).Clin Transl Sci. 2023;16:1445-57.
56. Hsu C et al. Immunotherapy in hepatocellular carcinoma: evaluation and management of adverse events associated with atezolizumab plus bevacizumab. Ther Adv Med Oncol. 2021;13:17588359211031141.
57. Finn RS et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894-905.
Platinum Resistance in Ovarian Cancer: Is This the End of the Line?
This industry symposium took place during the European Society for Medical Oncology (ESMO) Congress held in Barcelona, Spain, 13th–17th September 2024.
Chairperson: Ana Oaknin1
Speakers: Philipp Harter,2 Kathleen Moore3
1. Vall d’Hebron University Hospital, Barcelona, Spain
2. Evangelische Kliniken Essen-Mitte, Germany
3. Stephenson Cancer Center, University of Oklahoma, Norman, USA
Disclosure: Oaknin has received personal fees for advisory board membership from AbbVie, Agenus, AstraZeneca, Clovis Oncology, Corcept Therapeutics, Daiichi Sankyo, Debiopharm International, Deciphera Pharmaceuticals, Eisai, Exelisis, Genmab, GSK, ImmunoGen, Itheos, Merck Sharp & Dohme, Mersana Therapeutics, Myriad Genetics, Novocure, OncoXerna Therapeutics, PharmaMar, Regeneron, Roche, Sattucklabs, Seagen/Pfizer, Sutro Biopharma, TORL Therapeutics, Zentalis, and Zymeworks; and personal fees for travel/accommodation from AstraZeneca, PharmaMar, and Roche. Harter has been on the advisory board for AstraZeneca, BioNTech, Clovis, Corcept, Eisai, GSK, ImmunoGen, Merck Sharp & Dohme, Miltenyi, Novartis, and Roche; and has received honoraria from AbbVie, Amgen, AstraZeneca, Clovis, Daiichi Sankyo, Eisai, Exscientia, GSK, ImmunoGen, Karyopharm, Merck Sharp & Dohme, Mersana, Miltenyi, Roche, Sotio, Stryker, and Zai Lab; and research funding to the institute from AstraZeneca, Clovis, DFG, DKH, European Union, Genmab, GSK, ImmunoGen, Novartis, Roche, and Seagen. Moore has received fees for research, educational, or advisory activities from AbbVie, Aravive, AstraZeneca, Blueprint Medicines, Eisai/Serono, Elevar Therapeutics, Eli Lilly, Genentech/Roche, GSK/Tesaro, I-Mab Biopharma, ImmunoGen, InxMed, Jiangsu Hengrui Pharmaceuticals, Merck Sharp & Dohme, Mereo BioPharma, Mersana Therapeutics, Myriad Genetics, Novartis, Tarveda Therapeutics, Vavotar Life Sciences, and VBL Therapeutics.
Acknowledgements: Medical writing assistance was provided by Brigitte Scott, MarYas Editorial Services, Cowlinge, UK.
Disclaimer: The information/views presented are Authors’ own and not necessarily those of AbbVie Inc.
Support: The symposium and publication of this article was funded by AbbVie Inc.
PHARMA
Meeting Summary
Approximately 80% of females with ovarian cancer are diagnosed with advanced disease, and around 70% of these females relapse within 3 years of first-line treatment. Five-year survival for newly diagnosed advanced ovarian cancer is less than 50%. Platinum-based chemotherapy is the cornerstone of systemic treatment; however, it is not appropriate for patients with relapsed ovarian cancer who had disease progression during previous platinum treatment, early symptomatic progression post-platinum treatment, or who are platinum intolerant. New drugs are needed to address the unmet need in patients with relapsed ovarian cancer who are not eligible for platinum-based chemotherapy. This article presents highlights from a satellite symposium conducted as part of the European Society for Medical Oncology (ESMO) Congress 2024, which took place from 13th–17th September 2024 in Barcelona, Spain. The objectives of the symposium were to improve understanding of the current treatment pathways and unmet needs for patients with ovarian cancer who are ineligible for platinum-based therapies, to raise awareness of the rationale for targeting folate receptor α (FRα) in a variety of novel therapeutics for platinum-resistant ovarian cancer (PROC), and to review the efficacy outcomes and side effects from recent clinical trials involving antibody–drug conjugates (ADC) in PROC. In this symposium, Ana Oaknin, Vall d’Hebron University Hospital, Barcelona, Spain, described the current landscape in advanced ovarian cancer and the unmet need in relapsed disease; Philipp Harter, Evangelische Kliniken Essen-Mitte, Germany, explored FRα-targeted therapeutics in PROC; and Kathleen Moore, Stephenson Cancer Center, University of Oklahoma, Norman, USA, discussed ADC development beyond FRα in PROC. The symposium concluded with a lively discussion, including questions from the audience, key examples of which are included in this article.
The Unmet Need in Relapsed Ovarian Cancer: What Do We Need To Do Now?
Ana Oaknin
Approximately 80% of females with ovarian cancer are diagnosed with advanced disease (International Federation of Gynecology and Obstetrics [FIGO] Stage III or IV),1 and around 70% of these females relapse within 3 years of first-line treatment.2,3 Five-year survival for newly diagnosed advanced ovarian cancer is less than 50%.1,4 According to Oaknin: “Relapsed advanced ovarian cancer is typically incurable.” There is a significant need for better first-line treatment to improve outcomes for females with ovarian cancer.2,4,5-7
First-line treatment options in newly diagnosed ovarian cancer have expanded in recent years, especially with the
introduction of poly ADP ribose polymerase (PARP) inhibitors as maintenance treatment. There was a paradigm shift in 2018, with the publication of the SOLOL1 clinical trial with olaparib.8 This was followed by PAOLA-1 with olaparib plus bevacizumab,9 PRIMA with niraparib,10 and ATHENA with rucaparib.11 All four trials showed a substantial progression-free survival (PFS) benefit with PARP inhibitor as maintenance therapy;8-12 however, whether this benefit translated to overall survival (OS) benefit was not clear.13,14 A clinically meaningful, but not statistically significant, OS benefit compared with placebo was seen with olaparib at 7 years in SOLO1,13 and with olaparib plus bevacizumab in the patients with homologous recombination deficiency, but not in the intention-to-treat population as a whole, in PAOLA-1.14 The OS results from PRIMA10 were anticipated during ESMO Congress 2024.
Systemic therapy for recurrent ovarian cancer is based on platinum-containing or non-platinum-containing regimens.2 There are currently no molecular biomarkers to predict the efficacy of platinum re-challenge in patients with recurrent ovarian cancer.2 According to ESMO Guidelines, platinumbased therapy is not appropriate for patients with relapsed ovarian cancer who had disease progression during previous platinum treatment, early symptomatic progression post-platinum treatment, or who are platinum intolerant (Figure 1).2 These patients are recommended to receive early palliative care, trabectedin-pegylated liposomal doxorubicin (PLD; i.e., in patients with platinum intolerance who have relapsed >6 months from previous platinum, the combination of trabectedin and PLD may be recommended), or single-agent (non-platinum) treatment with conditional addition of bevacizumab (i.e., in patients
without contraindications to bevacizumab and in those not previously exposed to bevacizumab),2 based on the results of the AURELIA trial.15 However, the addition of bevacizumab to chemotherapy did not improve OS (HR=0.85; 95% CI: 0.66 - 1.08; p <0.174).15
In AURELIA, patients with platinum resistance received investigator’s choice chemotherapy (ICC; weekly paclitaxel, PLD, or topotecan) with or without bevacizumab.15 Addition of bevacizumab to ICC was associated with a clinically meaningful and statistically significant improvement in PFS (hazard ratio [HR]: 0.48; 95% CI: 0.38–0.60; p<0.001).15
In an exploratory analysis according to chemotherapy cohort in AURELIA, weekly paclitaxel emerged as a single-agent chemotherapy choice of interest, with
Figure 1: ESMO guidelines for patients with recurrent ovarian cancer who are ineligible for platinum-based therapy.2
Recurrent epithelial ovarian cancer
Assessment of the following factors [I-II, A]:
• Histotype
• BRCA1/2 status
• Number of prior lines
• Exposure and response to prior condition treatment
• TFIp
• Potential for surgery
• Residual toxicity
• Patient preference
• Patient’s general
Platinum is the best option when:
• Prior response to platinum
• No contraindication
Unfit or not willing to receive anticancer therapy
Best supportive care (BSC)
Platinum is not the best option when* [II-IV, A]:
• Progression during platinum
• Early symptomatic progression
• Platinum intolerance†
• Early palliative care [I, A]
• Single agent (non-platinum)‡ [I, B] + bevacizumab, if not contraindicated or previously exposed [I, A; MCBS 4]§
EOC: epithelial ovarian cancer; ESMO: European Society for Medical Oncology; MCBS: ESMO-Magnitude of Clinical Benefit Scale; PLD: pegylated liposomal doxorubicin; TFIp: treatment-free interval from last platinum.
* Patient choice and quality-of-life issues may also suggest that platinum is not the best option.
† In patients with platinum intolerance who have relapsed >6 months from previous platinum, the combination of trabectedin and PLD may be recommended [II, C; ESMO-MCBS v1.1 score: 2 for patients with platinum-sensitive disease; EMA approved, not FDA approved].
‡ Weekly paclitaxel, PLD, topotecan, or gemcitabine.
§ ESMO-MCBS v1.1104 was used to calculate scores for new therapies/indications approved by the EMA or FDA.
Figure adapted from González-Martin A et al.2
an objective response rate (ORR) of 28.8% versus 7.9% for PLD and 3.3% for topotecan.16,17
Phase III studies with novel therapies added to weekly paclitaxel compared to weekly paclitaxel alone have not shown a benefit in patients with platinum resistance.18-20 This includes the AGO OVAR 2.29 trial, in which the addition of atezolizumab, a humanised programmed death-ligand 1 (PD-L1) antibody, to single-agent, non-platinumbased chemotherapy (weekly paclitaxel or PLD) plus bevacizumab did not significantly improve PFS or OS.21
ADCs comprise an antibody with high affinity for tumour-associated antigens (the target for ADCs); a drug payload, which is a potent chemotherapy that induces tumour cell death when it is internalised
and released; and a linker that controls the release of the payload at the target site.22 According to Oaknin, ideally, the tumourassociated antigens should have a high level of expression on malignant cells, and no or negligible expression on non-malignant tissue. Moreover, the target tumourassociated antigens should be present on the cell surface, thus accessible to the ADC, and should be internalised, by which process the ADC is transported into the cell.
There are several ADCs in clinical development in ovarian cancer (Table 1). Mirvetuximab soravtansine (MIRV),23 targeting FRα, and trastuzumab deruxtecan (T-DXd),24 targeting human epidermal growth factor receptor 2 (HER2), are the only ADCs that have been approved by the U.S. FDA. There are no ADCs approved in Europe.
Table 1: Antibody–drug conjugates of clinical interest in ovarian cancer.
Raludotatug deruxtecan (R-DXd; DS-6000a)59 60 CDH6 Cleavable Topoisomerase I inhibitor
SGN-B7H4V61,62 B7-H4 Cleavable MMAE
SGN-ALPV63,64 ALPP; ALPPL2 Cleavable MMAE
Table compiled by Oaknin. MMAE: monomethyl auristatin E.
Oaknin concluded: “We need to develop new drugs to address the unmet need in relapsed ovarian cancer. Antibody–drug conjugates represent another paradigm shift in the treatment of this cancer.
ADCs enable us to deliver highly potent chemotherapy into tumour cells through targeting tumour-associated antigens.”
Exploring Folate Receptor-α-
Targeted Therapeutics in PlatinumResistant Ovarian Cancer
Philipp Harter
FRα is an attractive candidate for molecularly targeted approaches for ovarian cancer as it has almost ubiquitous expression on the surface of ovarian cancer cells, and can internalise large molecules containing a cytotoxic payload.65
Expression of FRα is characterised using immunohistochemistry (IHC) and classified into low, medium, and high based on the percentage of cells with staining and the intensity of staining. A study of FRα expression in pooled samples from patients with high-grade serous ovarian cancer showed that around two-thirds of samples had positive staining intensity ≥2 (PS2+) in ≥50% of cells, and around one-third had PS2+ in ≥75% of cells.66
MIRV is a first-in-class ADC comprising an FRα-binding antibody, a cleavable linker, and a maytansinoid DM4 payload (a potent tubulin-targeting agent).28,29 In the confirmatory Phase III MIRASOL trial, patients with PROC, highgrade serous histology, and FRα detected by IHC with PS2+ in ≥75% of viable tumour cells were randomised to MIRV or ICC (paclitaxel, PLD, or topotecan).31
Patients included in the trial were heavily pretreated, with approximately half the patients having received three prior lines of systemic therapy.31
PFS, the primary endpoint, was statistically significantly longer with MIRV versus ICC: median PFS was 5.62 months (95% CI: 4.34–5.95) with MIRV and 3.98
months (95% CI: 2.86–4.47) with ICC (p<0.001).31 In terms of key secondary endpoints, ORR was 42.3% with MIRV and 15.9% with ICC (ORR difference: 26.4%; 95% CI: 18.4–34.4), and OS results were statistically significant, with an HR of 0.67 (95% CI: 0.50–0.89; p=0.0046) (Figure 2).31
Harter emphasised that this was the first Phase III trial to show positive OS in PROC comparing a new drug versus ICC. Furthermore, no new safety signals were identified compared with ICC,31 and the discontinuation rate due to adverse events in the MIRV arm was 9% versus 16% with ICC.31
Ocular events, such as blurred vision, keratopathy, and dry eye, were more common with MIRV than with ICC.31 Harter stated that the ocular side effects are manageable, but clinicians will need to learn about and gain experience with these effects.
Several other drugs targeting FRα are in clinical development.67 For example, luveltamab tazevibulin was shown to be efficacious in a Phase I trial including patients with PROC (any level of FRα expression), with an overall response rate of 31.7% and a disease control rate of 78%.33 The most common treatment-emergent adverse events of any grade in the trial were neutropenia, nausea, fatigue, and arthralgia.33 Neuropathy was also noted as an important side effect in the trial.33 A Phase II/III trial with luveltamab tazevibulin is ongoing.68
Farletuzumab ecteribulin was evaluated in a Phase I trial including patients with high-grade serous ovarian cancer or other histologies.36 ORR was 25.0% and 52.4% for 0.9 mg/kg and 1.2 mg/kg farletuzumab ecteribulin, respectively.36 However, a limiting toxicity, interstitial lung disease/pneumonitis, was observed in this trial, occurring in 37.5% and 66.7% of patients receiving 0.9 mg/kg and 1.2 mg/kg, respectively.36
Another drug targeting FRα, rinatabart sesutecan, is currently being evaluated in a Phase I/II dose escalation and expansion trial,69 with initial data due at the ESMO Congress 2024.
Figure adapted from Moore et al.31 Data cut-off: 6 March 2023. Median follow-up time: 13.11 months.
There are already attempts to combine ADCs with other agents. For example, in the FORWARD II trial, a combination of MIRV and bevacizumab in patients with relapsed FRα-expressing ovarian cancer showed “very impressive response rates”, and was well tolerated, with adverse events as expected based on the side effect profiles of each drug, and there were no new safety signals.70
Harter concluded that MIRV has shown PFS benefit in PROC in the MIRASOL trial, and there are multiple further trials with drugs targeting FRα ongoing or under development. Areas of interest for future research include efficacy in a broader patient population regarding FRα status and effectiveness of ADCs in earlier lines of therapy.
Exploring Antibody–Drug Conjugate Development Beyond Folate Receptor-α in Platinum-Resistant Ovarian Cancer
Kathleen Moore
Moore summarised that there are numerous ADCs currently in development (including approximately 190 for solid tumours), with a variety of tumour-associated antigens and proprietary linkers, and a range of cytotoxics (mainly in two categories: microtubule inhibitors and camptothecins). ADCs differ chemically in terms of the drug-to-antibody ratio, the linkers, and the payloads; however, whether this makes a difference clinically is unknown.
According to Moore, the tumour-associated antigens aside from FRα that are of particular interest in gynaecological cancers include B7H4, claudin 6, cadherin-6 (CDH6), HER2, trophoblast antigen 2 (Trop-2), and mesothelin (MSLN).
Figure 2: Overall survival in the confirmatory Phase III MIRASOL trial in patients with platinum-resistant ovarian cancer.31
Mirvetuximab Soravtansine
B7H4 is a transmembrane protein widely expressed across many solid tumours.71 B7H4 expression has been reported in approximately 75% of patients with advanced epithelial ovarian cancer, and approximately 50% of patients had >50% of cells with any B7H4 staining intensity.71 However, in Moore’s opinion, the association of B7H4 with ovarian cancer prognosis has not been validated, there are no biomarkers for B7H4, and little is known about targeting B7H4.
A total of 75 patients with ovarian cancer were treated with SGN-B7H4V in a Phase I trial, and the overall response rate across all dose levels was 20%.62 Moore considered this a “modest signal” in these heavily pre-treated patients that merited further investigation. Other ADCs targeting B7H4 in clinical research include puxitaug samrotecan (AZD8205)71 and XMT-1660.72
Claudin 6, another transmembrane protein, is highly overexpressed in many solid tumours, including ovarian cancer, and has little to no expression in non-malignant tissues.73 Hence, Moore noted that claudin 6 may be the closest to a lineage marker (i.e., a ‘tumourspecific’ rather than a ‘tumour-associated’ antigen) among the solid tumour targets.
Initial results from a first-in-human study of TORL-1-23, an ADC targeting claudin 6, in epithelial ovarian cancer showed an ORR of 32%.73 Moore considered claudin 6 to be a “different and exciting target for gynaecological cancers”.
CDH6 is a transmembrane protein that is highly overexpressed in epithelial ovarian cancer.74 Moore noted that the function of CDH6 in ovarian cancer and the impact on prognosis has not yet been fully elucidated. Raludotatug deruxtecan (R-DXd), an ADC targeting CDH6, has been tested in a Phase I trial in patients with PROC, irrespective of biomarkers.75 The confirmed overall response rate was 48.6% (95% CI: 31.9–65.6) in the 4.8–6.4 mg/kg ovarian cancer cohort, duration of response was 11.2 months (95% CI: 3.1–not estimable [NE]), and median PFS was 8.1 months (95% CI: 5.3–NE).75
Moore described these results as “a very strong signal right out of the gates"; however, this is not sufficient to progress into Phase III trials versus standard-of-care treatments because dose optimisation and therapeutic windows for ADCs need to be understood. Therefore, the REJOICEOvarian01 randomised Phase II/III trial is being conducted to identify the optimal dose of R-DXd (4.8, 5.6, or 6.4 mg/kg) for efficacy and safety to inform the Phase III trial.76
CUSP06/AMT-707 is another ADC targeting CDH6 of interest for the treatment of ovarian cancer, and clinical data are awaited.77,78
HER2 has long been known to be a target in ovarian cancer; however, this target has received little ovarian cancer research attention until recently.79 HER2+ IHC 3+ is uncommon in high-grade serous ovarian cancer (2–5%),80-82 whereas HER2+ IHC 2+ occurs more frequently (8–18%).80,82,83 The incidence of HER2+ IHC 1+ is not yet known.
ORR in patients with PROC receiving T-DXd, which targets HER2, in the DESTINYPanTumor02 trial was 45% (18/40) for all patients, 63.6% (7/11) for IHC 3+, and 36.8% (7/19) for IHC 2+;80 however, these results need to be confirmed in a larger dataset.
The frequency of strongly positive cases of Trop-2 with immuno-staining is >40% of serous, approximately 40% of endometrioid, approximately 30% of mucinous, and approximately 20% of clear cell ovarian cancers, as well as >20% of carcinosarcomas of the ovary.84 There is growing interest in ADCs targeting Trop-2 in ovarian cancer.
Finally, MSLN is highly overexpressed in many solid tumours, and has recently reemerged as a target of interest in ovarian cancer following a confirmed ORR of 41.9% (95% CI: 24.5–60.9) with RC88, an ADC with a monomethyl auristatin E payload, in a platinum-resistant population.85
Moore concluded that ADCs are some of the most promising new agents in gynaecological cancers, and the variety of ADCs in development for ovarian cancer is exciting; however, better understanding of sequencing of ADCs, payload factors, and potential resistance mechanisms is needed.
Digesting the Data: Reflections on Antibody–Drug Conjugates
Panel discussion with all speakers
Four Key Questions from the Audience During the Panel Discussion are Covered Below
The experts discussed the potential use of technology, such as analysis of circulating tumour cells or circulating tumour DNA (ctDNA), to alleviate the delay in obtaining tissue biopsies in clinical trials. Moore considered that ctDNA technology in ovarian cancer is not sufficiently mature or reliable to warrant replacing tumour biopsies and biomarkers with a circulating marker. As technologies improve, this approach may be helpful as part of optimising treatment for patients.
The approach for the patient newly diagnosed with PROC was considered, including whether clinicians should start testing for FRα early in the patient journey. Harter noted that FRα testing does not necessarily have to be conducted early, unless it is important for the patient’s therapy; however, clinicians should be aware of the location of the
References
1. Torre LA et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284-96.
2. González-Martín A et al. Newly diagnosed and relapsed epithelial ovarian cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(10):833-48.
patient’s tissue specimens, with these specimens ideally kept in the centre where the patient is being treated, to allow timely testing, if and when required.
A further topic in the panel discussion was the management of ocular toxicity under MIRV therapy. Oaknin emphasised that clinicians should be aware of the type of eye events the patient might develop with this treatment, ensure that they follow their patients carefully from the beginning of therapy, and identify ocular events as early as possible. In addition, clinicians should regularly ask patients if they are suffering from any kind of ocular symptoms and refer patients to an ophthalmologist as necessary. Moore emphasised that patients should be encouraged to inform their doctor even if they are having only mild ocular symptoms so that mitigation strategies can be implemented. In addition, Moore stated that it is important that clinicians acknowledge the patient’s ocular symptoms and treat them early.
Whether treatments have an impact on the biology of the cancer cell, and the possibility of restoring platinum sensitivity with MIRV, was also considered by the panel. Moore outlined that research is required in this area, including assessing the efficacy of paclitaxel before and after MIRV, responses with subsequent lines of therapy, and treatment sequencing. Moore added that MIRV and weekly paclitaxel are two of the most active agents in the platinumresistant space, so these are the treatments of choice (in the USA) if the patient has not yet received these drugs and has no contraindications.
3. Ledermann JA et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi24-32.
4. du Bois A et al. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d'Investigateurs Nationaux Pour les Etudes des Cancers de l'Ovaire (GINECO). Cancer. 2009;115(6):1234-44.
5. Tewari KS et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J Clin Oncol. 2019;37(26):2317-28.
6. Bookman MA et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J Clin Oncol. 2009;27(9):1419-25.
7. Burger RA et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473-83.
8. Moore K et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495-505.
9. Ray-Coquard I et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416-28.
10. González-Martín A et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391-402.
11. Monk BJ et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENAMONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;40(34):3952-64.
12. González-Martín A et al. Progressionfree survival and safety at 3.5years of follow-up: results from the randomised phase 3 PRIMA/ENGOT-OV26/GOG3012 trial of niraparib maintenance treatment in patients with newly diagnosed ovarian cancer. Eur J Cancer. 2023;189:112908.
13. DiSilvestro P et al. Overall survival with maintenance olaparib at a 7-year follow-up in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: the SOLO1/GOG 3004 trial. J Clin Oncol. 2023;41(3):609-17.
14. Ray-Coquard I et al. Olaparib plus bevacizumab first-line maintenance in ovarian cancer: final overall survival results from the PAOLA-1/ENGOT-ov25 trial. Ann Oncol. 2023;34(8):681-92.
15. Pujade-Lauraine E et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302-8.
16. Poveda AM et al. Bevacizumab combined with weekly paclitaxel, pegylated liposomal doxorubicin, or topotecan in platinum-resistant recurrent ovarian cancer: analysis by chemotherapy cohort of the randomized Phase III AURELIA trial. J Clin Oncol. 2015;33(32):3836-8.
17. Poveda AM et al. Weekly paclitaxel (PAC), pegylated liposomal doxorubicin (PLD) or topotecan (TOP) ± bevacizumab (BEV) in platinum (PT)-resistant recurrent ovarian cancer (OC): analysis by chemotherapy (CT) cohort in the GCIG AURELIA randomised phase III trial. Ann Oncol. 2012;23(Suppl 17):LBA26.
18. Arend RC et al. Randomized controlled phase III trial of weekly paclitaxel ± ofranergene obadenovec (VB-111) for platinum-resistant ovarian cancer (OVAL Study/GOG 3018). J Clin Oncol. 2023;41(Suppl 16):5505.
19. Fuh KC et al. AXLerate-OC/GOG-
3059/ENGOT OV-66: Results of a phase 3, randomized, doubleblind, placebo/paclitaxel-controlled study of batiraxcept (AVB-S6-500) in combination with paclitaxel in patients with platinum-resistant recurrent ovarian cancer. J Clin Oncol. 2024;42(Suppl 17):LBA5515.
20. Vergote IB et al. Tumor treating fields (TTFields) therapy in platinumresistant ovarian cancer: results from the ENGOT-ov50/GOG-3029/ INNOVATE-3 phase 3 study. Abstract 1350. European Society of Gynaecological Oncology (ESGO) Congress, 7-10 March, 2024.
21. Marmé F et al. Atezolizumab versus placebo in combination with bevacizumab and non-platinum-based chemotherapy in recurrent ovarian cancer: final overall and progressionfree survival results from the AGOOVAR 2.29/ENGOT-ov34 study. J Clin Oncol. 2024;42(Suppl 17):LBA5501.
22. Marks S, Naidoo J. Antibody drug conjugates in non-small cell lung cancer: An emerging therapeutic approach. Lung Cancer. 2022;163:5968.
23. U.S. Food and Drug Administration (FDA). FDA grants accelerated approval to mirvetuximab soravtansine-gynx for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer. 2022. Available at: https:// www.fda.gov/drugs/resourcesinformation-approved-drugs/ fda-grants-accelerated-approvalmirvetuximab-soravtansine-gynxfra-positive-platinum-resistant. Last accessed: 25 September 2024.
24. U.S. Food and Drug Administration (FDA). FDA grants accelerated approval to fam-trastuzumab deruxtecan-nxki for unresectable or metastatic HER2-positive solid tumors. 2024. Available at: https:// www.fda.gov/drugs/resourcesinformation-approved-drugs/ fda-grants-accelerated-approvalfam-trastuzumab-deruxtecan-nxkiunresectable-or-metastatic-her2. Last accessed: 25 September 2024.
25. Vergote I et al. Tisotumab vedotin as second- or third-line therapy for recurrent cervical cancer. N Engl J Med. 2024;391(1):44-55.
26. Seagen Inc. A study of weekly tisotumab vedotin for patients with platinum-resistant ovarian cancer with safety run-in (innovaTV 208). NCT03657043. https://clinicaltrials. gov/study/NCT03657043.
27. Mahdi H et al. Phase 2 trial of tisotumab vedotin in platinumresistant ovarian cancer (innovaTV 208). J Clin Oncol 2019;37(Suppl 15):TPS5602.
28. Ab O et al. IMGN853, a folate
receptor-α (FRα)-targeting antibody–drug conjugate, exhibits potent targeted antitumor activity against FRα-expressing tumors. Mol Cancer Ther. 2015;14(7):1605-13.
29. Moore KN et al. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alphatargeting antibody–drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: a phase I expansion study. J Clin Oncol. 2017;35(10):1112-8.
30. Matulonis UA et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J Clin Oncol. 2023;41(13):2436-45.
31. Moore KN et al. Mirvetuximab soravtansine in FRα-positive, platinumresistant ovarian cancer. N Engl J Med. 2023;389(23):2162-74.
32. Li X et al. Discovery of STRO-002, a novel homogeneous ADC targeting folate receptor alpha, for the treatment of ovarian and endometrial cancers. Mol Cancer Ther. 2023;22(2):155-67.
33. Oaknin A et al. Luveltamab tazevibulin (STRO-002), an anti-folate receptor alpha (FolRα) antibody drug conjugate (ADC), safety and efficacy in a broad distribution of FolRα expression in patients with recurrent epithelial ovarian cancer (OC): Update of STRO002-GM1 phase 1 dose expansion cohort. Abstract 5508. American Society of Clinical Oncology (ASCO) Annual Meeting, 2-6 June, 2023.
34. Sutro Biopharma, Inc. Study of STRO002, an anti-folate receptor alpha (FolRα) antibody–drug conjugate in ovarian and endometrial cancers. NCT03748186. https://www. clinicaltrials.gov/study/NCT03748186.
35. Shimizu T et al. First-in-human phase 1 study of MORAb-202, an antibody–drug conjugate comprising farletuzumab linked to eribulin mesylate, in patients with folate receptor-α-positive advanced solid tumors. Clin Cancer Res. 2021;27(14):3905-15.
36. Nishio S et al. Safety and efficacy of MORAb-202 in patients (pts) with platinum-resistant ovarian cancer (PROC): results from the expansion part of a phase 1 trial. J Clin Oncol. 2022;40(Suppl 16):5513.
37. Eisai Inc. A study of MORAb-202 (herein referred to as farletuzumab ecteribulin) in participants with solid tumors. NCT03386942. https:// clinicaltrials.gov/study/NCT03386942.
38. Meric-Bernstam F et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: primary results from the
DESTINY-PanTumor02 phase II trial. J Clin Oncol. 2024;42(1):47-58.
39. National Cancer Institute (NCI). Testing the combination of DS8201a and olaparib in HER2expressing cancers with expansion in patients with endometrial cancer. NCT04585958. https://clinicaltrials. gov/study/NCT04585958.
40. AstraZeneca. A phase 2 study of T-DXd in patients with selected HER2-expressing tumors (DPT02). NCT04482309. https://www. clinicaltrials.gov/study/NCT04482309.
41. AstraZeneca. A study of T-DXd for the treatment of solid tumors harboring HER2-activating mutations (DPT01). NCT04639219. https://clinicaltrials. gov/study/NCT04639219.
42. Yao HP et al. Duocarmycin-based antibody–drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: pharmaceutical strategy and clinical progress. Drug Discov Today. 2021;26(8):1857-74.
43. Byondis BV. SYD985 in patients with HER2-expressing recurrent, advanced or metastatic endometrial carcinoma. NCT04205630. https://clinicaltrials. gov/study/NCT04205630.
44. Byondis BV. Phase I study of SYD985 with niraparib in patients with solid tumors. NCT04235101. https:// clinicaltrials.gov/study/NCT04235101.
45. Nicoletti R et al. T-DM1, a novel antibody–drug conjugate, is highly effective against uterine and ovarian carcinosarcomas overexpressing HER2. Clin Exp Metastasis. 2015;32(1):29-38.
46. Hoffmann-La Roche. A study evaluating the efficacy and safety of biomarker-driven therapies in patients with persistent or recurrent rare epithelial ovarian tumors (BOUQUET). NCT04931342. https://clinicaltrials. gov/study/NCT04931342.
47. National Cancer Institute (NCI). Targeted therapy directed by genetic testing in treating patients with advanced refractory solid tumors, lymphomas, or multiple myeloma (the MATCH screening trial). NCT02465060. https://clinicaltrials. gov/study/NCT02465060.
48. Rinnerthaler G et al. HER2 directed antibody–drug-conjugates beyond T-DM1 in breast cancer. Int J Mol Sci. 2019;20(5):1115.
49. Bliss Biopharmaceutical (Hangzhou) Co., Ltd. A first-in-human study of multiple doses of BB-1701 in subjects with locally advanced/metastatic HER2-expressing solid tumors. NCT04257110. https://clinicaltrials. gov/study/NCT04257110.
50. Ma F et al. A first-in-human, open label, multiple dose, dose escalation, and cohort expansion phase I study to investigate the safety, tolerability, pharmacokinetics and antitumor activity of BB-1701 in patients with locally advanced/metastatic HER2expressing solid tumors. J Clin Oncol. 2023;41(Suppl 16):3029.
51. DualityBio Inc. A phase 1/2a study of DB-1303/BNT323 in advanced/metastatic solid tumors. NCT05150691. https://www. clinicaltrials.gov/study/NCT05150691.
52. Moore KN et al. Safety and efficacy of DB-1303 in patients with advanced/ metastatic solid tumors: a multicenter, open-label, first-in-human, phase 1/2a study. J Clin Oncol. 2023;41(Suppl 16):3023.
53. Bardia A et al. Sacituzumab govitecan, a Trop-2-directed antibody–drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU132-01 basket trial. Ann Oncol. 2021;32(6):746-56.
54. Saxena A et al. TROPiCS–03: A phase II open-label study of sacituzumab govitecan (SG) in patients with metastatic solid tumors. J Clin Oncol 2020;38(Suppl 15):TPS3648.
55. Icahn School of Medicine at Mount Sinai. Sacituzumab govitecan in combination with cisplatin in platinum sensitive recurrent ovarian and endometrial cancer. NCT06040970. https://clinicaltrials.gov/study/ NCT06040970.
56. Fang W et al. SKB264 (TROP2-ADC) for the treatment of patients with advanced NSCLC: efficacy and safety data from a phase 2 study. J Clin Oncol. 2023;41(Suppl 16):9114.
57. Merck Sharp & Dohme LLC. Sacituzumab tirumotecan (MK-2870) as monotherapy and in combination with pembrolizumab in participants with advanced solid tumors (MK-2870008) (TroFuse-008). NCT06049212. https://clinicaltrials.gov/study/ NCT06049212.
58. Wang D et al. Safety and efficacy of sacituzumab tirumotecan (sac-TMT) in patients (pts) with previously treated advanced endometrial carcinoma (EC) and ovarian cancer (OC) from a phase II study. Annals Oncol. 2024;35:S548.
59. Hamilton EP et al. Phase I, two-part, multicenter, first-in-human (FIH) study of DS-6000a in subjects with advanced renal cell carcinoma (RCC) and ovarian tumors (OVC). J Clin Oncol. 2022;40(Suppl 16):3002.
60. Daiichi Sankyo. A study of DS6000a in subjects with advanced renal cell carcinoma and ovarian tumors. NCT04707248. https://www.
clinicaltrials.gov/study/NCT04707248.
61. Seagen Inc. A study of SGN-B7H4V in advanced solid tumors. NCT05194072. https://clinicaltrials.gov/study/ NCT05194072.
62. Perez C et al. First-in-human study of SGN-B7H4V, a B7-H4-directed vedotin ADC, in patients with advanced solid tumors: Preliminary results of a phase I study (SGNB7H4V-001). Abstract 660MO. European Society for Medical Oncology (ESMO) Annual Meeting, 2024 October, 2023.
63. Seagen Inc. A study of SGNALPV in advanced solid tumors. NCT05229900. https://clinicaltrials. gov/study/NCT05229900.
64. Lakhani NJ et al. Phase 1 study of SGN-ALPV, a novel, investigational vedotin antibody–drug conjugate directed to ALPP/ALPPL2 in advanced solid tumors (SGNALPV-001, trial in progress). J Clin Oncol. 2022;40(Suppl 16):TPS3159.
65. Chelariu-Raicu A et al. Integrating antibody drug conjugates in the management of gynecologic cancers. Int J Gynecol Cancer. 2023;33(3):4209.
66. Deutschman E et al. Folate receptor alpha prevalence and association with ovarian cancer patient and disease characteristics 36th European Congress of Pathology (ECP) 2024. PS-10-012. Available at: https://online.flippingbook.com/ view/677164284/98/. Last accessed: 25 September 2024.
67. Udofa E et al. Antibody drug conjugates in the clinic. Bioeng Transl Med. 2024;DOI: 10.1002/btm2.10677.
68. Oaknin A et al. Efficacy and safety of luveltamab tazevibulin vs investigator’s choice of chemotherapy in patients with recurrent platinum-resistant ovarian cancer (PROC) expressing folate receptor alpha (FRα): the REFRaME-01 (GOG-3086, ENGOT79ov, and APGOT-OV9) phase 2/3 study. Abstract TPS5637. ASCO Annual Meeting. 31 May-4 June, 2024.
69. Call J et al. Phase 1/2 study of PRO1184, a novel folate receptor alpha-directed antibody–drug conjugate, in patients with locally advanced and/or metastatic solid tumors. J Clin Oncol. 2023:41(Suppl 16):TPS3157.
70. Gilbert L et al. Safety and efficacy of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody–drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2023;170:241-7.
71. Meric-Berstam F et al. First-in-human study of the B7-H4 antibody–drug
conjugate (ADC) AZD8205 in patients with advanced/metastatic solid tumors. Abstract TPS3153. American Society of Clinical Oncology (ASCO) Annual Meeting. 3-7 June, 2022.
72. Hamilton E et al. XMT-1660: a phase 1B trial of a B7-H4 targeting antibody drug conjugate (ADC) in endometrial, ovarian, and breast cancers. Abstract 1420. IGCS Annual Global Meeting, 29 September-1 October, 2022.
73. Konecny GE et al. Initial results of dose finding in a first-in-human phase 1 study of a novel claudin 6 (CLDN6) targeted antibody drug conjugate (ADC) TORL-1-23 in patients with advanced solid tumors. Abstract 3082. ASCO Annual Meeting, 2-6 June, 2023.
74. Suzuki H et al. Raludotatug deruxtecan, a CDH6-targeting antibody–drug conjugate with a DNA topoisomerase I inhibitor DXd, is efficacious in human ovarian and kidney cancer models. Mol Cancer Ther. 2024;23(3):257-71.
75. Moore KN et al. Raludotatug deruxtecan (R-DXd; DS-6000) monotherapy in patients with previously treated ovarian cancer (OVC): subgroup analysis of a firstin-human phase I study. Abstract 745
MO. ESMO Annual Meeting, 20-24 October, 2023.
76. Daiichi Sankyo. A study of raludotatug deruxtecan (R-DXd) in subjects with platinum-resistant, high-grade ovarian, primary peritoneal, or fallopian tube cancer. NCT06161025. https:// clinicaltrials.gov/study/NCT06161025.
77. Lu W et al. CUSP06/AMT-707, a new CDH6-targeting antibody–drug conjugate, demonstrates potent antitumor activity in preclinical models. Cancer Res. 2023;83(Suppl 7):6320.
78. Spira AI et al. A phase 1, first-inhuman study of CUSP06, a cadherin-6 (CDH6)-directed antibody–drug conjugate, in patients with platinumrefractory/resistant ovarian cancer and other advanced solid tumors. J Clin Oncol. 2024:42(Suppl 16):TPS3166.
80. Makker V et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: biomarker and subgroup analyses from the cervical, endometrial, and ovarian cancer cohorts of the DESTINYPanTumor02 study. Abstract 460150.
SGO Annual Meeting on Women’s Cancer, 16-18 March, 2024.
81. Yan M et al. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34(1):157-64.
82. Tuefferd M et al. HER2 status in ovarian carcinomas: a multicenter GINECO study of 320 patients. PLoS One. 2007;2(11):e1138.
83. Ersoy E et al. HER2 protein overexpression and gene amplification in tubo-ovarian high-grade serous carcinomas. Int J Gynecol Pathol. 2022;41(4):313-9.
84. Dum D et al. Patterns of trophoblast cell surface antigen 2 (TROP2) and epithelial cell adhesion molecule (EPCAM) expression in human tumors: a tissue microarray study on 14,766 tumors. Abstract 83P. ESMO Annual Meeting, 9-13 September, 2022.
85. Liu Y et al. The efficacy and safety of RC88 in patients with ovarian cancer, non-squamous-non-small-cell lungcarcinoma and cervical cancer: results from a first-in-human phase 1/2 study. Abstract 5551. ASCO Annual Meeting, 31 May- 4 June, 2024.
Abstract Review
This abstract review is an example of some of the promising research presented at the European Society for Medical Oncology (ESMO) Congress 2024.
Predisposition, Clinical Characteristics, and Management of Immune Checkpoint
Inhibitor-Induced Nephritis: A Series of 190 Cases
Authors: *Anna Olsson-Brown,1 Danielle Aje,2 Nicholas Garbutt,3 Joseph J Sacco4,5
1. Sussex Cancer Centre, UK
2. Liverpool Medical School, University of Liverpool, UK
3. Immunotherapy Project Manager, Clatterbridge Cancer Centre, Liverpool, UK
4. University of Liverpool, UK
5. The Clatterbridge Cancer Centre, Liverpool, UK
*Correspondence to a.olsson-brown@nhs.net
Disclosure: Olsson-Brown has received educational honoraria from BMS, MSD, Johnson & Johnson, Ipsen, Eisai, BI, GSK, and Roche; payment for expert testimony from Immunocare; received support for attending meetings and travel from Ipsen, MSD, Chugai, and Roche; is a Charity Trustee for IO Clinical Network and UKASCC; and is a Director for OBM Perspective LTD. Garbutt has received payment or honoraria from BMS. Sacco has received consultancy fees from BMS, MSD, and Pierre Fabre; payment or honoraria from BMS and MSD; and payment for expert testimony from Immunocare and Replimmune.
Immune checkpoint inhibitors (ICI) use can lead to an array of immune-related adverse events (irAE). Nephritis is considered to be uncommon, with an incidence of 2–7%.1 The factors predisposing to developing ICIinduced nephritis remain unclear, so does the reversibility and management of steroid-
refractory toxicity. Additionally, the role of nephrotoxic drugs is unclear. It has been reported that some patients experience incomplete reversibility of toxicity,1 and the utility of additional immunosuppression is currently limited to case reports.2
MATERIALS AND METHODS
This was a retrospective review of a large, single centre with a population of 2.3 million people, consisting of all patients who had received ICIs in the management of multiple malignancies between January 2020–February 2023. There was a centralised referral service for all patients experiencing irAEs from ICIs. Grade 1 nephritis was considered as low-grade nephritis, and Grade 2 or above was defined as high-grade nephritis.
RESULTS
A total of 190 patients developed ICIinduced nephritis. In that same period, 2,645 patients commenced on ICIs with an incidence of 7.2%. Of these, 58% (111/190) developed low-grade nephritis and 42% (79/190) developed high-grade nephritis. Pre-nephritis features such as primary disease, immunotherapy regime, use of combined therapy or monotherapy, ethnicity, gender, or smoking status were not associated with nephritis outcome. The only pre-nephritis feature that was statistically significant was the use of
nephrotoxic drugs. Of the patients, 23.4% (26/111) with low-grade nephritis and 32.9% (26/79) of those with high-grade nephritis were on a proton pump inhibitor (PPI) and appeared to predispose patients (odds ratio [OR]: 2.250; 95% CI: 1.134–4.496; p<0.001), as well as the combination of a PPI and ACEi (OR: 7.677; 95% CI: 2.599–22.678; p<0.001). ACEi alone (OR: 1.29; 95% CI: 0.352–4.731) and NSAIDs or the addition of NSAIDs to other therapies (OR: 1.129; 95% CI: 0.0987–12.921) were not associated.
Those who had low-grade nephritis were less likely to require higher grades of treatment such as the use of steroids (P<0.001). However, if toxicity progressed and steroids were necessary, the patients would normally be associated with having a shorter course (P<0.001). There was also an association between the grade of nephritis outcomes and the return to baseline renal function (P<0.001), with patients that had a highgrade being less likely to return to their baseline renal function than those with lowgrade nephritis. Across all grades, only 51% of patients had a complete return to baseline.
Use of second-line immunosuppression with mycophenolate mofetil (MMF) was used in 13% (24/190) of patients to treat refractory nephritis not responding to steroids alone. Additional immunosuppression was well tolerated. All patients who were treated with second-line immunosuppression either
improved to Grade 1 or their baseline renal function, with 35% (8/23 monotherapy patients) returning to baseline/normal renal function despite being treatment refractory prior to the introduction of MMF.
CONCLUSION
This large series illustrated that there is a concomitant medication predisposition to ICI-induced nephritis, and the clinical need for PPIs should be considered prior to commencing ICI treatment. Additionally, clinical categorisation into low, high, and refectory nephritis is likely to be helpful in treatment. In line with previous studies, return to baseline function was seen in 51% and appears relative to the degree of nephritis; however, the majority of patients resolved to Grade 1 toxicity. Finally, MMF illustrated a benefit in managing refractory nephritis, with the majority resolving to clinically acceptable levels.
References
1. Miao J et al. Immune checkpoint inhibitor related nephrotoxicity: advances in clinicopathologic features, noninvasive approaches, and therapeutic strategy and rechallenge. Front Nephrol. 2022;2:1017921.
2. Moku P et al. Steroid refractory immune checkpoint induced acute interstitial nephritis salvaged by mycophenolate mofetil. Brown Hospital Medicine. 2023;2(2):74097.
The following abstracts showcase the top cutting-edge research presented at the European Society for Medical Oncology (ESMO) Congress 2024. Topics covered include the use of AI in breast cancer screening, racial disparities in clinical trial enrollment, and germline variants associated with non-small cell lung cancer, amongst others.
Genetic Variants in RNA Modification Genes Impact Non-small Cell Lung Cancer Prognosis
RNA modifications are crucial in regulating gene expression and cellular functions, with their dysregulation linked to cancer progression. This study, presented at ESMO Congress 2024, investigated the association between genetic variants in 25 RNA modification regulatory genes and the prognosis of 744 patients with non-small cell lung cancer (NSCLC). Among the 21 single nucleotide polymorphisms (SNP) analysed, three were significantly linked to overall survival (OS) outcomes.
The SNP rs10877013T>C in the METTL1 gene was associated with improved OS. Specifically, patients with the variant allele had a 33% lower risk of death (adjusted hazard ratio [aHR]: 0.67; 95% CI: 0.48–0.93; p=0.02). This SNP is located in the intronic region, and a linked variant, rs703842G>A, located in METTL1's 3' untranslated region, was identified. Functional assays demonstrated that the rs703842 A allele significantly increased promoter activity in lung cancer cells, suggesting a regulatory role in gene expression. Additionally, knocking down METTL1 using specific siRNA resulted in reduced cancer cell proliferation, invasion, and migration, while inducing apoptosis.
Genetic variations in RNA modification regulatory genes play a significant role in the prognosis of NSCLC patients
Conversely, two SNPs in the ADAR2 gene, rs3788152A>C and rs414743G>A, were associated with worse OS as patients with these variants had a higher risk of death (aHR: 1.46; 95% CI: 1.05–2.03; p=0.03 and aHR: 1.40; 95% CI: 1.02–1.93; p=0.04, respectively).
In conclusion, genetic variations in RNA modification regulatory genes, particularly METTL1 rs10877013T>C and ADAR2 rs3788152A>C and rs414743G>A, play a significant role in the prognosis of NSCLC patients, highlighting their potential as prognostic biomarkers.
Pathogenic Germline Variants Associated with Non-small Cell Lung Cancer
RESULTS from the INHERITY LC study have demonstrated the prevalence of pathogenic germline variants (PGV) in patients with non-small cell lung cancer (NSCLC), primarily affecting DNA damage repair pathway genes.
The INHERITY LC study, presented at ESMO Congress 2024, was a prospective, multicentre investigation designed to explore the presence of germline mutations in a cohort of patients with NSCLC, and to assess the genetic factors influencing lung cancer development. Historically, inherited predisposition to lung cancer has not been a routine consideration in clinical evaluations, and genetic testing has not been routinely established. However, preliminary studies have shown that the prevalence of PGVs in NSCLC ranges from 2.3–14.9%, prompting researchers to focus on a selected group of patients who might be at higher risk due to specific clinical or familial factors.
From May 2021–April 2023, 145 patients with NSCLC were enrolled in the study based on three selection criteria: a family history of lung cancer, diagnosis at an early age or with minimal tobacco exposure, and the presence of actionable somatic mutations in their tumour biopsies. Genetic testing was conducted using nextgeneration sequencing (NGS) with a 74gene panel. The study identified 15 patients (10.3%) with germline mutations classified as pathogenic or likely pathogenic, with most of the genes involved in the DNA damage repair pathway: BRCA2
(one patient), CHEK2 (two), ATM (two), PALB2 (one), BARD1 (one), XRCC2 (one), MRE11(one), NBN (three), FAN1 (one), MLH1 (one), and TP53 (one). Among the PVG carriers, 67% were women; 73% had adenocarcinoma, a common subtype of NSCLC; and they typically had low tobacco exposure, with a median of 9 pack-years. The analysis also revealed that patients who met all three selection criteria had a higher prevalence of PGVs (22%), highlighting the importance of comprehensive patient selection for genetic testing.
These findings suggest that whilst inherited predisposition has not traditionally been considered a significant risk factor for lung cancer, patients with specific clinical criteria may benefit from genetic testing to improve early detection and prevention, by identifying individuals at higher risk for lung cancer.
Patients with specific clinical criteria may benefit from genetic testing to improve
early detection and prevention
Racial Disparities in Cholangiocarcinoma Clinical Trials
A STUDY presented at ESMO Congress 2024 has revealed that clinical trials have significant racial disparities in participant enrolment, particularly a decline in the representation of Black patients over the past decade.
The study’s authors identified 57 clinical trials focused on cholangiocarcinoma, with 91.2% conducted in Europe or the USA. The majority of these trials (59.6%) were Phase II studies. Data on racial distribution was publicly available for 63.4% of the trials, and enrolment varied across phases, with 100% of Phase IV trials reporting race distribution, compared to 50% of Phase I trials.
The study analysed 1,946 participants across 52 clinical trials. The results revealed that 76% were White, 8.9% were Asian, 7.1% were Black, 3.8% were of other races, and 4.1% had unreported race. The results also revealed an enrolment factor of 0.87 for Black participants, 1.48 for Asians, and 0.97 for White, showing a stark underrepresentation of Black individuals compared to the prevalence of cholangiocarcinoma in the broader population.
Notable trends from 2009–2022 indicated a sharp decline in Black participant representation, dropping from 16% to 5.4%, while Asian enrolment increased significantly from 4% to 23%. This trend
Notable trends from 2009–2022 indicated a sharp decline in Black participant representation, dropping from 16% to 5.4%
suggests that while efforts may have been made to include Asian participants, the representation of Black patients has not only remained insufficient but has worsened over time.
The study concludes that urgent action is needed to address these disparities and implement strategies that promote equitable representation in cholangiocarcinoma clinical trials. Ensuring diverse participation is essential for developing treatment strategies that are effective across different populations and for improving health outcomes in minority communities.
K-index Could Predict Treatment Outcomes in Urologic Cancers
DESPITE the increased popularity of immune checkpoint inhibitor therapies, biomarkers utilising peripheral blood are not yet established. CD62LlowCD4+ T cells are thought to enhance the functions of cytotoxic CD8+ cells, and it is reported that the K-index (X2/Y; X: CD62LlowCD4+T cell %, Y: Treg % in peripheral blood CD4+ T cell population) could be augmented immune-reactive status and predict outcomes for anti-PD-1 therapy against non-small cell lung cancer.
Thus, research presented at ESMO Congress 2024 examined the clinical significance of the CD4+ T cell subsets in peripheral blood for anti-PD-1/PD-L1 therapy against other cancers except lung cancer.
Lead author, Yoshimichi Haruna, Osaka General Medical Center, Japan, utilised flow cytometry analysis analysed peripheral blood mononuclear cells obtained from patients with urological cancers or hepatocellular carcinoma before anti-PD-1/PD-L1 therapy. The correlation between K-index and therapy outcomes was assessed.
The objective response rate was significantly higher in patients with a high K-index
In total, 35 patients with urologic cancers (16 renal cell carcinoma, eight ureteral cancer, 11 bladder cancer) and 28 patients with hepatocellular carcinoma (HCC) participated in the study. Specifically considering urological cancers, the objective response rate (ORR) was significantly higher in patients with a high K-index (K-index ≥300, 75% versus K-index <300, 30.4%; P=0.012). On the other hand, patients with immune-related adverse events (irAE) often had a high K-index before treatment (P=0.017). In contrast, in patients with HCC, no relevant relationship was seen between the K-index and treatment outcome. Finally, the ORR of patients with a high
Treg population (≥3% of CD4+ T cells) was significantly higher than that of patients with low Treg (<3%; P=0.019).
Overall, the researcher suggests that the K-index could predict treatment outcomes and the possibility of irAE in urological cancers, and it is considered a valuable biomarker to select the anti-cancer regimens. Further study would be valuable to assess whether the K-marker could act as a predictive biomarker in other cancers. Alternatively, in patients with HCC, the Treg population could be a valuable biomarker.
AI for Ki67 Scoring in Breast Cancer
AI COULD limit inter-pathologist variability in Ki67 scoring for breast cancer, according to novel research presented at ESMO Congress 2024.
Whilst guidance to reduce the variability in breast cancer Ki67 immunohistochemistry scoring has been provided by working groups, no universal scoring method has yet been agreed upon.
Researchers sought to evaluate the potential impact that AI could have on Ki67 immunohistochemistry scoring by comparing results achieved by an AI deep learning image analysis platform, image analysis supervised software, and two independent pathologists, across a population of patients with breast cancer.
The authors stained a total of 114 breast tumours for Ki67. The deep learning image platform was trained for breast cancer and the Ki67 clone; the two independent pathologists were trained in accordance with the international Ki67 Working Group guidelines; and the image analysis supervised software was used to separate the image into tumour, non-tumour, and background, with pathologist approval. Ki67 positivity was assessed via threshing and the time to complete each analysis was recorded for each method.
The results showed that whilst there was high reproducibility at the intra-pathologist level (r2=0.95), matched pair analysis between the two pathologists was lower (r2=0.86). There was consistency between the AI deep learning model and Pathologist 1 (r2=0.92), Pathologist 2 (r2=0.90), and the image analysis supervised software (r2=0.93). Whilst within an acceptable range, the consistency between the image analysis supervised software and Pathologist 1 and 2 was lower (r2=0.79 and r2=0.84, respectively). The fastest approach was the AI deep learning model, followed by pathologists and then the image analysis supervised software.
The research concluded that the approaches to Ki67 tumour analysis yielded similar results, highlighting that AI could prove to be useful in aiding Ki67 scoring and could reduce inter-pathologist variability in scoring.
AI
could
prove
to be useful in aiding Ki67 scoring and could reduce inter-pathologist variability in scoring
The authors stained a total of breast tumours for Ki67
Congress Interview
An expert in the field of oncology, Jianjun Zhang discusses his career journey, highlights from the European Society for Medical Oncology (ESMO) Congress 2024, and more. Read on to learn about Zhang’s cutting-edge research on lung cancer, and his insightful advice for the next generation of oncologists.
Jianjun Zhang
Department of Thoracic/Head and Neck Medical Oncology, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA
Citation:
I think oncology has been at the bottom of my heart since I was in high school
Your journey into medicine is quite remarkable, having pursued both an MD and a PhD in China. What originally inspired you to take this path, and how did your early experiences shape your passion for oncology?
I think oncology has been at the bottom of my heart since I was in high school. When I was a young kid, my dream career was to become an astronaut. I was very good with physics and mathematics. But my interest turned towards biology and medicine when I was in high school when my father was diagnosed with cancer. I got to spend a year with him, and he unfortunately passed away. So that is the time, as a defensive mechanism, I started reading a lot of medical books, even before I went to medical school. Since then, I became more and more interested in the science associated with oncology. That's one of the reasons why I went on to pursue my PhD in oncology after medical school. I have always been interested in science, because of which I'm currently
doing three jobs: the first as an oncologist where I treat patients, the second as a scientist where I am trying to understand biology and find new treatment options, and the third as a teacher for the young generation; I enjoy that.
Q2
After moving to the USA, you completed a research fellowship at Memorial Sloan Kettering, focusing on cancer genomics. How did that experience influence your decision to specialise in this field, particularly in relation to lung cancer?
As mentioned earlier, I knew what I wanted to do from a very young age. So, my initial plan was to do research for 3–4 years and then go back to the medical field, resume my medical training, and then work as a physician scientist as I do right now; the plan was a little bit different than the reality. My project was not going on well for quite some time. So, I had to stay for longer in the research field, making me always the oldest in my class, in my residency training, and also in my fellowship training.
I chose lung cancer because lung cancer is the number one cancer killer in the world. When we consider making a major impact in a small field versus making a smaller impact on a bigger field, though my ideas may seem conservative to many, I think I would rather be the latter. I do think the major impact in small fields is still important, but I think the chances are lower if you want to make a bigger impact.
I chose lung cancer because lung cancer is the number one cancer killer in the world
Lung cancer is a very big problem, so even if you make a small improvement, you can actually make a very big impact in the community. That's one of the reasons why I went for lung cancer. Additionally, the active research opportunities within lung cancer research make it a particularly engaging field for someone passionate about science.
Q3
Since joining MD Anderson, you’ve worn many hats: clinician, researcher, and educator. How do you balance these roles, and what drives you to bridge the gap between research and patient care?
This is a very tough question. There are times when I wish a day had 27 hours or sometimes 30 hours. For me, patient care is my priority. On paper, I should spend 20–25% of my time on patient care, but most of the time I spend more time on it. Patients can call you or email you anytime with questions, questions that you
would have to answer. So, I think that is our number one goal. Our second goal would be research and education. I think education is key. There is only so much one can do individually, but when you train more people and work towards the same goal, the impact on the community becomes exponential.
I spend a lot of time with my trainees and fellows when it comes to research. When I was a junior faculty member, my research just stopped whenever I had to go into meetings, but now that I have a lot of trainees, I feel the research never stops; people are there to carry on your work. I think things are getting a little bit easier that way; of course, the more responsibilities you have, the busier you get.
Q4
Your lab’s work in cancer genomics and immunogenomics is groundbreaking. Could you share some of the most exciting findings from your research, especially in terms of how they’re impacting lung cancer treatment?
There are two major directions in my lab. One is looking into the intra-tumour heterogeneity as a mechanism of therapeutic resistance. We published our first paper in Science in 2014 with Charlie Swanton from The Francis Crick Institute, London, UK, and we are continuing this work. Now, we’re not only looking at intra-tumour heterogeneity in resected tumours but also in primary and metastatic tumours with and without treatment. We've identified specific mutations or genomic changes linked to the development of metastatic disease, which could help inform adjuvant treatments to prevent micrometastatic disease.
The second direction is the precancer space. Over the past two decades, lung cancer treatment has improved significantly. The 5-year survival rate has increased from around 5–10% to 30–40%, which is a major advancement. However, 60% of patients still die within 5 years, so there's much more to be done. Since staging is the most important prognostic factor, there's a strong focus on early detection and lung cancer screening. That's partly why I’ve shifted some focus in that direction. But as an oncologist, not a pulmonologist or radiologist, lung cancer screening isn’t my primary expertise.
I’m particularly interested in the approach taken when precancer is detected. Typically, patients are referred for surgery or radiation to address the localised issue. However, many patients present with multiple lung nodules, some of which are not cancerous and may not be suitable for biopsy. Our goal is to understand how we can utilise pre-cancerous or pre-invasive lesions, which are theoretically simpler on a molecular level. Laboratory studies have shown that these lesions often maintain a better-preserved immune response. We are exploring how to leverage these two factors: the simpler molecular makeup and the stronger immune response.
The 5-year survival rate has increased from around 5–10% to 30–40%, which is a major advancement
How can we effectively kill precancerous cells to prevent the progression to invasive cancer, which is much more challenging to treat? This question led us to explore the molecular evolution of precancers into invasive cancers in our lab. We are investigating the immune characteristics associated with this evolution and how we can use that information to prevent lung cancer. We have developed animal models in collaboration with John Heymach, MD Anderson Cancer Center, and are utilising human specimens through international collaborations, which has allowed us to initiate two clinical trials based on our research. I’m proud that we have successfully translated our findings from the laboratory to clinical practice.
Q5Drawing now on the European Society for Medical Oncology (ESMO) Congress 2024, what sessions are you most excited to attend?
I think the oncology field overall has been moving so fast, in a way, you know, we're spoiled. We always expect big things to happen. Many years ago, when I started practice in the USA, we used the National Comprehensive Cancer Network (NCCN) guideline as one of the major guidelines. Usually, you don't get a major change, maybe one major change every 1 or 2 years. But a couple of years ago, we started seeing 10–12 major
changes within a year. I have now noticed that this has slowed down a bit. It's very reasonable after the initial success of immunotherapy that has raised the bar high. We probably need another basic scientific breakthrough to more clinical breakthroughs.
So, this year at the American Society of Clinical Oncology (ASCO) Annual Meeting just couple months ago, we had a good time for lung cancer with the presentation of several practicechanging trials. Another highlight was a new drug presented at the World Conference on Lung Cancer (WCLC) 2024 and ASCO, a bispecific antibody called ivonescimab, which targets programmed death ligand 1 and vascular endothelial growth factor. This drug was compared to pembrolizumab (the most commonly used immunotherapy drug for lung cancer), and the results obtained were statistically significant, and clinical meaningful. This ESMO, there are several interesting studies with trial updates and some translational aspects related to lung cancer, but none were changing practice. For me, I was thrilled to chair a session that finally included lung cancer prevention and interception/stop progression of precancers to prevent invasive cancer in a major conference. I’ve been advocating for this for many years, and it’s exciting to see it happen.
Q6For the next generation of oncologists, what advice would you like to instil in them, to ensure they are as successful as yourself?
Success can be defined in different ways, but for me, it means enjoying what I do and feeling fulfilled, even if it keeps me busy. I believe the work I do has a positive impact on people's lives, and that's how I define success. So, I think for the younger generation, the first thing would be to find something you really love; otherwise, you will suffer. Even if you are successful, standing on the podium, teaching, with all the glories, you're still going to suffer if you don't like what you do. I think that's the first important thing one should do. The second thing is to love science; oncology or medicine in general is driven by science. If you want to be an academic physician, a passion for science is essential. While treating patients is important, pursuing academia requires a deep love for scientific research. And the last thing would be to have an inclination towards AI. Having knowledge of mathematics and computer science is a significant advantage in many fields. Even if you don't directly work with these technologies, understanding the process can make your work much more effective.
From Podium into Practice: Working Together to Revolutionise Cancer Care in the Real World
Interviewee: Greg Rossi1
1. Senior VP, Head of Oncology, AstraZeneca, Europe and Canada
Disclosure: Rossi is employed by AstraZeneca and holds stock/other ownership in AstraZeneca/MedImmune.
Disclaimer: All opinions in this article reflect the views of the author.
Keywords: Cancer, clinical trials, early detection, equitable access, partnership, patient care sustainable healthcare.
Support: This article has been funded and authored by AstraZeneca.
Interview Summary
Cancer is on course to be the leading cause of death in the EU by 2035. Europe's population is ageing rapidly, and obesity rates continue to climb; meanwhile, healthcare systems struggle with delayed diagnoses and unequal access to treatments. Despite these challenges, the oncology community has many reasons to be optimistic. Our increased understanding of cancer has enabled us to create potentially transformative treatments, which could deliver life-changing outcomes that were unimaginable 20 years ago. Much of this emerging science has been showcased at this year’s European Society for Medical Oncology (ESMO) Congress, leading to wellwarranted excitement across the community.
However, it will take more than early research and positive clinical trial data to transform cancer care. We must embed new technologies and approaches in real health systems, looking 10 or 20 years ahead, to truly redefine cancer care.
Greg Rossi, Senior VP, Head of Oncology, Europe and Canada, AstraZeneca, shares his thoughts on the critical approaches and concrete steps needed for progress and the importance of collaboration to achieve these goals.
PHARMA PARTNERSHIP
INTRODUCTION
Based on current projections, cancer will be the leading cause of death in the EU by 2035.1
Why? Firstly, demographic factors associated with cancer are on the rise, especially age and obesity. Europe’s population is ageing rapidly, with the over65 population doubling from 11% to 22% between 1970–2023.2 At the same time, obesity rates across Europe have climbed from around 12% in the early 1980s to well over 20% by 2016.3 These demographic factors are compounded by a combination of delays in presentation and diagnosis, inequitable access to medicines, lack of clarity around expected treatment responses, and suboptimal care pathways, which can ultimately lead to progression and mortality. Too many people wait too long to access potentially lifesaving diagnostics and treatments, while pressures continue to grow on already stretched healthcare systems.
Sadly, an individual’s experience of cancer and chances of survival depends very much on who they are and where they live. Differences between countries are stark; for example, based on the EUROCARE-3 study published in 2018, 5-year survival in lung cancer ranged from under 10% in Bulgaria to over 20% in Latvia.4
There is also immense inequality within countries in Europe. For example, cancer mortality rates are 75% higher for men than women across the EU27, a pattern which pertains in every European country.5 People with lower levels of education have higher mortality rates for nearly all types of cancer compared with their highly educated counterparts.6 Across the globe, there are also poorer cancer survival rates for people living in rural areas compared with those in urban areas.7
TRANSLATING PROMISING PODIUM PRESENTATIONS INTO BETTER CARE FOR PATIENTS
The cancer community has many reasons to be optimistic. Over the last few decades, our increased understanding of cancer has enabled us to take significant strides forward in tackling the disease. We now have transformative treatments with the potential to deliver life-changing outcomes that would have been unimaginable 20 years ago. Many people with cancer are now living longer, healthier lives.8
Looking ahead, we can see critical trends for how the future of cancer treatment will continue to evolve. For example, the next wave of immuno-oncology therapies has the potential to empower the immune system to better recognise and kill cancer cells, overcoming immunosuppressive mechanisms that cancers can develop as they evolve. In addition, the next generation of cell and gene therapies, multi-specific biologics, or radioconjugates have the potential to change the lives of more patients than ever before. Collaborative research partnerships, such as the joint AstraZeneca–Cancer Research UK Functional Genomics Centre9 that uses CRISPR technology, big data, and clinical insights to discover new targets and disease pathways in oncology, are critical for continuing to advance the science and develop innovative cancer medicines.
Much of this emerging science has been showcased at this year’s ESMO Congress. It has been exciting to see scientific research continue to elevate treatment expectations across many different cancer types, including those that have traditionally been tough to treat and have seen little progress in decades.
We must, however, recognise that it will take so much more than positive clinical trial data to transform cancer care. Embedding new personalised treatments, technologies, and clinical approaches in real health systems and patient pathways is incredibly complex and intertwined with multifactorial issues. Together, across healthcare, research, patients, governments, and industry, we
need to look ahead to the next 10–20 years and seize the opportunities to redefine cancer care.
Rossi sees four key areas of focus:
Early Detection and Screening
An essential first step is to detect cancers earlier. A 20-year follow-up of the International Early Lung Cancer Action Plan (ELCAP) found that when lung cancer is detected at an early stage, up to approximately 80% of people live for 10 years or more;10 the 5-year survival rate drops to a mere 10% if the cancer is detected at a late stage.11 This pattern is mirrored in other cancers.12
Screening is a core component of early detection, but its full potential is yet to be realised across Europe. In 2023, the European screening recommendations were expanded to include lung and prostate cancer.13 However, while many European countries have already seen promising results from pilot screening programmes for lung cancer, very few have made the transition to implementing a national programme. Furthermore, “we need to accelerate the use of blood tests alongside screening to detect cancer earlier and expand population-scale initiatives, like the NHS-Galleri study, which randomised 140,000 people in just 1 year,” Rossi emphasised.14 “In future, I would like to see all people at high risk of cancer benefit from early detection and screening.”
Timely and Equitable Access to Diagnostics and Treatment
In the past 10 years or so, incredible advances in genomics, imaging, and other fields have transformed the ability to characterise cancer based on genetic markers or individual risk profiles. This has opened the door for personalised care approaches, where treatments can be tailored, improving people’s survival and quality of life and reducing their exposure to side effects from treatments that are likely to be ineffective.11
Unfortunately, not everyone has access to the diagnostic tools needed to achieve personalised cancer care. There are significant disparities in access even for some of the more conventional biomarker tests that have been around for years.15 “We need to challenge health systems to integrate these new diagnostics into models of care, with appropriate and up-to-date regulatory and funding pathways to make sure this happens,” said Rossi. Ambitious pilots should be considered to increase the national adoption rate of these new technologies; for example, AstraZeneca (Cambridge, UK) has established a consortium with three pathology laboratories in France that are completely digitising their activities to prepare for AI algorithm implementation. Over the next 5 years, Rossi mentioned that they aim to establish best practices, demonstrate value, and build the case for nationwide investment.
Even when patients are diagnosed, Europe still lags behind in terms of access to medicines. Recent data regarding access to medicines in the EU shows that only 50% of EMA-approved indications are available in all EU member states, with the average EU patient waiting over 500 days after EMA approval to gain access.16 This can have devastating consequences for patients, and improvement is needed. Rossi emphasised that: “We must work together to remove these barriers and ensure that Europeans receive timely access to treatments.”
The EU regulation on Health Technology Appraisals (HTA) is one initiative that aims to address some of these challenges. However, for this to be successful in accelerating timely access across the entire region, member states must work with the relevant stakeholders to align their clinical evidence reviews and, in many countries, significantly speed up their access decision processes.
Equitable Access to Clinical Trials
Clinical trials are fundamental to bringing new treatments into practice, so it is important to make Europe an environment that promotes research and innovation as recognised in the Accelerating Clinical Trials in the EU (ACT EU) initiative.17 Rossi
said: “I despair seeing obstacles hindering a productive environment for clinical trials in Europe, including a slow approvals process and conflicting application requirements between countries.18 This needs to change if we are to make Europe a hub of innovation and research in health.” He added that “data must flow for research, and healthcare records must be used to bring new efficiencies to trials. This includes digital tools for patient identification, consent, treatment, and follow-up. The European Health Data Space19 offers a promising framework, and we must ensure that this is implemented in a way that benefits our ability to do effective research.”
Another area of concern is inequity in clinical trials. In Europe and globally, only a minority of people with cancer participate in clinical trials, and participation is particularly low among those from underserved communities, who are often disenfranchised from health systems and have the poorest outcomes.20,21 Ensuring that participants in clinical trials accurately represent the entire population these treatments are designed for is thus an important goal and one that, if not achieved, risks developing ineffective and inappropriate interventions.
Sustainable and Resilient Healthcare Systems
“We will not progress in cancer care without strengthening health systems as a whole
References
1. European Commission. Europe’s Beating Cancer Plan. 2021. Available at: https://health.ec.europa.eu/system/ files/2022-02/eu_cancer-plan_en_0. pdf. Last accessed: 14 October 2024.
2. World Bank Group. Population ages 65 and above (% of total population) - European Union. Available at: https://data.worldbank.org/indicator/ SP.POP.65UP.TO.ZS?locations=EU. Last accessed: 14 October 2024.
3. World Health Organization (WHO). WHO European Regional Obesity Report. 2022. Available at: https:// iris.who.int/bitstream/hand le/10665/353747/9789289057738eng.pdf. Last accessed: 14 October 2024.
and tackling the challenges facing them,” Rossi said. Probably the most urgent among these is the workforce crisis; the number of people diagnosed with cancer in Europe is projected to increase by 21% in 2040,22 while the total healthcare workforce is expected to grow by only 5%.23 This imbalance could lead to a shortage of 4.1 million healthcare workers in Europe by 2030; “a scary thought”.23
Rossi added: “Of course, clinically trained staff will not materialise overnight, so creative thinking is needed to optimise models of care. This is where AI and digital technologies can help cancer care professionals deliver faster, more accurate, and resource-efficient care. Better integration of patient care pathways will also be key, building on Europe’s growing network of comprehensive cancer centres.”
CONCLUSION
“Now, more than ever,” Rossi emphasised, “we have the opportunity to change the course of cancer care for future generations. Forging even stronger partnerships between clinicians, governments, industry, and patients is the right thing to do; it will enable us to take concrete action. People living with cancer need us to take on this challenge, and I’m confident that by working together, we can deliver.”
4. The Swedish Institute of Health Economics (IHE). Tackling inequalities in cancer care in the European Union. Available at: https://efpia.eu/media/ cnygfywo/tackling-inequalities-incancer-care-in-the-european-union. pdf. Last accessed: 14 October 2024.
5. European Cancer Inequalities Registry (ECIR). ECIR data tool. Available at: https://cancer-inequalities.jrc. ec.europa.eu/. Last accessed: 14 October 2024.
6. Vaccarella S et al. Socioeconomic inequalities in cancer mortality between and within countries in Europe: a population-based study. Lancet Reg Health Eur. 2022;25:100551.
7. Afshar N et al. Rural-urban residence and cancer survival in high-income countries: a systematic review. Cancer. 2019;125(13):2172-84.
8. Botta L et al. Changes in life expectancy for cancer patients over time since diagnosis. J Adv Res. 2019;20:153-9.
9. Cancer Research Horizons. Functional Genomics Centre. Available at: https:// www.cancerresearchhorizons.com/ our-translational-science/functionalgenomics-centre. Last accessed: 14 October 2024.
10. Henschke CI et al. A 20-year follow-up of the International Early Lung Cancer Action Program (I-ELCAP). Radiology. 2023;309(2):e231988.
11. The Health Policy Partnership (HPP). Powering the future of cancer care with advanced diagnostics. 2024. Available at: https://www. healthpolicypartnership.com/app/ uploads/Powering-the-futureof-cancer-care-with-advanceddiagnostics_report.pdf. Last accessed: 14 October 2024.
12. United States National Institutes of Health (NIH), National Cancer Institute (NCI); Surveillance, Epidemiology, and End Results (SEER) Program. Cancer stat facts. Available at: https://seer. cancer.gov/statfacts/. Last accessed: September 2024.
13. Lung Cancer Policy Network. What can imaging-based screening programmes learn from each other? 2024. Available at: https://www. lungcancerpolicynetwork.com/ what-can-imaging-based-screeningprogrammes-learn-from-each-other/. Last accessed: 14 October 2024.
14. NHS Galleri Trial. About the trial. Available at: https://www.nhs-galleri. org/about-the-trial. Last accessed: 14 October 2024.
15. The European Federation of Pharmaceutical Industries and Associations (EFPIA). Unlocking the potential of precision medicine in Europe. 2021. Available at: https:// www.efpia.eu/media/589727/
unlocking-the-potential-of-precisionmedicine-in-europe.pdf. Last accessed: 14 October 2024.
16. EFPIA Patients W.A.I.T. Indicator 2022 survey. Available at: https://www.efpia. eu/media/s4qf1eqo/efpia_patient_ wait_indicator_final_report.pdf. Last accessed: 14 October 2024.
17. European Medicines Agency (EMA). Clinical trials in the EU - improving the clinical research environment. 2021. Available at: https://www.ema.europa. eu/en/annual-report/2021/clinicaltrials-eu-improving-clinical-researchenvironment.html. Last accessed: 14 October 2024.
18. European Federation of Pharmaceutical Industries and Association (EFPIA). Breaking down barriers: making cross-border access to clinical trials a reality. 2024. Available at: https://www.efpia.eu/ news-events/the-efpia-view/efpianews/breaking-down-barriers-makingcross-border-access-to-clinical-trialsa-reality/. Last accessed: 14 October 2024.
19. European Commission. European Health Data Space. Available at: https://health.ec.europa.eu/ehealthdigital-health-and-care/europeanhealth-data-space_en. Last accessed: 14 October 2024.
20. Dunn C et al. Older cancer patients in cancer clinical trials are underrepresented. systematic literature review of almost 5000 meta- and pooled analyses of phase III randomized trials of survival from breast, prostate and lung cancer. Cancer Epidemiol. 2017;51:113-7.
21. U.S Food & Drug Administration (FDA). 2019 drug trials snapshots summary report. Available at: https://www.fda. gov/media/135337/download. Last accessed: 14 October 2024.
22. European Commission. European Cancer Information System: 21% increase in new cancer cases by 2040. 2022. Available at: https:// joint-research-centre.ec.europa. eu/jrc-news-and-updates/ european-cancer-informationsystem-21-increase-new-cancercases-2040-2022-03-16_en. Last accessed: 14 October 2024.
23. Vintura. Innovation for sustainable cancer care. Addressing urgent workforce shortages. 2023. Available at: https://www.efpia.eu/media/ qdknvwve/addressing-urgentworkforce-shortages.pdf. Last accessed: 14 October 2024.
Veeva ID: Z4-68575
Date of Preparation: October 2024
Interviews
Two renowned female oncologists discuss the latest advancements in oncology, specifically breast cancer and translational research. Conversations centre on their career journey, current challenges in the field, and the potential of treatments such as CDK4/6 inhibitors and antibody-drug conjugates.
Featuring: Sara Tolaney and Sarah Blagden
Sara Tolaney
Chief of the Division of Breast Oncology, Dana-Farber
Your academic journey spans prestigious institutions like Princeton, University of California San Francisco, and Johns Hopkins. What motivated you to pursue a career in medicine, and how did your experiences during your education shape your focus on oncology?
I started having an interest in medicine from a young age. Part of what drove my interest was having various family members go through complicated medical journeys. When I was in high school, my dad was diagnosed with prostate cancer at an early age, and my mom was actually diagnosed with lupus, so I saw the challenges that my own parents went through. I realised that it's such an important relationship that patients have with their physicians, and what a difference it can make to have a physician whom you feel that you can trust and rely on through that challenging journey. I felt like it was a privilege to be in that kind of a role and thought it would be a nice thing to be able to pursue. I was also very interested in
science when I was in college and did some research during my summers.
At Princeton, they make you do a senior thesis, so I had the opportunity to spend some time in the laboratory. However, I did realise that the lab probably wasn't the place for me. While I enjoyed the science behind things, I really felt that I loved the interaction with people the most, and I loved trying to see a more immediate impact on patients. Through medical school, I started getting more involved in clinical research and also had an opportunity to spend time doing an oncology rotation. I think that's when I realised that's where my passion lay because I had a chance to see the journey that physicians take with their patients when they have a cancer diagnosis, and how close that relationship is. These are patients whom you're often seeing, sometimes weekly, over years, and you get to know the challenges that they're going through. You get to know their mom, their brother, and their sister. You really get a different
experience with that individual patient at a very challenging time in their life.
Then, during residency at Johns Hopkins, I did an internal medicine residency. However, I still did spend time starting to do some clinical research in breast cancer with Antonio Wolff there, and I even learnt how to write a clinical trial. When I started my fellowship at Dana-Farber Cancer Institute, I had the honour of working with Eric Winer, who is a world leader in the field of breast oncology. From an early time point, I realised his vision was to always put the patient first.
That vision really carried over into everything he did. Not just what wonderful care he took of patients, where it was clear that, when he was in the room with the patient, all that mattered was making sure that they felt heard and that he could do all that he could for them. This vision also influenced the work he did in his research, where I could see that the design of clinical trials also took that into account. It really influenced me to put the patient at the forefront of
everything that I did, not just to make outcomes better but also to improve the quality of life, and it has carried over into the work that I've done ever since.
Q2After completing your residency and fellowships, you obtained a Master’s in Public Health from Harvard University. How has this additional training in public health influenced your approach to cancer research and patient care?
During fellowship, we have an opportunity to take what they call a ‘clinical effectiveness course’ at the Harvard School of Public Health. I realised how much of a gap in my knowledge there was during that time because you get to take statistics and you have an opportunity to take more epidemiology. There was also a course that I really loved, which was the fundamentals of clinical trial design. I realised I did need more formal training to be able to do the work I wanted to do well, so I decided to actually complete the Master's in Public Health so that I felt that I had the fundamentals that I needed to be able to do clinical research.
Q3 You've been instrumental in advancing treatment approaches for breast cancer. Could you describe your role in the development of novel therapies, particularly in the context of HER2+ breast cancer?
When I started on faculty at Dana-Farber, I started not just working within the breast oncology centre but also within our Phase I clinical trials unit. This is where I had the opportunity to be able to work with a lot of drugs that were being tested for the very first time in humans, and that was a good learning lesson for how drug development was done. It really taught me how you take a drug from the first time being given to humans and figure out what the optimal dose is, what the side effects are, how to be able to understand pharmacokinetics and pharmacodynamics, and how that then could play a role in trying to move that drug forward. During that time, I had an opportunity to see that Phase I work is tough because a lot of drugs, unfortunately, will not move forward and end up not panning out.
But every once in a while, you get to find a drug that is not just super effective, but also well tolerated. So, during my time doing Phase 1 work, one agent that was just being tested for the first time was CDK4/6 inhibitors. We did the work first in human studies, and then eventually combined it with endocrine therapy, and even combined it with anti- HER2 therapy. It was really such a wonderful experience to be able to see a drug move from Phase I to Phase II and Phase III, and get registered, not just in the metastatic setting, but now also in the curative earlystage setting.
I really enjoyed that aspect of drug development, and I continued to do a lot of Phase I work for many years. But in that work, taking the lesson Winer had taught me, which was putting the patient first, I wanted to make sure that we weren't just developing drugs that were going to be super effective for people, but we also needed to figure out how to give the right amount of therapy to the right patient.
In that time, one of the major questions was how do we treat people who had smaller HER2+ cancers, because most of the large registration trials had not addressed this population. We knew, for example, that trastuzumab was highly effective. In essence, it could reduce recurrence almost by half if added to chemotherapy, but the way it was given back then was with a lot of chemotherapy. So, if you had a patient who had a slightly lower-risk disease, it seemed a little excessive to give them all that chemotherapy with trastuzumab, but it also seemed like they needed some level of systemic therapy because their risk was not insignificant. We tried to figure out how to appropriately treat these patients by trying to give a little bit of chemotherapy with trastuzumab and found that you could give, in essence, 12 weeks of paclitaxel and trastuzumab, and that these patients with small HER2+ cancers rarely recurred if you did that.
Since then, I've been trying to figure out how we can do even better. When trying to tailor therapy for this group of patients, could we give agents that have less side effects? Could we use antibody-drug conjugates? We did a study where we used trastuzumab emtansine (TDM1) in this population, which also looked very effective, but it was given for a year, and we thought that maybe that's too much, so now we're doing more work trying to shorten the duration. We are also doing other work where we're trying to get rid of chemotherapy, even completely. All of this work really stems from the idea of optimising therapy, and we've even taken this concept and applied it to triple-negative breast cancer, where we are looking at optimising immunotherapy. We are not sure that we need to be giving patients a full year of checkpoint inhibition, so we're now running a trial through the cooperative group system, trying to see if patients who achieve a good response to chemotherapy and a checkpoint inhibitor may not need additional checkpoint inhibition. So again, a lot of my work has been trying to tailor therapy to the individual patient.
A lot of my work has been trying to tailor therapy to the individual patient
Q4
Antibody-drug conjugates (ADC) have been a significant focus of your research. Can you explain how ADCs are revolutionising breast cancer treatment, and what potential they hold for the future?
ADCs are the new wave of the future. The idea that we could give targeted chemotherapy into cancer cells has proven to be certainly more effective than standard chemotherapy has been to date. Doing all that Phase I work early on in my career, I got to see one of the very first trials with the Phase I study for sacituzumab govitecan and saw the tremendous responses. I remember a patient that I had on the very first study of sacituzumab, where they had an expansion cohort in triple-negative breast cancer, who had had seven prior lines of chemotherapy for metastatic triple-negative breast cancer and went on to this trial. You could imagine her cancer probably was highly resistant to multiple therapies, and yet, she went on to this and was on therapy for almost 5 years with a tremendous response. That really just highlighted the powers of an ADC, because you could see a patient who was, in essence, resistant to almost every drug that we have in breast cancer and yet could have such a response to an ADC. I think one of the challenges with ADCs, though, is initially we would have thought that if you're giving targeted delivery of chemotherapy, that maybe you're not going to see chemotherapy side effects. But unfortunately, we do see side effects from these agents that are not that dissimilar from standard chemotherapy.
Right now, we've seen how ADCs can transform outcomes for patients. We're trying to move these drugs even earlier in the
metastatic setting, and with many trials now ongoing to try to move it as the first chemotherapy and, maybe even more importantly, also trying to move it into the curative setting. There are trials that are looking to see if we can replace part or all of standard chemotherapy in the preoperative or adjuvant setting, and I think this will be really important to see. But we haven't quite figured out which patient is going to benefit the most from which drug or if they can be used sequentially. That's going to really need to be the wave of the future as more of these agents come out. They’re truly transformative drugs, and hopefully we'll not just figure out how to sequence and order them but also how to optimise the toxicities of these agents.
Q5De-escalation of therapy is an emerging trend aimed at reducing treatment intensity without compromising outcomes. Could you elaborate on the criteria and considerations that guide the decision to deescalate therapy in patients with breast cancer?
Yes, I think this idea of deescalation really stems from the same concept of personalising therapy or optimising treatment for the patient. This means trying to give the right amount of treatment to the right patient at the right time. One would hope that we could think about doing this in a biomarker-driven approach; however, I don't think we are quite there yet. Instead, much of the work with optimisation has initially focused on trying to use the basic clinical anatomic stage. So, if someone had a tiny cancer, for example, they probably could get away with less therapy. If someone has a big cancer, they probably need more therapy. That’s the way, I think,
crudely, things were tailored, with the initial optimisation studies purely based on risk, based on anatomic stage.
One of the first studies that did this looked at paclitaxel and trastuzumab in Stage 1 HER2+ cancers predominantly. It found that people at lower clinical anatomic risk could get away with less therapy, but I think we're trying to become even more sophisticated, and so another way to do that could be to tailor based on response to treatment. For example, if someone gets preoperative therapy and they have a complete pathologic response, they're probably going to do pretty well. But then there's the flip side of people who don't have a good response, who have residual disease, who probably need more, and so, there are escalation trials for the people with residual disease that are ongoing. We are moving from tailoring therapy based on anatomic risk to tailoring based on response to therapy, and the next step is to be able to use biomarkers. There are efforts trying to develop more novel assays that could help us tailor therapy. For example, there's a novel assay called HER2DX, which is trying to use gene expression on a tumour that's HER2+ to hopefully be able to help us tailor therapy. So, if you have a HER2DX result that suggests a very high probability of pathological complete response, maybe you get away with single-agent chemotherapy with dual HER2directed therapy; whereas, if you have not such a good probability of pathological complete response, maybe you need more intensification of chemotherapy with HER+ directed treatment. We need these kinds of biomarkers. I also hope ctDNA could help us tailor in the future, helping
understand if minimal residual disease, for example, can tell you if someone needs more versus less. That's the direction a lot of these tailoring studies are going to move in breast cancer over the next several years.
Q6
What drives your passion for breast cancer research, and how do you stay motivated despite the challenges associated with developing new therapies?
You know, I think just seeing patients in clinic every day is what keeps us motivated. It is honestly the most thrilling thing in the world when you see a patient who gets a new drug and has a home run response; that's what keeps us all going. It's what drives us to do more. I think it's also what makes this job such a wonderful one.
Q7
You’ve mentored many young professionals in oncology. What advice do you offer to those entering the field, particularly those interested in breast cancer research?
It’s a privilege to get to mentor a lot of our young fellows and faculty members. It's fun to see them grow in their careers and be able to succeed. What I've found to be very useful for young faculty and mentors is to make sure that they have a mentor that they feel comfortable with and feel like it is giving them opportunities. One challenge that I see, though, is that breast oncology has become really complicated, and sometimes one mentor can't mentor you in everything. For example, I may be able to mentor someone in clinical research, but I don't have the skill set to help them understand computational biology
or to address very fundamental basic science questions. So, what I've done for a lot of our mentees is to make sure they all have a senior mentor who is their primary mentor, but they also have a mentorship team that meets with them regularly. We also have mentorship committee meetings on a regular basis. I think it's so important that they feel that they're surrounded by a team of individuals who can help address all the things that they need to be able to learn and be able to develop their own interests and careers and give them the opportunities to do what they think is going to be the most gratifying for them and fulfilling. It does take a village, and it's always important to surround yourself with those people who are going to help you succeed.
It does take a village, and it's always important to surround yourself with those people who are going to help you succeed
Sarah Blagden Professor of Experimental Oncology, University of Oxford, UK
Q1Can you share a bit about your journey into oncology research?
It is a dynamic relationship with proteins altering both RNA and DNA
expression
I decided to be an oncologist when I was in medical school, mainly because of the inspiring oncologists I met in training and also my grandmother’s diagnosis of advanced cancer at the same time. It was humbling to see how Bob Phillips, a clinical oncologist, interacted with his patients. He combined a comprehensive understanding of cancer science with emotional intelligence in a fabulous bedside manner. Medical oncology appealed to me not only for its academic focus on cancer, but also for its anarchic approach. However, as a junior doctor, I didn’t feel particularly academic. With notso-subtle encouragement from my mentor Helena Earl, I agreed to visit some labs in Cambridge, UK, and was intrigued by the marble-lined Department of Genetics and its strong aroma of fruit fly media. With support from Cancer Research UK (CRUK), I started working on a PhD characterising a gene LARP (then with the whimsical name ‘meteor’) in fruit flies. This was in 1999, just before the human genome was sequenced, so it was possible to dedicate an entire PhD to uncovering a single gene. And with only four pairs of chromosomes in a fruit fly, how hard could it be? Turns out it was a very complex few years of fruit fly crossing schedules, failed western blots, and endless days of time-lapse confocal microscopy. I look back on those lab years as my personal ‘tempus horribilis’, but somewhere along
the way I made a small scientific discovery and felt a euphoria I had never experienced before. That was the beginning of my ‘discovery addiction’ and I’ve been chasing those highs ever since.
Q2
What motivated you to transition from clinical practice to a focus on translational research?
After my stint in Cambridge, I was left with a feeling of unfinished business. I needed to clone the human homologue of LARP and understand what it did. I couldn’t convince funders to support me as a clinicianscientist, so I switched to working in early-phase trials, which was the nearest I could get to basic research without having my own lab. I was fortunate to spend a year at the Institute of Cancer Research’s (ICR) Early Trials Unit under the leadership of Stan Kaye and Johann de Bono. It was at the dawn of precision oncology, and we were testing the first poly (ADP-ribose) polymerase (PARP) inhibitors, abiraterone, and later immunotherapy on patients with advanced cancers. One of the first studies I worked on was a clinical trial of a polo kinase inhibitor; polo had been discovered in my old fruit fly lab, and it left me convinced that good basic research can quickly move into clinical trials. However, the path from Phase I trials to changing clinical practice was slow, so slow, and an urgent need to bring innovations to patients has motivated me ever since.
Q3
Your career has spanned several prestigious institutions, from Addenbrooke’s Hospital to Oxford University. How have your experiences at these various institutions influenced your approach to oncology research and clinical trials?
I had my two children comparatively late, so I was able to be fairly footloose during my oncology training. I grew up as an army child, so I have an innate wanderlust acquired from our three-yearly posting schedule. The advantage of working in so many places (if I count from house jobs onwards, Oxford is my 12th hospital so far) is you meet many inspiring people and learn a lot about organisational cultures. Some things hold true to all: there are pockets of brilliance, IT systems are slow, some staff are disgruntled and others are ruthless political movers, and most hate change. The leadership team is massively influential on the institution’s culture. Moving around teaches you about yourself as well. I realise I enjoy innovating, and I need to work somewhere that gives me space to do this.
Q4
Your research began with a CRUK Junior Clinician Scientist PhD fellowship focused on fruit fly genetics. How has your background in genetics shaped your current work in oncology, particularly in the area of posttranscriptional gene regulation?
The conclusion at the end of my PhD was that LARP was an RNA binding protein (RBP), a class of proteins that were assumed at the time to be “boring chaperones”. I found it intriguing that these proteins could bind to mRNA and literally thwart them from synthesising proteins. RBPs directly contradict the linear central dogma that “DNA makes RNA, and RNA makes protein”. It is a dynamic relationship with proteins altering both RNA and DNA expression. This idea was counterculture to the majority of research in the early 2000s, in which cancer was solidified under the definition of being a “disease of the genome”. The recent evolution of RNA sequencing and proteomics is challenging that, but in my opinion, we are still too fixated on the genome in clinical practice and are missing what is actually going on in the cell. My own contribution to challenging that
This got me thinking about preventing cancer, using our collective knowledge to stop cancers from starting and redesigning clinical trials to provide a modern infrastructure for testing them
reductionism has been to launch a lab exploring RNA biology in cancer and founding the LARP Society, an unapologetically nerdy collective for people working on the LARP gene family around the world.
Q5 At Imperial College, you launched the Early Phase Trial portfolio and established a laboratory focused on the dysregulation of mRNA translation in cancer. Could you elaborate on the significance of mRNA translation in cancer progression and how your work has advanced this field?
We know that cancer starts with a driver genetic mutation in an individual cell, and we know that other events like pollution are added into the mix to make that cell transition to cancer, but we still don’t know how those two events conspire. It is a bit like laying down a fire; you put the kindling in and you add the firelighters, but still need to light the match. Our research shows that, in some contexts, RNAbinding proteins provide the flames that ignite and maintain the fire. These findings have led my lab to switch focus to researching tumorigenesis (the process of normal cells becoming cancerous). However, at that time I was still testing drugs on people with advanced cancer. Targeted therapies can seem depressingly futile in someone with an advanced cancer that is complex, molecularly messy, and progressing at pace. This got me thinking about preventing cancer, using our collective knowledge to stop cancers from starting and redesigning clinical trials to provide a modern infrastructure for testing them.
Q6Your research has a particular focus on Precision Prevention studies. How do you see this area evolving in the next decade, and what role do you anticipate it will play in the future of cancer treatment?
Having dipped my toe into tumorigenesis research, I was surprised to learn just how many cancers take years or even decades to develop in our bodies. The problem is most precancers are clinically invisible while they are undergoing this transition to cancer, they are too small to be seen on X-rays or CT scans and no biomarkers have yet been developed to reliably detect them. This 5–10-year window is a massive, currently missed opportunity to intercept and prevent cancers from starting. But there is a chicken and egg problem; how can you design drugs to treat precancers if you don’t understand the biology, and how can you test drugs to prevent cancer if you cannot measure what you are treating? We held a consultation with the scientists in Oxford to measure the level of enthusiasm for tackling this challenge, and the room was full. Scientists from across the city had made important discoveries around tumorigenesis and were keen to see them tested in the clinic. We see a similar levels of enthusiasm at national and international meetings. Sometimes, a clinical plan can galvanise and focus research. Precision Prevention studies are small, very biologically focused trials of drugs in people at high risk of cancer. Their design makes them quicker than conventional population-based studies that need to recruit tens of thousands of patients and wait for decades to get an answer. I
don’t think Precision Prevention trials should (or could) dominate research in the next decade, but they certainly have a place in the pantheon of clinical drug development and have a realistic chance of accelerating biology into the clinic.
Q7 Given your extensive experience in both clinical and experimental oncology, what advice would you give to young oncologists and researchers looking to make a meaningful impact in the field of cancer research?
Early in my career, I was too influenced by the opinions of others, and it took me a long time to believe in myself. Now, I’m delighted if someone tells me: “It can’t be done”; it’s a green light! I look back and see my career has transitioned from being mostly National Health Service (NHS) to almost completely academic but has never been boring or predictable. I’d advise young researchers to go with the flow and be prepared to switch career direction if opportunities arise. My father had quite a strong influence
on me, and he left the army as a Brigadier in his 50s to become a mine clearance expert. He described a mixture of stage fright and imposter syndrome before entering a minefield. He taught me that you will always be surrounded by internal and external critics, but if you sincerely believe something needs to be done, you have no choice but to do it anyway.
Q8
What do you believe are the biggest challenges in translating research findings into clinical practice?
Our biggest and most exciting challenge in oncology is to make cancer a predictable and preventable disease. To achieve this, we need to tear up the rule book and work together rather than in silos, embracing multiple disciplines, and involving the opinions of the patients and the public themselves. It feels like we are assembling our very own Mathematical Bridge (like the one in Cambridge), the interlocking of many straight beams so that they collectively arch, connecting where we are today to where we want to be in the future.
We need to tear up the rule book and work together rather than in silos, embracing multiple disciplines, and involving the opinions of the patients and the public themselves
Occurring in more than four in 10 new cases of cancer, common types include:
Current cancer vaccines are important because they can prevent certain cancers and enhance the immune system's ability to target and treat cancer cells.
Mechanism and Types2,3
Two main classes of tumour antigens targeted by T cell immunotherapies: and Tumour-specific antigens (TSAs).
Tumour-associated antigens (TAAs)
TSAs are expressed only in tumour cells, produced by mutations that create novel peptide sequences. TAAs may be present in normal cells but are expressed at higher levels or are more accessible in cancer cells. The main types of cancer vaccines are DNA, RNA, peptides, whole cells, dendritic cells, and viral vaccines.
Current and Emerging
Selected Approved Therapeutic Cancer Vaccines4
Melanoma Cancer Vaccine
Talimogene laherparepvec is approved to treat advanced melanoma.4
Prostate Cancer Vaccine
Sipuleucel-T is approved to treat metastatic prostate cancer4
Bladder Cancer Vaccine
Bacillus Calmette-Guérin approved to treat early-stage cancer. It is made of inactivated tuberculosis-like bacteria.
Nadofaragene fireadonevec approved for early-stage cancer that has progressed BCG therapy.4
Future of Cancer
NHS Cancer Vaccine Launch Pad8
The NHS Cancer Vaccine Launch Pad (CVLP) is a program aiming to accelerate the access to personalised cancer vaccine clinical trials for people with This initiative aims to provide up to 10,000 patients personalised cancer treatments in the UK by 2030.
Emerging Cancer Vaccines
Selected Ongoing and Emerging Clinical Trials4-7
Head and Neck Cancer Vaccine
Phase I/II clinical trial for HPV-associated head and neck cancer vaccine.4 Vaccine
Calmette-Guérin (BCG) is early-stage bladder inactivated bacteria.4
fireadonevec is early-stage bladder progressed despite
LungVax:
Researchers at the University of Oxford, the Francis Crick Institute, and University College London have received funding from Cancer Research UK and the CRIS Cancer Foundation to develop a preventative lung cancer vaccine, ‘LungVax’.7
Pancreatic Cancer Vaccine
Autogene cevumeran is a personalised mRNA vaccine that is being tested to prevent recurrence of pancreatic cancer after surgery.6
Melanoma
A Phase III study has just started for mRNA cancer immunotherapy for melanoma. Including 1,089 patients with melanoma, the study is testing mRNA-4157 and pembrolizumab against the use of pembrolizumab alone, in individuals with high-risk Stage II-IV melanoma.5
Vaccines
and Potential Impact
Future Outlooks9,10
program access vaccine cancer. patients with treatments
Many factors can improve the clinical efficacy of cancer vaccines, including finding new cancer antigens, optimising delivery methods, and strategic patient selection for example.
Lipid nanoparticles have shown promise as a carrier for mRNA cancer therapies.
Four potential research hotspots and frontiers for future research could be, ‘immune checkpoint inhibitors’, ‘tumour microenvironment’, ‘T-cell suppressor’, and ‘dendritic cells’.
References
1. Cancer Research UK. Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/ worldwide-cancer#heading-Zero. Last accessed: 6 September 2024.
2. Cancer Research UK. Available at: https://www.cancerresearchuk.org/about-cancer/treatment/immunother apy/types/vaccines-to-treat-cancer. Last accessed: 6 September 2024.
3. Fan T et al. Sig Transduct Target Ther. 2023;8:450.
4. Memorial Sloan Kettering Cancer Center. Available at: https://www.mskcc.org/cancer-care/diagnosis-treatment/cancer-treatm ents/immunotherapy/cancer-vaccines. Last accessed: 6 September 2024.
5. A Clinical Study of V940 Plus Pembrolizumab in People With High-Risk Melanoma (V940-001). Available at: https://clinicaltrials.gov/study/NCT05933577. Last accessed: 16 October 2024.
6. A Study of the Efficacy and Safety of Adjuvant Autogene Cevumeran Plus Atezolizumab and mFOLFIRINOX Versus mFOLFIRINOX Alone in Participants With Resected Pancreatic Ductal Adenocarcinoma (IMCODE003). Available at: https://clinicaltrials.gov/study/NCT05968326. Last accessed: 16 October 2024.
7. Cancer Research UK. 2024. Available at: https://news.cancerresearchuk.org/2024/03/22/1-7-million-for-the-worl ds-first-vaccine-to-prevent-lung-cancer/. Last accessed: 6 September 2024.
8. National Health Service (NHS). Available at: https://www.england.nhs.uk/cancer/nhs-cancer-vaccine-launch-pad/. Last accessed: 6 September 2024.
9. Zong Y et al. Advanced Materials. 2023;35(51):2303261.
A type of breast cancer that tests positive for a protein called human epidermal growth factor 2 (HER2).
15 out of every 100
breast cancers are HER2+.
HER2+ means presence of gene mutation of ERBB2, causing overexpression of HER2, the protein encoded by this gene.
Future of HER2+ Breast Cancer
Selected ongoing clinical trials for HER2+ cancer:7–9
CompassHER2 RD
This Phase III trial is testing the efficacy of T-DM1, combined with tucatinib, in preventing breast cancer relapse in patients with high risk, HER2+ breast cancer.9
DESTINY-Breast11
A Phase III trial testing the efficacy and safety of trastuzumab deruxtecan as a neoadjuvant therapy in a neoadjuvant setting, in high-risk, HER2-positive early non-metastatic breast cancer.8
HER2Climb-02
A randomised, double-blind, placebo-controlled Phase III study to test the efficacy and safety of tucatinib and ado-trastuzumab emtansine (T-DM1) in patients with unresectable locally advanced or metastatic HER2+ breast cancer.7
Diagnosis, Treatment Options, and Prognosis2
Techniques used for HER2+ breast cancer diagnosis:2
Mammogram X-ray of the breast.
Immunohistochemistry (IHC test)
The IHC test gives a score of 0–3, where 0–1 is HER2-negative; 2 is a borderline diagnosis, and the amplification of the ERBB2 gene by ISH or FISH needs to be performed for positivity confirmation; whilst 3 is HER2+.
Current treatment options4–6
Patients will commonly receive a combination of the following:
Chemotherapy
Cytotoxicity of cancer cells.
Targeted
therapy
Drugs that specifically interfere with the ERBB2 signalling pathway. Types include monoclonal antibodies (e.g., trastuzumab, trastuzumab+pertuzumab), tyrosine kinase inhibitors (e.g., lapatinib), and antibody-drug conjugates (e.g., trastuzumab deruxtecan).
Fluorescence in situ hybridisation (FISH)
It is a laboratory technique that uses fluorescent probes to identify extra copies of the HER2 gene in breast cancer cells.
Endocrine therapy
Use medications that block oestrogen (e.g., tamoxifen), required for breast tumour growth. Endocrine therapy is administered to patients bearing tumours with hormone receptors.
Radiation therapy
Uses high-energy X-rays to slow or stop tumour.
References
Cleveland Clinic. 2023. Available at: https://my.clevelandclinic.org/health/diseases/25213-her2-positive-breast-cancer. Last accessed: 19 September 2024.
MD Anderson Cancer Center. 2022. Available at: https://www.mdanderson.org/cancerwise/her2-positive-breast-cancer--what-it-is--diagnosis-and-tr eatment.h00-159542112.html. Last accessed: 19 September 2024. Cancer Research UK. 2023. Available at: https://www.cancerresearchuk.org/about-cancer/breast-cancer/getting-diagnosed/tests-breast-canc er-cells#:~:text=HER2%20receptor%20tests,-Some%20breast%20cancers&text=About%2015%20out %20of%20every,control%20the%20growth%20of%20cancer. Last accessed: 19 September 2024.
Click here to download
4. WebMD. 2023. Available at: https://www.webmd.com/breast-cancer/her2-positive-treatments. Last accessed: 19 September 2024.
5. Zhu K et al. Biomarker Research. 2024;12(1):16.
6. UpToDate. 2022. Available at: https://www.uptodate.com/contents/treatment-of-early-her2-positive-breast-cancer-beyond-th e-basics/print. Last accessed: 19 September 2024.
7. Agostinetto E et al. Cell Reports Medicine. 2024;5:101575.
9. Yale Medicine. 2024. Available at: https://www.yalemedicine.org/clinical-trials/t-dm1-and-tucatinib-compared-with-t-dm1-alone-in -preventing-relapses-in-people-with-high-risk-her2. Last accessed: 16 October 2024.
Novel Approaches to Treat Glioblastoma Multiforme
Authors: Mark G. Malkin,1 Laurence Booth,2 Jane L. Roberts,2 Andrew Poklepovic,3 *Paul Dent2
1. Cleveland Clinic, Brain Tumour and Neuro-oncology Center, Ohio, USA
2. Department of Biochemistry and Molecular Biology, Virginia Commonwealth University, Richmond, USA
3. Department of Medicine, Virginia Commonwealth University, Richmond, USA
*Correspondence to paul.dent@vcuhealth.org
Disclosure: The authors have declared no conflicts of interest.
Glioblastoma multiforme (GBM), central nervous system (CNS) WHO Grade 4 astrocytoma, is an almost uniformly lethal disease with survival measured in months. Despite improved radiotherapeutic technologies and the use of the alkylating agent temozolomide, the majority of GBM patients succumb to their disease within approximately 17 months. This value has not been significantly altered in the past 20 years. Novel ‘outside the box’ therapeutic ideas will be required to prolong progression-free and overall survival in patients with an acceptable quality of life. This article discusses two recently completed clinical trials in primary diagnosed and recurrent GBM that were supported by the author’s earlier pre-clinical studies.
THE APPROACH
GBM is a particularly lethal malignancy when compared to many other types of solid tumours. The primary treatment option for GBM, surgery followed by radiotherapy
and temozolomide chemotherapy, was established 20 years ago by Stupp R et al.1 Recurrent GBM is most often treated with the anti-angiogenic agent bevacizumab and a nitrogen mustard such as lomustine, which enhances 6-month progression-free survival.2 None of these approaches are curative. Clearly, better treatment regimens, using previously untried novel cell biology concepts, need to be developed for patients in the primary diagnosis setting and in recurrent disease.
GBM is a heterogeneous disease, with multiple driving mutations present in different populations of cells simultaneously within the same tumour.3-5 This largely defeats the modern 'personalised medicine' concept of one drug for one target. Common driving mutations in GBM include loss of the lipid phosphatase PTEN, expression of the truncated mutant activated epidermal growth factor receptor variant III (EGFR vIII), loss of p53, and overexpression of platelet-derived growth factor receptor A (PDGFRa).6-8 More rarely, activating mutations in B-RAF are observed. Unlike other solid tumour types such as pancreatic and colon cancers,
GBM tumours do not express mutant RAS proteins. Thus, if an individual GBM tumour contains groups of cells, e.g., some expressing EGFR vIII and others expressing PDGFRa, any novel therapeutic approach will have to be developed that has a broad inhibitory spectrum against both primary driving oncogenes as well as any potential evolutionary escape/survival mechanisms.
An additional complication of treating GBM is the privileged environment within the CNS both restricting entry of therapeutic agents and by its unique cellular environment of neurons, astrocytes, microglia, and other cell types.9-12 GBM, like all tumours, requires a supporting cast of different cell types to facilitate its growth, invasion, and therapeutic resistance. For GBM, one vital supporting player is CNS-localised macrophages, the microglia.13,14 Minimally transformed astrocytes and microglia enter a symbiotic relationship where the transformed astrocyte releases growth factors, e.g., IL6, which activate the microglia. The activated microglia secrete additional growth factors and cytokines, which promote an inflammatory environment as well as the growth of the now well-established transformed astrocytes. As the transformed astrocytes progress through multiple cell cycles, genomic instability increases to the point where additional mutations/loss of tumour suppressors/gain of tumour promoters occur, and the transformed astrocyte eventually converts to become a malignant GBM tumour cell.15
Two novel therapeutic concepts have been developed by the authors' group, which attempted to address both the supporting role of microglia in GBM and interdiction of the multiple oncogenic drivers within any GBM tumour. Their initial concept was to suppress the actions of activated microglia, and for this they repurposed the multiple sclerosis drug dimethyl fumarate (DMF; NCT02337426).16 Subsequently, to simultaneously attack GBM cells regardless of their oncogenic drivers, the group developed a combination of the liver cancer drug sorafenib, the anti-seizure medication sodium valproate, and the erectile dysfunction agent sildenafil (NCT01817751).17
DMF is approved for the treatment of multiple sclerosis. DMF breaks down in plasma to the active agent monomethyl fumarate (MMF).18,19 The drug can inactivate T cells, but its mechanisms of action remain poorly understood. DMF has been shown to suppress the activities of microglia and astrocytes.20,21 Microglia and astrocytes have important roles in the biology and progression of glial tumours, and repurposing DMF could be useful, changing glial cell viability and prolonging survival. For example, using microglia freshly isolated from glial tumours, they found that MMF significantly and rapidly reduced their production of IL-6, TNF-α, and TNF-β.22,23
Treatment of microglia with MMF as a single agent for 24 hours killed glioma cells, and in tumours MMF, dramatically reduced the levels of microglia within the GBM tumour.
Based on the group's pre-clinical findings with DMF, they performed a Phase I trial to evaluate its safety and toxicity when combined with the standard Stupp protocol of concurrent radiotherapy and temozolomide followed by maintenance temozolomide.16 Twelve patients were treated at three dose levels. No doselimiting-toxicities were observed. The most common related adverse observations were haematologic (and typically seen with temozolomide alone): lymphopenia (58%), decreased CD4 count (17%), and thrombocytopenia (17%). The median progression-free survival (PFS) for all patients was 8.7 months, with no difference in PFS between those with stable disease (seven patients) or a partial response (four patients). The median overall survival (OS) was 13.8 months. For the six patients treated at the highest dose level, the median PFS was 11.8 months, and the median OS was 14.5 months. Due to the lack of sufficient power in the Phase I trial design, the group still does not definitively know whether DMF delivers any significant survival benefit to GBM patients.
The team then performed an additional series of pre-clinical studies in GBM cells where they combined DMF with another drug approved for MS, fingolimod.23 Fingolimod is an analog of sphingosine1-phosphate (S1P). Cells phosphorylate
fingolimod before it then, in an autocrine fashion, activates S1P receptors causing their proteolytic destruction. Immune cells lacking S1P receptors cannot migrate from lymph nodes to sites of myelin destruction.23 MMF and fingolimod combined to kill, more than either drug alone, primary GBM cells and activated microglia, and their synthesis of cytokines. In mice treated with DMF and fingolimod for 14 days, no obvious normal tissue damage was noted. The drugs radiosensitised cells and enhanced the efficacy of temozolomide. Due to financial issues (i.e., the very high cost of purchasing dimethyl fumarate and fingolimod by the authors' institution) no translational studies in GBM patients have yet been proposed.
Sorafenib was originally developed to inhibit the proto-oncogenes RAF-1 and B-RAF in the ERK1/2 MAP kinase pathway.24,25 The kinase domain of the S/T kinase RAF-1 has similarities to that of Y kinase SRC family proteins, and subsequently it was discovered that sorafenib inhibited Class III receptor tyrosine kinases such as PDGFRs and vascular endothelial growth factor receptors.26 More recently the group demonstrated that sorafenib is a low affinity inhibitor of Hsp90 and Hsp70 family chaperones. Hence, the biological activities of sorafenib result in the drug having a complex mechanism of anti-tumour action.27
The authors' initial studies with sorafenib combined the drug with the epigenetic modulator family of drugs, histone deacetylase inhibitors (HDACi), in gastrointestinal tumour cells. This research initially resulted in clinical trials in liver cancer and in pancreatic cancer (NCT01075113; NCT02349867).28 As would be expected with two agents that have a broad spectrum of action, the mechanisms by which tumour cells were killed are complex: death receptor signalling, ceramide generation, macroautophagy, reactive oxygen species generation, and calcium fluxes. Additional work demonstrated that sorafenib could interact with the HDACi and the antiseizure medication sodium valproate to kill CNS tumour cells, including those derived from primary GBM and primary medulloblastoma. The mechanism of tumour
cell killing was identical to that observed in gastrointestinal tumour cells. The sorafenib/ HDACi combinations radiosensitised GBM cells, and the group demonstrated using molecular tools that at least a portion of sorafenib’s anti-tumour activity was by inhibiting PDGFRa.29-31
One of the authors' earliest observations regarding the biology of sorafenib was that it caused an endoplasmic reticulum (ER) stress response.31 Subsequently, they demonstrated that clinically relevant free concentrations of sorafenib inhibited the ATPase activity of the key chaperone regulating the ER stress response, GRP78 (BiP, HSPA5).32 Under resting conditions GRP78 binds to PERK and IRE1, preventing them from signaling. When the levels of misfolded proteins in the ER increase, GRP78 dissociates from PERK and IRE1 to act as a chaperone, causing activation of PERK and IRE1. PERK, by the phosphorylation of serine 51, inactivates eIF2a resulting in the translation of approximately 90% of all mRNAs not being translated. Thus, proteins with particularly short half-lives, for example, the mitochondrial protective protein MCL1, have their expression reduced. For some genes, such as those for Beclin-1 and autophagy protein 5 (ATG5), eIF2a S51 phosphorylation increases their translation. Enhanced levels of Beclin1 and ATG5 would act to facilitate macroautophagy and the degradation of misfolded proteins. Once the misfolded proteins are degraded via autophagy, GRP78 re-associates with PERK, shutting of the ER stress response, and eIF2a is dephosphorylated by PP1.
Sildenafil is a phosphodiesterase 5 (PDE5) inhibitor.33 It is used both as a medication for blood pressure and for erectile dysfunction. Inhibition of PDE5 results in elevated levels of its substrate cyclic GMP and activation of protein kinase G (PKG).34 The biological actions of PKG are pleiotropic. PKG can act to increase expression of nitric oxide synthase, whose production of NO causes relaxation of smooth muscle in the cardiovascular system.35 In tumour cells, which generally produce orders of magnitude greater levels of reactive oxygen species than non-transformed cells, NO can
interact with oxygen radicals and hydrogen peroxide to generate peroxynitrite (ONOO¯), a short-lived but lethal free radical.36 Peroxynitrite causes lipid peroxidation, protein oxidation, and protein nitration, as well as DNA damage. Sildenafil synergises with sorafenib to kill tumour cells in vitro and in vivo, and this requires PKG signalling and the production of NO. This work resulted in a Phase I trial combining regorafenib and sildenafil in solid tumour patients (NCT02466802).37 Sildenafil has also been recognised as an inhibitor of plasma membrane drug-efflux pumps responsible for the blood–brain barrier and chemotherapy resistance, e.g., ABCB1 and ABCG2.38,39 Hence, the three-drug combination of sorafenib, valproate, and sildenafil has multiple overlapping two-drug combination synergies that collectively will act to kill tumour cells.
In the Phase II trial 'Sorafenib, Valproic Acid, and Sildenafil in Treating Patients with Recurrent High-Grade Glioma' (NCT01817751), the authors' combined sorafenib, valproate, and sildenafil based on validated concepts from their preclinical data, and that sildenafil may prevent the efflux of drugs out of the GBM cells.17 At the end of the trial, 33 patients were available for evaluation. The most frequently observed negative sequela, as a priori expected, was skin rash. A statistical difference in OS was seen between patients with ECOG PS of 1 versus 2. Based on the authors' pre-clinical studies, the trial also examined the expression of PDGFRa and GRP78 in each patient’s tumour as both are targets of sorafenib. OS was not different between tumours expressing high levels of PDGFRa compared to those with low levels (p<0.07). However, for the chaperone GRP78, OS was significantly higher for patients expressing low levels of the chaperone (p<0.0026). Tumours expressing high levels of GRP78 were associated with a shorter survival time than those expressing low levels of GRP78.
Of perhaps greater interest to the wider field, with respect to the authors' findings, is the extended survival of five patients (~15%) in the group who had the lowest protein levels of GRP78.17 For all patients
the median PFS was 3.7 months and OS 10.0 months. For the surviving five patients in the Kaplan-Meier tail, the mean value for PFS was 24.9 months (~2 years) and the mean OS value was 73.6 months (~6.1 years). The authors do not have a complete understanding as to how and why these five patients have extended survival, other than that they all had the lowest GRP78 levels. The five patients with lower GRP78 levels (15.2%) presently have a mean PFS of over 2-years and mean OS of over 6-years, and they all remain alive. This is three-times as many long-term survivors as the team would have predicted. The long-term survivors were four Europeanheritage males and one African American, both representing 16.7% of their respective populations within the trial.
GRP78 is predominantly found in the ER and PM, where it is known to act as a chaperone and as a regulatory protein required to maintain signalling through multiple intracellular signal transduction proteins, and to a lesser extent GRP78 is in the nucleus, where it acts as a cotranscription factor.40 At present the authors do not know which sub-population of this chaperone plays the most important role in mediating resistance to sorafenib plus valproate plus sildenafil, though they postulate it is likely to be the GRP78 populations both in the ER and in the PM. High levels of GRP78 are significantly associated with lower PFS and OS, but it is not known whether other chaperones in the HSP70 family, like GRP78, or those in the HSP90 family, also play a role in modulating ER stress signalling by PERK/PKR-eIF2a and are additional significant correlates to be examined in future studies.
The essential autophagosome-regulatory protein ATG16L1 has two isoforms. Although adult glioma survival rates for people of European and African heritage are similar, the team know that Europeans trend to express the ATG16L1 A300 isoform and those of African heritage trend to express the ATG16L1 T300 isoform.41 People homozygous for the T300 isoform are more able to facilitate autophagosome formation and digest antigenic materials in the gastrointestinal tract, which is a reason
why African Americans are less frequently diagnosed with Crohn’s disease.41,42 Hence, as a future long-term goal, the authors will need to statistically define whether the expression of GRP78 correlates with the isoform status of ATG16L1 to influence PFS and OS. This may provide additional clues for future studies as part of grant submissions in adult glioma.
CONCLUSION
The team at Virginia Commonwealth University has been very fortunate to not only translate into the clinic two investigator-initiated trials in GBM, but also perform many other trials in a variety of solid tumour types. Cancer developmental therapeutics by individual investigative teams, scientists, and physicians often encounter Himalayan sized obstacles preventing the team from taking any concept from the bedside to the bench and back to the bedside. First, there are 'technical issues' of obtaining drugs from drug companies who may not wish to collaborate with each other or further their development of a particular drug, with compromises therefore having to be made by investigators to use generic drugs, e.g., sodium valproate, over more expensive proprietary drugs in the same class such as vorinostat. Second, receiving regulatory approval from the FDA and local IRB can delay translation, particularly if the drugs
References
1. Stupp R et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20(5):1375-82.
2. Lin P et al. Increased infiltration of CD8 T cells in recurrent glioblastoma patients is a useful biomarker for assessing the response to combined bevacizumab and lomustine therapy. Int Immunopharmacol. 2021;97:107826.
3. Mathur R et al. Glioblastoma evolution and heterogeneity from a 3D whole-tumor perspective. Cell. 2024;187(2):446-63.e16.
4. Nóbrega AHL et al. Neuroinflammation in glioblastoma: the role of the
have overlapping normal tissue toxicities that will require careful lead-in dose escalation approaches in the clinical protocol.
Third, with the first two issues paling into insignificance, is that translating a concept from the bench to the bedside must consider the cost of patient care and having all the necessary protocols overseen by regulatory committees within the academic cancer center. For example, a small 3x3 two-agent Phase I trial, with gratis supply of drugs, still has regulatory and healthcare costs that run into the many hundreds of thousands of dollars. The cost of a twodrug Phase I trial where both drugs must be commercially purchased can run into many millions of dollars, which is prohibitively expensive for any academic cancer centre. Hence, in an ideal world for GBM patients, who experience rapid morbidity and mortality, conceptually, scientists and physicians need to 'think outside the box' to increase the number of rapidly deployable therapeutic options. Perhaps, in the future, novel drug combinations that are known to have benign toxicity profiles and have exhibited broad anti-cancer effects in other solid tumour types could be more rapidly processed through the standard approvals process, including new laws that facilitate the billing of trial drug and healthcare costs to insurance, resulting in the more rapid delivery of new GBM therapeutic options.
microenvironment in tumour progression. Curr Cancer Drug Targets. 2024;24(6):579-94.
5. Verduin M et al. Patient-derived glioblastoma organoids reflect tumor heterogeneity and treatment sensitivity. Neurooncol Adv. 2023;5(1):vdad152.
6. Eisenbarth D, Wang YA. Glioblastoma heterogeneity at single cell resolution. Oncogene. 2023;42:2155-65.
7. Verdugo E et al. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun (Lond). 2022;42:1083-111.
8. Decraene B et al. Cellular and molecular features related to exceptional therapy response and extreme long-term survival in glioblastoma. Cancer Med. 2023;12(10):11107-26.
9. Sevenich L. Turning "cold" into "hot" tumours-opportunities and challenges for radio-immunotherapy against primary and metastatic brain cancers. Front Oncol. 2019;9:163.
10. Van Gool S et al. Dendritic cell therapy of high-grade gliomas. Brain Pathol. 2009;19:694-712.
11. Jiang S et al. Cathepsin B-responsive programmed brain targeted delivery system for chemo-immunotherapy combination therapy of glioblastoma. ACS Nano. 2024;18(8):6445-62.
12. Zhao C et al. Lipid-based nanoparticles to address the limitations of GBM therapy by overcoming the bloodbrain barrier, targeting glioblastoma stem cells, and counteracting
the immunosuppressive tumour microenvironment. Biomed Pharmacother. 2024; 171:116113.
13. Weyer MP et al. Repurposing of pexidartinib for microglia depletion and renewal. Pharmacol Ther. 2024;253:108565.
14. Genoud V et al. Therapeutic targeting of glioblastoma and the interactions with its microenvironment. Cancers (Basel). 2023;15:5790.
15. Trevisi G, Mangiola A. Current knowledge about the peritumoural microenvironment in glioblastoma. Cancers (Basel). 2023;15:5460.
16. Shafer D et al. Phase I trial of dimethyl fumarate, temozolomide, and radiation therapy in glioblastoma. Neurooncol Adv. 2020;2:vdz052.
17. Poklepovic AS et al. Phase 2 study of sorafenib, valproic acid, and sildenafil in the treatment of recurrent highgrade glioma. medRxiv. 2024:DOI: 10.1101/2024.04.23.24304634.
18. Schmidt MM, Dringen R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem Int. 2010;57:460-7.
19. Linker RA et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678-92.
20. Wilms H et al. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;19:7-30.
21. Ghods AJ et al. Beneficial actions of the anti-inflammatory dimethyl fumarate in glioblastomas. Surg Neurol Int. 2013;4:160.
22. Booth L et al. Regulation of dimethylfumarate toxicity by proteasome inhibitors. Cancer Biol Ther. 2014;15:1646-57.
23. Dent P et al. Fingolimod augments monomethylfumarate killing of GBM Cells. Front Oncol. 2020;10:22.
24. Beeram M et al. Regulation of c-Raf-1: therapeutic implications. Clin Adv Hematol Oncol. 2003;1:476-81.
25. Bollag G et al. Raf pathway inhibitors in oncology. Curr Opin Investig Drugs. 2003;4:1436-41.
26. Riely GJ, Miller VA. Vascular endothelial growth factor trap in non-small cell lung cancer. Clin Cancer Res. 2007;13:s4623-7.
27. Booth L et al. Multi-kinase inhibitors can associate with heat shock proteins through their NH2-termini by which they suppress chaperone function. Oncotarget. 2016;7:12975-96.
28. Gordon SW et al. Phase I study of sorafenib and vorinostat in advanced hepatocellular carcinoma. Am J Clin Oncol. 2019;42:649-54.
29. Tavallai M et al. Nexavar/stivarga and viagra interact to kill tumour cells. J Cell Physiol. 2015;230:2281-98.
30. Tang Y et al. Sorafenib and HDAC inhibitors synergize to kill CNS tumour cells. Cancer Biol Ther. 2012;13:567-74.
31. Park MA et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumour cells through a Ca(2+)-de novo ceramide-PP2Areactive oxygen species-dependent signaling pathway. Cancer Res. 2010;70:6313-24.
32. Roberts JL et al. GRP78/Dna K Is a Target for nexavar/stivarga/votrient in the treatment of human malignancies, viral infections and bacterial diseases. J Cell Physiol. 2015; 230:2552-78.
34. Booth L et al. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8:1449-68.
35. Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280:12944-55
36. Fujii J, Osaki T. Involvement of nitric oxide in protecting against radical species and autoregulation of m1polarized macrophages through metabolic remodeling. Molecules. 2023;28:814.
37. Poklepovic AS et al. A Phase 1 Study of Regorafenib and Sildenafil in Adults with Advanced Solid Tumours. Anticancer Drugs. 2024;35(5):450-8.
38. Shi Z et al. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011;71:3029-41.
39. Agarwal S et al. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223-33.
40. Liu Z et al. ER chaperone GRP78/BiP translocates to the nucleus under stress and acts as a transcriptional regulator. Proc Natl Acad Sci U S A. 2023;120:e2303448120.
41. Grimm WA et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut. 2016;65:456-64.
42. Messer JS et al. The Crohn's disease: associated atg16l1 variant and salmonella invasion. BMJ Open. 2013;3:e002790.
Comparison of Breast Sensibility Following Breast Reconstruction with Two Different Techniques: Deep Inferior Epigastric Perforator Flap and Implant
Editor's Pick
I have selected this article as an Editor's Pick for its critical exploration of breast sensibility outcomes following two widely used reconstruction techniques: deep inferior epigastric perforator flap and implant-based reconstruction. Sensory recovery is a key yet often underreported factor that significantly impacts patient satisfaction, safety, and long-term quality of life after mastectomy. This study highlights important clinical outcomes, examining sociodemographic and clinical variables that influence recovery.
Prof Ahmad Awada
Université Libre de Bruxelles, Belgium
Author: *Soraya
Tadimi Tazi1,2
1. School of Medicine, Faculty of Biomedical Science and Health, Universidad Europea de Madrid, Spain
*Correspondence to soraya.tt@live.fr
Disclosure: The author has declared no conflicts of interest.
Received: 08.05.24
Accepted: 02.10.24
2. Department of Plastic and Reconstructive Surgery, University Hospital of Getafe, Madrid, Spain
Keywords: Breast sensibility, breast reconstruction, deep inferior epigastric perforator (DIEP) abdominal flap, implant, Semmes-Weinstein monofilaments, thermal discrimination, quality of life, BREAST-Q.
Introduction: Breast sensibility following reconstruction surgery, though often overlooked, holds significant importance due to its widespread occurrence and profound effects on patients’ well-being and safety. The author’s objective is to compare the sensory outcomes between deep inferior epigastric perforator (DIEP) flap microsurgery reconstruction and implant-based reconstruction. Additionally, the potential influence of sociodemographic and clinical factors on sensory recovery is explored, along with assessing temperature discrimination abilities and evaluating quality of life.
Material and methods: An ambispective descriptive-analytical study was conducted involving women who underwent mastectomy with reconstruction using either DIEP flap
and implants at the author’s hospital between 1990–2021. Data were collected from medical records, patient histories, physical examinations, and validated quality of life questionnaires (BREAST-Q). Spearman’s or Pearson’s correlation coefficients were employed for the analysis of quantitative variables, while the Student’s T test or Mann-Whitney U test were used to compare quantitative and qualitative variables.
Results: A total of 99 women with breast reconstruction were included, 47 with DIEP flap and 52 with implants. Sensory recovery in implant-based reconstruction was found to be superior to DIEP flap reconstruction (5.03 and 5.18, respectively; p<0.005). A direct correlation was observed between sensory improvement and thermal discrimination (Spearman coefficient 0.9; p<0.001). Factors such as radiotherapy, delayed reconstruction with DIEP flap, and height with implants were associated with poorer sensory recovery in the breast. Women experiencing better breast sensibility reported higher satisfaction with their surgeon, medical team, and psychosocial quality of life.
Conclusion: Women reconstructed with implants or DIEP abdominal flap exhibit suboptimal overall sensory recovery of the reconstructed breast, with slightly better outcomes observed in women reconstructed with implants. The development of novel surgical techniques aimed at enhancing sensibility after breast reconstruction could significantly benefit these patients.
Key Points
1. The study compares sensory outcomes between deep inferior epigastric perforator flap and implant-based breast reconstruction, with implant-based reconstruction showing slightly better sensory recovery.
2. The research provides valuable insight into the factors influencing sensory recovery, which can guide surgeons in making more informed decisions about the best reconstruction technique for each patient.
3. The findings emphasise the importance of considering both aesthetic and functional outcomes, aiming to enhance patient satisfaction and quality of life after breast reconstruction surgery.
INTRODUCTION
Breast cancer is the most common cancer worldwide, accounting for 12.5% of all new cancer cases per year.1 According to the Spanish Society of Medical Oncology (SEOM), 34,750 women were diagnosed with breast cancer in Spain in 2022.2 Furthermore, it is the leading cause of cancer-related death among Spanish women. Due to its significant prevalence and mortality, the modernisation of breast reconstruction techniques is necessary to offer better results and quality of life for those women.3
At the hospital where this study was conducted, the two main procedures performed were microsurgical reconstruction with the deep inferior
epigastric perforator (DIEP) abdominal flap and reconstruction with implants.
Microsurgical breast reconstruction with DIEP involves taking an abdominal flap with its skin, fat, and blood vessels, and extracting it from the body for transplantation to the breast of the same patient.4,5 This technique was described by surgeon Koshima in 1989 and has been used since then, currently being one of the most popular techniques for breast reconstruction.6 Based on several clinical trials, the DIEP flap has proven to be a valuable reconstructive method for many women, with low complications and a significant satisfaction with the result, as it creates a natural appearance, identical touch, and consistency to the healthy breast. Unlike the transverse rectus abdominus myocutaneous (TRAM) flap, which uses
muscle, the DIEP flap spares muscle, resulting in shorter recovery times and fewer complications, such as hernias.7-9 Therefore, nowadays reconstruction with the DIEP abdominal flap is preferred.10,11 Furthermore, innervation to the flap is increasingly being included in DIEP surgical procedures to improve sensory recovery in that area. Research using sensory segments of the intercostal nerve has shown promising results, as it has been demonstrated that sensory recovery in innervated flaps is superior and develops earlier compared to non-innervated flaps.12,13 Another study has confirmed improved breast sensibility with innervated flaps, as well as a higher physical well-being with breast appearance in the BREAST-Q questionnaire.14
Reconstruction using implants is a breast reconstruction alternative, which was first described in the 1960s and is realised by placing a submuscular implant below the pectoralis major muscle. At the author’s hospital, reconstruction with an expander is used initially to allow breast tissue expansion. The expander, equipped with a valve for fluid inflation, is gradually enlarged over several months until the desired breast size is reached, followed by a second surgery to insert the permanent implant.15 The main drawbacks are the requirement of twostage surgery and the less natural result. However, the main complication is capsular contracture, which is the formation of fibrotic scar tissue around the implant, resulting in hardness, discomfort, and pain in the breast area. This complication is often associated with radiotherapy, making implant-based reconstruction unsuitable. Radiotherapy thins the skin, limits breast projection, hampers healing, and can cause implant perforation or extrusion.16,17
Both reconstruction techniques can be performed simultaneously with mastectomy or in a second-stage surgery. Delayed reconstruction allows histopathological analysis of the tumour and accomplishment of oncological treatment before reconstruction.18 Both techniques are valid, but studies have shown better recovery and fewer complications in patients undergoing two-stage reconstruction, although it is always necessary to evaluate each
patient’s individual preference. However, immediate breast reconstruction is increasingly performed because several studies have not shown a higher risk of complications or worse patient satisfaction with breast appearance.18-20
Regarding breast sensibility, it is a poorly investigated matter because research has prioritised achieving better aesthetic and long-lasting results after surgery.21,22 However, it is important to assess breast sensibility, as it has an impact on women’s intimate life, and having good sensibility prevents injuries, such as traumas or burns. Sensibility is measured by the cutaneous pressure threshold in nine areas of the breast using Semmes-Weinstein monofilaments, being held in each area for approximately 1.5 seconds. The measurement takes place in a consultation room with appropriate temperature and humidity, with the patient lying down, the breast exposed, and their eyes closed.23,24 Therefore, measuring the degree of breast sensory recovery with these two techniques will allow data and conclusions to be established to better understand which factors influence breast sensibility and compare which procedure offers greater sensory recovery. This study will help apply these results in surgical procedures or future research, to improve the quality of life of women who are breast cancer survivors.25
MATERIAL AND METHODS
Objectives
Main objective
To compare the level of breast sensibility with microsurgical reconstruction and implant reconstruction in women undergoing breast reconstruction with DIEP and implants at the Plastic Surgery Department of the author’s hospital from 1990–2021.
Secondary objectives
• To compare thermal discrimination to heat and cold between DIEP reconstruction and breast
implant reconstruction.
• To assess the aesthetic and sensory satisfaction of patients using the Breast-Q questionnaire, thereby evaluating their quality of life and postoperative satisfaction.
• To determine whether previous neuropathies, cardiovascular risk factors (such as diabetes or smoking), weight, or age influence sensibility.
• To investigate differences in sensory recovery among women who have received chemotherapy, radiotherapy, or hormonotherapy, and whether it was administrated in a neoadjuvant or adjuvant setting.
• To compare differences in sensory recovery between immediate and delayed breast reconstruction.
Study Population
A study involving 99 women who underwent breast reconstruction surgery at the Plastic Surgery Department of the University Hospital of Getafe from 1990–2021 was conducted. The procedures included microsurgical reconstruction with the DIEP flap and reconstruction with implants.
Inclusion criteria
All women aged 18 years and above who underwent primary breast reconstruction with implants or DIEP following breast cancer mastectomy.
Exclusion criteria
Women who underwent mastectomy of the contralateral breast were excluded from the study. Additionally, women who experienced any complications or implant/ flap rejection resulting in its removal were excluded. Those who later experienced tumour recurrence were also excluded.
Data Collection
Each patient was selected from a database provided by the Plastic Surgery Department at the University Hospital of Getafe, Madrid, Spain, ensuring adherence to the inclusion and exclusion criteria. Their medical records were accessed to gather comprehensive information regarding their intervention, surgical procedure, recovery, oncological treatment, and medical history. Subsequently, selected
patients were scheduled for a consultation to perform sensibility measurement, thermal discrimination test, medical history review, and physical examination. To avoid biases, appropriate temperature and humidity levels were maintained in the consultation room.
For quantifying breast sensibility, SemmesWeinstein monofilaments were utilised. These nylon filaments facilitate the assessment of tactile sensibility in specific areas. The filaments bend under applied pressure, maintaining a constant pressure of typically 10 g regardless of the force exerted by the examiner. Different types of monofilaments, identified by numbers ranging from 1.65–6.65, were used, with higher values indicating larger diameters and poorer skin sensibility in the area being studied.
The breast was divided into nine different points, starting from the nipple areola.
Semmes-Weinstein monofilaments were applied to each point on both breasts, while patients lay on the consultation bed with their eyes closed.12 Patients were asked to indicate whether they felt each monofilament, and the smallest monofilament with which skin sensibility was perceived was recorded for each area. Following the sensory examination, a thermal discrimination test was conducted using bottles of physiological saline heated to 60 °C and cooled to 4 °C, assessing the ability to discriminate temperature at the nine points of both breasts.
After the sensory and thermal examinations, patients were given the BREAST-Q questionnaire, enabling evaluation of satisfaction levels across various aspects of the breast reconstruction process. This questionnaire addressed satisfaction with the breast, overall outcome, quality of care received, and quality of life (including physical, psychological, and sexual wellbeing of the patient).
Statistical Analysis
In the descriptive analysis, qualitative data were presented as relative frequencies (n) and absolute percentages (%). For
quantitative variables, the team first verified normal distribution using the ShapiroWilk test. If the variables exhibited normal distribution, they computed the mean and standard deviation. Alternatively, if the normal distribution was not observed, they reported the median and interquartile range.
In the bivariate analysis, various tests were employed to evaluate the relationship between breast sensibility and potential influencing factors. Specifically, for analysing the association of quantitative variables, the team utilised Pearson correlation when normal distribution was met and Spearman correlation otherwise. To examine the association between a quantitative variable and a dichotomous qualitative variable, they employed Student’s t-test for independent samples under normality assumptions, or the MannWhitney U test otherwise. In cases where the qualitative variable was polytomous, ANOVA for independent samples was applied if the variables followed a normal distribution, while the Kruskal-Wallis test was used if not.
A p-value <0.05 was considered statistically significant.
RESULTS
Population Description
Between 1990–2021, the team identified 877 mastectomised patients who had undergone breast reconstruction at the hospital. Among them, 103 underwent reconstruction with DIEP flap and 774 underwent reconstruction with implants.
Of the 103 patients who underwent DIEP abdominal flap reconstruction between 2005–2020, all met the inclusion criteria, but some were excluded for the following reasons: two due to death, 20 because they did not want to participate in the study, and 34 due to lack of time and availability.
Of the 774 patients who underwent prosthetic reconstruction, 88 were excluded due to death, 85 because they did not
want to participate in the study, and 174 due to lack of time and availability. Another 427 patients were excluded due to lack of data and or not meeting the criteria due to the following reasons: having subsequent reconstructions with other surgical techniques (DIEP, TRAM), removal of the prosthesis, or having undergone bilateral mastectomy. Ultimately, the sample consisted of 47 patients with DIEP reconstruction and 52 patients with implant reconstruction, being the sample with both types of reconstruction, 99 patients.
Out of the 99 women included in the study, the age range was 38–82 years old, with a significant age gap between the two reconstruction techniques, indicating that women with implants were, on average, 13.7 years older.
Regarding the clinical history of the studied patients, the pack years index had a higher median in women reconstructed with DIEP flap (11.3) compared to those with implants (8.97). Similarly, the relative frequency of smokers was higher among women reconstructed with DIEP flap compared to those with implants (19.1% and 9.6%, respectively). Moreover, the relative frequency of smokers during the reconstruction process, spanning from reconstructive surgery to the following 18 months, was slightly higher in women reconstructed with DIEP flap.
There were notable differences in the oncologic treatment received between both reconstructions. While tumour resection prior to mastectomy was common overall (25.3%), it was slightly more prevalent in women with implants than in those with DIEP flap (28.8% versus 21.3%, respectively). However, women reconstructed with DIEP flap received more chemotherapy, immunotherapy, and hormonotherapy than those with implants. In both types of reconstructions, chemotherapy and radiotherapy were administrated adjuvant rather than neoadjuvant. Additionally, there were mostly no postoperative complications, with touch-ups being the most frequent complication.
The surgeries performed on the contralateral breast differed significantly between the two reconstruction techniques. For instance, most women with flap reconstruction did not undergo any intervention on the other breast (38.3%), and among those who did, breast reduction was the most common procedure (31.9%), followed by mastopexy (25.5%). Conversely, among women with implant reconstruction, the vast majority underwent mastopexy on the contralateral breast (65.4%), followed by breast reduction (21.2%).
In the study, patients who underwent implant-based reconstruction had been reconstructed for a longer period than those reconstructed with DIEP flap, with a statistically significant difference of 8.87 years.
Comparison Of Breast Sensibility
Breast skin sensibility is better with lower Semmes-Weinstein monofilament values depicted in the graphs. Reference values are based on measurements from areas 1–4 and 5–9, which are 4.54±1.16 and 5.69±0.93
with DIEP compared to 4.48±0.97 and 5.47±0.80 with implants, respectively. Consequently, skin sensibility is slightly higher in breasts reconstructed with implants than in those reconstructed with DIEP. The effect size (Cohen’s d) for this difference is 0.18, indicating a small effect. The difference in skin sensibility between DIEP and implant can be observed visually in Figure 1, where a positive difference indicates higher sensibility with abdominal flap compared to implants, suggesting poorer sensibility with DIEP.
The sensibility differences illustrate the reduced sensibility of the reconstructed breast in comparison to the healthy one (Figure 2). Across most points (excluding 1 and 4), differences are more significant with DIEP than with implants, signifying better sensitivity recovery with implantbased reconstruction.
Thermal Discrimination Ability
Regarding heat sensitivity, breast reconstructions with implants demonstrate superior responsiveness to hot temperatures
Figure 1: Sensibility differences between deep inferior epigastric perforator and implant reconstructions across nine points.
p-value is not significant p-value is significant
P value of the paired samples T-test.
Figure 2: Sensibility differences between reconstructed and healthy breasts with deep inferior epigastric perforator and implants.
Article
P value of the paired samples T-test (p<0.001).
DIEP: deep inferior epigastric perforator.
in seven of the evaluated areas compared to those with DIEP. Additionally, across both reconstruction methods, the capacity to detect heat is notably higher in areas 1–4 compared to areas 5–9, with frequencies falling below 30% in the latter. For cold sensitivity, eight out of the nine studied areas exhibit better responsiveness to cold temperatures in implant-based reconstruction compared to DIEP flap reconstruction. Similarly, it is observed that in both reconstruction techniques, the ability to detect cold is notably higher in areas 1–4 than in areas 5–9. However, it is worth mentioning that frequencies in detecting cold in areas 5–9 are relatively higher in implant reconstruction.
The correlation between the mean breast sensibility and thermal discrimination with each reconstruction technique in the nine points was assessed using Pearson’s rank correlation. Statistically significant associations were found for both reconstruction techniques. Thus, a lower mean sensibility calculated with monofilaments corresponded to a greater heat and cold sensibility.
BREAST-Q Questionnaire
The Breast-Q questionnaire assesses satisfaction levels across various aspects of the reconstruction process, with a higher score indicating greater personal satisfaction with each scale. In both reconstruction techniques, scores are consistently above 50% across all scales, except for physical well-being with the postoperative chest in both reconstructions and satisfaction with the effects of radiation in microsurgical reconstruction.
To compare DIEP and implant reconstructions, the score difference is calculated for scales present in both BREAST-Q questionnaires. The satisfaction scores for breasts in the preoperative period, and with information and surgeon, are higher in DIEP reconstruction than in implant-based reconstruction (p<0.005). The effect sizes (Cohen’s d) for these differences are 0.62 for preoperative breast satisfaction, 0.58 for satisfaction with information, and 0.55 for satisfaction with the surgeon, indicating medium effects.
The correlation between BREAST-Q questionnaire scores and sensibility was
statistically analysed for each breast reconstruction. Higher breast sensibility with implant-based reconstruction corresponds to higher scores in satisfaction
with their breasts preoperatively, satisfaction with the surgeon, and satisfaction with the medical team (p<0.005).
Table 1: Bivariate analysis of the correlation between sensibility and qualitative and quantitative variables for each type of reconstruction.
treatment
Table 1 continued.
P value for dichotomous variables with Student's t-test or Mann-Whitney U test and for non-dichotomous variables with ANOVA or Kruskal-Wallis test.
*P value is stastically significant (<0.05).
BMI: body mass index; DIEP: deep inferior epigastric perforator; NAC: nipple-areola complex; PYI : pack years index; r: Spearman’s correlation coefficient; SLNB: sentinel lymph node biopsy.
Sensibility and SociodemographicClinical Variables
The association between breast sensibility and the studied sociodemographic and clinical variables is observed in Table 1 In implant-based reconstruction, a statistically significant yet mild correlation is found between sensibility and height (r=0.36; p=0.008), indicating that higher height corresponds to lower monofilament values and therefore better cutaneous sensibility. This represents a medium effect size (Table 1).
In DIEP flap reconstruction, statistically significant differences are noted between sensibility and radiotherapy (p=0.007), indicating worse sensibility in women treated with radiotherapy. The effect size
(Cohen’s d) for this difference is 0.82, representing a large effect. Additionally, better breast sensibility is associated with adjuvant radiotherapy received after reconstruction rather than neoadjuvant (p<0.005). Furthermore, statistically significant differences are observed regarding the timing of reconstruction in relation to mastectomy (p=0.03), revealing worse breast sensibility when reconstruction is delayed. The effect size (Cohen’s d) for this difference is 0.64, indicating a medium effect (Table 1).
DISCUSSION
The aim of this study was to compare breast sensibility between the two most
prevalent breast reconstruction methods: DIEP flap and breast implants. Generally, sensibility with implants is superior due to minimal skin modifications during reconstruction. Conversely, with DIEP microsurgical reconstruction, transplanted autologous abdominal skin tissue may lead to insensitivity in the breast area due to skin modifications.
However, the difference in breast sensibility between the two techniques was smaller than expected.22 The mean sensibility measured at points 1–9 was 5.03±1.01 with implants and 5.18±1.18 with DIEP, varying by only 0.21 monofilament units. This slight difference may be attributed to insensitivity caused by radical mastectomy, irrespective of the reconstruction method employed.
Breast sensibility is generally better with implants, except at points 1 and 4, where DIEP reconstruction exhibits higher sensitivity. These two points correspond to the upper quadrant of the breast, where the skin is preserved during the DIEP flap reconstruction process. Plus, a significant loss of sensibility in flap skin leads to higher sensibility with implants at points 5–9.
Secondary objectives focused on evaluating thermal discrimination capacity, patient satisfaction with the reconstruction process, and other clinical and sociodemographic factors potentially related to sensibility. In the thermal discrimination test, the ability to discern cold and heat was generally better in implant-based reconstruction, except at point 4, possibly due to the presence of healthy skin in DIEP reconstruction at this point. However, both reconstructions exhibited difficulty in determining heat at points 5–9, increasing susceptibility to accidental burns. Higher scores on the Breast-Q questionnaire were found in satisfaction with the surgeon, the medical team, and other staff, as well as in psychosocial well-being. Women with DIEP reconstruction reported greater satisfaction with the information and with their breasts preoperatively compared to those with implants. Additionally, higher breast sensibility correlated with increased satisfaction with the surgeon and medical team in women with implants.
Although the BREAST-Q scores provided useful insights into patient satisfaction, a deeper analysis of the qualitative feedback would enrich the understanding of its impact on quality of life. Some patients were unable to respond to open-ended questions due to time constraints, and while others provided detailed feedback, this qualitative data has not yet been fully analysed. However, during clinical examinations, patients frequently expressed high satisfaction with their care, which underscores the importance of examining this qualitative feedback in future studies for a more comprehensive view of patient experience.
Limitations of the study include a lack of research comparing breast sensibility between microsurgical reconstruction and implants, constraining comprehensive literature review, and results comparison. Time constraints also affected data collection, as being a prospective study, scheduling with patients depends on availability, leading to potential biases in patient selection.
One of the critical limitations of this study was the time bias between the two groups, where women who underwent implant-based reconstruction had their surgeries, on average, nearly a decade earlier than those who received DIEP flaps. This difference in time since surgery is a significant factor when considering sensory recovery outcomes, as multiple studies have shown that sensory recovery after breast reconstruction improves gradually over time. Since the implant group had significantly more time to heal, their sensory recovery may appear better simply because they have had more time for sensory reinnervation and adaptation. In contrast, women in the DIEP group, with less time since surgery, might still be in earlier phases of sensory recovery, leading to a lower measured sensitivity at the time of assessment. This temporal discrepancy could confound the results, making it difficult to attribute the differences in sensory recovery solely to the reconstruction technique. Women who underwent implant reconstruction may have benefited from having more post-surgery years, giving nerves more time to regenerate, whereas the sensory
outcomes for the DIEP group might improve further with additional follow-up time.26 Thus, the study’s finding that implantbased reconstruction results in superior sensory recovery should be interpreted with caution, as it may partially reflect this difference in recovery timelines rather than a true advantage of one technique over the other. To mitigate this bias in future research, controlling for time since surgery in statistical analyses could provide a more accurate comparison of sensory outcomes between the two groups.
The role of radiotherapy in influencing breast sensibility outcomes is nuanced, with several factors playing a key role. This study highlighted that timing, particularly the distinction between neoadjuvant and adjuvant radiotherapy, has a significant impact on sensory recovery. Women who received adjuvant radiotherapy demonstrated better sensory outcomes compared to those who received it in a neoadjuvant setting. This suggests that delaying radiotherapy until after reconstruction may allow for improved nerve regeneration or reduce the adverse effects of radiation on the healing process.27,28 However, while timing appears to be a critical factor, this study is limited in its exploration of specific radiotherapy dosage and cycle variations. These variables could also influence sensory recovery but were not extensively examined in this dataset. Future research with a more detailed focus on the radiotherapy regimen, including dosage and the number of cycles, would be necessary to fully understand their relationship with breast sensibility.
In addition to sensibility outcomes, it’s important to consider other factors that influence the choice between DIEP flap and implant-based reconstructions. Cost-effectiveness analysis shows that while DIEP flap procedures have higher initial costs due to longer operating times and hospital stays, they may be more cost-effective in the long term due to fewer complications and revision surgeries compared to implant-based reconstructions.29 Recovery times also differ significantly; patients undergoing DIEP flap reconstruction typically require
a hospital stay of 3–5 days and 4–6 weeks before returning to normal activities, whereas implant-based reconstruction patients often have shorter hospital stays of 1–2 days and can resume normal activities within 2–4 weeks.30 However, implant-based reconstructions may require more frequent follow-up visits and potential revision surgeries over time due to complications such as capsular contracture or implant rupture.31,32 These factors should be carefully considered alongside sensibility outcomes when deciding between reconstruction techniques.
CONCLUSION
Breast sensibility at points 1–4 in women reconstructed with implants averaged 4.48±0.97, while in women with DIEP it averaged 4.54±1.16, indicating a minimal difference of 0.06 monofilament units. At points 5–9, sensibility with implantbased reconstruction averaged 5.47±0.8, compared to 5.69±0.93 with microsurgical reconstruction, showing a difference of 0.23 monofilament units. Overall, sensitivity recovery was slightly better in implantbased reconstruction, although sensibility is higher with DIEP than with implants at point 4. Moreover, sensibility was constantly better at points 1–4 than at points 5–9 of the breast. The difference between DIEP and implant reconstruction across all points of the breast (1–9) was 0.21 units, indicating superior sensibility in breasts reconstructed with implants.
The capacity for thermal discrimination, both for heat and cold is generally better in implant reconstruction compared to DIEP reconstruction, except at point 4. Thermal discrimination with implant-based reconstruction constantly exceeded 50% for points 1–4. In both breast reconstruction techniques, the inability to discriminate thermal stimuli is associated with poorer skin sensibility.
In DIEP microsurgical reconstruction, treatment with radiotherapy is associated with decreased breast sensibility, and adjuvant radiotherapy is linked to better sensibility compared to neoadjuvant
therapy. Additionally, women undergoing delayed reconstruction have poorer sensibility than those with immediate reconstruction post-mastectomy. In implant reconstruction, increased sensibility is associated with greater patient height. However, the number and duration of oncological treatments do not seem to influence breast sensibility in either reconstruction method.
Regarding the BREAST-Q questionnaire, most scale scores exceed 50%, indicating high satisfaction levels, particularly in psychosocial well-being. However, scores were lower than 50% in physical well-being with the postoperative chest and in adverse effects of radiotherapy. In implant-based reconstruction, higher sensibility values are correlated with greater satisfaction with breasts preoperatively, the surgeon, and the medical team.
This study provided valuable insights into breast reconstruction techniques, comparing sensory recovery between DIEP flap and implant-based methods. While implant-based reconstruction showed slightly better overall sensory recovery, DIEP flap offered long-term costeffectiveness and fewer complications. Importantly, this research can aid in patient care and decision-making, guiding personalised reconstruction choices based on individual needs. Furthermore, as microsurgical techniques continue to evolve,
References
1. Breastcancer.org. Datos y estadísticas sobre el cáncer de mama. 2023. Available at: https://www.breastcancer. org/es/datos-estadisticas. Last accessed: 5 April 2024.
2. SEOM. Las cifras del cáncer en España. 2022. Available at: https:// seom.org/images/LAS_CIFRAS_DEL_ CANCER_EN_ESPANA_2022.pdf. Last accessed: 5 April 2024.
3. Kaya B, Serel S. Breast reconstruction. Exp Oncol. 2013;35(4):280-6
4. Breastcancer.org. Colgajo DIEP. 2023. Available at: https://www. breastcancer.org/es/tratamiento/ cirugia/reconstruccion-mamaria/tipos/ colgajo-autogeno/diep. Last accessed: 5 April 2024.
the potential of innervated flaps presents a promising avenue for improving sensory recovery, which could significantly enhance patient outcomes. Future studies should focus on this emerging area to further refine breast reconstruction techniques.
ETHICAL STATEMENT
The research study was approved by the Committee of Ethics in Drug Research (CEIm) of the University Hospital of Getafe on 30 January 2023. The research study was conducted in accordance with bioethics regulations following the Helsinki Declaration, the Belmont Report; the Oviedo Convention on Human Rights and Biomedicine; and Law 14/2007, of July 3, on biomedical research. It also complied with EU legislation on personal data, specifically Organic Law 3/2018, of December 5, on the Protection of Personal Data and guarantee of digital rights, Royal Decree 1720/2007, Law 41/2002, of November 14, which regulates the basic rights of patients and obligations regarding information and clinical documentation. All study patients were identified by a case code in a database, through pseudonymisation of the initial database. Confidentiality of patients’ personal data was respected at all times, and data could not be copied or used for purposes other than those determined, nor transferred to other individuals not involved in the study.
5. American Cancer Society. Reconstrucción del seno con sus propios tejidos (procedimientos de colgajo). 2019. Available at: https:// www.cancer.org/es/cancer/cancer-deseno/cirugia-reconstructiva/opcionesde-reconstruccion-del-seno/ reconstruccion-del-seno-usando-suspropios-tejidos.html. Last accessed: 5 April 2024.
6. Lozano JÁ et al. Reconstrucción mamaria con colgajos microquirúrgicos de perforantes. An Sist Sanit Navar. 2005;28(Suppl 2):73-9
7. Breastcancer.org. Colgajo TRAM. 2023. Available at: https://www. breastcancer.org/es/tratamiento/ cirugia/reconstruccion-mamaria/ tipos/colgajo-autogeno/tram. Last accessed: 5 April 2024.
8. Anureet K B et al. Comparison of donor-site complications and functional outcomes in free musclesparing TRAM flap and free DIEP flap breast reconstruction. Plast Reconstr Surg. 2006;117(3):737-46.
9. Uda H et al. Clinical and quantitative isokinetic comparison of abdominal morbidity and dynamics following DIEP versus muscle-sparing free TRAM flap breast reconstruction. Plast Reconstr Surg. 2017;140(6):1101-9.
10. MD Anderson Cancer Center. Reconstrucción mamaria con colgajo DIEP. Available at: https://mdanderson. es/elhospital/cuadromedico/ serviciosmedicos/reconstruccionmamaria-con-colgajo-diep. Last accessed: 5 April 2024.
11. Macadam SA et al. Quality of life
and patient-reported outcomes in breast cancer survivors: a multicenter comparison of four abdominally based autologous reconstruction methods. Plast Reconstr Surg. 2016;137(3)75871.
12. Beugels J et al. Sensory Recovery of the Breast following Innervated and noninnervated DIEP flap breast reconstruction. Plast Reconstr Surg. 2019;144(2):178-88.
13. Beugels J et al. Nerve coaptation improves the sensory recovery of the breast in DIEP flap breast reconstruction. Plast Reconstr Surg. 2021;148(2):273-84.
14. Bijkerk E et al. Clinical relevance of sensory nerve coaptation in DIEP flap breast reconstruction evaluated using the BREAST-Q. Plast Reconstr Surg. 2022;150(5):959-69.
15. Regan J-P, Schaffner AD. Breast Reconstruction Expander Implant [Internet] (2023) Treasure Island: StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/ NBK431062/. Last accessed: 5 April 2024.
16. López MA et al. Radioterapia y técnicas de reconstrucción mamaria. Revista de Senología y Patología Mamaria. Revista de Senología y Patología Mamaria. 2013;26(1):25-32.
17. Thione A, Jarillo LL. Radioterapia y reconstrucción de mama. Available at: https://reconstrucciondemama. com/reconstruccion-mama/efectoradioterapia/#:~:text=Efecto%20 de%20radioterapia%20sobre%20 prótesis&text=La%20complicación%20 más%20frecuente%20asociada,la%20 mama%20y%20el%20hombro. Last
accessed: 5 April 2024.
18. pFrey M et al. Immediate breast reconstruction – a review of indications, techniques and results. Eur Surg. 2007;39:238-48.
19. He S et al. Comparison of outcomes between immediate implant-based and autologous reconstruction: 15-year, single-center experience in a propensity score-matched Chinese cohort. Cancer Biol Med. 2022;19(9):1410-21.
20. L Prantl et al. Heidekrueger. Immediate versus secondary DIEP flap breast reconstruction: a multicenter outcome study. Arch Gynecol Obst. 2020;302(6):1451-9.
21. Hwang YJ et al. A comparative study of breast sensibility and patient satisfaction after breast reconstruction: autologous, 2-stage implant-based, and prepectoral direct-to-implant reconstruction. Ann Plast Surg. 2022;88(3):262-70.
22. Hein N et al. Comparison of skin sensitivity following breast reconstruction with three different techniques: Autologous fat grafting, DIEP flap and expander/implant1. Clin Hemorheol Microcirc. 2022;80(4):38997.
23. Haloua MH et al. Semmes-weinstein monofilaments: influence of temperature, humidity, and age. J Hand Surg Am. 2011;36(7):1191-6.
24. Chikai M et al. Evaluation of the variation in sensory test results using Semmes-Weinstein monofilaments. Annu Int Conf IEEE Eng Med Biol Soc. 2015;2015:1259-62.
25. Petrou IG et al. Defining the ideal breast reconstruction procedure after mastectomy from the patient perspective: a retrospective analysis. Breast Cancer (Auckl) 2022;16:11782234221089597.
26. Shridharani SM et al. Breast sensation after breast reconstruction: a systematic review. J Reconstr Microsurg. 2010;26(5):303-10.
27. Garvey PB et al. Muscle-sparing TRAM flap does not protect breast reconstruction from postmastectomy radiation damage compared with the DIEP flap. Plast Reconstr Surg. 2014;133(2)223-33.
28. Kelley BP et al. A systematic review of morbidity associated with autologous breast reconstruction before and after exposure to radiotherapy: are current practices ideal? Ann Surg Oncol. 2014;21(5):1732-8.
29. Matros E et al. Cost-effectiveness analysis of implants versus autologous perforator flaps using the BREAST-Q. Plast Reconstr Surg. 2015;135(4):93746.
30. Mioton LM et al. Tracking the aesthetic outcomes of prosthetic breast reconstructions that have complications. Plast Surg (Oakv). 2014;22(2):70-4.
31. Spear S et al. Inamed silicone breast implant core study results at 6 years. Plast Reconstr Surg. 2007;120(7 Suppl 1):8-16S.
32. Handel N et al. A long-term study of outcomes, complications, and patient satisfaction with breast implants. Plast Reconstr Surg. 2006;117(3):757-67.
BRCA Mutation in Ovarian Cancer: Implications for Screening, Diagnosis, and Preventive Measures
Ovarian cancer is the most common gynaecological malignancy and the seventh most common malignancy in women. Inherited ovarian cancer is caused by mutations in certain genes, such as BRCA1 and BRCA2, as well as many minor genes. The pathology of ovarian cancer involves damage to the cell cycle mechanism secondary to mutations in BRCA1/2 protective genes. These mutations provide a meaningful marker for screening and diagnosing hereditary ovarian cancer. Classification of ovarian cancer is based on histology, depending on which layers of the ovary are affected.
The authors conducted an electronic search using keywords and selected the included studies based on pre-established inclusion criteria. To avoid bias in the data extraction process, three reviewers extracted information independently. Risk assessment models provided by the National Comprehensive Cancer Network (NCCN) and American College of Obstetricians and Gynecologists (ACOG) are mostly used in clinical practice. The combination of serial serum cancer antigen-125 (CA-125) levels and transvaginal ultrasound is the only evidence-based screening approach available to patients at increased risk for ovarian cancer.
Strong evidence has made salpingo-oophorectomy the gold standard for risk-reducing surgery. Bilateral salpingectomy, in contrast, is restricted to clinical trials currently. The protective effects of oral contraceptives have made them suitable agents for chemoprevention. Whilst the potential benefits of aspirin and certain other drugs have been investigated, further research is required to address the gap in data for them to be used in clinical practice for the purpose of ovarian cancer prevention.
Key Points
1. Increased risk of developing breast cancer conferred by BRCA1 and BRCA2 gene mutation is the principle on which current screening test relies upon.
2. Primary prevention by genetic testing has been gaining ground in recent years with the National Comprehensive Cancer Network putting forward guidelines and criteria.
3. Along with surgical management, certain drugs and substances have also been found to reduce risk by various mechanisms.
INTRODUCTION
Ovarian cancer is a serious type of cancer that affects women. It has high morbidity and mortality and is the seventh biggest cause of cancer deaths in women.1
According to Global Cancer Statistics 2022, ovarian cancer shows a 3.4% chance of occurring among almost 10 million new cancer cases, with a 4.8% mortality rate in approximately 4 million female patients with cancer.1 In India, northeastern areas like Arunachal Pradesh (specifically Papumpare District) along with Delhi have a notably high number of ovarian cancer cases.2 More developed countries, like the USA and those in the European Union, are expected to see a 42% rise in various cancers by 2050.1 Meanwhile, countries with medium development levels, like India, might face a dramatic rise of up to 100% in cancer cases during the same time frame.1 This means that the incidence of ovarian cancer could increase by approximately 10–15% in the coming decades.1
Since Mary-Claire King, University of Washington, USA, and her team identified the BRCA1 gene linked to ovarian and breast cancers, there have been huge advances in prevention and treatment.3
The BRCA2 gene was located by Michael Stratton, Wellcome Sanger Institute, Hinxton, UK, and Richard Wooster, Haddow Laboratories, Sutton Surrey, UK.4 In India, mutations in the BRCA1/2 genes account for around 25.69% of hereditary breast and ovarian cancer cases.5 Other genes like TP53, PALB2, BRIP1, and ATM also play a part, contributing to another 3.47% of hereditary breast cancers.5 Because of this high occurrence of BRCA-related ovarian cancer in India, it is important for health professionals to check risk factors and take action to detect, diagnose, and treat it effectively.
Role of BRCA Genes
The BRCA1 gene is activated upon detection of DNA damage and when there are irregularities in the cell cycle. BRCA1 is located on chromosome 17q. It is composed of 22 coding exons distributed over 100 KB of DNA. It becomes hyperphosphorylated in response to DNA damage, and it relocates to the site of replication forks marked by proliferating cell nuclear antigen (PCNA). In response to ionising radiation, BRCA1 is bound and phosphorylated by ATM kinase.6 There are proteins that regulate the action of BRCA1 during DNA repair, transcription, and cell cycle. Ionising radiation potentially destroys the interactions of proteins with the BRCA1 gene. The number of proteins that interact with BRCA1 and the arrangement of proteins on BRCA1 is highlighted in Figure 1. Similarly, BRCA2 helps to repair damaged DNA more sensitively. It is present on chromosome 13q12 and repairs chromosomal breaks and aberrant mitotic exchanges that occur during the cell cycle.6 Studies show that BRCA2 and RAD51 are fundamental for the maintenance of cell division and chromosomal structure. Proteins interacting with BRCA2 and the arrangement of proteins on BRCA2 are highlighted in Figure 1. Proteins interacting with BRCA1 and BRCA2 and the arrangement of proteins on them are discussed in Figure 1.
The mechanism of action of both BRCA1 and BRCA2 indicate the significance of BRCA in the cell cycle and cell regulation, as these genes encode proteins that function to limit proliferation. However, if the tumour suppressor genes are inactivated by a point mutation, deletion, or loss of expression, there is no longer any restraint on tissue growth. The loss of heterozygosity (LOH) is observed in ovarian cancer in the BRCA1 or BRCA2 allele. Such cases are exquisitely sensitive to DNA damaging agents like
platinum and poly(ADP-ribose) polymerase (PARP) inhibitors.7 Inactivation of proteins interacting with BRCA or deletion of a protein on BRCA may substantially lead to development of breast- and ovarian-related malignancy. It is crucial that patients with breast cancer undergo genetic screening to help prevent and manage BRCA-related ovarian cancer.
Ovarian cancer is also histologically classified into five categories.8 Among these, BRCA mutation is mostly seen in high-grade serous carcinoma.
METHODS
The authors conducted an electronic search using various databases, including Medline through Ovid, PubMed, Embase, and Google Scholar. The goal was to find articles related to keywords such as ‘ovarian cancer’, ‘ovarian neoplasm’, ‘ovarian carcinoma’, ‘ovarian malignancies’, and terms like ‘BRCA1’, ‘BRCA2’, or ‘BRCA’, in conjunction with ‘genetic screening’, ‘genetic testing’,
and ‘preventive measures’. This search focused solely on studies involving humans, without any language restrictions. Moreover, the authors manually checked reference lists from relevant studies to see how the information applied to their research. To prevent overlap in patient groups, e.g., if authors wrote about the same cohorts in multiple publications, the study authors only included the most recent or detailed study in their analysis. For a study to be eligible for inclusion, it had to meet the following set of criteria: address ovarian cancer, note BRCA mutations, discuss preventive measures, and be published after January 2010.
Data Extraction and Methodological Assessment
The reports the authors retrieved contained information such as authorship, publication year, journal name, sample size, methodologies used, histology types, and preventive strategies employed. To minimise bias during data extraction, three reviewers independently gathered the required data.
Figure 1: The arrangement of protein on BRCA1 and BRCA2
RISK ASSESSMENT MODELS
The main goal of genetic testing for harmful mutations in the BRCA1/2 genes is to pinpoint women who face the highest risk of developing ovarian cancer. Doing this means that effective preventive measures can be taken. It is important to note that pathogenic BRCA1/2 mutations are quite rare; they occur in about 1/800–1/1000 individuals for each gene.9 These mutations tend to occur more frequently among individuals or families with certain risk factors, including early onset (<50 years of age), prior cases of breast cancer or ovarian cancer within the family tree, multiple cancers (either similar or related), family history of male breast cancer, or among those at higher risk for founder mutations linked to specific ancestries like Ashkenazi Jewish or Swedish backgrounds.10
Advancements in cancer genetics have raised awareness about tailored risk assessments concerning primary cancer prevention.11 A thorough risk evaluation requires detailed family histories plus comprehensive medical insights from patients. Understanding both clinical prediction models and the underlying pathophysiology and aetiology of breast and ovarian cancer risks are crucial. Given these complexities, professional organisations suggest that only trained healthcare providers in the field of genetics should carry out risk assessments. This way they can accurately counsel patients while minimising potential harms.12,13 Currently utilised models include those provided by the NCCN and ACOG. Furthermore, models like BRCAPRO and BOADICEA have been created to estimate the likelihood that a person might have a pathogenic BRCA1/2 mutation based on their family history.14
GUIDELINE ON GENETIC TESTING
Patients often look into genetic testing either after learning about a mutation from a relative or qualifying through personalised screening following a diagnosis of breast or ovarian cancer at a young age (≤50 years). NCCN guidelines provide clear criteria for referring individuals to genetic specialists
based on both personal and family cancer histories 15
SCREENING TOOLS AND ALGORITHM
Currently available guidelines do not recommend routine ultrasound for ovarian cancer screening unless it is before undertaking risk-reducing prophylactic bilateral salpingo-oophorectomy (RRSO) at age 35–40 years or once childbearing is completed.16,17 Still, some carriers of BRCA mutations may opt for screenings so that they can manage their fertility and overall quality of life. Organisations like the NCCN and ACOG now consider image-based screenings reasonable for short-term monitoring in women younger than 35 years until they proceed with prophylactic surgeries.18 Conventional practices for screening high-risk women include using serial serum CA-125 tests paired with annual transvaginal ultrasound featuring Doppler evaluation.19 Research indicates that frequent CA-125 tests offer more value compared to one-time measurements while noting that CA-125 levels can be normal or only slightly elevated during earlystage ovarian cancer.20 That is why relying solely on serum CA-125 measurements is not sufficient for proper screening. The combination approach remains the only evidence-based option currently accessible to at-risk patients; however, better screening methods are still being explored.
COUNSELLING
Genetic counselling for BRCA1/2 mutation testing should only be performed by qualified health professionals, including well-trained primary care providers. The genetic counselling process involves detailed analyses concerning families and assessing the risks associated with potentially harmful mutations. It also focuses on identifying suitable candidates for testing alongside providing education about the process itself. This includes discussing the benefits and drawbacks linked with genetic testing and interpreting results post-testing, as well as reviewing
management options moving forward. Preand post-test genetic counselling is also supported by the NCCN guidelines.15
PREVENTIVE MEASURES
Increasing incidence of ovarian cancer warrants the implementation of appropriate preventive strategies. This becomes even more important for BRCA mutation carriers who are at an increased risk of ovarian cancer. The current preventive gold standard is RRSO. Another surgical alternative to this is prophylactic salpingectomy with delayed oophorectomy (PSDO).
Chemoprevention, a developing area, deals with the preventive efficacy of certain drugs, including but not limited to oral contraceptive pills. Other prospective candidates include retinoids and other phytochemicals, anti-angiogenic agents, and vitamin D analogues.
Risk-Reducing Salpingo-Oopherectomy
This is a surgical procedure to remove both fallopian tubes and ovaries, which substantially reduces the risk of ovarian and fallopian tube cancer in BRCA mutation carriers.21 The specific protocol for high-risk women involves exploring the pelvic organs for any evidence of cancer, peritoneal wash, and removal of the ovaries and fallopian tubes in their entirety.22 A prospective study of 80 women in China enrolled for RRSO had an overall 4.1% rate of cancer in mutation carriers, out of which 74 had deleterious gene mutations: 58.1% BRCA 1 and 35.1% BRCA2. 23 Analysis by Eleje at al.,22 showed that RRSO may improve overall survival and reduce mortality from high-grade serous carcinomas (HGSC) and breast cancer. Overall quality of life is not affected by RRSO, but lower cancer-related anxiety is reported, with most women very satisfied with their choice of risk-reducing surgery.24
A standardised histopathological sectioning and extensively examining the fimbria (SEE-FIM) protocol should be used after RRSO for identifying invasive cancers, which, if detected, should be referred to a
tertiary gynaecological oncology centre for appropriate management.25
Though RRSO remains the gold standard for reduction of the risk of ovarian cancer in BRCA carriers, it is associated with significant side-effects. The acute loss of oestrogen exposure induces premature menopause with short-term effects including vasomotor complaints such as hot flushes, sleep disturbances, and impaired sexual functioning, as well as long-term effects, which include a risk of cardiovascular disease, osteoporosis, and cognitive impairment.24,26
Prophylactic Salpingectomy with Delayed Oophorectomy
Even though advanced stages of the disease often appear in the ovaries, sometimes right at the clinical presentation, it has been suggested that the ovarian surface epithelium might not be where it all starts.27
The idea of the fallopian tube or the Müllerian model was initially put forth by Louis Dubaeu.28 Now, it is well known that many ovarian HGSCs likely come from the distal fimbrial end of the fallopian tube. This is linked to a precursor known as serous tubal intraepithelial carcinoma (STIC). In contrast, low-grade serous carcinomas (LGSC) generally arise within the ovary from benign or borderline serous tumours.29 The most common histological subtype is HGSC, more than 95% of which is characterised by mutations in the tumour suppressor gene TP53, known as the p53 signature.30-33
For those with BRCA mutations, if a p53 signature is found in the fallopian tubes, it suggests HGSC may likely come from there.34,35 Consequently, PSDO is being floated as a less morbid alternative to RRSO in BRCA mutation carriers.36-39 This novel strategy has been demonstrated to have positive effects on menopause-related quality of life and sexual health when compared to RRSO.40 This strategy involves two steps.41 First, all salpingeal tissue is surgically removed after childbearing is complete (or sooner if assisted reproductive technology is anticipated), followed by
oophorectomy at a later point. When performing salpingectomy, doctors inspect the peritoneum and take peritoneal washings for cytology. They also use SEEFIM processing on the fallopian tube to check for precursor lesions and sometimes hidden cancers.42
This technique is, however, discouraged in clinical practice outside of clinical trials due to a lack of sufficient evidence on oncological safety. The UK Cancer Genetics Group (CGG) and British Gynaecological Cancer Society (BGCS) pointed out several main concerns: limited data showing benefits (83%), increased surgical risks (79%), loss of breast cancer risk reduction (68%), need for long-term follow-up (61%), and a percentage that may not proceed with the second procedure (66%).43
Oral Contraceptives
Oral contraceptives (OCP) are associated with a significant reduction in risk of ovarian cancer and are an important preventive factor for most histological types.44 A metaanalysis by Iodice et al.,45 provides evidence that in women with an ascertained germ line mutation in BRCA1 or BRCA2, OCPs reduce ovarian cancer risk and found no evidence that formulations after 1975 increased the risk of breast cancer.45 The risk of ovarian cancer was more strongly reduced with longer durations of use, and the relative risks remain low for a long period, attenuating 20 years after stopping.46
The exact mechanism by which OCPs confer long-lasting protection against ovarian cancer is not well understood, but a causal association can be inferred from its ability to suppress ovarian activity.47
Among women who use a combined oestrogen–progesterone therapy, the annual risk of breast cancer increases with the duration of use and dissipates within 2 years of cessation of therapy.48 However, data on breast cancer risk associated with OCP use in BRCA mutation carriers is heterogeneous, and hence, a theoretical risk should be considered when giving clinical recommendations.49
Non-steroidal Anti-inflammatory Drugs
Analysis of observational epidemiological studies suggests an at least 10% relative risk reduction of ovarian cancer with aspirin use.50 Aspirin has been found in vitro to reverse the metabolic derangements such as the increased activation of hexokinase–2 activation, resulting in increased glycolysis caused by loss of BRCA1 51 Animal experiments conducted by Guo et al.,52 concluded that the anti-cancer effect of aspirin is due to increased p53 activation in a concentration-dependent manner, which enhances sensitivity to a combination of cisplatin and aspirin.52
Non-aspirin NSAIDs like acetaminophen and ibuprofen have statistically nonsignificant relationships with the risk of ovarian cancer.53
Potential gastrointestinal adverse effects and heterogeneity in data, coupled with the absence of data on frequency and dose of NSAIDs and the role of age, discourage regular use of NSAIDs as chemoprotective agents.54,55
Others
Retinoids
Retinoids have a wide range of pleiotropic effects, but monotherapy is not likely to be an effective prevention strategy, and analysis of combination therapies to address toxicity and resistance issues is needed.56 Cytoplasmic retinol binding protein (CRBP)-1, the most diffuse CRBP isoform, is frequently downregulated or lost in ovarian cancer.57,58
Fenretinide is a synthetic analogue of alltrans retinoic acid that was first produced in the late 1960’s.59 It is somewhat protective against ovarian cancers in women with germline BRCA mutations, and further investigation is required.60
Anti-angiogenic agents
Targeting multiple points in the angiogenesis pathway can help in the prevention of cancer in high-risk individuals, especially with BRCA 61 Anti-angiogenic
strategies mainly aim to inhibit the early stages of angiogenesis where there are leaky intercellular connections with irregular blood flow.62,63
Wang et al.,64 evaluated 10 phytochemicals and found that oleanolic acid, silibinin, curcumin, epigallocatechin gallate, melatonin, and resveratrol have antitumourigenic effects in almost all cancer hallmarks, along with strong antitumour/anti-angiogenic action. They suggested further experimental tests for developing these as complements or alternative agents.64
Curcumin
Curcumin has been shown to inhibit the expression of MMP-9, VEGF, and HER2.65,66 The anti-cancer activity is believed to be exerted through inhibiting proliferation and inducing apoptosis in a wide array of cancer cell types in vitro, including ovarian cells.67
References
1. Bray F et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229-63.
2. Chaturvedi M et al. Descriptive epidemiology of ovarian cancers in India: a report from national cancer registry programme. Indian J Gynecol Oncolog. 2023;21:25.
3. Schubert C. Mary-Claire King. Nat Med. 2003;9(6):633.
4. Wooster, R et al. Identification of the breast cancer susceptibility gene BRCA2 Nature. 1995;378(6559):78992. Corrected and republished from: Nature. 1996;379(6567):749.
5. Kadri MSN et al. Mutational landscape for Indian hereditary breast and ovarian cancer cohort suggests need for identifying population specific genes and biomarkers for screening. Front Oncol. 2021;10:568786.
6. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866-71.
7. Maxwell KN et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat Commun. 2017;8(1):319.
8. Adhikari L, Hassell LA. WHO classification. PathologyOutlines.
Limited bioavailability is a limitation, but its safety profile and nominal cost make it a candidate to explore as an agent for chemoprevention.68
Vitamin D
Meta-analyses recommend a converse relationship between circulating Vitamin D3 levels with rates of ovarian and other cancers.69,70 It has been demonstrated that vitamin D biosynthesis and signalling is compromised in the ovarian epithelium of women with BRCA1 mutation.71
Growing evidence suggests that vitamin D deficiency is correlated to ovarian cancer and that calcitriol and its analogues could be promising agents for cancer prevention.72
The active metabolite of vitamin D, calcitriol, has been found to inhibit proliferation of the ovarian cancer cell line OVCAR4.73
com website. Available at: https:// www.pathologyoutlines.com/topic/ ovarytumorwhoclassif.html. Last accessed: 7 July 2024.
9. Balmaña J et al. ESMO Guidelines Working Group. BRCA in breast cancer: ESMO Clinical Practice Guidelines. Ann Oncol. 2011;22(Suppl 6):vi31-vi34.
10. Berliner JL et al. NSGC practice guideline: risk assessment and genetic counseling for hereditary breast and ovarian cancer. J Genet Couns. 2013;22(2):155-63.
11. Desmond A et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. 2015;1(7):943-51.
12. Moyer VA; U.S. Preventive Services Task Force. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2014;160(4):271-81.
13. National Comprehensive Cancer Network. Clinical practice guidelines in oncology. Genetic/familial high risk assessment: breast and ovarian cancer syndromes (version 1.2018). Available at: https://www.nccn. org/professionals/physician_gls/ pdf/genetics_screening.pdf. Last accessed: 2 January 2018.
14. Talwar V, Rauthan A. BRCA mutations: implications of genetic testing in
ovarian cancer. Indian J Cancer. 2022;59(Supplement):S56-67.
15. National Comprehensive Cancer Network (NCCN) Guidelines, Inc. Genetic/Familial High Risk Assessment: Breast and Ovarian (v3.2024). 2024. Available at: nccn. org/professionals/physician_gls/pdf/ genetics_bop.pdf. Last accessed: 4 July 2024.
16. Committee on Practice Bulletins–Gynecology, Committee on Genetics, Society of Gynecologic Oncology. Practice bulletin No 182: hereditary breast and ovarian cancer syndrome. Obstet Gynecol. 2017;130(3):e110-26.
17. Walker JL et al. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer. 2015;121(13):2108-20.
18. Elezaby Mai et al. BRCA mutation carriers: breast and ovarian cancer screening guidelines and imaging considerations. Radiology. 2019;291(3):554-69.
19. National Comprehensive Cancer Network. Clinical practice guidelines in oncology. Ovarian cancer (version 2.2022). Available at: Ovarian Cancer/ Fallopian Tube Cancer/Primary Peritoneal Cancer - Guidelines Detail (nccn.org). Last Accessed: 2 July 2024.
20. Bristow RE et al. Ovarian malignancy risk stratification of the adnexal mass using a multivariate index assay.
Gynecol Oncol 2013;128(2):252-9.
21. Tschernichovsky R, Goodman A. Riskreducing strategies for ovarian cancer in BRCA mutation carriers: a balancing act. Oncologist. 2017;22(4):450-9.
22. Eleje GU et al. Risk-reducing bilateral salpingo-oophorectomy in women with BRCA1 or BRCA2 mutations. Cochrane Database Syst Rev. 2018;8(8):CD012464.
23. Feng, Z et al. Risk-reducing salpingooophorectomy among Chinese women at increased risk of breast and ovarian cancer. J Ovarian Res. 2023;16(1):125.
24. Vermeulen RFM et al. Impact of riskreducing salpingo-oophorectomy in premenopausal women. Climacteric. 2017;20(3):212-21.
25. Sideris M et al. Screening and prevention of ovarian cancer. Med J Aust. 2024;220(5):264-74.
26. Rocca WA et al. Accelerated accumulation of multimorbidity after bilateral oophorectomy: a populationbased cohort study. Mayo Clin Proc. 2016;91(11):1577-89.
27. Klotz DM, Wimberger P. Cells of origin of ovarian cancer: ovarian surface epithelium or fallopian tube? Arch Gynecol Obstet. 2017;296(6):1055-62.
28. Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecol Oncol. 1999;72(3):437-42.
29. Cheung AN et al., WHO classification of tumours: female genital tumours, 2020, 5th edition, Switzerland: WHO press.
30. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma Nature. 2011;474(7353):609-15. Corrected and republished from: Nature. 2012 Oct 11;490(7419):298.
31. Ahmed AA et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221(1):49-56.
32. Singer G et al. Patterns of p53 mutations separate ovarian serous borderline tumors and low- and high-grade carcinomas and provide support for a new model of ovarian carcinogenesis: a mutational analysis with immunohistochemical correlation. Am J Surg Pathol. 2005;29(2):218-24.
33. Hofstetter G et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br J Cancer. 2011;105(10):1593-9.
34. Piek JM et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol.
2001;195(4):451-6.
35. Piek JM et al. Intraperitoneal serous adenocarcinoma: a critical appraisal of three hypotheses on its cause. Am J Obstet Gynecol. 2004;191(3):718-32
36. Greene MH et al. Does bilateral salpingectomy with ovarian retention warrant consideration as a temporary bridge to risk-reducing bilateral oophorectomy in BRCA1/2 mutation carriers? Am J Obstet Gynecol. 2011;204:19.e1-e6.
37. Anderson CK et al. Riskreducing salpingectomy as preventative strategy for pelvic serous cancer. Int J Gynecol Cancer 2013;23:417-21.
38. Schenberg T, Mitchell G. Prophylactic bilateral salpingectomy as a prevention strategy in women at high-risk of ovarian cancer: a mini-review. Front Oncol. 2014;4:21
39. Nebgen DR et al. Prophylactic salpingectomy with delayed oophorectomy (PSDO): Feasibility study in women with BRCA mutations. Abstract 1506. ASCO Annual Meeting I, 3-7 June, 2016.
40. Steenbeek MP et al. TUBectomy with delayed oophorectomy as an alternative to risk-reducing salpingooophorectomy in high-risk women to assess the safety of prevention: the TUBA-WISP II study protocol. Int J Gynecol Cancer. 2023;33(6):982-7.
41. J.W.H. Wong et al. Completeness of salpingectomy intended for ovarian cancer risk reduction. Gynecol Oncol. 2019;155(2):280-2.
42. Boerner T, Long Roche K. Salpingectomy for the risk reduction of ovarian cancer: is it time for a salpingectomy-alone approach? J Minim Invasive Gynecol. 2021;28(3):403-8.
43. Chandrasekaran D et al. Risk reducing salpingectomy and delayed oophorectomy in high risk women: views of cancer geneticists, genetic counsellors and gynaecological oncologists in the UK. Fam Cancer. 2015;14(4):521-30.
44. Wentzensen N et al. Ovarian cancer risk factors by histologic subtype: an analysis from the ovarian cancer cohort consortium. J Clin Oncol. 2016;34(24):2888-98.
45. Iodice S et al. Oral contraceptive use and breast or ovarian cancer risk in BRCA1/2 carriers: a meta-analysis. Eur J Cancer. 2010;46(12):2275-84.
46. Schrijver LH et al. Oral contraceptive use and ovarian cancer risk for BRCA1/2 mutation carriers: an international cohort study. Am J Obstet Gynecol. 2021;225(1):51.e1-51. e17.
47. Collaborative Group on
Epidemiological Studies of Ovarian Cancer et al. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet. 2008;371(9609):303-14.
48. Narod SA. Hormone replacement therapy and the risk of breast cancer. Nat Rev Clin Oncol. 2011;8(11):669-76.
49. Park J et al. Oral contraceptives and risk of breast cancer and ovarian cancer in women with a BRCA1 or BRCA2 mutation: a meta-analysis of observational studies. Carcinogenesis. 2022;43(3):231-42.
50. Zhang D et al. Is aspirin use associated with a decreased risk of ovarian cancer? A systematic review and meta-analysis of observational studies with dose-response analysis. Gynecol Oncol. 2016;142(2):368-77.
51. Chiyoda T et al. Loss of BRCA1 in the cells of origin of ovarian cancer induces glycolysis: a window of opportunity for ovarian cancer chemoprevention. Cancer Prev Res (Phila). 2017;10(4):255-66.
52. Guo J et al. Aspirin inhibits tumor progression and enhances cisplatin sensitivity in epithelial ovarian cancer. PeerJ. 2021;9:e11591.
53. Baandrup L et al. Nonsteroidal anti-inflammatory drugs and risk of ovarian cancer: systematic review and meta-analysis of observational studies. Acta Obstet Gynecol Scand. 2013;92(3):245-55.
54. Wang P et al. The relationship between nonsteroidal anti-inflammatory drugs and cancer incidence: an umbrella review. Heliyon. 2024;10(2):e23203.
55. Sadeghi J et al. Impact of analgesics on the risk of ovarian cancer; a systematic review and metaanalysis of cohort and case-control studies. Immunopathologia Persa. 2023;DOI:10.34172/ipp.2023.40572.
56. Uray IP et al. Retinoids and rexinoids in cancer prevention: from laboratory to clinic. Semin Oncol. 2016;43(1):49-64.
57. Cvetković D et al. Loss of cellular retinol-binding protein 1 gene expression in microdissected human ovarian cancer. Clin Cancer Res. 2003;9(3):1013-20.
58. Doldo E et al. CRBP-1 expression in ovarian cancer: a potential therapeutic target. Anticancer Res. 2014;34(7):3303-3312.
59. Hail N Jr et al. Mechanisms of fenretinide-induced apoptosis. Apoptosis. 2006;11(10):1677-94.
60. Veronesi U et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast
cancer. Ann Oncol. 2006;17(7):106571.
61. Bisacchi D et al. Anti-angiogenesis and angioprevention: mechanisms, problems and perspectives. Cancer Detect Prev. 2003;27(3):229-38.
62. Sogno I et al. Angioprevention with fenretinide: targeting angiogenesis in prevention and therapeutic strategies. Crit Rev Oncol Hematol. 2010;75(1):214.
63. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62.
64. Wang Z et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35 Suppl(Suppl):S224-43.
65. Lin YG et al. Curcumin inhibits tumor growth and angiogenesis in ovarian
carcinoma by targeting the nuclear factor-kappaB pathway. Clin Cancer Res. 2007;13(11):3423-30.
66. Korutla L. Inhibition of ligand-induced activation of epidermal growth factor receptor tyrosine phosphorylation by curcumin. Carcinogenesis. 1995;16(8):1741-5.
67. Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23(1A):363-98.
68. Kunnumakkara AB et al. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269(2):199-225.
69. Yin L et al. Circulating 25-hydroxyvitamin D serum concentration and total cancer
incidence and mortality: a systematic review and meta-analysis. Prev Med. 2013;57(6):753-64.
70. Webb PM et al. Circulating 25-hydroxyvitamin D and survival in women with ovarian cancer. Am J Clin Nutr. 2015;102(1):109-14.
71. Pejovic T et al. Association between vitamin D and ovarian cancer development in BRCA1 mutation carriers. Oncotarget. 2020;11(45):4104-14.
72. Guo H et al. The role of vitamin D in ovarian cancer: epidemiology, molecular mechanism and prevention. J Ovarian Res. 2018;11(1):71.
73. Mueller PR et al. Vitamin D and docosahexaenoic acid inhibit proliferation of the ovarian cancer cell line OVCAR4. Nutr Health. 2023;DOI:10 .1177/02601060231202565.
MammaPrint Genomic Assay Providing Prognostic Information in Early Breast Cancer: 10-Year Follow-Up From a Retrospective German Breast Cancer Registry Analysis
Authors: *C. Jackisch,1 D. Pronin,2 Christa Dreezen,2 T. Dimpfl,3 R. Büttner,4 G. Kunz,5 C. Langwieder,6 M. Rees,6 K. Lerchl7
1. KEM Evangelische Kliniken Essen Mitte gGmbH, Germany
2. Medical Affairs, Agendia NV, Amsterdam, the Netherlands
3. Klinikum Kassel, Germany
4. University of Cologne, Germany
5. St. Johannes Hospital Dortmund, Germany
6. Institute of Pathology at Josefshaus, Dortmund, Germany
7. Patients Tumor Bank of Hope (PATH), Germany
*Correspondence to c.jackisch@kem-med.com
Disclosure: The authors declare no conflicts of interest.
Introduction: Gene expression assays, such as the MammaPrint® (Agendia, Amsterdam, the Netherlands) 70-gene signature, are increasingly used by oncologists to understand breast cancer biology and improve treatment planning. This study assesses the utility of MammaPrint genomic risk in predicting treatment outcomes for women with breast cancer in a retrospective German cohort with a 10-year follow-up, treated based on clinicopathological features alone.
Methods: The sample set of 117 tumours from the ‘Patients Tumour Bank of Hope’ (PATH) biobank with 10-year follow-up were classified using MammaPrint into high or low risk of distant metastasis. Patients were previously treated according to St. Gallen and Adjuvant! Online high- or low-risk criteria. Statistical analyses compared overall survival (OS) and treatment outcomes between clinical and genomic risk groups.
Results: Among the 78 patients with clinically high-risk tumours, 50% (39) were reclassified as MammaPrint low risk. In total, 57.3% (67/117) patients with MammaPrint low-risk tumours demonstrated a significantly higher 10-year OS of 93.4%, irrespective of nodal status, compared to patients with MammaPrint high-risk tumours (71.2%; p=0.001). Chemotherapy improved OS in patients with MammaPrint high-risk tumours by 29.4%, but not for patients with MammaPrint low-risk tumours (p=0.016).
Discussion: The findings confirm the prognostic utility of MammaPrint for identifying genomically low-risk patients who may safely omit chemotherapy while suggesting genomically high-risk cases may benefit from chemotherapy. By providing a more precise assessment of cancer risk than traditional clinicopathological methods alone, MammaPrint may help reduce unnecessary treatments and improve long-term quality of life for patients diagnosed with early-stage breast cancer.
Key Points
1. The prognosis in early breast cancer is based on the underlying personal risk of recurrence.
2. To receive more detailed information on the breast cancer biology of a given patient, more information is needed for individualised treatment recommendations to avoid unnecessary over- and undertreatment.
3. Multi-gene assays such as the MammaPrint 70-gene signature provide this information to allow classical prognostic information such as ‘clinical risk’ into ‘genomic risk’ to be transformed to gain more information on tumour biology and optimise the early breast cancer therapy.
INTRODUCTION
In modern oncology, the utilisation of gene expression assays has driven the development of personalised treatment strategies for patients with breast cancer, offering a deeper understanding of the molecular underpinnings of the disease. The most frequently used genomic breast cancer tests in clinical practice include MammaPrint® (Agendia, Amsterdam, the Netherlands), Oncotype DX® (Exact Sciences, Madison, Wisconsin, USA), Prosigna™ (NanoString Technologies, Inc., Seattle, Washington, USA), and EndoPredict (Eurobio Scientific, Essonnes, France).1 Among these, only MammaPrint and Oncotype DX are validated by prospective randomised Phase III trials for hormone receptor positive (HR+) and HER2-negative (HER2-) disease.2,3,4
Prospective non-inferiority trials designed to validate these tests include TAILORx and RxPONDER, which assess the deescalation of chemotherapy using the 21gene Oncotype DX assay in breast cancer treatment. The TAILORx trial showed that, at 9-year follow-up, women with Oncotype intermediate scores often do not benefit from additional chemotherapy when they have HR+, HER2-, and lymph node-negative (LN-) breast cancer.4 The RxPONDER trial’s 5-year follow-up data extended the
assay’s evaluation to node-positive (LN+) patients, suggesting that some might avoid chemotherapy based on their gene expression profiles.2
MammaPrint, a 70-gene recurrence risk signature assay, evaluates the expression of cancer-related genes to classify earlystage breast cancer tumours into highrisk or low-risk categories for developing distant recurrence. This prognostic tool was specifically designed for patients with both HR+ and HR-, HER2- breast cancer with either LN- or LN+ disease (up to three positive nodes).5-9
The prospective, randomised, non-inferiority trial designed to validate MammaPrint, MINDACT (NCT00433589),10 identified patients with a low genomic risk of breast cancer recurrence who might safely omit chemotherapy from their treatment plan. With a median followup of 8.7 years, the trial demonstrated the capability of MammaPrint to identify clinically high-risk patients who exhibit low genomic risk and can achieve excellent outcomes with endocrine therapy alone, without the need for chemotherapy. The study found comparable distant metastasisfree survival rates between patients who received endocrine therapy alone and those who underwent chemotherapy, with a negligible difference of 0.9% at the 5-year
mark, regardless of whether the cancer had spread to lymph nodes.3,6
Complementing the findings of the MINDACT trial, a German retrospective multicentre pilot study using samples from the ‘Patients Tumour Bank of Hope’ (PATH) further explored the concordance between treatment recommendations based on MammaPrint and those based on clinical risk classifications at the time of diagnosis. The study found that MammaPrint reclassified 40% of the patients in the PATH study population, resulting in different treatment recommendations for these patients compared to the initial plan.11
Building on these initial results, the authors analysed the impact of MammaPrint genomic classification compared to clinical risk assessment on treatment planning and 10-year outcomes in patients from the PATH cohort. The rationale of this analysis was to evaluate the predictive utility of MammaPrint for long-term treatment planning and therapeutic efficacy of adjuvant chemotherapy and/or endocrine therapy 10 years after presentation for early-stage breast cancer.
MATERIALS AND METHODS
Patient Population
Tumour samples of 140 German patients who had been diagnosed with Stage I and II early-stage breast cancer between November 2005–April 2008 were obtained from the PATH biobank. Biopsies of the primary tumour were snap-frozen at the clinical site and stored at the PATH Tumour Bank of Hope under specific conditions. All patients had undergone standard surgical procedures, including modified radical mastectomy or breast-conserving surgery including axillary clearance and radiotherapy according to national guidelines. Initially, the necessity for adjuvant therapy was determined based on clinical risk factors, including factors such as tumour size, nodal status, histological grade, and hormone receptor status.
Clinical Risk Classification
Histopathological data, including tumour grading, were assessed according to the Elston and Ellis method.12 Oestrogen receptor (ER) and progesterone receptor (PR) status were determined through immunohistochemistry, with tumours classified as ER/PR-positive if more than 10% of the cell nuclei stained positive for these receptors. The prognostic value of MammaPrint was assessed in comparison with the St. Gallen criteria, which were defined at the 9th St. Gallen Consensus Meeting in 2005. These criteria take several clinicopathologic factors into account, including the size of the primary tumour, patient age, histological grade, hormone receptor status, peritumoiral vascular invasion, and HER2 status. A significant number of the patient cohort was in the intermediate-risk category according to the St. Gallen criteria. The clinical risk of developing distant metastases was further assessed using Adjuvant! Online (site inactive since 2015) and included patient age,13 ER status, nodal status, tumour size, and histological grade. Adjuvant! Online provided estimates of overall survival (OS), breast cancer-specific survival, and eventfree survival.
MammaPrint Laboratory Assay
As described in the original publication RNA extraction from the tissue provided by the PATH study bank was conducted at Agendia, with the process blinded to clinical-pathologic data. Custom-designed arrays (Agilent Technologies, Santa Clara, California, USA) were utilised to generate microarray gene expression data according to established protocols.14 The 70-gene MammaPrint signature identified genomically low-risk tumours by the previously established threshold, defined as the patient’s likelihood of 5-year distant metastasis-free survival surpassing 90%.8 All other tumours were classified as high-risk.
Statistical Analysis
The primary objective was to analyse patient outcomes 10 years after an earlystage breast cancer diagnosis, focusing on the prognostic impact of MammaPrint.
The impact of MammaPrint risk, lymph node status, and adjuvant chemotherapy treatments were considered.
Differences in clinical characteristics were analysed by Chi-Squared or Fisher’s Exact tests. Differences in OS and death by any cause were assessed using KaplanMeier survival analysis. Log-rank tests were conducted to determine significant differences in 10-year survival rates between groups, denoting p-values of 0.05 or less as statistically significant.
RESULTS
Patient Population
The study population consisted of 117 patients with 10-year follow-up and a median age of 62.5 years (Table 1). The majority of patients were over 55 years old, accounting for 64.1% of the total cohort.
The median tumour size was 15.0 mm, and nuclear grading was most frequently Grade 2 (63.2%). In addition, 67.5% of patients presented with negative nodes (LN-) and 76.9% of tumours were classified as invasive ductal cancers. The MammaPrint testing revealed that 57.3% (67) patients had tumours classified as genomically low risk, while 42.7% (50) patients were classified as having MammaPrint high-risk tumours. Age, tumour stage, and nodal status were similar between both genomic risk categories. In contrast, patients with MammaPrint highrisk tumours exhibited a significantly higher frequency of Grade 3 tumours (44.0%) compared to those with MammaPrint lowrisk tumours (11.9%; p<0.001). Among PATH participants with 10-year outcomes data, 42.7% (50/117) of tumours were genomically discordant with clinical risk, with 50.0% of clinically high-risk tumours reclassified as MammaPrint low risk and 28.0% of clinically low-risk tumours reclassified as MammaPrint high risk (Supplementary Figure 1).
Systemic Therapy
Adjuvant systemic therapy was indicated in 95 of the 117 patients, consisting of either chemotherapy (10/95; 10.5%), endocrine therapy (42/95; 44.2%), or both (43/95;
45.3%). One patient did not receive any adjuvant systemic therapy, and in 21 cases, treatment was unknown. Out of 67 MammaPrint low-risk patients, 43.3% (29) were treated with chemoendorince therapy (chemotherapy plus endocrine therapy), and 43.3% (29) only received endocrine therapy. Within the MammaPrint high-risk group, 13 out of 50 patients (26.0%) only received endocrine therapy, 10 (20.0%) received chemotherapy only, and 14 (28.0%) received chemotherapy plus endocrine therapy. In a cohort treated based on clinical risk assessments only, 44.8% (43/96) of patients with recorded treatments received a therapy not in line with MammaPrint recommended treatments (Table 2).
10-Year Survival
The median follow-up period for OS was 10 years. At 10 years of follow-up, there were a total of 17 recorded death events. Among these, four deaths occurred in patients with MammaPrint low-risk tumours (6.0%; 4/67), while 13 deaths were observed in patients with MammaPrint high-risk tumours (26.0%; 13/50). Patients with MammaPrint low-risk tumours displayed a remarkable 10-year OS rate of 93.4% (95% CI: 87.1–99.7), which was significantly higher when compared to those with MammaPrint high-risk tumours (71.2%; 95% CI: 57.9–84.5; p=0.001) (Figure 1A). Among patients with MammaPrint low-risk tumours, 10-year OS was the same regardless of nodal status (93.3%; low-risk LN-: 95% CI: 85.9–100; and node-positive: 95% CI: 80.8–100) (Figure 1B). In contrast, the MammaPrint high-risk LN+ group exhibited a significantly lower 10-year OS rate of 40.4% (95% CI: 16.3–64.5) compared to the high-risk LNgroup, with a rate of 89.0% (95% CI: 77.2–100), and to MammaPrint low-risk groups (p<0.001).
Among patients with MammaPrint low-risk tumours, OS was similar over time between those who received endocrine treatment only and those who received chemotherapy with or without endocrine therapy. At 10 years, OS rates among the low-risk group were 92.5% (95% CI: 82.5–100) for endocrine-only treated and 96.3% (95%
Table 1: Clinical characteristics.
Data represented as n (%), unless otherwise specified. Differences in groups were assessed by using Pearson’s Chisquared tests or Fisher’s exact test. Statistical significance was defined as P<0.05.
of participants;
Table 2: Adjuvant treatments for clinical and genomic risk groups.
Data represented as n (%), unless otherwise specified. Differences in groups were assessed by using Pearson’s Chisquared tests. Statistical significance was defined as P<0.05.
*Indicates recommended treatments based on MammaPrint result. Patients received treatments based on clinical risk factors, according to German national guidelines and standard of care treatments at the time of diagnosis.
CT: chemotherapy; ET: endocrine therapy; MP: MammaPrint; n: number of participants.
Figure 1: 10-year overall survival stratified by mammaprint risk, lymph node status, and chemotherapy treatment.
10-Year Overall Survival (OS) by MammaPrint
10-Year OS by MammaPrint and Lymp Node (LN) Status
Figure 1 continued.
Chemotherapy (CT) Improved 10-Year OS for Patients with High Risk Tumors
Low risk, no CT
Low risk, CT
High risk, no CT
High risk, CT
A) 10-year OS stratified MammaPrint low (green) and high (red) risk of distant metastasis.
B) 10-year OS stratified MP low risk/LN- (purple), MP low risk/LN+ (green), MP high risk/LN- (light blue), and MP high risk/LN+ (orange).
C) 10-year OS stratified MP low risk no CT (green), MP low risk with CT (yellow), MP high risk no CT (red), and MP high risk with CT (blue grey). Significance between groups was assessed by a log-rank test. Statistical significance was defined as p<0.05.
CI: 89.2–100; p=0.572) for chemotherapytreated patients (Figure 1C).
In contrast, patients with MammaPrint high-risk tumours who did not receive any chemotherapy had significantly poorer survival at 10 years with an OS rate of 61.5% (95% CI: 31.5–91.5) compared to patients with MammaPrint high-risk tumours that received chemotherapy (10-year OS rate: 90.9%; 95% CI: 78.9–100; p=0.016) and to MammaPrint low-risk groups.
DISCUSSION
This study evaluated the prognostic utility of MammaPrint genomic classification at 10 years of follow-up in patients with early-stage breast cancer tumour samples donated from the German PATH cohort (N=117). These findings reinforce existing studies demonstrating the utility of MammaPrint for treatment planning and prognosis. The observed 10-year OS for all patients with MammaPrint low-risk tumours was more than 93%, regardless of lymph node involvement. This aligns with the MINDACT trial, which reported excellent 8-year OS of >94% for MammaPrint lowrisk groups irrespective of treatment and
lymph node status.3 Here, the authors observed no significant differences in 10-year OS of patients with MammaPrint low-risk tumours regardless of the use of chemotherapy. Knauer et al.15 also showed favourable outcomes of 97% breast cancer-specific survival (BCSS) 5 years after diagnosis for the MammaPrint lowrisk group treated with endocrine therapy alone.15 Additionally, the authors found that patients with MammaPrint low-risk tumours had the same 10-year outcomes with either LN+ or LN- disease (93.3% OS). The MINDACT trial, which included patients with LN+ disease, found that women with clinically high/MammaPrint low-risk tumours (approximately 47% LN+ samples) demonstrated similar 8-year OS of 94.7%, compared to 96.5% for women with clinically low/MammaPrint low-risk tumours (approximately 6% LN+ samples).3 These data are in alignment with existing research suggesting that the omission of chemotherapy and treatment with endocrine therapy alone are sufficient to sustain excellent survival outcomes for patients with MammaPrint low-risk tumours, even in cases of LN+ or clinically high-risk status.
With strong evidence of high survival probability for MammaPrint low-risk patients, increased utilisation of MammaPrint for treatment planning can help spare patients with genomically low-risk tumours from adverse effects of chemotoxicity and improve long-term quality-of-life for survivors. Previously, other studies have observed large proportions (30–50%) of patients with clinically high-risk features identified as low risk by MammaPrint;3,16 and therefore genomically eligible for chemotherapy omission. In the PATH cohort, it was originally observed that approximately 40% of patients had discordant clinical and MammaPrint Risk results.11 In this 10-year follow-up population, MammaPrint identified 50% (39/78) of clinically high-risk tumours as genomically low risk. Among this subset, most patients (24/39) were treated with chemotherapy but could have had equally favourable outcomes, lower cytotoxicity, and reduced treatment costs with endocrine therapy alone.17
In contrast to MammaPrint low-risk 10-year outcomes, the MammaPrint high-risk group had significantly lower OS (71.2%; p=0.001). These data are consistent with worse 10-year BCSS outcomes observed in MammaPrint high-risk (<82%) compared to low-risk (>92%) in the STO-3 trial for patients with node-negative disease.18 This study also observed the poorest survival for MammaPrint highrisk LN+ tumours. Similarly, Mook et al.19 observed a 20% decrease in 10-year BCSS for patients with MammaPrint highrisk compared to MammaPrint low-risk tumours with 1–3 positive lymph nodes.19 Additionally, the MINDACT trial observed a worse 8-year OS of 90.1% for women with clinically high/MammaPrint highrisk tumours (approximately 26% LN+ samples) compared to 94.7% for clinically high/MammaPrint low-risk tumours (approximately 47% LN+ samples). These results suggest that MammaPrint risk analysis provides a more accurate prognosis for LN+ disease compared to relying on clinical risk alone. These data highlight the importance of treating patients with MammaPrint high-risk tumours and LN+ with chemotherapy, while patients with MammaPrint low-risk tumours and LN+ may be safely spared from the toxicities associated with chemotherapy.
Moreover, the authors observed poorer 10year survival in patients with MammaPrint high-risk tumours who did not receive chemotherapy, with a notable 29% drop in survival compared to patients with MammaPrint high-risk tumours who received chemotherapy. A meaningful chemotherapy benefit was also reported by Knauer et al.,15 in which adjuvant chemotherapy treatment led to significantly better 5-year distant disease-free survival of 88% for patients with MammaPrint high-risk tumours who received chemotherapy compared to 76% for those not treated with chemotherapy.15 In support of these findings, neoadjuvant studies have also demonstrated an increase in chemosensitivity in MammaPrint highrisk tumours. In the NBRST trial, greater chemosensitivity in the form of pathological complete response (pCR) was observed for patients with MammaPrint high-risk tumours.20 Of the patients who achieved
pCR with neoadjuvant chemotherapy, 97% had tumours characterised as MammaPrint high risk. Further investigation into the mechanisms of chemosensitivity in MammaPrint high-risk tumours is warranted.21
While this retrospective observational analysis is non-randomised and limited by the small sample size of 117 patients receiving standard-of-care treatments based on clinical-pathological features, the 10-year outcomes provide valuable longterm survival data that reflect existing study results for patients with early-stage breast cancer with HR+HER2- tumours.
Furthermore, since its inception, clinical studies utilising MammaPrint have identified cohorts of women with genomically low-risk disease with indolent disease. 18,22,23 Unlike Oncotype DX, MammaPrint’s ultra low-risk
References
1. Buus R et al. Molecular drivers of oncotype DX, prosigna, endopredict, and the breast cancer index: A TransATAC study. J Clin Oncol. 2021;39(2):126-35.
2. Kalinsky K et al. 21-gene assay to inform chemotherapy benefit in node-positive breast cancer. New England Journal of Medicine. 2021;385(25):2336-47.
3. Piccart M et al. 70-gene signature as an aid for treatment decisions in early breast cancer: updated results of the phase 3 randomised MINDACT trial with an exploratory analysis by age. Lancet Oncol. 2021;22(4):476-88.
4. Sparano JA et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. New Engl J Med. 2018;379(2):111-21.
5. Buyse M et al. Validation and clinical utility of a 70-gene prognostic signature for women with nodenegative breast cancer. JNCI. 2006;98(17):1183-92.
6. Cardoso F et al. 70-gene signature as an aid to treatment decisions in earlystage breast cancer. New Engl J Med. 2016;375(8):717-29.
7. van de Vijver et al. A gene-expression signature as a predictor of survival in breast cancer. The New Engl J Med. 2002;347(25):1999-2009.
8. Van’t Veer LJ. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530-6.
9. Weigelt B et al. Gene expression profiles of primary breast tumors maintained in distant metastases.
category offers a more distinct and clinically relevant stratification of patients that show excellent survival outcomes without extended endocrine therapy, underscoring the potential over-treatment of these patients, not only with chemotherapy but also with extended endocrine therapy.
Here, the authors demonstrated the utility of MammaPrint for treatment planning based on 10-year outcomes and reinforced previous evidence advocating for chemotherapy benefits when treating MammaPrint high-risk tumours. The data further strengthens the rationale for omitting chemotherapy in the management of MammaPrint low-risk tumours, aiming to reduce the detrimental effects of chemotoxicity while maintaining favourable long-term survival outcomes.
PNAS. 2003;100(26):15901-5.
10. European Organisation for Research and Treatment of Cancer (EORTC). Genetic testing or clinical assessment in determining the need for chemotherapy in women with breast cancer that involves no more than 3 lymph nodes (MINDACT). NCT00433589. https://clinicaltrials. gov/study/NCT00433589.
11. Gevensleben H et al. Comparison of MammaPrint and TargetPrint results with clinical parameters in German patients with early stage breast cancer. Int J Mol Med. 2010;26(6): 837-43.
12. Elston CW et al. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19(5):403-10.
13. Ravdin PM et al. Computer program to assist in making decisions about adjuvant therapy for women with early breast cancer. Am J Clin Oncol. 2001;19(4):980-91.
14. Glas AM et al. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006;7:278.
15. Knauer M et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat. 2010;120(3):655-61.
16. Drukker CA et al. A prospective evaluation of a breast cancer prognosis signature in the observational RASTER study. International Journal of Cancer. 2013;133(4):929-36.
17. Retèl VP et al. Cost-effectiveness analysis of the 70-gene signature compared with clinical assessment in breast cancer based on a randomised controlled trial. Eur J Cancer. 2020;137:193-203.
18. Esserman LJ. Use of molecular tools to identify patients with indolent breast cancers with ultralow risk over 2 decades. JAMA Oncol. 2017;3(11):1503-10.
19. Mook S et al. The 70-gene prognosissignature predicts disease outcome in breast cancer patients with 1-3 positive lymph nodes in an independent validation study. Breast Cancer Res Treat. 2009;116(2):295302.
20. Whitworth P et al. Chemosensitivity and endocrine sensitivity in clinical luminal breast cancer patients in the prospective neoadjuvant breast registry symphony trial (NBRST) predicted by molecular subtyping. Ann Surg Oncol. 2017;24(3):669-75.
21. Ríos-Hoyo A et al. Neoadjuvant chemotherapy and immunotherapy for estrogen receptor–positive human epidermal growth factor 2–negative breast cancer. J Clin Oncol. 2024;41(22):2632-6.
22. Bottosso M et al. Gene-expression assays to tailor adjuvant endocrine therapy for HR+/HER2- breast cancer. Clinical Cancer Research. 2024;30(14):2884-94.
23. Delahaye LJ et al. A breast cancer gene signature for indolent disease. Breast Cancer Res Treat. 2017;164(2):461-6.
Congenital Dermatofibrosarcoma Presenting as an Atrophic Plaque
Authors: *Jenna Koblinski,1 Katarina Lequeux Nalovic2
1. Department of Dermatology, Emory University, Atlanta, Georgia, USA
2. Atlanta Skin Cancer Specialists, Georgia, USA *Correspondence to jennakoblinski@gmail.com
Disclosure: The authors disclose no conflicts of interest. The patient and the patient’s guardian consented to her case being shared.
Dermatofibrosarcoma protuberans (DFSP) is a rare sarcoma that most commonly occurs on the trunk of adult patients. However, DFSP can present congenitally, and this rarity may lead to misdiagnosis or late diagnosis. The authors present a case of a congenital DFSP that was not diagnosed until the patient was 17 years old. Her tumour was successfully treated with Mohs micrographic surgery. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age.
Key Points
1. Dermatofibrosarcoma protuberans (DFSP) can present congenitally, though their rarity may lead to misdiagnosis or late diagnosis. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age.
2. Mohs micrographic surgery is a well-described treatment modality for DFSP that can be used for all ages.
3. It is important to evaluate DFSP for fibrosarcomatous change as these tumours are more aggressive.
INTRODUCTION
Dermatofibrosarcoma protuberans (DFSP) is a rare, locally aggressive sarcoma that most commonly occurs on the trunk of patients.1,2 The lesion typically presents as either an indurated plaque or red-brown nodules.1 DFSP occurs most often in adults during the third to fifth decades of life; however, there are reports of DFSP in childhood and even fewer reports of congenital DFSP.2-4
The authors present a case of congenital DFSP that had a delayed diagnosis and subsequent treatment with Mohs micrographic surgery (MMS).
CASE
An otherwise healthy 17-year-old female presented for evaluation of a congenital plaque on her chest. Her mother noted the
plaque had been present since birth and had grown with the patient. The patient reported this previously asymptomatic lesion had recently become sore, leading her to seek evaluation. There was no family history of immunosuppression. On examination, there was a freely movable red-brown atrophic plaque 3.1x2.4 cm on the right chest (Figure 1). The remainder of her examination was unremarkable. Given the unusual congenital plaque with the new-onset of symptoms, shared decisionmaking was utilised to perform a punch biopsy of this lesion. The differential at this time included atrophoderma, DFSP, and medallion-like dermal dendrocyte hamartoma. The results of this biopsy demonstrated a cellular dermal and subcutaneous spindle cell proliferation (Figure 2). On staining, there was strong positivity for the CD34 antigen (Figure 3). A fluorescence in situ hybridisation (FISH) analysis was performed, which detected a platelet-derived growth factor beta (PDGFB) gene rearrangement. These
Figure 1: Red-brown atrophic plaque over the right chest.
constellations of findings were consistent with a diagnosis of DFSP, and MMS was elected for treatment.
On the day of the procedure, the patient continued to report pain from the lesion. On exam, there was a 3.1x2.4 cm atrophic plaque on the right chest with retained sutures from the punch biopsy. There was no lymphadenopathy. MMS required three stages to clear the tumour, and the site was closed with a complex primary closure. The patient has since been healing well without complications.
DISCUSSION
DFSP is a rare sarcomatous malignancy that has an overall estimated incidence of 0.8 per million persons per year.2-4 Previous reports demonstrate that children less than 16 years old account for approximately 6% of total DFSP cases, with congenital cases being even scarcer.4 The authors’
Figure 2: Haematoxylin and eosin at 4x demonstrates a dense spindle cell neoplasm infiltrating dermis and subcutis.
Figure 3: Immunohistochemistry for CD34 at 4x is diffusely positive in the spindle cell neoplasm.
report highlights another rare case of congenital DFSP and a striking delay of diagnosis until the age of 17. A review of the literature on congenital DFSP from 2009 found that the most common presentation of congenital DFSP are nodular plaques of varying colours, but they noted that its presentation can be variable.3 This variation was demonstrated by the authors’ patient presenting with an atrophic plaque initially thought to be atrophoderma given its appearance. It is important for clinicians to recognise that congenital DFSP can occur and have differing features.3 There should be a low threshold to biopsy suspicious congenital lesions, as delay in diagnosis or misdiagnosis may lead to the continued growth of the malignancy and initiation of symptoms (as with this patient), as well as the potential for inappropriate treatments for incorrectly suspected benign congenital lesions; these impacts of delayed diagnosis may also complicate later definitive surgical treatment for a DFSP.3
On histopathology, DFSPs are composed of spindle cells with scant cytoplasm and little pleomorphism, and they are often CD34 positive.2 They are arranged in a storiform configuration and extend into the subcutaneous fat in a 'honeycomb' pattern.5 The aetiology of both acquired and congenital DFSP is unknown; however, many DFSPs also demonstrate a chromosomal translocation between chromosomes 17 and 22, which may be a pathogenic factor.2,3 This rearrangement leads to the fusion of PDGFB and collagen type 1A1 genes (COL1A1).2,3 This rearrangement in the authors’ patient could be evaluated by utilising fluorescence in situ hybridisation analysis.2 In the aforementioned review, they found that of 13 congenital DFSP cases tested for a PDGFB translocation, nine were positive (69%).3 The authors’ case illustrates another example of congenital DFSP having PDGFB translocation positivity and exhibits the importance of performing this analysis if there is diagnostic uncertainty in order to potentially support a diagnosis of DFSP.
Further, when evaluating DFSP histopathologically, it is important to evaluate the tumour for fibrosarcomatous (FS) change. FS under the microscope will demonstrate high-grade features including cellular atypia, increased mitoses, and a 'herring-bone' or fascicular growth pattern.5 It is important to evaluate for FS change of DFSP as these tumours are more aggressive with lower recurrencefree survival and increased potential for metastasis.5 The authors’ patient had no evidence of FS change on initial biopsies or final MMS procedure.
MMS is the preferred treatment modality for DFSP of all ages given lower recurrence rates, greater amounts of tissue sparing, and less overall number of surgeries.2-4 DFSP often have an unpredictable subclinical extension, and MMS provides real-time direct visualisation of the tumour over wide local excision. This is exemplified by the authors’ patient requiring three stages of MMS for clearance. In the 2009 review, they found that for congenital DFSP treated with MMS, the clearance rate was 100% (average follow-up: 4.3 years), and for the tumours treated with wide local excision, the clearance rate was 89% (average follow-up: 1.9 years).3 They also found that excised margins were smaller with MMS over wide local excision.3 The authors’ adolescent patient was able to have tumour clearance with the greatest amount of tissue sparing in a sensitive site, demonstrating the benefit of MMS in these cases.
CONCLUSION
DFSP can present congenitally, though their rarity may lead to misdiagnosis or late diagnosis. It is important to have DFSP on the differential for atrophic plaques or violaceous to red-brown nodules, even at a young age. Biopsy and subsequent histopathological testing are key in the diagnosis of DFSP. MMS is the preferred treatment of choice for congenital DFSP given higher clearance rates and smaller excised margins.
2. Gopal M et al. Dermatofibrosarcoma Protuberans [Internet] (2023) Treasure Island: StatPearls. Available at: https://www.ncbi.nlm.nih.gov/ books/NBK513305/. Last accessed: 6 June 2024.
3. Love WE et al. Surgical management of congenital dermatofibrosarcoma protuberans. J Am Acad Dermatol. 2009;61(6):1014-23.
4. Blaser JL et al. Using the “feed and wrap” technique and mohs surgery to eradicate congenital dermatofibrosarcoma protuberans in a 4‐month‐old. Derm Surg. 2011;37(6):862-6.
5. Hoesly PM et al. Prognostic impact of fibrosarcomatous transformation in dermatofibrosarcoma protuberans: a cohort study. J Am Acad Dermatol. 2015;72(3):419-25.
Effectiveness of Vinorelbine in the Management of Pseudomyogenic Hemangioendothelioma: A Case Report
Authors: *Safae Toumi,1 Youssef Mahdi,2 Ismail Mohamed Halfi,3 Sarah Naciri,1 Hind Mrabti,1 Rachida Latib,² Fouad Zouaidia,⁴ Basma El Khannoussi,³ Sanae Amalik²
1. Medical Oncology department, National Institute of Oncology, Mohammed V University, Rabat, Morocco
2. Pathology department, National Institute of Oncology, Mohammed V University, Rabat, Morocco
3. Radiology department, National Institute of Oncology, Mohammed V University, Rabat, Morocco
4. Pathology department, Ibn Sina University Hospital, Mohammed V University, Rabat, Morocco
*Correspondence to safae.toumi19@gmail.com
Disclosure: The authors have declared no conflicts of interest. Written informed consent for publication was obtained from the patient.
Pseudomyogenic hemangioendothelioma is an uncommon vascular neoplasm that has recently been identified as a distinct entity. Despite being classified as a malignant tumour and its often-worrisome clinical presentation, the progression typically involves relapses after surgery and a rather infrequent potential for metastasis.
The authors present the clinical case of a pseudomyogenic hemangioendothelioma of the lower limb, its evolution to lymph node involvement and distant metastasis, and the effectiveness of vinorelbine in its management. Through this case report, the authors underscore the significance of a precise histological and immunohistochemical assessment considering the usual misdiagnosis as sarcoma or metaplastic carcinoma. They also emphasise the significance of observing the evolutionary aspects of this indolent tumour while outlining the therapeutic strategy and systemic therapies’ sequences to enhance the quality of life for these long survivors.
Key Points
1. Pseudomyogenic hemangioendothelioma is a scarce tumour with distinct clinicopathological features that raise diagnostic and therapeutic challenges.
2. Advances in molecular genetics and mostly immunohistochemistry have allowed better diagnostic criteria and hence an earlier diagnosis.
3. A series of individual chemotherapy treatments over time appears to be the most effective approach for metastatic or recurrent pseudomyogenic hemangioendothelioma.
INTRODUCTION
Pseudomyogenic hemangioendothelioma (PMH) is an uncommon vascular tumour with challenges often in its diagnosis and treatment. It is classified as an endothelial neoplasm that rarely metastasises and typically exhibits a slow-growing, indolent nature. No universally accepted standard of care has been defined for such cases; nevertheless, the literature suggests a wide range of treatment options varying from local control surgeries and cryotherapy all the way to systemic therapies such as tyrosine kinase inhibitors and bone resorptive agents. The aim of this case report is to describe a frequent instance of PMH and to examine its clinical and pathological features, treatment options, evolutionary aspects, and prognosis.
CASE REPORT
The authors report the case of a 66-yearold man with no notable prior medical history who presented with a growing lump and right knee pain for over 6 months. Physical examination revealed a 30 mm multinodular mass of the crural stump of the right knee.
A first excisional surgery was performed, and no specimen was provided for pathological analysis at that time. This was followed by a second surgery for local recurrence within a year.
The surgical wound exhibited impaired healing, and the patient underwent an amputation of the lower limb. He was then eventually referred to the authors’ department for continued management and care.
The histologic examination each time revealed a tumour proliferation made of sheets and short fascicles. Tumour cells were spindle to epithelioid with vesicular
nuclei and abundant eosinophilic cytoplasm. Hemangiopericytoma-like vasculature was observed (Figure 1). Immunohistochemical (IHC) study showed strong expression of ERG and focal expression of AE1-AE3. Tumour cells were negative for h-caldesmon, smooth muscle actin, desmin, epithelial membrane antigen (EMA), CD34, and S100 protein (Figure 2). Based on all these results, a diagnosis of PMH was made.
An MRI of the right thigh showed a subcutaneous mass of right crural bump with lobulated contours, heterogeneous tumour-like tissue. Through the MRI, a second similar mass on the track of the sciatic nerve was identified (Figure 3). CT scan showed a subdiaphragmatic lymph node associated with secondary pulmonary lesions. An ultrasound biopsy was performed to confirm the nature of the mass. The biopsy and IHC appearance were in favour of PMH.
Given the multifocal nature of the disease, the patient was deemed unsuitable for surgical resection and was started on a gemcitabine 900 mg/m2 and docetaxel 75 mg/m2 regimen with no notable adverse events.
The evaluation after the third session showed clinical and radiological progression, and vinorelbine as a second line was recommended by an expert in the European Soft Tissue and Bone Sarcoma Group (STBSG). The patient showed good clinical response to treatment, demonstrating a volume reduction of primary and secondary sites and a stable disease within 8 months of follow-up.
The patient is currently on treatment with a good overall performance status ranging from 2 to 1. Ongoing monitoring for disease progression or potential complications is being maintained.
Figure 1: Representative micrographs of the tumour.
Tumour cells are arranged in sheets or in short fascicles. A) Hemangiopericytoma-like vasculature is observed. B) Tumour infiltrates surrounding adipose tissue. C and D) Tumour cells are plump spindle to epithelioid with vesicular nuclei and abundant eosinophilic cytoplasm. (haematoxylin-eosin; A: x40, B: x100, C and D: x200).
Figure 2: Immunohistochemical profile of the lesion.
A) The tumour cells are positive for ERG. B) They are negative for h-caldesmon (with blood vessels as positive control).
A B
MRI of the right amputated leg shows a tissue mass in the posterior region of the right thigh located opposite the femoral amputation stump, with an isointense signal on T1 relatively to the adjacent muscles and a high signal intensity on T2 with restriction in diffusion, and an intense heterogeneous enhancement after gadolinium injection. Locally, the mass invaded the muscular fascia with extension to the facing subcutaneous soft tissues. MRI identified a second mass with identical characteristics to the previous one located on the distal portion of the sciatic nerve (yellow square).
DISCUSSION
PMH, alternatively referred to as epithelioid sarcoma-like hemangioendothelioma, which exhibits morphological characteristics resembling those of a myoid neoplasm or an epithelioid carcinoma, is now regarded as a unique endothelial neoplasm with minimal propensity for metastasis.1
In a series of 50 cases of PMH, the majority of patients (82%) were male and under 40 years of age. Lower extremity involvement was prevalent (54%), with a predominance of soft-tissue lesions. Multifocal lesions were present in 66% of the cases.2
Diagnosis of PMH is usually based on clinical presentation, histological examination, and verifying the
Figure 3: MRI of the right amputated leg.
manifestation of both keratins and endothelial markers via IHC analysis.
Most patients remain asymptomatic up to 10 years before the diagnosis,3 while 60% of patients with PMH will experience local recurrence,2 which indicates its locally aggressive but relatively indolent profile.
PMH typically presents as lobular, clearly demarcated lesions. Cross-sectional imaging modalities are vital for diagnosing and locating PMH, given its manifestation across diverse anatomical sites.
On conventional radiographic imaging, PMH of bone presents as osteolytic, lobular, and proliferative lesions, with well-defined borders and a sclerotic rim, but without periosteal reaction. These lesions can lead to cortical destruction and pathological fractures. CT imaging shows similar characteristics, while soft tissue lesions, as exemplified by the case of the authors’ patient, manifest as indeterminate, lobular formations with well-defined demarcations.
On MRI, PMH-indicative lesions will usually exhibit low-to-intermediate signal intensity on T1-weighted sequences and will appear hyperintense on T2-weighted sequences. T1 fat-saturated post-gadolinium enhanced MRI aids in the recognition of intramuscular localisations, which appear as clearly delineated, hyperintense foci with notable contrast enhancement.
A comprehensive whole-body PET-CT scan is advised owing to the disease’s prevalent diffused manifestation.
Although whole-body MRI has great sensitivity, PET-CT is preferred for detecting metabolic activity in lesions, showing high fluorodeoxyglucose uptake. To date, all medical practitioners have employed PET-CT for lesion localisation as opposed to utilising whole-body MRI.4-5
Immunohistochemically, these tumours show widespread expression of cytokeratin (AE1/AE3), primarily in focal areas.6 In some instances, there is occasional staining for
epithelial membrane antigen and smooth muscle actin.7 In this study, the tumour cells were negative for anti-PS100, desmin, EMA, and CD34, but all showed focal expression of cytokeratin (AE1-AE3), which was consistent with the literature.
The main distinguishing characteristics of PMH in contrast to other forms of hemangioendothelioma are the variability in CD31 expression, the nonexistence of CD34 expression, and the absence of discernible histological indicators of endothelial differentiation.3
The primary differential diagnosis of PMH includes other vascular tumours (epithelioid haemangioma, and epithelioid hemangioendothelioma), epithelioid sarcoma, and metaplasic carcinoma with sarcomatoid features.8
Surgical removal with clear margins remains the primary treatment option for PMH; however, achieving negative margins can be challenging due to the tumour’s infiltrative nature and multifocal growth pattern.
Due to the rare occurrence of PMH, clinical investigations are absent. However, a range of systemic therapeutic modalities have been documented in clinical case studies.
Given the recurrent local relapses and disseminated distribution of PMH, a series of individual chemotherapy treatments over time appears to be the most effective approach, such as weekly gemcitabine, doxorubicin, oral vinorelbine, and oral cyclophosphamide, which has ensured satisfactory local tumour control.
Sirolimus and everolimus, known as mTOR inhibitors, have shown significant efficacy, especially for tumours where the DNA sequencing revealed a TSC1 mutation. While mTOR inhibitors are not currently established as definitive curative treatments, they can be utilised for longterm and maintenance therapy with minimal side effects.9
Patients with extensive disease under tyrosine kinase inhibitors such as pazopanib have demonstrated a positive clinical and radiological response, with sustained disease regression observable within a 6-month follow-up period.10
The current National Comprehensive Cancer Network (NCCN) guidelines recommend the use of pazopanib (mostly for patients unfit for intravenous systemic therapy or patients who are not candidates for anthracycline-based regimens), sirolimus, and anthracyclinebased regimens as first-line treatment for advanced or metastatic disease.1
Brance ML et al.11 reported a favourable response to pamidronate (90 mg/monthly) for a locally advanced PMH with osseous and soft tissue dissemination, and a PETCT that showed no metabolically active site in a 3-year follow-up.
In a comparable manner, the two case reports have shown encouraging results where 4 weekly doses of denosumab have resulted in stable disease for up to 4 years and mostly an alleviation of pain even in multifocal osseous lesions.6-12
References
1. National Comprehensive Cancer Network (NCCN). Practice guidelines: Soft Tissue Sarcoma. Available at: https://www.nccn.org/professionals/ physician_gls/pdf/sarcoma.pdf. Last accessed: 27 September 2024.
2. Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often multicentric tumor with indolent behavior. Am J Surg Pathol. 2011;35(2):190-201.
3. Fan C et al. Pseudomyogenic hemangioendothelioma/epithelioid sarcoma-like hemangioendothelioma of the lower limb: report of a rare case. Diagn Pathol. 2015;10:150.
PMH is a scarce tumour with distinct clinicopathological features that raise diagnostic and therapeutic challenges. Advances in molecular genetics and immunohistochemistry have improved the understanding of its pathogenesis and diagnostic criteria. This advancement has allowed for a reduction in the aggressive surgical approach, which typically leans towards amputation, especially when the tumour affects limbs.
Further studies are needed to elucidate the underlying mechanisms driving its aggressive behaviour and to identify a more effective standard of care for this challenging entity.
PATIENT PERSPECTIVE
"After my diagnosis, I experienced significant anxiety; however, the thorough explanations from the healthcare team were instrumental in alleviating my concerns. After the second line of treatment, I noticed substantial improvements in my symptoms, mostly in the alleviation of pain, which also improved my emotional health."
5. Dianat S et al. Pseudomyogenic hemangioendothelioma-a case report and review of the literature. Radiol Case Rep. 2019;14(10):1228-32.
6. Choi ME et al. A case of pseudomyogenic hemangioendothelioma of the lower extremity. Ann Dermatol. 2020;32(5):426-9.
7. Sassi F et al. Pseudomyogenic hemangioendothelioma: a misleading vascular tumor. Int J Surg Case Rep. 2022;99:107639.
8. Pasricha S et al. Multifocal primary pseudomyogenic hemangioendothelioma of bone managed with denosumab: a rare case with diagnostic and therapeutic
challenge. J Cancer Res Ther. 2022;18(3):817-9.
9. Ozeki M et al. Everolimus for treatment of pseudomyogenic hemangioendothelioma. J Pediatr Hematol Oncol. 2017;39(6):e328-31.
10. Alhanash A et al. Pazopanib as treatment option for pseudomyogenic hemangioendothelioma: a case report. Cureus. 2022;14(5):e25250.
11. Brance ML et al. Pseudomyogenic hemangioendothelioma with bone and soft tissue involvement with favorable response to pamidronate: a case report and systematic review of the literature. Arch Osteoporos. 2022;17(1):28.
12. Otani S et al. Pseudomyogenic hemangioendothelioma of bone treated with denosumab: a case report. BMC Cancer. 2019;19(1):872.
Ependymoma Alongside Hereditary Paraganglioma-Phaeochromocytoma Syndrome Due to a SDHB Mutation: A Case Report
Authors: Renee-Marie Ragguett,1 Seth A Climans,2,3 Gabe Boldt,4 Jacob Houpt,5 *Vivian S Tan6
1. Schulich School of Medicine and Dentistry, Western University, London, Ontario, Canada
2. Department of Clinical Neurological Sciences, Western University, London, Ontario, Canada
3. Department of Oncology, Western University, London, Ontario, Canada
4. London Regional Cancer Program, London Health Sciences Centre, London, Ontario, Canada
5. Department of Pathology and Laboratory Medicine, Western University, London, Ontario, Canada
6. Department of Radiation Oncology, Western University, London, Ontario, Canada
*Correspondence to vivian.tan@lhsc.on.ca
Disclosure: The authors have declared no conflicts of interest. Written informed consent was obtained from the patient’s family for the publication of this case report and the accompanying images.
Received: 16.08.24
Accepted: 10.10.24
Keywords: Ependymoma, case report, paraganglioma, phaeochromocytoma, SDHB
Mutations in the SDHB gene cause a characteristic syndrome that includes paragangliomas (PGL) and phaeochromocytomas (PCC). Herein we present a rare case of an ependymoma in a patient with a germline SDHB mutation.
A 41-year-old man with a positive family history of PGL/PCC syndrome was found to have the familial SDHB mutation. Screening imaging for paragangliomas revealed an incidental presumed ependymoma originating from the fourth ventricle. He was followed with serial imaging to assess for progression of the lesion. Due to slow, substantial growth of the tumour, and increasing symptoms which included diplopia, unsteadiness, and wide-based gait, he underwent a resection 5 years after the lesion’s identification. Following resection of the tumour, the pathology confirmed the tumour as a posterior fossa type B (PFB) ependymoma of the fourth ventricle. Unfortunately, on Day 26 post-operatively, the patient had a pulmonary embolism and died. His family consented to an autopsy, which revealed the presence of a clinically-silent pituitary neuroendocrine tumour (PitNET).
Though ependymomas are not commonly seen in PGL/PCC syndrome, they can occur. This case represents the first molecularly-characterised ependymal tumour described in this tumour predisposition syndrome. Clinicians ought to be aware of the risk of ependymoma in patients with PGL/PCC syndrome and consider this tumour when conducting their screening and follow-up of asymptomatic SDHx mutation carriers.
Key Points
1. Individuals with a family history of paragangliomas (PGL) and phaeochromocytomas (PCC) syndromes often carry genetic mutations, such as the SDHB mutation, which can increase the risk of various tumours.
2. A 41-year-old male with a known familial SDHB mutation underwent screening for PGL/PCC syndrome. During this process, an incidental ependymoma originating from the fourth ventricle was discovered. This case is the first to report a molecularly categorised ependymoma found alongside an SDHB mutation reported in literature.
3. This case suggests that ependymomas may potentially be part of the tumour spectrum caused by PGL/PCC syndrome, which could have implications for expanding screening protocols in patients with SDHB mutations.
INTRODUCTION
Succinate dehydrogenase (SDH) is a mitochondrial enzyme with a role in both the Krebs cycle and the electron transport chain. The SDH genes which encodes SDH’s four subunits have been characterised as tumour suppressor genes.1 Specifically, germline mutations in SDH genes can result in hereditary paraganglioma (PGL)/phaeochromocytoma (PCC) syndrome, where a recent study shows penetrance of 21% at 50 years old, though the rate does vary throughout the literature.2 Moreover, mutations in the SDH complex iron sulfur subunit B (SDHB) are not only associated with PGLs and PCCs, but also tumours including renal cell carcinomas, gastrointestinal stromal tumours (GIST), and pituitary neuroendocrine tumours (PitNETs).3-5
Ependymomas are relatively uncommon, constituting approximately 4% of neuroepithelial tissue tumours in those over 40 years of age. Relative to other primary malignant neuroepithelial tissue tumours, Grade 2 or 3 ependymomas have a favourable 5-year survival rate of 81.2% between the ages of 40–64 years, and 56.9% over the age of 65 years, in Canada.6
Ependymomas are primary central nervous system tumours that putatively originate from ependymal cells lining the cerebral ventricles and spinal cord.7 The clinical symptoms associated with ependymomas often include headaches, vomiting, and diplopia.8 Gold standard treatment is maximal safe surgical resection followed by radiotherapy depending on tumour location
and residual and molecular features. Chemotherapy has a relatively limited role in the management of adult ependymomas.9
Ependymomas are not known to be caused by germline SDHB mutations. Herein, the authors present a case of a patient with a germline SDHB mutation who developed a posterior fossa type B (PFB) ependymoma and was also incidentally found to have a PitNET. The patient did not have PCC or PGL.
CASE PRESENTATION
A 41-year-old male presented for presymptomatic genetic testing. His maternal aunt was found to have an SDHB mutation and was diagnosed with tumours characteristic of PGL/PCC syndrome in her thirties. This led to the rest of the family having genetic testing. The patient was found to be a carrier of the familial mutation in the SDHB gene c.137G>A (p.Arg46Gln), which is pathogenic for hereditary PGL/ PCC syndrome.10 The patient’s mother, as well as a maternal first cousin, were both also confirmed to have the SDHB mutation. Furthermore, the patient also noted that two maternal aunts had developed brain tumours; however, further details including their diagnoses or whether they were carriers of the SDHB mutation were unavailable.
The patient was started on a presymptomatic screening plan, which involved serial monitoring of urinary fractionated metanephrines and catecholamines, and imaging, which included MRI of the head, neck, abdomen, and pelvis; thorax CT; and oesophagogastroduodenoscopy.
The initial screening imaging series showed no signs of PCCs or PGLs. However, the brain MRI, which was meant to look for a glomus jugulare, instead revealed a posterior fossa tumour measuring 27 mm x 39 mm × 47 mm, and was thought to be an ependymal tumour. The mass originated in the fourth ventricle, extending into the foramen of Magendie and foramen of Luschka bilaterally. There was mild dilatation of the cerebral aqueduct and lateral ventricle, as well as some flattening of the medulla and lower pons. Imaging also identified a small right adrenal nodule, likely a myelolipoma, and a small left adrenal presumed adenoma.
Following the MRI scan, the patient was reviewed by neurosurgery. He reported an approximate 2-year history of dizziness, predominately with neck extension and
going from standing to supine quickly. However, his neurological examination revealed only subtle horizontal nystagmus on extremes of lateral gaze.
Follow-up imaging and laboratory tests were performed for 5 years. The ependymoma slowly grew and the patient’s symptoms progressed (Figure 1).
After 4 years of clinical follow-up, he developed increased dizziness with head motion, occasional nausea and vomiting in the morning, gait unsteadiness when turning, and increased fatigue. There was diplopia on left gaze and left-beating nystagmus on primary gaze and gazeevoked nystagmus. The hydrocephalus progressed slightly and imaging revealed obvious dilation of the lateral ventricles, as well as the third ventricle.
-6 years
Confirmed carrier of clinically significant mutation in SDHB gene (p.R46Q)
Dizziness (-2015)
Ependymoma
Progression:
-5 years
Imaging findings: 4th ventricular tumour
0 (Surgery)
Surgery: 4th ventricular tumour resection
Postoperative diagnosis: posterior fossa ependymoma type B
Incoordination, unsteadiness, distortion in taste, slightly wide gait
Fatigue, unsteady gait when turning Nausea, vomiting
+26 days
Death due to pulmonary embolism
Ependymoma progression is reported in millimeters in the superior-inferior, anterior-posterior, and medial-lateral directions. Time 0 represents the date that the patient had surgery.
Figure 1: Timeline of significant events including symptom onset and ependymoma progression in a 41-year-old male with a germline SDHB mutation.
When evaluated almost 5 years after the tumour was discovered, the patient reported hypergeusia, unsteadiness, incoordination, wide-based gait, morning nausea, and occasional vomiting. On examination, the patient was unable to perform the tandem gait test and demonstrated dysmetria on the left side. Imaging revealed that the mass was filling the fourth ventricle, extending caudally through the foramen of Magendie (Figure 2) and measured 32 mm x 43 mm x 55 mm. Given the increase in symptoms, at this point, the patient was referred to surgery with the goal for treatment to decrease symptoms and prevent progression.
He underwent a suboccipital craniotomy and C1 laminectomy for tumour resection approximately 5 years after the tumour was first discovered on MRI imaging. The pathology revealed a PFB ependymoma of the fourth ventricle and was classified as World Health Organization (WHO) Grade 2. Immunohistochemistry was used to characterise the tumour as a PFB ependymoma and showed that H3K27me3 nuclear expression was retained (Figure 3G), H3K7M mutation was immunonegative, and the Ki-67 index was 1–3%. Other
immunohistochemical markers are seen in Figure 3. The postoperative MRI showed an apparent gross total resection, as well as bilateral inferomedial cerebellar infarcts. Fourteen days postoperatively, the patient had ongoing diplopia. There were bilateral abducens nerve palsies (worse on the left) and horizontal nystagmus, which likely stemmed from a perioperative cerebellar ischemic stroke. Otherwise, the patient moved his extremities well and was considered stable from a neurological perspective. There were plans for a staging lumbar puncture and an MRI complete spine.
While rehabilitating in hospital, 26 days postoperatively, he collapsed suddenly and could not be resuscitated. The patient had been receiving pharmacological deep vein thrombosis prophylaxis. An autopsy was performed, which confirmed a saddle pulmonary embolism as the cause of death.
Bilateral cerebellar infarcts were also visualised during the autopsy (Figure 3D) and the patient was also found to have residual ependymoma in the foramina of Luschka (Figure 3H) and subarachnoid space. There was also a pituitary neuroendocrine tumour, which was 5 mm in size. This tumour was of
2: Final preoperative post-gadolinium T1 MRI imaging of the head in A) axial; B) coronal; and C) sagittal orientations.
Figure
Ependymoma (red arrow) filling the fourth ventricle.
3:
A, B) The tumour consists of uniform round-to-ovoid cells in a fibrillary matrix with perivascular pseudorosettes (H, E).
C) immunohistochemistry for glial fibrillary acidic protein demonstrates diffuse strong cytoplasmic expression in tumour cells.
D) posterior view of infratentorial area with bilateral cerebellar infarcts.
E, F) diffuse cytoplasmic immunopositivity for D2-40 (D) and CD99 (F), with occasional dot-like perinuclear expression.
G) H3 K27me3 expression is entirely retained in tumour cell nuclei. H) axial slice of the rostral medulla and cerebellum with residual tumour (*) at the left foramen of Luschka.
somatotroph subtype, Pit-1 positive, growth hormone expressing, and densely granulated (immunohistochemical markers seen in Figure 4). No phaeochromocytomas or paragangliomas were identified at autopsy.
DISCUSSION
Mutations in the SDH genes are associated with specific PGL syndromes. These syndromes are inherited in an autosomal dominant fashion and have characteristic clinical features depending on the SDH mutation present.3 Notably, ependymomas are not commonly seen with any of the SDHx mutations, and from the authors' literature search, up to June 2024, there has only been one report of an SDHD mutation alongside an ependymoma.11 The study examined the impact of higher altitudes on the development of more severe PGL syndrome 1 (PGL1), and unfortunately, no details were provided about the patient
that developed the ependymoma. While the occurrence of concurrent PGLs/PCCs and ependymomas is relatively uncommon, it has been reported previously; however, these did not occur alongside SDHx mutations. One occurrence had an unknown genetic status, and the other occurred alongside heritable multiple endocrine neoplasia type 2A caused by a D631YRET mutation.12,13 The authors have presented a patient with an ependymoma and SDHB mutation, and to their knowledge, following a literature conducted in June 2024, this combination has not otherwise been reported in the literature.
Specifically, mutations in the SDHB gene have been associated with PGL syndrome 4 (PGL4). Compared to other syndromes, PGL4 generally presents with higher rates of PCCs, thoracoabdominal PGLs, renal cell carcinomas, and distant metastases. Furthermore, like the syndromes occurring with SDHD mutations (i.e., PGL1) and
Figure
A B C D
Figure 4:
A,B) H&E section of pituitary gland with a round well-circumscribed hypercellular lesion (A) featuring tumour cells (B) with predominantly eosinophilic cytoplasm, densely-arranged in sheets (B, left) relative to the adjacent uninvolved pituitary (B, top-right).
C,D) tumour cells are immunopositive for the pituitary lineage factor Pit1 (C), while being immunonegative for SF1 (D).
E) strong synaptophysin expression is supportive of a pituitary adenoma/pituitary neuroendocrine tumour.
H,I,J,K,L) expression of adrenocorticotropic hormone (H), thyroid-stimulating hormone (I), prolactin (J), luteinizing hormone (K), and follicle-stimulating hormone (L) is restricted to the uninvolved pituitary gland.
M) reticulin silver stain demonstrates a loss of normal acinar architecture in the tumour.
N,O) tumour cells diffusely express cytokeratin 8/18 stronger than background pituitary (N), with rare fibrous bodies (O).
SDHA mutations (i.e., PGL5), PGL4 is also associated with GISTs and pituitary neuroendocrine tumours. In contrast, relative to many other PGL syndromes, PGL4 is associated with lower rates of head and neck PGLs, and lower rates of multifocal disease.3 The patient in the authors' article did not present with many of the characteristic neoplasms of PGL4. Furthermore, the specific growth hormonesecreting pituitary neuroendocrine tumour
that the patient presented with can cause acromegaly, which has been associated with a hypercoagulable state, and as a result, massive pulmonary embolisms.14 Interestingly, whilst the patient had a positive family history of PCCs/PGLs, he also had a positive family history for other brain tumours. Although ependymoma has only been reported with a SDHx mutation once, the relationship between SDHx mutations and other tumours, including
ependymomas, has been explored, as SDH genes have proposed roles in tumourigenesis in general, and there are occasionally reports of various non PGL syndrome–like tumours occurring in those with SDHx mutations.15
The SDH gene has been suggested to have a possible genotype–phenotype–environment relationship on PGL syndromes. This has been explored by Astrom et al.,11 who have reported the only other occurrence of an ependymoma alongside an SDH mutation. The influence of higher altitude on PGL1 disease (i.e., those with SDHD mutations) was explored given the increased incidence of PGLs found in those living at high altitudes and the hypothesis that mutations in SDHD disrupt oxygen sensing in the carotid bodies, which may result in tumour formation. They found that, indeed, high altitude plays a negative role in the PGL1 phenotype, wherein those at higher altitudes had, for example, an increased number of tumours at first diagnosis and had symptoms at earlier ages. A number of the participants in this study were also reported to have developed non-paraganglionic tumours, including an ependymoma; however, these non-paraganglionic tumours did not have an association with altitude.11
Because SDH genes have role in DNA hypermethylation and hypoxia-inducible factor activation modification, their role in tumourigenesis has been explored.16,17 It has been demonstrated that tumours associated with SDHB mutations, such as GISTs, stain negative for SDHB immunohistochemistry; however, if the GIST was sporadic, in most cases there was positive SDHB immunohistochemistry.18 In studies that have assessed SDHB immunohistochemistry in ependymomas, SDHB expression was preserved, suggesting SDHB did not play a role in the tumour’s genesis.19 These findings
References
1. Bardella C et al. SDH mutations in cancer. Biochim et Biophys Acta. 2011;1807(11):1432-43.
2. Rijken JA et al. The penetrance of paraganglioma and
were further corroborated by another study which found that ependymomas had preserved SDHB expression, though other central nervous system tumours, such as hemangioblastomas, did show loss of SDHB expression.20 Taken together, while SDHB expression may be altered in some tumours, expression is normal in most ependymomas, suggesting that SDHB may not play a large role in formation of ependymomas.
In 2021, a consensus statement was published, outlining recommendations for screening and follow-up of asymptomatic carriers of SDHx mutations. Recommendations for screening include blood pressure monitoring; an assessment of possible symptoms and signs of a jugular foramen paraganglioma (e.g., dysphagia, palpitations, and neck mass); measure of urine or plasma levels of metanephrines and normetanephrines; an MRI of the head, neck, abdomen, and pelvis; and a PET-CT. Given the continued risk of developing PCCs/PGLs, it has been suggested that individuals should have a regular follow-up following a negative workup, with an annual blood pressure measurement, biennial plasma or urine metanephrines, and an imaging series every 3 years.21
CONCLUSION
The authors present a unique case of a patient with an incidentally discovered Grade 2 PFB ependymoma and a small PitNET in the context of a known germline, pathogenic SDHB mutation. The patient did not have PCCs or PGLs, which are often seen in the syndrome. Although there is a chance the ependymoma was unrelated to the SDHB mutation, based on this report and one other report in the literature, the authors suspect that the spectrum of tumours caused by PCC/PGL syndrome includes ependymomas too.
pheochromocytoma in SDHB germline mutation carriers. Clin Genet. 2018;93(1):60-6.
3. Benn DE et al. 15 Years of paraganglioma: clinical manifestations of paraganglioma syndromes
types 1–5. Endocr Rel Cancer. 2015;22(4):T91-103.
4. Gimenez-Roqueplo A-P et al.; COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant
phaeochromocytomas. Cancer Res. 2003;63(17):5615-21.
5. Benn DE et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827-36.
6. Walker EV et al. The incidence and prevalence of primary central nervous system (CNS) tumours in Canada (2010–2017), and the survival of patients diagnosed with CNS tumours (2008–2017). Curr Oncol. 2023;30(4):4311-28.
7. Low JT et al. Primary brain and other central nervous system tumors in the United States (2014-2018): a summary of the CBTRUS statistical report for clinicians. Neurooncol Pract. 2022;9(3):165-82.
9. Chai Y-H et al. Ependymomas: prognostic factors and outcome analysis in a retrospective series of 33 patients. Brain Tumor Res Treat. 2017;5(2):70-6.
10. Landrum MJ et al. ClinVar: improving access to variant interpretations and
11. Astrom K et al. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113(3):228-37.
12. de Tersant M et al. Pheochromocytoma and paraganglioma in children and adolescents: experience of the French Society of Pediatric Oncology (SFCE). J Endocr Soc. 2020;4(5):bvaa039.
13. Ospina NS et al. Clinical features of a family with multiple endocrine neoplasia type 2A caused by the D631Y RET mutation. Thyroid. 2017;27(10):1332-4.
14. Elarabi AM et al. Massive pulmonary embolism as the initial presentation of acromegaly: is acromegaly a hypercoagulable condition? Am J Case Rep. 2018;19:1541-5.
15. Pasini B, Stratakis CA. SDH mutations in tumourigenesis and inherited endocrine tumours:lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med. 2009;266(1):19-42.
16. Killian JK et al. Succinate dehydrogenase mutation underlies
REPRINT QUERIES PLEASE CONTACT:
global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648-57.
17. Guzy RD et al. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen speciesdependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28(2):718-31.
18. Gaal J et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney–Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol. 2011;24(1):147-51.
19. Ballester LY et al. Expression of IDH1 mutant protein R132H and SDHB in adult and pediatric gliomas. Int J Neuropathol. 2014;2(1):17-23.
20. Roh TH et al. The loss of succinate dehydrogenase B expression is frequently identified in hemangioblastoma of the central nervous system. Sci Rep. 2019;9(1):5873.
21. Amar L et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol. 2021;17(7):43544.