Saskia Middeldorp, Elizabeth Macintyre, and 2024 EHA President
Antonio Almeida
Exploring CAR-T cell therapy in leukaemia, and therapies for platelet disorders Infographics:
10 Review of the 2024 European Hematology Association (EHA) Congress, 13th-16th June 2024
Congress Features
20 Ageing and Haematology: The Clone Wars
Aleksandra Zurowska
25 A Debate on the Use of Gene Therapy in Patients with Haemophilia
Katrina Thornber
Symposium Review
29 Normalisation of Haemostasis in Haemophilia A
Abstract Reviews
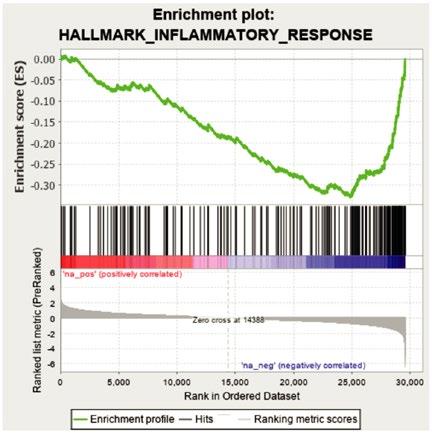
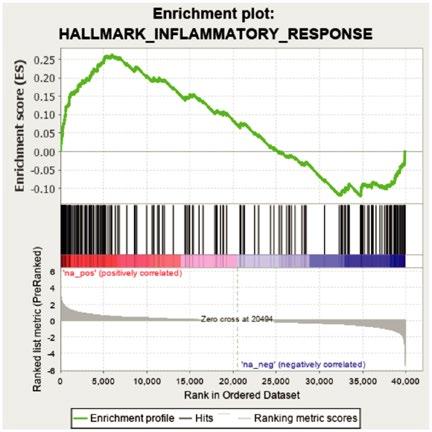
38 Impact of TET2 Mutations on Haematopoietic Stem Cell Resilience to Immune-Related Stress
Encabo et al.
42 KMT2A-MLLT3-Induced Leukaemia Changes During Ontogenic Stages
Almowaled et al.
44 Advanced-Stage Hodgkin Lymphoma International Prognostic Index (A-HIPI) in Turkish Patients with Classical Hodgkin Lymphoma: A Single-Center Retrospective Study
Koca et al.
46 Five-year Outcome of CD19 Combined With CD22 CAR-T Cell Therapy in Patients With B-ALL Relapsed After Allo-transplantation
Liu et al.
Interviews
64 Niamh O'Connell
68 Pier Mannuccio Mannucci
Infographics
72 Leukaemia: CAR-T Cell Therapy Innovations
74 Therapeutic Approaches for Platelet Disorders Articles
76 Editor's Pick: Extensive Left Sided Venous Thrombi and Bilateral Pulmonary Emboli in the Context of May-Thurner Syndrome for a Patient Presenting with Acute Flank Pain: A Case Report
Scadding and Ramoutar
82 Spinal Plasmacytoma Transformed Into Solitary Sacral Amyloidoma: A Case Report
Neupane et al.
87 Filgrastim Used for Infection Prophylaxis for Moderate Neutropenia Related to Primary Myelodysplasia Prior to Elective Surgery: A Case Report
Mannala and Harrison
91 Introducing the Concept of Patient Blood Management and Haemovigilance in Government Sector Hospitals of Karachi, Pakistan
Waheed et al.
98 Diffuse Large B Cell Lymphoma of Spleen: An Important Differential of a Nodular Splenomegaly: A Case Report
Punia et al.
104 The Impact of ‘Pre-conception’ on Conception: An Inadvertent Form of Infertility
Lipton
"Today we come together as a community of haematologists, bound by our collective commitment to advancing the field of haematology, and enhancing patient care"
Editorial Board
Editor-in-Chief
Emanuele Angelucci
Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Genova, Italy
Chair, Hematology and Cellular Therapy Unit, Ospedale Policlinico
San Martino; Transplant Program Director, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Genova, Italy
Dr Dimitar Efremov
International Centre for Genetic Engineering & Biotechnology, Italy
Dr David Gómez Almaguer
Hospital Universitario Dr. Jose E. González Universidad Autónoma de Nuevo León, México
Prof Loredana Bury
University of Perugia, Italy
Prof Ahmet Muzaffer Demir
Trakya University, Türkiye
Prof Sabri Kemahli
Yeditepe University, Türkiye
Dr Dominique Bonnet
Francis Crick Institute, UK
Dr Utkarsh Acharya
Brigham & Women’s Hospital, Massachusetts, USA
Aims and Scope
EMJ Hematology is an open access, peer-reviewed eJournal committed to publishing the highest quality medical research concerning all aspects of diseases of the blood and bone marrow to help advance the development of this field.
The journal is published annually, approximately six weeks after the European Hematology Association (EHA) Congress, and features highlights from this congress, alongside interviews with experts in the field, reviews of abstracts presented at the congress, as well as in-depth features on congress sessions. The journal also covers advances within the clinical and pharmaceutical arenas by publishing sponsored content from congress symposia, which is of high educational value for healthcare professionals. This undergoes rigorous quality control checks by independent experts and the in-house editorial team.
EMJ Hematology also publishes peer-reviewed research papers, review articles, and case reports in the field. In addition, the journal welcomes the submission of features and opinion pieces intended to create a discussion around key topics in the field and broaden readers’ professional interests. The journal is managed by a dedicated editorial team that adheres to a rigorous double-blind peer-review process, maintains high standards of copy editing, and ensures timely publication.
EMJ Hematology endeavours to increase knowledge, stimulate discussion, and contribute to a better understanding of blood disorders. Our focus is on research that is relevant to healthcare professionals in this field. We do not publish veterinary science papers or laboratory studies not linked to patient outcomes. We have a particular interest in topical studies that advance research and inform of coming trends affecting clinical practice in haematology.
Further details on coverage can be found here: www.emjreviews.com
Editorial Expertise
EMJ is supported by various levels of expertise:
• Guidance from an Editorial Board consisting of leading authorities from a wide variety of disciplines.
• Invited contributors who are recognised authorities in their respective fields.
• Peer review, which is conducted by expert reviewers who are invited by the Editorial team and appointed based on their knowledge of a specific topic.
• An experienced team of editors and technical editors.
Peer Review
On submission, all articles are assessed by the editorial team to determine their suitability for the journal and appropriateness for peer review.
Editorial staff, following consultation with a member of the Editorial Board if necessary, identify three appropriate reviewers, who are selected based on their specialist knowledge in the relevant area.
All peer review is double blind. Following review, papers are either accepted without modification, returned to the author(s) to incorporate required changes, or rejected.
Editorial staff have final discretion over any proposed amendments.
Submissions
We welcome contributions from professionals, consultants, academics, and industry leaders on relevant and topical subjects. We seek papers with the most current, interesting, and relevant information in each therapeutic area and accept original research, review articles, case reports, and features.
We are always keen to hear from healthcare professionals wishing to discuss potential submissions, please email: editorial.assistant@emjreviews.com
To submit a paper, use our online submission site: www.editorialmanager.com/e-m-j
Submission details can be found through our website: www.emjreviews.com/contributors/authors
Reprints
All articles included in EMJ are available as reprints (minimum order 1,000). Please contact hello@emjreviews.com if you would like to order reprints.
Distribution and Readership
EMJ is distributed through controlled circulation to healthcare professionals in the relevant fields across Europe.
Indexing and Availability
EMJ is indexed on DOAJ, the Royal Society of Medicine, and Google Scholar®
EMJ is available through the websites of our leading partners and collaborating societies. EMJ journals are all available via our website: www.emjreviews.com
Open Access
This is an open-access journal in accordance with the Creative Commons Attribution-Non Commercial 4.0 (CC BY-NC 4.0) license.
Congress Notice
Staff members attend medical congresses as reporters when required.
All information obtained by EMJ and each of the contributions from various sources is as current and accurate as possible. However, due to human or mechanical errors, EMJ and the contributors cannot guarantee the accuracy, adequacy, or completeness of any information, and cannot be held responsible for any errors or omissions. EMJ is completely independent of the review event (EHA2024) and the use of the organisations does not constitute endorsement or media partnership in any form whatsoever. The cover photo is of Madrid, Spain, the location of EHA2024.
Victoria Antoniou, Helena Bradbury, Ada Enesco, Laith Gergi, Katrina Thornber, Katie Wright, Aleksandra Zurowska
Creative Director
Tim Uden
Design Manager
Stacey Rivers
Senior Designers
Roy Ikoroha, Steven Paul
Designer
Owen Silcox
Junior Designers
Dillon Benn Grove, Shanjok Gurung
Senior Business
Unit Leader
Nabihah Durani
Senior Performance & Insight Lead
Darren Brace
Marketing Director
Kristina Mestsaninova
Chief Content Officer
Justin Levett
Chief Commercial
Officer
Dan Healy
Founder and Chief Executive Officer
Spencer Gore
Welcome
Dear Readers,
Welcome to the 2024 issue of EMJ Hematology, bringing you highlights from this year’s European Hematology Association (EHA) Congress, alongside exclusive interviews with experts and peer-reviewed articles covering a plethora of topics in the field.
It was a pleasure to attend this year’s EHA Congress, and I want to take this opportunity to talk about my highlight from the event. I attended a debate session between two opinion leaders on gene therapy for patients with haemophilia, and whether this should be available to all, or only the “happy few”, a subset of patients with a severe haemophilia B phenotype.
Gene therapy, in theory, promises that, with a single infusion, the need for patients with haemophilia to receive continuous prophylaxis is eliminated, with fewer bleeding episodes, increased clotting factor expression, and a potentially lifelong response duration. Nonetheless, there are side effects and limitations in real-life scenarios, as there is no guarantee that the need for prophylaxis after treatment is completely removed. High numbers of patients have reactions to treatment (in some cases, severe allergic reactions) and liver function abnormalities. Importantly, the cost of treating one single patient is 2.8 million EUR, making it potentially inaccessible for several healthcare systems, particularly in limited-resource settings.
Ultimately, for me, the key element in favour of gene therapy comes from the patients themselves, who have emphasised the value of having a few haemophilia-free years, even if they eventually do need prophylaxis again. This, in my opinion, is the strongest argument for making gene therapy available to all: putting the patient first!
I do hope you enjoy reading through this issue and learning about the pertinent topics in haematology!
Editorial enquiries: editor@emjreviews.com
Sales opportunities: salesadmin@emjreviews.com
Permissions and copyright: accountsreceivable@emjreviews.com
Evgenia Koutsouki
Reprints: info@emjreviews.com
Media enquiries: marketing@emjreviews.com
THE PREMIER MEETING IN THE FIGHT AGAINST BLOOD CANCERS
We look forward to welcoming you in Houston, Texas at SOHO 2O24, the premier meeting focused solely on the latest advances and practical clinical applications in the field of hematologic oncology. SOHO has no parallel general sessions so that delegates may attend all sessions in sequential order. View the program and details at https://soho.click/2O24.
Don’t miss out on this one-of-a-kind opportunity to network with fellow attendees from across the globe and learn the latest updates in hematologic oncology from a multidisciplinary group of internationally recognized experts!
Register today for the 12th Annual Meeting of the Society of Hematologic Oncology. The meeting is hybrid so you may attend in-person or online!
SEPTEMBER 4-7, 2O24
George R. Brown Convention Center
Houston, Texas USA
soho.click/2O24
Foreword
I am delighted to present the latest edition of EMJ Hematology. This issue is packed with a wealth of content designed to engage and inform our readers. Among the standout pieces are several compelling case reports, insightful reviews, and interviews with leading experts in the field.
In this issue, you will find an in-depth case report on the use of filgrastim for infection prophylaxis in moderate neutropenia related to primary myelodysplasia prior to elective surgery. Another notable case report examines extensive left-sided venous thrombi and bilateral pulmonary emboli in the context of May–Thurner syndrome. We also feature a transformative case report on spinal plasmacytoma evolving into solitary sacral amyloidoma, among many other articles and abstract reviews.
Our infographic section includes captivating pieces on innovations in chimeric antigen receptor T-cell therapy in leukaemia, and therapeutic approaches for platelet disorders, which are guaranteed to be informative reads.
The level of expertise from this year’s interviewees was remarkable. We feature Congress interviews with distinguished
figures such as Elizabeth Macintyre, former president of the European Haematology Association (EHA), and president-elect of Biomedical Alliance Europe; Saskia Middeldorp; and 2024 EHA President Antonio Almeida. Other interviews include enlightening conversations with Niamh O’Connell and Pier Manucci, offering personal insights and expert commentary on current haematology practices.
The level of expertise from this year’s interviewees was remarkable
I hope you enjoy the variety of content offered as much as we enjoyed curating it. I extend my gratitude to all the authors, peer reviewers, and interviewees for their invaluable contributions, and to the Editorial Board and team at EMJ for their unwavering commitment to delivering exceptionally high-quality content.
Emanuele Angelucci
Chair, Hematology and Cellular Therapy Unit, Ospedale Policlinico San Martino; Transplant Program Director, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Genova, Italy
Today we come together as a community of haematologists, bound by our collective commitment to advancing the field of haematology, and enhancing patient care
Review of the 2024 European Hematology Association (EHA) Congress Congress Review
THE 29th annual Congress of the European Hematology Association (EHA) took place this year in the vibrant city of Madrid, Spain, from 13th–16th June. It was a unique opportunity for haematologists from around the world to gather and discuss cutting-edge innovations in the field, advancing patient care in the process.
“Today we come together as a community of haematologists, bound by our collective commitment to advancing the field of haematology, and enhancing patient care,” announced Antonio Almeida, 2024 EHA President. He drew attention to EHA’s ambitious agenda for 2024: advancing haematology technology, personalised medicine, markers in diagnostics, equitable access to medicine, and improved sustainability. Finally, Almeida extended thanks to all the contributors of the Congress, and welcomed Brian Huntley, Chair of the EHA Scientific Committee, to the stage.
“As we gather here in Madrid, I am reminded of the profound impact that our collective efforts can have on the lives of our patients and their families,” began Huntley. He highlighted the vast quantity of sessions prepared this year (180, to be exact), and the diversity of topics covered. Additionally, over 3,500 abstract were submitted, a staggering, record-breaking figure for EHA. Continuing on, Huntley stressed EHA’s commitment to diversity, equity, and inclusion, praising the 40 countries represented in the faculty, and the introduction of their new EHA Diversity, Equity, and Inclusion award.
Winners of the EHA Research Grants were subsequently commended. For the junior research grants, aimed at supporting those starting out in their research endeavours, the winners were Lakshmi Sandhow, Institut Cochin, Paris, France; Helga Simon Molas, Amsterdam University Medical Center, the Netherlands; Femke Hormann, Karolinska Institutet, Stockholm, Sweden; Václav Šeda, CEITEC Masaryk University, Brno, Czech Republic; and Sigrún Thorsteinsdóttir, University of Iceland, Reykjavík, Iceland.
Moreover, the Advanced Research Grants, aimed at individuals 4–8 years post-PhD, were awarded to Alba Maiques-Diaz, IDIBAPS, Barcelona, Spain; Serena Scala, Ospedale San Raffaele, Milan Italy; and Mathijs Sanders, Erasmus University Medical Center, Rotterdam, the Netherlands. The Physician Scientist Research Grants were awarded to Camille Bigenwald, Institut Gustave Roussy, Villejuif, France; Simon Richardson, University of Cambridge, UK; and Delfim Duarte, Instituto de Biologia Molecular e Celular, Porto, Portugal. Enrica Federti, University of Verona, Italy, was awarded the ‘Topic-in-Focus’ Advanced Research Award.
Spotlighting global collaboration, the EHA Bilateral Collaborative Grant winners were praised and welcomed to the stage. Countries represented among the awardees included the UK, Germany, Spain, Italy, and Switzerland. Other grants additionally awarded included the 2023 and 2024 Research Mobility and Innovation grants.
Following the grant ceremony, Almeida retook the stage, focusing the audience’s attention to the José Carreras Award. Named after the famous opera singer, Carreras himself underwent a haematopoietic transplant procedure in Seattle, in 1988, and since set up the José Carreras Foundation, honouring those actively contributing to the field. The 2024 winner was John Gribben, Barts Cancer Institute, London, UK, who subsequently shared an insightful lecture on the impact of haematologic malignancies on the immune system.
“Education and mentoring are a fundamental pillar of EHA activity,” reminded Almeida, as he handed the 2024 Education and Mentoring Award to Jan Trnka, Charles University and University Hospital Motol, Prague, Czech Republic. Finally, Ivo Touw, Erasmus University Medical Center, was awarded the prestigious David Grimwade Award for his research into bone marrow failure and leukaemia predisposition syndromes, and presented an informative talk detailing the complex biology of congenital neutropenia.
Education and mentoring are a fundamental pillar of EHA activity
Stay tuned for more insights from this incredible Congress, including late-breaking clinical trial data and an interview with the EHA President himself, Antonio Almeida.
Benefit of Isa-VRd for Transplant-Ineligible Patients with Multiple Myeloma
FIRST line treatment is crucial for patients with newly diagnosed multiple myeloma (NDMM), especially in patients who are not eligible for transplant due to age or comorbidities. The current standard of care includes a combination of bortezomib, lenalidomide, and dexamethasone (VRd).
The randomised trial was conducted at
102 sites across
21 countries with a total of
446 patients with active, measurable NDMM
The Phase III study, presented at EHA2024 by Thierry Facon, University of Lille and French National Academy of Medicine in Paris, France, aimed to assess the clinical benefit, efficacy, and safety of adding isatuximab (Isa), an anti-CD38 monoclonal antibody, to the VRd regimen (Isa-VRd), compared to VRd alone, in transplantineligible patients with NDMM.
The global IMROZ study, an open-label, global, prospective, randomised trial was conducted at 102 sites across 21 countries, with a total of 446 patients with active, measurable NDMM. The patients were randomised in a 3:2 ratio to receive either Isa-VRd or VRd. Patients over 80 years of age were excluded from the study.
The primary endpoint was progression-free survival (PFS), with key secondary endpoints being complete response rate, minimal residual disease negativity, very good partial response or better, and overall survival. Adverse events were graded according to NCI CTCAE v4.03 standards.
Results showed that, at the time of data cutoff on 26th September 2023, 265 patients received Isa-VRd, and 181 received VRd. The median treatment duration was 53.2 months for Isa-VRd compared to 31.3 months for VRd. At a median follow-up of 59.7 months, median PFS was not reached for Isa-VRd vs 54.3 months for VRd. The hazard ratio (HR) for PFS was 0.596 (98.5% CI: 0.406–0.876), indicating a significant reduction in the risk of progression or death by 40.4% with Isa-VRd. The PFS benefit was consistent across subgroups and maintained through subsequent lines of therapy and the addition of Isa did not significantly affect the relative dose intensity of VRd.
The study results demonstrate that Isa-VRd significantly reduces the risk of disease progression or death by 40.4%, compared to VRd alone, while providing deep and sustained responses. Additionally, the safety profile of Isa-VRd was consistent with the addition of Isa, and the observed numerical differences in treatment-emergent adverse events were largely due to longer exposure in the Isa-VRd arm. These results support Isa-VRd as a potential new standard of care for transplant-ineligible patients with NDMM.
APOLLO Trial: New Hope for High-Risk Acute Promyelocytic Leukaemia
HIGH-RISK acute promyelocytic leukaemia (HR-APL) is a very rare form of acute myeloid leukaemia, defined by a white blood cell count at diagnosis >10,000/µL. Only one-third of patients with APL have HR-APL, which is accompanied by a high risk of complications, including bleeding and thrombosis, especially during the first stage of treatment.
1 in 3
patients with APL have HR-APL
Chemotherapy combined with all transretinoic acid (ATRA-CHT) has become the gold standard for treatment of APL. A recent trial presented at EHA2024 assessed a new chemotherapy-free regimen of ATRA and arsenic trioxide (ATO), supplemented with two-shots of idarubicin, for treatment of patients with HR-APL compared to conventional ATRA-CHT treatment.
In the APOLLO Trial, an open-label, prospective, multicentre multinational Phase III trial, patients were randomised 1:1 to receive ATRA-ATO plus two shots of idarubicin, or ATRA-CHT. The study found that the 2-year survival rate in the ATRA-ATO group (88%; 95% CI: 80–96%) was
significantly higher than in the ATRACHT group (70%; 95% CI: 59–83%; P=0.02). The main factors explaining the improved event-free survival were incidence of molecular relapse and molecular resistance.
The authors concluded that first-line therapy with ATRA-ATO with two initial doses of IDA results in superior eventfree survival compared to conventional ATRA-CHT in patients with HR-APL, which may support implementation of this regimen as the new standard of care for patients with HR-APL.
Low-Molecular-Weight Heparin Ineffective for Preventing Thrombosis in Leukaemia
RECENT data presented at EHA2024 revealed that primary thromboprophylaxis using a fixed intermediate-dose low-molecular-weight heparin (LMWH) does not effectively prevent thrombosis in adults undergoing remission-induction treatment for acute lymphoblastic leukaemia (ALL).
Adults with ALL face a significant risk of VTE, which can lead to increased morbidity and decreased survival. This comprehensive study, a prospective side-study within the HOVON-100 trial, aimed to evaluate whether thromboprophylaxis with LMWH could mitigate this risk.
The trial included adults aged 18–70 years with newly diagnosed ALL, with patients receiving LMWH (nadroparin 5700 anti-Xa IU) from the start of their ALL treatment until Day 35 of the first remission-induction cycle (RI1). The study analysed data from 369 eligible patients, of whom 49% were aged 18–40 years, and 51% were aged 41–70 years. Patients were divided into two groups: 253 received LMWH thromboprophylaxis, while 116 did not.
The primary outcome measured was the incidence of first venous or arterial thrombosis within the first 60 days of ALL treatment. Secondary outcomes included overall thrombosis during treatment and follow-up, major bleeding events, and event-free survival.
Results indicated that 15% of patients experienced their first thrombosis within 60 days of treatment. However, LMWH did not significantly reduce this risk. Specifically, 17% of patients receiving LMWH experienced thrombosis, compared to 11% without thromboprophylaxis. The adjusted subdistribution hazard ratio (SHR) was 1.52, indicating no significant protective effect of LMWH.
Additionally, age-dependent disparities were observed, with older patients (41–70 years) showing a higher risk when receiving LMWH (SHR: 2.47) compared to younger patients (18–40 years; SHR: 0.88).
Furthermore, no significant differences in bleeding rates were observed between the LMWH group (7%) and the nonthromboprophylaxis group (3%) during the pre-phase and RI1. Overall, 33% of patients receiving LMWH experienced thrombosis compared to 22% without thromboprophylaxis, with an adjusted SHR of 1.59. LMWH also did not impact event-free survival, with an adjusted hazard ratio of 0.82.
These findings underscore the need to re-evaluate the use of LMWH in preventing thrombosis in patients with ALL. The study concluded that thromboprophylaxis with LMWH may not only be ineffective, but potentially disadvantageous for older patients, especially those not receiving PEG-asparaginase in RI1, calling for randomised trials to confirm these results and guide future treatment protocols.
Results indicated that of patients experienced their first thrombosis within 60 days of treatment 15%
Novel CAR-T Cell Therapy for Myelofibrosis
A NOVEL chimeric antigen receptor (CAR)-T cell therapy has been developed to treat myelofibrosis by targeting calreticulin mutant neoplasms, according to findings presented at EHA2024.
Currently, the median survival time for myelofibrosis is 5 years
In vitro, there was a 60–70% elimination of malignant cells, with no off-target toxicity against JAK2-mutated stem cells
Currently, the median survival time for myelofibrosis is 5 years, and the only curative treatment option available is allogeneic stem cell transplantation, which is highly toxic (with a 25% treatment-related mortality) and only available to 10–20% of patients. CAR-T cell therapy offers a new curative treatment option, as CAR-T cells specifically target malignant stem cells, and restore normal haematopoiesis.
To develop the novel CAR-T cell therapy, researchers first identified a binder that selectively binds to mutated calreticulin, and incorporated it into a CAR-T structure. The function and persistence of the CAR-T cells were analysed with repeated stimulation from calreticulin-mutated cancer cells.
Efficacy of treatment was tested in patient samples in vitro, and then in vivo using mouse models. In vitro, there was a 60–70% elimination of malignant cells, with no offtarget toxicity against JAK2-mutated stem cells, and there was no unexpected toxicity in the mouse models.
CAR-T cells specifically target malignant stem cells, and restore normal haematopoiesis
The pre-clinical results show that this novel CAR-T cell therapy can selectively target calreticulin mutant neoplasms, without toxicity, highlighting its potential as a new curative treatment option to induce long-lasting remission in patients with myelofibrosis. The researchers are continuing to develop the therapy, including identifying a suitable lymphodepleting strategy, and plan to conduct a clinical trial in humans in the future.
New Treatment Regimen for Hodgkin’s Lymphoma
A NEW study presented at EHA2024 has analysed a new intensive regimen, BrECADD, for treating advanced-stage Hodgkin's lymphoma in adult patients.
2 in 3
patients completing treatment within 97%
The study achieved better outcomes than expected, with a remarkable result in clinical trials and a 4-year progression-free survival rate of
The BEACOPP regimen, while highly effective and offering impressive progression-free survival rates, has raised concerns due to its intense shortand long-term side effects, prompting questions about its risk-to-benefit ratio. Globally, many physicians opt for the chemotherapy drug combination ABVD, including doxorubicin, bleomycin, vinblastine, and dacarbazine; which is less intensive but also less effective.
In response to these concerns, the German Hodgkin Study Group developed a strategy that adapts to the individual risk of patients using interim PET scans. Patients showing a strong response to treatment after four cycles continue with just four cycles, while those not responding as well receive six cycles. In the HD21 study, researchers aimed to improve the regimen by significantly reducing its duration and intensity. The traditional BEACOPP regimen, which involved 2-week cycles and 8-day infusions, was modified with brentuximab vedotin, an antibody-drug conjugate with an improved risk-benefit profile, to create a more manageable 3-day regimen.
The HD21 study enrolled 1,500 patients from nine countries, including those in Western Europe, Australia, and New Zealand.
This investigator-initiated trial addressed two primary questions: improving tolerability and maintaining efficacy. The new BrECADD regimen showed significantly reduced treatment-related morbidity compared to BEACOPP, with fewer transfusions and lower incidences of neuropathy. Notably, the recovery of gonadal function in females and males was significantly better.
Regarding efficacy, BrECADD exceeded expectations. Unlike ABVD, which has a more tolerable toxicity profile, BrECADD maintained high efficacy rates. The study achieved better outcomes than expected, with two-thirds of patients completing treatment within 12 weeks, and a 4-year progression-free survival rate of 97%, a remarkable result in clinical trials.
The research team concluded by highlighting that the novel BrECADD regimen is not only better tolerated, but also more effective. It significantly reduces treatment duration and toxicity while achieving unprecedented progression-free survival rates. The riskto-benefit ratio is highly favourable, making it a recommended treatment approach for patients with Hodgkin's lymphoma.
Glofitamab-GemOx Therapy Significantly Improves Outcome in Relapsed Lymphoma
RESEARCHERS presented a promising new combination therapy for patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Glofitamab (Glofit), a CD20 bispecific antibody, had previously shown durable responses as a monotherapy for DLBCL.
The STARGLO trial aimed to evaluate the efficacy and safety of Glofit in combination with GemOx compared to the conventional Rituximab-GemOx regimen. Findings from the global Phase III STARGLO were presented in a plenary abstract at EHA2024.
The study enrolled 274 patients who had undergone at least one prior line of therapy. Participants were randomly assigned to receive either Glofit-GemOx or RituximabGemOx, with the study population stratified by the number of prior therapies and refractoriness to the last treatment.
The trial comprised adults aged 18–70 years, with many participants ineligible for autologous stem cell transplant due to age, organ dysfunction, or other comorbidities. In the study, patients received eight cycles of their assigned combination therapy. Those in the Glofit-GemOx group continued with four additional cycles of glofitamab monotherapy. The primary endpoint was overall survival (OS), while secondary endpoints included progression-free survival (PFS) and complete remission rates, assessed by an independent review committee.
At the primary analysis cut-off date of 29th March 2023, Glofit-GemOx demonstrated a significant OS benefit with a hazard ratio of 0.59. With a median followup of 11.3 months, the median OS for the Glofit-GemOx group was not reached, while
it was 9 months for the Rituximab-GemOx group. The Glofit-GemOx group also showed a marked improvement in PFS with a hazard ratio of 0.37 and a significantly higher complete remission rate (50.3%) compared to the Rituximab-GemOx group (22.0%).
At the primary analysis cut-off date of 29th March 2023, Glofit-GemOx demonstrated a significant OS benefit with a hazard ratio of 0.59
A follow-up analysis with a median follow-up of 20.7 months reinforced these findings, showing continued superiority of Glofit-GemOx in median OS (25.5 versus 12.9 months) and PFS (13.8 versus 3.6 months). Adverse event rates were higher in the Glofit-GemOx group, including serious adverse events such as cytokine release syndrome. However, when adjusted for exposure differences, the rates were comparable between the groups.
The study authors concluded that Glofit-GemOx offers a statistically significant and clinically meaningful improvement in survival outcomes for patients with relapsed/ refractory DLBCL who are ineligible for autologous stem cell transplant.
Vitamin C Supplementation for Clonal Cytopenia or Low-Risk Myeloid Malignancies
STINE Ulrik Mikkelsen, Biotech Research and Innovation Centre, University of Copenhagen, Denmark, presented novel data at EHA2024 regarding vitamin C supplementation in patients with low-risk myeloid malignancies and the precursor condition clonal cytopenia of undetermined significance (CCUS).
A significant improvement in overall survival was observed in patients taking vitamin C supplements
The purpose of the EVI-2 study was to investigate if oral vitamin C supplementation was safe and could alter disease characteristics and health outcomes in patients with low-risk myeloid malignancies (low-risk myelodysplastic syndromes and myeloproliferative neoplasms) and CCUS. The study was an international, multicentre, randomised, placebo-controlled, double-blind Phase II study that enrolled 109 patients from four sites in Denmark and the USA. Patients were randomised 1:1 to receive oral vitamin C 1,000 mg (n=55) or placebo (n=54) every day for 12 months. The primary study endpoint was change in mean allele variant frequency (clone size), and key secondary endpoint was vitamin C plasma concentration, safety, and overall survival.
Results showed that vitamin C deficiency was effectively overcome in all patients in the vitamin C group. Notably, a significant improvement in overall survival was observed in patients taking vitamin C supplements compared to the placebo group. The preliminary primary endpoint showed that clone size did not differ between treatment groups.
As the first study to report on vitamin C supplementation in patients with low-risk myeloid cancers and CCUS, it is a significant step forward in understanding this association. Mikelsen stressed that additional larger studies will be required to fully understand the effect of oral vitamin C in patients with low-risk myeloid cancers.
IN THIS year’s European Hematology Association (EHA) Congress, held in Madrid, Spain between the 13ᵗʰ–16ᵗʰ June 2024, an insightful session explored the link between ageing, mutant clones, and the development of haematological malignancies and cardiovascular disease.
TARGETING INFLAMMAGEING AGAINST LEUKAEMIC TRANSFORMATION
Alba Rodriguez-Meira, Dana Faber Cancer Institute, Boston, Massachusetts, USA, began her talk by discussing the origins and evolution of myeloid malignancies, and how these malignancies develop into aggressive diseases. Myeloid malignancies are known to originate in the haematopoietic stem and progenitor cell compartment during initial clonal expansion. The acquisition of genetic mutations like TET2 and JAK2 leads to the development of proliferitic neoplasms, as well as other extrinsic factors such as inflammageing, and progresses slowly over decades. Often beginning in utero, and typically manifesting in individuals over 50–60 years of age, it can acquire additional mutations, such as TP53, leading to the rapid development of acute myeloid leukaemia (AML), a highly aggressive disease with a median survival of less than 3 months.
Rodriguez-Meira’s research aims to prevent this clonal expansion before it becomes malignant, and to understand which patients are at risk of disease progression. To achieve this, Rodriguez-Meira explained that, firstly, it is necessary to understand the molecular mechanisms driving clonal expansion in the haematopoietic system. To dive into this, she explained a model of clonal expansion that begins with JAK-STAT signalling mutations
leading to myeloproliferative neoplasms (MPN). Upon acquisition of further mutations, particularly in TP53, patients can either develop secondary AML, or acquire the mutation without undergoing disease transformation. This is an ideal model for understanding where the non-genetic factors might be promoting the progression from MPN to secondary AML, highlighting the important role of non-genetic factors in disease progression.
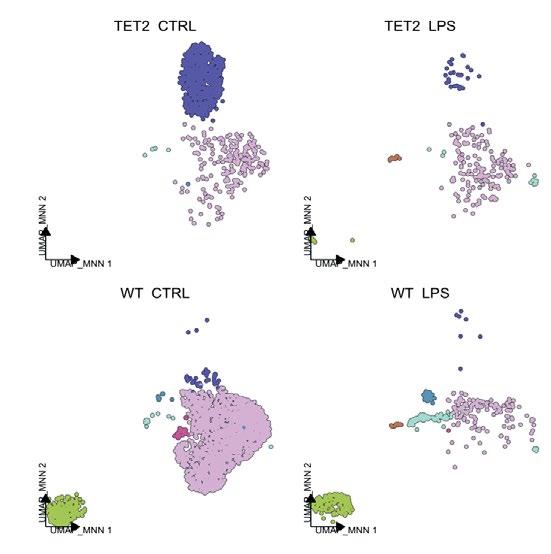
To do this, Rodriquez-Meira utilised a large cohort, including age-matched control donors, patients with MPN with and without TP53 mutations, and patients with TP53 mutations who developed secondary AML. Positive cells were extracted from these patients with TARGET-seq, a method for the high-sensitivity detection of multiple mutations within single cells from both genomic and coding DNA, in parallel with unbiased whole-transcriptome analysis.1 As this method has a >95% allelic resolution, it accurately identifies cells with various TP53 mutations. RodriquezMeira’s analysis revealed a population of TP53 mutant leukaemic stem cells overexpressing inflammatory signatures, alongside TP53 wild-type (WT) cells in the same microenvironment, also showing inflammation-associated transcription. This suggests that chronic inflammation may drive disease progression.
To test the hypothesis that chronic inflammation promotes leukaemic transformation, Rodriguez-Meira and her team conducted a competition model in mice by injecting mice with 50:50 TP53-WT cells and TP53 mutant cells, and subjecting them to inflammatory stimuli, leading to a 2.5-fold expansion of TP53 mutant cells compared to controls. This expansion was accompanied by suppression of WT haematopoiesis, demonstrating that inflammation promotes disease development.
Further investigation revealed that, under stress conditions, TP53 plays a role in apoptosis and DNA damage repair
Further investigation revealed that, under stress conditions, TP53 plays a role in apoptosis and DNA damage repair. Inflammatory stimuli increased apoptotic resistance and DNA damage in TP53 mutant cells, confirmed by M-FISH analysis showing a higher number of karyotypic abnormalities in these cells.
Rodriquez-Meira concluded her talk by proposing a TP53-mediated transformation model in MPN, a model that aligns well with existing literature on inflammationpromoted clonal expansion in the context of TET2hr mutant, ASXL1 mutant, and DNMT3A mutant. Future research will explore the sources of inflammatory molecules and their epigenetic encoding and memorisation.
HAEMATOPOIESIS, INFLAMMATION, AND CARDIOVASCULAR DISEASE
Jose Fuster, National Center for Cardiovascular Research (CNIC), Madrid, Spain, introduced the topic of clonal haematopoiesis of indeterminate potential (CHIP), a condition characterised by the presence of somatic mutations in haematopoietic cells, leading to the expansion of mutant cell clones in the absence of overt haematological abnormalities.
Fuster also mentioned that one of the most profound implications of CHIP is its strong association with CVDs
CHIP is defined by the presence of mutations in certain genes, typically those involved in tumour suppression or DNA repair, such as DNMT3A, TET2, and ASXL1. These mutations can be detected when the proportion of mutant cells in the bone marrow or peripheral blood exceeds 2%, but more commonly above 4%, assuming the mutation is monolithic. The condition is considered a pre-leukaemic state, raising the risk of haematological malignancies and other diseases over time.
A critical aspect of CHIP is its association with chronic inflammation. The expansion of mutant haematopoietic stem cells includes immune cells, which can alter inflammatory responses. This is significant because inflammation is central to many age-related diseases, such as cardiovascular disease (CVD).
Fuster mentioned several research studies, which utilised high-sensitivity sequencing approaches, where results have shown that the prevalence of CHIP increases with age. This highlights the role of CHIP as a risk factor for various age-related diseases. Fuster also mentioned that one of the most profound implications of CHIP is its strong association with CVDs. Studies have demonstrated that individuals with CHIP mutations have a substantially higher risk of developing cardiovascular conditions, such as coronary heart disease and stroke.
Fuster explained results from several experimental studies, including his own, where mouse models provided more insights into the mechanisms by which CHIP mutations contribute to CVD. These studies have shown that CHIP-associated mutations can lead to the accumulation of mutant macrophages, which exhibit heightened inflammatory activity. This exacerbated inflammation can accelerate atherosclerosis, contributing to the development and progression of CVDs.2 The expansion of TET2-deficient cells
is associated with a 60% increase in the size of atherosclerotic plaques. Fuster conducted a similar analysis in the context of heart failure, where studies showed a worse clinical progression of the disease, hospitalisations, and mortality in patients with TET2 mutations.
Fuster stressed that the human and experimental studies in mouse models support the hypothesis that TET2 mutations that lead to haematopoiesis are associated with the development of atherosclerosis and cardiac disease. Fuster backed up his claims by citing clinical evidence from the CANTOS clinical trial, where participants with CHIP mutations showed a nine-fold greater response to the anti-inflammatory drug canakinumab, an anti-IL-1β antibody;
a 60% risk reduction of atherosclerotic CVD in TET2 mutation carriers compared to a 7% risk reduction of atherosclerotic CVD in patients without CHIP; and a decreased risk of recurrent ischaemic CVD events (myocardial infarction, stroke, and CVDrelated death) in post-myocardial infarction patients with elevated C-reactive protein. Targeted anti-inflammatory therapies might therefore be particularly beneficial for this group. However, this was associated with an increased risk of fatal infections,3 and Fuster stressed the importance of developing personalised preventive care strategies.
Fuster concluded his talk by explaining that future research aims to develop tailored interventions for individuals with CHIP, focusing on mitigating the enhanced inflammatory response and reducing the risk of disease progression. This will pave the way for significant advancements in prevention and therapies for a range of conditions.
WHAT HAEMATOLOGISTS SHOULD KNOW ABOUT CLONAL HAEMATOPOIESIS OF INDETERMINATE POTENTIAL
Carsten Müller-Tidow, University Hospital Heidelberg, Germany, gave a presentation on the clinical implications and management strategies for patients with CHIP, discussing key points that haematologists need to consider for patient care, and future directions in the field.
CHIP is characterised by the presence of somatic mutations in haematopoietic stem cells, leading to the expansion of these mutated clones. It is distinguished from other haematological conditions by the absence of significant blood abnormalities, and is often discovered incidentally during DNA sequencing for other purposes.
Müller-Tidow explained that the condition is prevalent among the elderly, with variant allele frequencies (VAF) typically above 2%. Higher VAFs can indicate a higher risk of progression to haematological malignancies.
Patients with existing haematological malignancies who also have CHIP pose a unique challenge
Müller-Tidow explained that the clinical management of CHIP should focus on assessing the risk of progression to myeloid malignancies and the associated cardiovascular risks. Patients with highrisk mutations, such as those in TP53 or splicing factors, require more frequent monitoring. However, for most patients, particularly those with low VAFs and no significant blood abnormalities, extensive interventions like bone marrow analysis are often unnecessary. Instead, a focus on cardiovascular health is crucial, given the higher incidence of cardiovascular events in patients with CHIP.
Risk assessment involves evaluating the types of mutations, number of mutations, VAFs, and other clinical parameters. For instance, DNMT3A mutations are generally benign, whereas TP53 mutations signal a higher risk of malignancy. The clonal haematopoiesis risk score (CHRS) incorporates these factors, and helps guide the monitoring and management of decisions.4
Patients with existing haematological malignancies who also have CHIP pose a unique challenge. Studies show that CHIP can influence outcomes post-treatment, for example after autologous stem cell transplantation. CHIP-positive donors might have lower relapse rates but higher inflammation and graft-versus-host disease risks.5 Müller-Tidow emphasised that highrisk CHIP mutations, such as TP53, are particularly concerning in this context.
Müller-Tidow acknowledged the ongoing debate around screening for CHIP in stem cell donors. While CHIP-positive donors
might offer some proliferation advantages, they also carry risks of donor cell leukaemia, particularly with high-risk mutations. Therefore, careful consideration is needed when selecting donors, especially from older populations.
Müller-Tidow mentioned recent advances in sequencing technologies, such as single-cell sequencing and multi-omics approaches, which promise to improve the diagnosis and risk stratification of CHIP. These methods could lead to better individualised patient care by accurately identifying high-risk clones, and tailoring monitoring and treatment strategies accordingly.
In his concluding remarks, Müller-Tidow reiterated that CHIP is a common condition in the elderly that necessitates careful risk assessment and management. For most patients, cardiovascular risk management is paramount. He stressed that haematologists should focus on identifying high-risk mutations and closely monitoring affected patients, particularly those with concurrent haematological malignancies. Future research and technological advancements will likely refine these strategies, enhancing patient outcomes and care.
CONCLUSION
These sessions delivered in-depth insights on the role of chronic inflammation in the progression of haematological diseases. Experts shed light on the significant roles of clonal expression and inflammageing in the development of aggressive conditions such as AML, and the impact of CHIP on cardiovascular health.
References
1. Rodriguez-Meira A et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell. 2019;73(6):1292-305.e8.
2. José J Fuster et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842-7.
3. Ridker et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. NEJM. 2017;377(12):1119-31.
4. Weeks et al. Prediction of risk for myeloid malignancy in clonal hematopoiesis. NEJM Evid. 2023;DOI:10.1056/evidoa2200310
5. Fick et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2018;37(5):375-85.
A Debate on the Use of Gene Therapy in Patients with Haemophilia
GENE therapy is an innovative approach to treating haemophilia A and haemophilia B, with the potential to increase quality of life, promote prophylaxis, and even achieve curative factor levels in some cases. Despite early success in recent clinical trials, gene therapy for treating haemophilia is a relatively new area of research, and the long-term safety and efficacy are yet to be determined. Additionally, the current high price limits access for most patients. The suitability, safety, and accessibility of gene therapy for patients with haemophilia were discussed during a highly engaging debate session at the European Haematology Association (EHA) Congress 2024, titled ‘Haemophilia: Gene Therapy Access for Patients?’.
Brian O'Mahony, Chief Executive of the Irish Haemophilia Society, and President of the European Haemophilia Consortium, Dublin, Ireland, began the session with a poll to the audience. This revealed that 28.58% of clinicians are ‘unlikely’ or ‘very unlikely’ to recommend gene therapy to their patients with haemophilia A. Similarly, 38.46% are ‘unlikely’ or ‘very unlikely’ to recommend gene therapy to patients with haemophilia B. Thus commenced a lively debate on the use of gene therapy for treating haemophilia; is there access for all, or only ‘a happy few’?
IN FAVOUR OF GENE THERAPY FOR ALL PATIENTS
Ana Boban, Haemophilia Centre, University Hospital Centre Zagreb, Croatia, began by presenting data from recent clinical trials that demonstrate the early successes of gene therapy. Boban explained that, with gene therapy, there is a sustained and durable expression of endogenous factor VIII and factor IX from a single intravenous administration, which subsequently controls bleeding and eliminates continuous prophylaxis. She therefore argued that gene therapy improves quality of life by reducing the frequency of hospital visits, and creates
a ‘haemophilia-free mind’. Whilst not intended to be a curative treatment, gene therapy infusion can reach curative levels in some patients. Moreover, the efficacy of gene therapy for haemophilia can be readily assessed via measurement of circulating factor levels produced by the liver, meaning disease trajectory can be easily assessed. She explained that the bleeding phenotype in patients with haemophilia is responsive to a wide range of factor levels, and precise regulation is unnecessary.
CLINICAL TRIAL SUCCESSES
Boban presented the latest results from a clinical trial in which 134 adult males with severe haemophilia A received a gene therapy called valoctocogene roxaparvovec, which aims to increase factor VIII levels.1 She highlighted that after 3 years, 28.4% of patients achieved factor VIII activity levels above the upper limit of normal. Additionally, at 3 years, the safety profile of valoctocogene roxaparvovec remained unchanged from previous reports in the cohort. It was noted that 23.7% of patients had mild alanine aminotransferase elevations, and one patient developed B cell acute lymphoblastic leukaemia. However, this was considered unrelated to treatment.
Compared to standard therapy of prophylactic factor VIII, participants receiving valoctocogene roxaparvovec gene therapy experienced lower annualised bleeding rates, and a higher proportion of patients had zero bleeds.2 Similarly, in patients with haemophilia B, the gene therapy etranacogene dezaparvovec demonstrated significantly lower bleeding rates, and a higher percentage of patients with zero bleeds, compared to standard, extended half-life factor IX therapies.3
RISKS OF GENE THERAPY
Frank Leebeek, Department of Hematology, Erasmus University Medical Center, Rotterdam, the Netherlands, contended the argument that gene therapy is ‘for all’ by highlighting its potential risks, side effects, and the barriers that limit access to all patients. Leebeek brought to attention that gene therapy infusion is a one-time, irreversible treatment; therefore, if there is a lack of response after a few years, patients will not have the opportunity to try new strategies within the gene therapy landscape. This is an important consideration given the continuous advancements in this innovative field.
Is there access for all, or only ‘a happy few’?
Regarding the improved quality of life, Leebeek argued that there is a lack of long-term data, beyond 3 or 4 years. He highlighted the risks of malignancy and liver damage, which would require long-term steroid use. In response, Boban argued that
whilst we don’t know the long-term risks of gene therapy for haemophilia, this is the same for many new, innovative treatments; is this a reason not to trial them? Boban proposed that effective data collection and management of patients can help mitigate any potential side effects that may arise in the future.
Leekbeek further argued against gene therapy for all patients with haemophilia by highlighting the lack of female patients in the gene therapy clinical trials presented by Boban. O'Mahony emphasised this point by stating that the European Medicines Agency (EMA) has licensed gene therapy treatment for severe and moderately severe haemophilia B in adults, without distinguishing between sexes, despite the lack of female clinical trial data. O'Mahony subsequently asked the speakers if they would give this treatment to a female patient, and Boban replied that she would, only if the patient has not given birth for at least 1 year, due to the potential risk of transmission to offspring.
Gene therapy improves quality of life by reducing the frequency of hospital visits
THE PRICE TO PAY
The next topic of debate was the high cost of gene therapy. Leebeck revealed that a 30-minute-long gene therapy infusion costs 2.8 million Euros in the Netherlands. Boban contended that is preferable to having no treatment at all, as is the case in 85% of the world. In response, Leebeck argued: “If 80% of the population can’t afford the, let’s say ‘cheap’ coagulation factors, how on earth could they get gene therapy of 2.8 million?”
However, Leebeck did admit that in some specific cases, gene therapy may be very suitable. He endorses gene therapy for patients with haemophilia B who are male, have access to treatment despite the high cost, and have poor venous access, as this would eliminate the need for frequent infusions. Leebeck emphasised the need for shared decision-making in which the benefits and long-term risks are weighed
out with patients, allowing an informed decision to be made. O'Mahony articulated that this decision will differ between countries, as the risks associated with gene therapy may be interpreted differently in a country with fewer treatment options.
CONCLUDING THE DEBATE
Towards the end of the debate, Boban admitted that whilst an advocate for gene therapy, if a patient is responding well to standard treatment, has no bleeding, and is living with a ‘haemophilia-free mind’, then gene therapy may not be worth the risk at this current time. Addressing the title of the session ‘Haemophilia: Gene Therapy Access for Patients?’, Boban concluded the debate with a balanced view that “maybe in the future gene therapy will be a treatment for all, but I don't think so at this moment.”
References
1. Madan B et al. Three-year outcomes of valoctocogene roxaparvovec gene therapy for hemophilia A. J Thromb Haemost. 2024;22(7):1880-93.
2. Oldenburg J et al. Comparative effectiveness of valoctocogene roxaparvovec and prophylactic factor VIII replacement in severe hemophilia A. Adv Ther. 2024;41(6):2267-81.
3. Klamroth R et al. Indirect treatment comparisons of the gene therapy etranacogene dezaparvovec versus extended half-life factor IX therapies for severe or moderately severe haemophilia B. Haemophilia. 2024;30(1):75-86.
Normalisation of Haemostasis in Haemophilia A
This Sobi™-sponsored non-promotional satellite symposium on the impact of the normalisation of haemostasis for people with haemophilia A took place on 13th June 2024, as part of the European Hematology Association (EHA) 2024 Hybrid Congress in Madrid, Spain
Chairperson: Cédric Hermans1
Speakers: Maria Elisa Mancuso2, Rubén Berrueco3
1. Division of Adult Haematology, Haemostasis and Thrombosis Unit and Haemophilia Centre, Cliniques Universitaires Saint-Luc, Brussels, Belgium
2. Centre for Thrombosis and Haemorrhagic Diseases, IRCCS
Humanitas Research Hospital & Humanitas University, Milan, Italy
3. Paediatric Haematology Department, Hospital Sant Joan de Déu, Institut de Recerca Sant Joan de Déu de Barcelona (IRSJD), Spain
Disclosure: Hermans has received research support and grant funding from Bayer, Pfizer, and Takeda; has acted as consultant for Bayer, CAF-DCF, CSL Behring, LFB, Novo Nordisk, Octapharma, Pfizer, Sanofi, and Sobi; received speaker’s fees from Bayer, CSL Behring, LFB, Novo Nordisk, Octapharma, Pfizer, Sanofi, and Sobi; and has participated as an advisory board member for Bayer, CSL Behring, LFB, Novo Nordisk, Octapharma, Pfizer, Sanofi, and Sobi. Mancuso has acted as consultant for Bayer, BioMarin, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, and Sobi; has received speaker’s fees from Bayer, BioMarin, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, Sobi, and Spark Therapeutics; has participated as an advisory board member for Bayer, BioMarin, CSL Behring, Grifols, Kedrion, LFB, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, Sobi, Takeda, and UniQure; and has received grant funding from Bayer, CSL Behring, Novo Nordisk, Takeda. Berrueco has acted as a consultant for Bayer, Novo Nordisk, Sobi, Roche, Amgen, Novartis; received speaker’s fees from Bayer, Novo Nordisk, Sobi, Roche, Pfizer, Novartis, and Werfen; has participated as an advisory board member for Bayer, Novo Nordisk, Sobi, Roche, Amgen, Novartis; and received grant funding from Sobi.
Acknowledgements: Medical writing assistance was provided by Kristina Standeven, DNA Communications, London, UK.
Disclaimer: The views and opinions expressed are those of the authors and not necessarily of Sobi.
Keywords: Factor VIII, haemophilia A, haemophilia-free mind, haemostasis, health equity, normalisation, innovation.
Support: The symposium and the publication of this article were funded by Sobi. Approval number: NP-35965-July 2024.
Erratum: This article was first published online on the 25th July 2024. Since then, an erratum was made. The erratum can be seen here.
Meeting Summary
Haemophilia A (Factor VIII [FVIII] levels ≤40 IU/dL) is a chronic condition with consequences beyond bleeding complications. Many people with haemophilia A (PwHA) experience pain, joint damage, psychosocial impacts, restrictions in daily activities, and limitations in physical activities. Cédric Hermans, Professor at the Cliniques Universitaires Saint-Luc, Brussels, Belgium, outlined how ambitious treatment goals, beyond converting severe haemophilia A into a more moderate or mild form of the condition, are required. With new treatments, it will be possible to target FVIII activity levels in the non-haemophilia range (>40 IU/ dL), allowing PwHA to reach freedom from bleeds, leading to a haemophilia-free mindset, and comparable quality of life (QoL) with their peers. Maria Elisa Mancuso, Senior Haematology Consultant at IRCCS Humanitas Research Hospital, Milan, Italy, highlighted the evolution of haemophilia A treatments; she showed clinical evidence that a zero-bleed goal may require sustained FVIII activity levels >40 IU/ dL for complete protection against all types of bleeds and joint damage. Rubén Berrueco, Paediatric Haematologist at the Sant Joan de Déu Barcelona Children's Hospital, Spain, described the haemophilia paediatric patient journey, and how uncertainties related to bleeds and treatment burden pose unique challenges for children and their caregivers. He presented his perspectives on challenges with current treatments (e.g., delayed inhibitor development, subclinical bleeds, and lack of skills for intravenous administration) and the need to improve self-autonomy and decrease hospital dependency. New treatments to achieve the non-haemophilia range of FVIII could address current unmet needs. The experts discussed that treatments for many diseases (e.g., diabetes, hypertension) aim to restore normal values (blood sugar, blood pressure), which was not the case until now for haemophilia. A more patient-centred approach with treatments targeting normal values of FVIII could allow all PwHA to become mentally and physically liberated from the constraints of their condition, and to live with optimised health and well-being.
Welcome and Introduction
Cédric Hermans
Haemophilia A is an inherited deficiency in coagulation FVIII. Based on their residual FVIII activity levels, PwHA are considered to have either severe (FVIII <1 IU/dL), moderate (FVIII 1–5 IU/dL), or mild (FVIII >5–40 IU/dL) disease.1 People with mild disease tend to bleed only after trauma, whereas those with severe or moderate disease also experience spontaneous bleeds into joints and muscles.1,2 However, residual FVIII activity levels do not always correlate with bleeding manifestations, and people with moderate or mild disease can have a similar bleeding phenotype to that associated with severe haemophilia A.3 The World Federation of Hemophilia (WFH) currently recommends regular replacement therapy (prophylaxis) with clotting factor concentrates or other
haemostasis products for all PwH with a severe bleeding phenotype, regardless of their laboratory-assigned severity, with a recommended FVIII target trough level of >3–5 IU/dL or higher.1 Hermans explained how the consequences of haemophilia go beyond bleeding complications, with many PwHA experiencing joint damage, acute and chronic pain, limitations in physical activities, restrictions in daily lives, and psychosocial impacts (Figure 1).4-9
In the past, haemophilia treatment aimed to convert severe disease into a moderate form (FVIII levels around 1–2%) to prevent life-threatening bleeds; however, this is not sufficient to protect all PwHA from bleeds and specifically joint damage.10 More sophisticated treatment options, such as extended half-life (EHL) FVIII products are maintaining FVIII levels around 3-5% (FVIII trough levels in moderate range and FVIII
Figure 1: Consequences of haemophilia: more than bleeds.4-9
Bleeding
• Joint and muscle bleeds
• Life-threatening bleeds
Pain
• Chronic pain
• Acute pain
Joints
• Chronic arthropathy and disability
• Functional impairment
• Need for orthopaedic surgery
Physical impacts
• Limitations in physical activities/sports
peak levels in non-haemophilia range), and non-factor therapies target an equivalent haemostatic activity in the mild haemophilia range, with the aim of providing improved protection.11-13 Now for the first time, with gene therapy and ultra-long FVIII, it is feasible to reach a new ambitious goal for PwHA: the normalisation of haemostasis (i.e., maintaining FVIII activity levels in the non-haemophilia range [>40 IU/dL]) without increasing treatment burden for PwHA (Figure 2).14-16
As haemophilia therapies are evolving, the expectations and aspirations of PwHA have moved beyond controlling symptomatic bleeds towards mental and physical liberation from the constraints of haemophilia and its treatment.16,18 By achieving sustained FVIII levels in the non-haemophilia range, health equity is becoming a realistic possibility for PwHA.16
Expanding Possibilities for People with Haemophilia A
Maria Elisa Mancuso
Mancuso acknowledged the tremendous success in the development of haemophilia therapies over the past five decades. Each
Daily life
• School
• Work productivity
• Career choice
• Financial burden
Psychosocial impacts
• Family
• Mental health
• Quality of life
innovation stemmed from the design of new molecules that addressed different unmet needs and allowed for the inclusion of new outcome measures. These ranged from preventing death, preventing joint disease, and improving QoL and other patient-reported outcomes, to targeting the non-haemophilia range of FVIII and heading towards health equity with people who do not have haemophilia.16,19-21 Prophylaxis is recognised as the standard of care for people with haemophilia and a severe bleeding phenotype; 1 however, to optimise a treatment regimen, a dynamic and patientcentric approach is needed, taking individual needs into account.22 Evidence is emerging that the guideline-recommended FVIII trough levels of 3–5 IU/dL are not enough to prevent subclinical bleeding and joint damage in all patients, and that for a zero joint bleed goal (including silent bleeds), FVIII levels in the non-haemophilia range may be required.23 Results from the Phase III GENEr8-1 trial with the gene therapy valoctocogene roxaparvovec in haemophilia
A showed that, overall, good bleed control was achieved in the majority of participants, but only people with FVIII levels >40 IU/ dL were 100% bleed-free.24 However, FVIII activity levels achieved with valoctocogene roxaparvovec declined over time, with 10.6% of patients maintaining levels in the nonhaemophilia range 3 years after infusion.15
Figure 2: The evolving goals of haemophilia therapy: from saving life to normalising life.16-18
The Evolving Goals of Hemophilia Therapies: From Saving
Life to Normalizing Life1–3
Non-factor replacement therapies such as emicizumab, fitusiran, and anti-tissue factor pathway inhibitor agents (concizumab and marstacimab) generate peak-and-troughfree steady states that likely achieve a correspondence to coagulation activation in the range of mild haemophilia.25 The ultralong FVIII product efanesoctocog alfa has a three-fold longer half-life compared to conventional EHL FVIII products,26 allowing maintenance of FVIII levels >40 IU/dL with a once-weekly dose of 50 IU/kg for up to 4 days,14, 26 thereby providing significant bleed protection with reduced treatment burden for PwHA.14
People with non-severe haemophilia are not uniformly protected from the development of arthropathy, with a proportion requiring prophylaxis.3 Mancuso mentioned that, to protect all PwHA from arthropathy, it is necessary to look beyond annualised bleeding rates (ABR) and employ imaging techniques such as ultrasound and MRI to monitor joints for subclinical bleeding and early evidence of joint damage.27,28 In a Dutch study of 43 people with severe haemophilia A, MRI detected haemosiderin deposits in 16% of all screened people; of these, 43% had synovial hypertrophy and/
or osteochondral changes.29 Without regular monitoring, synovial proliferation may be overlooked, as demonstrated in a recent ultrasound study of 79 people with severe haemophilia A without recent joint bleeds.30 In this cohort, ultrasound detected active synovial proliferation in 22% of patients; of these, 82% had no clinical signs of inflammation (swelling or warmth).30
Synovitis is the first step towards irreversible joint damage, but it can be reversed by intensified prophylaxis with FVIII and anti-inflammatory drugs, as recognised by the guidelines of the German Thrombosis Society.1,31,32 These guidelines recommend 1–2 daily doses of 40–60 IU/kg coagulation factor initially for acute synovitis, and a 6-month trough level target of ≥30 IU/dL to treat chronic synovitis (Figure 3).32
Patient Case 1
Mancuso presented the case of ‘Paul’, a 34-year-old physically active person who has moderate haemophilia with no history of inhibitors (neutralising antibodies against FVIII concentrates) and a baseline FVIII activity level of 2.3 IU/dL. ‘Paul’ works,
Acute joint bleed
Diagnosis Therapy
Acute synovitis
Coagulation factor product:
• Initial 40–60 IU/kg, 1–2× daily
• Children: Individual higher doses
Biopsy if required
Rest joint Pain therapy
Anti-inflammatory therapy
Physiotherapy
NSAID: non-steroidal anti-inflammatory drug.
travels often, and participates in sports on a regular basis. Secondary prophylaxis was started with thrice-weekly standard half-life recombinant FVIII at 4 years of age before switching to EHL FVIII every fifth day in 2016 due to time constraints impacting on the ability to perform thrice-weekly infusions. The patient started to experience ankle pain, especially in the morning, and minor bleeding, and the prophylaxis schedule was intensified to a twiceweekly infusion of 45 IU/kg. In their joint evaluation, a haemophilia joint health score of 6, and a haemophilia early arthropathy detection with ultrasound score of 4 were noted; the ultrasound investigation found evidence of synovitis in their right ankle that had not presented 12 months previously. The prophylaxis intensification was discussed with ‘Paul’, whose priorities were to stay active and independent, and be free of pain whilst maintaining a feasible treatment schedule. Mancuso highlighted the importance of discussing the clinical meaning of synovitis and the need for regular joint monitoring with patients,
Chronic synovitis
Coagulation factor product:
• Trough level of ≥30% for 6 months
If response is insufficient, quickly consider radiosynoviorthesis or synovectomy
Anti-inflammatory therapy with NSAID, if necessary, intra-articular cortisone
Physiotherapy
along with giving advice on how to manage synovitis with physiotherapy and antiinflammatory medication in addition to high-factor treatment, whilst keeping an open mind regarding treatment and schedule changes.
Realising New Opportunities in Children
Rubén Berrueco
Berrueco described the human-centred process carried out at his centre, aimed to better understand the unmet needs of children with haemophilia A (CwHA) and their caregivers. This qualitative research consisted of semi-structured interviews with healthcare professionals (HCP), CwH, and their caregivers, and other stakeholders, as well as observation during clinical appointments at the centre. The information allowed them to draw the paediatric haemophilia journey and
Figure 3: German Thrombosis Society treatment algorithm for synovitis in haemophilia.32
to describe the unmet needs that CwH experience through time. Potential moments to improve CwH’s and their caregivers’ experience are (i) treatment initiation, when caregivers tend to be keen to learn about haemophilia but, at the same time, experience many uncertainties related to the condition, and (ii) when CwH are 7–8 years old, and first notice that they are different to their peers and begin to ask, “why me?”
Apart from the unmet needs described by the patients, Berrueco believes that it is important to explain to parents that, even with treatment, HCPs are aware of other unmet needs that must be addressed. Haemophilia A is a chronic disease with a high treatment burden that impacts on daily activities and QoL.33 Moreover, CwHA are still at risk of life-threatening bleeds such as intracranial haemorrhage,34 a risk that can be significantly mitigated by prophylaxis.35 Importantly, compared with on-demand treatment, prophylaxis with FVIII is associated with a lower rate of inhibitors,36 and early primary prophylaxis with FVIII has been related to better joint health results.37,38 However, the Joint Outcome Study and subsequent Joint Outcome Continuation Study demonstrated that, despite prophylaxis with FVIII, joint damage can also occur in the absence of recognised bleeds.28 Nowadays, many CwHA are treated with non-factor treatment (emicizumab), but it is important to highlight that there is a paucity of data for this treatment regarding the risk of inhibitor development following on-demand FVIII exposure,39,40 and regarding the predictability of joint health.41
For prophylaxis to be successful, adherence is crucial at any age, but can be problematic in adolescents and young adults.42,43 Adolescents often consider prophylaxis infusions a time-consuming and inconvenient interference with their daily lives. They may lack the necessary skills to self-infuse, be phobic of needles, be forgetful, or lack family support.44,45 Educating adolescents about the consequences of non-adherence is crucial, and self-injection should be encouraged as early as possible in the
patient journey to ensure young people with haemophilia gain autonomy and good selfmanagement skills.43,44
Berrueco discussed how sports participation is an important way to improve QoL in CwHA;46 at the same time, this needs to be balanced with the associated increased bleed risk.46 Achieving normalisation of haemostasis and a ‘haemophilia-free mind’ could enable CwHA to live full lives with the same aspirations as their peers without haemophilia.47 In addition to considering the unmet needs in CwHA (Figure 4), Berrueco urged HCPs not to forget that caregivers continue to need support through all stages of their child’s development.
Treatment of CwHA has changed over the years, and tremendous progress has been achieved.21 A decade ago, FVIII prophylaxis was started at around 2 years of age, with a twice-to-three-times weekly infusion regimen,50 and tolerisation was achieved within 6 months.41 HCP concerns were focused on the type of FVIII product they should administrate. Now, prophylaxis is started even earlier, and worries are related to the protection against subclinical bleeds and the potentially increased risk to develop inhibitors at later age.
Recent and new treatment approaches offer opportunities to address unmet needs in CwHA. Emicizumab allows for either once-weekly, every 2-, or every 4-week subcutaneous injections,39,51 achieving FVIII equivalence of between 9–20 IU/dL.52-55 Whilst the recently published HAVEN 7 study of emicizumab in infants with severe haemophilia A showed no new safety signals in this age group, with a modelbased ABR of 0.4 and 54.5% of participants reaching zero treated bleeds,51 real-world data in 314 young people with severe haemophilia A with and without inhibitors found that 15 participants experienced at least one severe muscle bleed.56 The pharmacokinetic profile of the ultra-long FVIII efanesoctocog alfa supports a onceweekly dosing schedule, maintaining FVIII in the non-haemophilia range for up to 3 days in children.57 In the Phase III XTENDKids study in boys younger than 12 years
Figure 4:
Subclinical joint bleeds and haemophilic arthropathy
Predictability of joint status in later life
Intracranial bleeds
Breakthrough bleeds
Caregiver’s uncertainties
with severe haemophilia who had previously been treated with FVIII, the estimated mean ABR in the sensitivity population (n=73) was 0.61, and 64% of patients reached zero bleeds;58 crucially, no inhibitors were detected.59
Patient Case 2
Berrueco presented the case of ‘Valentín’, a 7-year-old boy with severe haemophilia A who started emicizumab at the age of 3 years (after >50 ED with FVIII), and recently started playing soccer. In a recent evaluation of the patient’s joint health, there was evidence of right knee swelling and a new synovitis in the suprapatellar recess. During further discussion with the parents, it was remembered that ‘Valentín’ had experienced knee pain for the past few weeks, suggesting that the synovitis was active (symptomatic). The clinical goal for this child was to achieve and then maintain optimal joint health, whilst allowing him to have a QoL of life comparable with his peers. For ‘Valentín’ and his parents, maintaining physical activity and having a low treatment burden were priorities. It was agreed that close clinical monitoring was necessary, and that the treatment schedule and other treatment options should be reviewed to achieve the desired clinical goal for this child and his caregivers.
Tolerance/inhibitors
Treatment burden
Pain of administration
Lack of adherence
Desire for greater quality of life (more physically active lives)
Panel Discussion and Q&A: Resetting Goals and Approaches in Haemophilia A
All Faculty
Speaker presentations were followed by a combined panel discussion and audience Q&A session. Mancuso explained that, to date, no treatment for haemophilia A is curative, but if joint health is already poor, a higher level of protection is required. Hermans noted that, for other acquired conditions (e.g., hypertension, diabetes, hypercholesterinaemia), the treatment goal is to restore normal physical or biological parameters;16 in haemophilia A, the treatment goal of trough levels of 3–5% merely converts severe-tomoderate disease, which Hermans argued is insufficiently ambitious. The goal of normalisation has simply not been considered achievable or affordable, but with new treatment options, it is now becoming realisable.16 Berrueco stressed the importance of looking beyond the impact of haemophilia on children and their parents, and also consider other family members, teachers, and healthcare providers. It was agreed that families need to be continuously educated on the treatment options and long-term treatment effects. With adequate monitoring, it is
expected that sustaining FVIII activity levels in the non-haemophilia range will protect joint health and thereby reduce sequelae of haemophilia later in life; monitoring with ultrasound also increases patient education and adherence. In adult PwHA, FVIII activity levels in the non-haemophilia range can offer a level of protection that allows treatment for comorbidities, such as cardiovascular pathologies, without having to prioritise bleed prevention or prevention
References
1. Srivastava A et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158. Erratum in: Haemophilia. 2021;27(4):699.
2. Rejto J et al. Bleeding phenotype in nonsevere hemophilia by International Society on Thrombosis and Haemostasis bleeding assessment tool, bleeding frequency, and the joint status. Res Pract Thromb Haemost. 2023;7:100047.
3. Castaman G et al. Mild and moderate hemophilia A: neglected conditions, still with unmet needs. J Clin Med. 2023;12:1368.
4. Kurnik K et al. How do I counsel parents of a newly diagnosed boy with haemophilia A? Hamostaseologie. 2020;40:88-96.
5. Auerswald G et al. Pain and pain management in haemophilia. Blood Coagul Fibrinolysis. 2016;27:845-54.
6. Smith N et al. Vocational experiences and career support opportunities among Canadian men with moderate and severe haemophilia. Haemophilia. 2019;25:441-6.
7. D'Angiolella LS et al. The socioeconomic burden of patients affected by hemophilia with inhibitors. Eur J Haematol. 2018;101:435-56.
8. Mehta P, Reddivari A, Haemophilia [Internet] (2024) Treasure Island: StatPearls. Available at: https://www. ncbi.nlm.nih.gov/books/NBK551607/. Last accessed: 25 June 2024.
9. Nomura S. Current status and challenges in delivering comprehensive care for patients with hemophilia. J Blood Med. 2023;14:629-37.
10. den Uijl I et al. Turning severe into moderate haemophilia by prophylaxis: are we reaching our goal? Blood Transfus. 2013;11:364-9.
11. Hermans C, Pierce GF. Bispecific antibodies mimicking factor VIII in hemophilia A: converting innovation
of thrombotic events. The ‘haemophilia-free mind’ is an aspirational concept, and with current treatments, PwHA will still have a life-long serious disease,19 but a reduction of the burden of treatment and better haemostatic protection can contribute to alleviating the burden of haemophilia. All panel members agreed that clinical studies should address outcomes beyond the prevention of bleeds, and include a focus on patient-related outcomes.60
to an essential medicine. Res Pract Thromb Haemost. 2023;7:100173.
12. Klamroth R et al. Rurioctocog alfa pegol PK-guided prophylaxis in hemophilia A: results from the phase 3 PROPEL study. Blood. 2021;137:181827.
13. Oldenburg J et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809-18.
14. von Drygalski A et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A. N Engl J Med. 2023;388:310-8.
15. Madan B et al. Three-year outcomes of valoctocogene roxaparvovec gene therapy for hemophilia A. J Thromb Haemost. 2024;22(7):1880-93. [Epub ahead of print].
16. Skinner MW et al. Achieving the unimaginable: health equity in haemophilia. Haemophilia. 2020;26:1724.
17. Carcao M et al. How much prophylaxis is enough in haemophilia? Haemophilia. 2024;30(Suppl 3):86-94.
18. Hermans C, Pierce GF. Towards achieving a haemophilia-free mind. Haemophilia. 2023;29:951-3.
19. Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105:545-53.
20. Hermans C, Pierce GF. Ultra-long FVIII: A major step forward to a hemophiliafree mind. J Thromb Haemost. 2024;22(7):1844-6. [Epub ahead of print].
21. Mannucci PM. Hemophilia treatment innovation: 50 years of progress and more to come. J Thromb Haemost. 2023;21:403-12.
22. Hermans C et al. Hemophilia treatment in 2021: choosing the"optimal" treatment using an integrative, patient-oriented approach to shared decision-making between patients and clinicians. Blood Rev. 2022;52:100890.
23. Malec L, Matino D. Targeting higher
factor VIII levels for prophylaxis in haemophilia A: A narrative review. Haemophilia. 2023;29:1419-29.
24. Mahlangu J et al. Efficacy and safety of valoctocogene roxaparvovec gene transfer for severe haemophilia A: results from the GENER8-1 two year analysis. Virtual Congress 2022.
25. Berntorp E et al. Optimising prophylaxis in haemophilia A: The ups and downs of treatment. Blood Rev. 2021;50:100852.
26. Lissitchkov T et al. Pharmacokinetics of recombinant factor VIII in adults with severe hemophilia A: fixedsequence single-dose study of octocog alfa, rurioctocog alfa pegol, and efanesoctocog alfa. Res Pract Thromb Haemost. 2023;7:100176.
27. Gualtierotti R et al. Assessing joint health in haemophilia patients: The combined value of physical examination and ultrasound imaging. Haemophilia. 2024;DOI:10.1111/ hae.15030. [Epub ahead of print].
28. Daffunchio C et al. The hidden joint in children with haemophilia on prophylaxis. Thromb Res. 2023;226:86-92.
29. van Leeuwen FHP et al. Magnetic resonance imaging evidence for subclinical joint bleeding in a Dutch population of people with severe hemophilia on prophylaxis. J Thromb Haemost. 2023;21:1156-63.
30. van Bergen EDP et al. Subclinical synovial proliferation in patients with severe haemophilia A: the value of ultrasound screening and biochemical markers. Haemophilia. 2023;29:15808.
31. Mancuso ME et al. Synovitis and joint health in patients with haemophilia: statements from a European e-Delphi consensus study. Haemophilia. 2023;29:619-28.
32. GTH. S2k-Leitlinie Synovitis bei Hämophilie, Langfassung. August 2022 update. 2022. Available at: https://register.awmf.org/assets/ guidelines/086-005l_S2k_Synovitis-
bei-Haemophilie_2023-02.pdf Last accessed: 9 July 2024.
33. Brod M et al. Understanding treatment burden in hemophilia: development and validation of the Hemophilia Treatment Experience Measure (Hemo-TEM). J Patient Rep Outcomes. 2023;7:17.
34. Zanon E et al. Intracranial haemorrhage in haemophilia patients is still an open issue: the final results of the italian EMO.REC Registry. J Clin Med. 2022;11:1969.
35. Hu J et al. Risk of intracranial hemorrhage in persons with hemophilia A in the United States: real-world retrospective cohort study using the ATHNdataset. J Blood Med. 2024;15:191-205.
36. Gouw SC et al. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109:4648-54.
37. Warren BB et al. Young adult outcomes of childhood prophylaxis for severe hemophilia A: results of the joint outcome continuation study. Blood Adv. 2020;4:2451-9.
38. Manco-Johnson MJ et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535-44.
39. Ranta S et al. Dilemmas on emicizumab in children with haemophilia A: a survey of strategies from PedNet centres. Haemophilia. 2023;29:1291-8.
40. Mason JA, Young G. Emicizumab prophylaxis in infants with severe haemophilia A without inhibitors: illustrative real-world cases to support shared decision-making. Haemophilia. 2021;27:724-9.
41. Young G. Management of children with hemophilia A: how emicizumab has changed the landscape. J Thromb
Haemost. 2021;19:1629-37.
42. Hoefnagels JW et al. The perspectives of adolescents and young adults on adherence to prophylaxis in hemophilia: a qualitative study. Patient Prefer Adherence. 2020;14:163-71.
43. Berube S et al. Motivational techniques to improve self-care in hemophilia: the need to support autonomy in children. BMC Pediatr. 2016;16:4.
44. Brand B et al. Challenges in the management of haemophilia on transition from adolescence to adulthood. Eur J Haematol. 2015;95(Suppl 81):30-5.
45. Lee Mortensen G et al. Adherence to prophylactic haemophilic treatment in young patients transitioning to adult care: a qualitative review. Haemophilia. 2018;24:862-72.
46. Khair K et al. The impact of sport on children with haemophilia. Haemophilia. 2012;18:898-905.
47. Krumb E, Hermans C. Living with a "hemophilia-free mind" - the new ambition of hemophilia care? Res Pract Thromb Haemost. 2021;5:e12567.
48. Banchev A et al. A cross-national survey of people living with hemophilia: impact on daily living and patient education in Central Europe. Patient Prefer Adherence. 2021;15:871-83.
49. Chowdary P et al. Disease burden, clinical outcomes, and quality of life in people with hemophilia A without inhibitors in Europe: analyses from CHESS II/CHESS PAEDs. TH Open. 2024;8(2):e181-93.
50. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125:2038-44.
51. Pipe SW et al. Emicizumab prophylaxis in infants with hemophilia A (HAVEN 7): primary analysis of a phase 3b open-
label trial. Blood. 2024;143:1355-64.
52. Ferriere S et al. A hemophilia A mouse model for the in vivo assessment of emicizumab function. Blood. 2020;136:740-8.
53. Lenting PJ. Laboratory monitoring of hemophilia A treatments: new challenges. Blood Adv. 2020;4:2111-8.
54. Pipe SW et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, nonrandomised phase 3 study. Lancet Haematol. 2019;6:e295-305.
55. Schmitt C et al. Pharmacokinetics and pharmacodynamics of emicizumab in persons with hemophilia A with factor VIII inhibitors: HAVEN 1 study. Thromb Haemost. 2021;121:351-60.
56. Batsuli G et al. Severe muscle bleeds in children and young adults with hemophilia A on emicizumab prophylaxis: real-world retrospective multi-institutional cohort. Am J Hematol. 2023;98:E285-7.
57. Peyvandi F et al. Once-weekly efanesoctocog alfa prophylaxis provided high sustained factor VIII activity levels, independent of blood group, in children <12 Years of Age with severe hemophilia A. Blood. 2023;142:506-8.
58. Malec L et al. Treatment of bleeding episodes with efanesoctocog alfa in children with severe hemophilia A in the XTEND-Kids phase 3 study. Blood. 2023;142:3993.
59. Malec L et al. Efanesoctocog alfa prophylaxis for previously treated patients <12 Years of Age with severe hemophilia A. Res Pract Thromb Haemost. 2023;7(Suppl 2):1-290.
60. Manco-Johnson MJ et al. Outcome measures in haemophilia: beyond ABR (Annualized Bleeding Rate). Haemophilia. 2021;27(Suppl 3):87-95.
Abstract Reviews
Cutting-edge research, presented at the 2024 European Hematology Association (EHA) Congress. The following abstract reviews spotlight exciting new developments in the field.
Impact of TET2 Mutations on Haematopoietic Stem Cell Resilience to Immune-Related Stress
Authors: Hector Huerga Encabo,1 Giuseppe D’Agostino,2 Alessandra Ferrelli,1 Aneesh Sharma,1 Syed Mian,1 Manuel Garcia-Albornoz,1 Linda Ariza-McNaughton,1 *Dominique Bonnet1
1. Haematopoietic Stem Cell Laboratory, The Francis Crick Institute, London, UK
2. Plasticell Limited, Stevenage, UK
*Correspondence to Dominique.bonnet@crick.ac.uk
Disclosure: Encabo has received funding from the European Hematology Association (EHA) and Cancer Research UK, and received a Kay Kendall postdoctoral fellowship. Bonnet has received funding from the Medical Research Council (MRC), Wellcome Trust, and Cancer Research UK, with payments made to the lab. The authors have declared no conflict of interest.
Acknowledgements: The authors would like to thank the Scientific and Technological Platforms at the Francis Crick Institute for providing help and expertise, especially the Advance sequencing and FACS facilities. This work has been supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CC1045), the UK Medical Research Council (CC1045), the Wellcome Trust (CC1045), and the Kay Kendall Leukaemia Foundation (KKL1397 to HHE).
Understanding how loss of function mutation of TET2 in haematopoietic stem cells (HSC) changes their response to
stress is crucial to elucidating the dynamics of clonal haematopoiesis throughout our lifespan. Over time, as we age, we encounter episodes of infection and hematopoietic stresses, yet how these complex episodes influence the expansion of the mutant clones needs to be fully understood. Previous work in mouse models has delineated the contribution of specific proinflammatory cytokines in the competitive advantage of TET2-KO HSCs;1-3 but the impact of multifactorial episodes, such as viral or bacterial infections, into the clonal evolution at the human cell level is scarce. Characterising when and how TET2 mutant human HSCs are favoured could help design therapeutic strategies to reduce the expansion of clonal haematopoiesis (CH) and the risk of individuals with CH developing haematological diseases and inflammatory pathologies.
AIMS
To provide new evidence of how TET2-derived clonal haematopoiesis evolves during our lifespan, and which environmental cues are more likely to favour TET2 mutant HSCs.
METHODS
The authors introduced CRISPR TET2 mutations into primary human HSCs (hHSC), allowing for a comprehensive examination of their behaviour under stress conditions. First, the authors characterised
the response of TET2-mutant hHSCs and differentiated progeny upon different immune challenges in long-term culture assays. The authors reproduced a TET2derived clonal haematopoiesis scenario in humanised mice,4 and performed functional, transcriptomic, and epigenomic analyses within the haematopoietic stem and progenitor cells (HSPC) compartment.
RESULTS
The authors unveil that hHSCs carrying a loss of function (Lof) TET2 mutations exhibit heightened resistance to haematopoietic stress compared to their wild-type counterparts. Interestingly, using long-term culture assays (Figure 1A), the authors tested the behaviour of TET2-mutant HSCs under different sources of stress (Figure 1B), and identified lipopolysaccharide (LPS) as the immune threat that leads to higher expansion of mutant clones (Figure 1C). Bulk RNA sequencing was performed to analyse cell-specific transcriptional response and the impact of TET2 LoF on LPS in both primitive CD34+ cells and differentiated myeloid CD14+ cells (Figure 1D). Interestingly, while TET2-mutant HSCs have a mitigated transcriptional upregulation of inflammatory pathways upon LPS response (Figure 1E), TET2-mutant myeloid cells showed an exacerbated inflammatory response (Figure 1F), consistent with previous reports characterising the impact of TET2 mutations in human myeloid progeny.4 The refractoriness of TET2mutant HSC to LPS-induced stress was
further validated by the increased long-term culture-initiating cell frequency (Figure 1G). Furthermore, employing a humanised mice model, transplanted with a mixture of wildtype and TET2 HSPCs, with or without LPS treatment (Figure 1H), and subsequently investigating the transcriptomic profile of the human HSPC compartment present in these mice by single-cell RNA sequencing, the authors identified two distinct subsets of HSCs within the human HSC pool characterised by differing properties in their response to haematopoietic stress (Figure 1I). Notably, within the subset that retained high HSC signature, while wild-type HSCs show a clear activation of transcriptional pathways, TET2 mutant HSCs show no significant changes in transcriptional programmes associated with LPS response.
DISCUSSION
The authors’ work underscores the cellspecific influence of TET2 mutations on the composition and functionality of HSCs and their immune progeny. This study showed the opposite consequence of the TET2 Lof between HSC and myeloid progeny upon an inflammatory challenge. TET2 mutant myeloid cells show exacerbated production of inflammatory cytokines, which is consistent with previous reports.4-8 In sharp contrast, TET2 mutant HSCs show a protective behaviour to LPS-mediated stress. Therefore, this cell-specific TET2mediated phenotype provides novel perspectives into the clonal haematopoiesis dynamics upon environmental stress.
Figure 1: Opposite and cell-specific impact of TET2 mutations in haematopoietic stem cells and myeloid progeny to inflammatory stress.
(CD34+ cells) PC2:
HSPCs (CD34+ cells) PC2:
PC1: 49% variance
PC1: 49% variance
TET2Mut HSPCs
PC2: 4% v a r iance
(CD14+ cells)
Monocytes (CD14+ cells)
PC1: 89% variance
TET2Mut Monocytes
Monocytes
PC1: 49% variance
Monocytes (CD14+ cells) PC2:
LPS (1ng/ml)
PC1: 89% variance
PC1: 89% variance
TET2Mut HSPCs TET2Mut Monocytes
TET2Mut HSPCs TET2Mut Monocytes
(1 / 43)
scRNA-sequencing sorted CD34+CD38-CD45RA- from humanised mice
Day 0 TET2 frequency in primary hHSCs
TET2 frequency in humanised mice after the treatment
scRNA-sequencing
scRNA-sequencing
* P value <0.05 and ** P value <0.01 using Anova test.
1. Pan W et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity. 2017;47(2):28497.e5.
2. Yura Y et al. clonal hematopoiesis: a new step linking inflammation to heart failure. JACC Basic to Transl Sci. 2020;5(2):196-207.
3. Cai Z et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stressinduced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018;23(6):833-49.e5.
4. Huerga Encabo H et al. Loss of TET2 in human hematopoietic stem cells alters the development and function of neutrophils. Cell Stem Cell.
2023;30(6):781-99.e9.
5. Calcagno DM et al. The myeloid type I interferon response to myocardial infarction begins in bone marrow and is regulated by Nrf2-activated macrophages. Sci Immunol. 2020;5(51):eaaz1974.
6. Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389-93.
7. Liu W et al. Blockade of IL-6 signaling alleviates atherosclerosis in Tet2-deficient clonal hematopoiesis. Nat Cardiovasc Res. 2023;2(6):572-86.
8. Cull AH et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56-70.e13.
KMT2A-MLLT3-Induced Leukaemia Changes During Ontogenic Stages
1. Paul O’Gorman Leukaemia Research Centre, Wolfson Wohl Cancer Research Centre, School of Cancer Sciences, University of Glasgow, UK
2. College of Applied Medical Sciences, King Saud Bin Abdulaziz University for Health Sciences, Jeddah, Saudi Arabia
3. King Abdullah International Medical Research Centre, Jeddah, Saudi Arabia
4. Department of Biomedicine, University Children's Hospital (UKBB), University of Basel, Switzerland
5. Cancer Research UK (CRUK) Scotland Centre, Glasgow, UK
*Correspondence to karen.keeshan@glasgow.ac.uk
Disclosure: TENOVUS Scotland and the Government of the Kingdom of Saudia Arabia provided funding for this research. Horne received support to attend conferences from Jass Pharmaceuticals and Novartis. Schwaller received grants from the Swiss National Science Foundation (SNF-3100A_173224/1 and SNF310030_215023/1), and is a member of the grant committee and associate editor for HemaSphere. The other authors declared no conflicts of interest.
Acknowledgements: The authors would like to thank TENOVUS Scotland and the Government of the Kingdom of Saudi Arabia for funding. They would also like to thank all the staff in the Biological Services Unit facility of the Cancer Research UK Scotland Institute and Flow
Cytometry facility in the School of Cancer Sciences.
The KMT2A-MLLT3 (also known as MLLAF9) fusion is associated with acute lymphoid leukaemia (ALL), acute myeloid leukaemia (AML), and mixed phenotype acute leukaemia (MPAL) in infants and children. Infant leukaemia originates in utero, yet this has never been formerly shown for KMT2A-MLLT3-driven disease. Multiple mouse models invariably revealed that KMT2A-MLLT3 can induce myelo-monocytic AML; however, they failed to recapitulate the ALL and MPAL phenotypes as presented in paediatric patients. To explore the impact of the developmental stage for KMT2AMLLT3+ leukaemic burden and phenotypes, the authors established a new inducible (‘Tet-off’), reversible, transplantable mouse model in which expression of the fusion is controlled by a stem cell leukaemia (SCL)enhancer element (Figure 1).
The authors crossed SCL-tTA mice with inducible KMT2A-MLLT3 mice1 to generate double transgenic SCLtTA/i KMT2A-MLLT3 mice. To determine the impact of the developmental stage, KMT2A-MLLT3 was induced from the conceptional stage for prenatal (E0), in neonates for postnatal (P0), and at 20 weeks for adult (≥20 weeks) mouse cohorts. Following induction for 6–8 weeks in vivo KMT2A-MLLT3 expression was measured in haematopoietic stem cells (HSC), haematopoietic progenitor cell-1/ lymphoid-myeloid primed progenitors (HPC-1/LMPP), and granulocyte-monocyte progenitors (GMP). Notably, the KMT2AMLLT3 expression levels were comparable within each population across the age groups.
RESULTS
Upon postnatal KMT2A-MLLT3 induction, all the mice developed leukaemia with a short median latency of 13 weeks and complete penetrance. In contrast, E0 prenatal- or adult-induction resulted in longer latencies of 32 and 24 weeks, respectively. Despite sustained KMT2A-MLLT3 expression, not all the mice developed leukaemia in prenatal- and adult-induced cohorts. Postnatal-induced mice developed myelomonocytic, megakaryoblastic, or MPAL, whereas prenatal and adult induction of KMT2A-MLLT3 developed myelomonocytic disease exclusively. Regardless of leukaemia phenotypes, all diseased mice displayed splenomegaly, and only postnatal and adult sick mice developed thrombocytopenia. Independent from the age at which KMT2A-MLLT3 was induced, all myelo-monocytic AML mice had clonal expansion of GMPs. Only postnatal-induced myelo-monocytic mice had a significant
Figure 1: Tet-Off system.
increase in HPC-1/LMPPs and reduction in megakaryocyte erythrocyte progenitor and multi-potent progenitor populations, relative to age-matched controls. There was expansion of GMPs and HPC-1/LMPP populations in the postnatal-induced MPAL disease; whereas, in the postnatal-induced megakaryoblastic disease, there was clonal expansions of HSCs, LK (lineage negative c-kit positive), and MEP populations with no expansion in GMP. Thus, the age when the KMT2A-MLLT3 fusion is induced does not only determine the disease latency and penetrance, but also affects lineage determination and the amount of leukaemic HSPCs.
Leukaemic-GMP and leukaemicHSC have been characterised in adult models as leukaemic stem cells. Indeed, transplantation of bulk and leukaemicGMPs from adult-induced myelo-monocytic AML cells led to secondary disease with complete penetrance. In all age cohorts, bulk myelo-monocytic AML cells formed
serially plateable large and compact colonies and were reversible. Collectively, leukaemic stem cells exist within a GMPlike population of adult-induced KMT2AMLLT3 AML, they may however reside in different HSPC populations in prenatal- and postnatal-induced disease.
CONCLUSION
In summary, the authors generated a novel inducible model that recapitulates several features of infant, paediatric, and adult KMT2A-MLLT3 leukaemia, demonstrating that the ontogenic stage is a determining factor in differences in disease biology.
References
1. Stavropoulou V et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell. 2016;30(1):43-58.
Advanced-Stage Hodgkin Lymphoma International Prognostic Index (A-HIPI) in Turkish Patients with Classical Hodgkin Lymphoma: A Single-Center Retrospective Study
Authors: *Oguzhan Koca,1 Berk Ozyurt,2 Aysenur Umar,2 Deniz Ozmen,3 Tugrul Elverdi,3 Ayse Salihoglu3, Muhlis Cem Ar,3 Zafer Baslar,3 Ahmet Emre Eskazan3
1. Department of Internal Medicine, Cerrahpasa Faculty of Medicine, Istanbul UniversityCerrahpasa, Türkiye
2. Cerrahpasa Faculty of Medicine, Istanbul University-Cerrahpasa, Türkiye
3. Division of Hematology, Department of Internal Medicine, Cerrahpasa Faculty of Medicine, Istanbul University-Cerrahpasa, Türkiye
*Correspondence to ogkoca@gmail.com
Disclosure: The authors have declared no conflicts of interest.
Keywords: Advanced-stage Hodgkin lymphoma international prognostic index (A-HIPI), clinical
prediction models, Hodgkin lymphoma, international prognostic score, prognosis, prognostic score.
The historical International Prognostic Score (IPS-7), which was developed with patient data from the 1980s, and the more recent IPS-3 clinical prediction models are thought to be less powerful in distinguishing good- and poor-risk groups among patients with advanced-stage classical Hodgkin lymphoma (cHL).1,2 Recently, the advancedstage Hodgkin lymphoma international
prognostic index (A-HIPI) model was created, and it was found to be superior when compared to IPS-7 and IPS-3 in terms of predicting prognosis in patients with advanced-stage cHL.2 It has also been validated in a few studies coming from different countries. The authors, for the first time, aimed to validate the A-HIPI model among Turkish patients with cHL, and to compare A-HIPI with other clinical prediction models.
MATERIALS AND METHODS
Patients with advanced-stage (2B, 3, and 4) cHL who were diagnosed between 2005–2018 were evaluated retrospectively. IPS-7, IPS-3, and A-HIPI scores were calculated, and the authors compared the performance of these models in terms of predicting progression-free survival (PFS) and overall survival (OS). To compare their calibration and their power to discriminate between low-risk and highrisk patients, discrimination and calibration
analyses were performed. The C-index (Harrell’s Concordance Index) method for discrimination, visual examination of calibration curves, calibration intercepts and calibration slope were evaluated for calibration. Models were compared with Akaike’s information criteria (AIC).
RESULTS
The authors included 207 patients with a median age of 37 years (range: 18–82 years). There were 19 patients (9.2%) aged >65 years. Fifty eight percentage of patients were male, and the percentages of patients with stages 2B, 3, and 4 disease were 28%, 37.2%, and 34.8%, respectively. All but five patients (97.6%) received a combination of doxorubicin, bleomycin, vinblastine, and dacarbazine as first-line therapy. The percentages of patients with IPS-7 scores 0, 1, 2, 3, 4, and ≥5 were 4.3%, 16.4%, 27.5%, 30%, 15%, and 6.8% respectively and 30.4%, 38.7%, 24.6%, and 6.3% for IPS-3 scores 0, 1, 2,
Figure 1: Kaplan-Meier survival analysis for the advanced-stage Hodgkin lymphoma international prognostic index (A-HIPI) quartiles.
Patients were divided into four groups (quartiles) according to their A-HIPI expected PFS and OS scores. Q1 represents the group with the lowest expected survival, and Q4 represents the group with the highest expected survival with the A-HIPI model.
and 3 respectively. With the A-HIPI model, the expected 5-year median PFS and OS were 76.6% and 92.3%, respectively. With a median follow-up of 75 months, 37 patients (17.9%) died. The 5-year PFS and OS were 67% and 84.9%. respectively. With regression analyses, all 3 models were found to be prognostic for PFS and OS for the entire cohort. In terms of OS, the model with the highest C-index in the entire sample and in patients ≤65 years of age is A-HIPI (C-index for the entire sample: 0.740, 95% CI: 0.642–0.838; C-index for patients ≤65 years of age of which it is validated: 0.726, 95% CI: 0.613–0.840). In terms of PFS, the model with the highest C-index is IPS-3 (C-index: 0.619, 95% CI: 0.535–0.703). In terms of PFS, the A-HIPI model had the lowest C-index (0.591, 95% CI: 0.506–0.677). The A-HIPI model showed a better fit with the authors’ study data in determining the prognosis in terms of OS by AIC analysis (A-HIPI, IPS-7 ΔAIC: 12.8; A-HIPI, IPS-3 ΔAIC: 9.7), and IPS-3 model showed a better fit in terms of PFS. In the patient group >65 years of age, all three models were not prognostic. In terms
of PFS and OS, the A-HIPI model appears to be prognostic and well-calibrated except for the patient group aged >65 years (Figure 1).
CONCLUSION
The authors’ study validated the A-HIPI model among homogeneously treated patients from a tertiary referral center in Türkiye. The A-HIPI model was observed to have a better performance than other models, except for patients aged >65 years. A new clinical prediction model is needed for patients >65 years as A-HIPI has not been validated for this population.
References
1. Moccia AA et al. International prognostic score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol. 2012;30:3383-8.
2. Rodday AM et al. The advanced-stage Hodgkin lymphoma international prognostic index: development and validation of a clinical prediction model from the HoLISTIC consortium. J Clin Oncol. 2023;41:2076-86.
Five-year Outcome of CD19 Combined With CD22 CAR-T Cell Therapy in Patients With B-ALL Relapsed After Allo-transplantation
1. Department of Hematology, Beijing Boren Hospital, China
2. Cytology Laboratory, Beijing Boren Hospital, China
3. Medical Laboratory, Beijing Boren Hospital, China
4. Department of Bone Marrow Transplantation, Beijing Boren Hospital, China
5. Engineering Research Center of Gene Technology, Institute of Genetics, School of Life Sciences, Fudan University, Shanghai, China
*Correspondence to liusy@gobroadhealthcare.com
Disclosure: Chang is a founding member of Shanghai YaKe Biotechnology Ltd., a biotechnology company focusing on research and development of tumour cellular immunotherapy. The remaining authors declare no conflicts of interest. This work was supported by Creative Research Project of Gaobo Healthcare Group (No. GBHG-CR2017003), and the National Key Basic Research Program of China (No. 2016YFC1303403).
Acknowledgements: The authors would like to thank all medical staff and patients who participated in the study.
The authors have previously reported their Phase I clinical trial regarding a combination treatment of CD19- and CD22-specific chimeric antigen receptor (CAR)-T cells for post-transplant relapsed B-cell acute lymphoblastic leukaemia (B-ALL). In the trial, which included both paediatric and adult patients, the first CD19 CAR-T therapy resulted in a complete remission (CR) rate of 85% (23 out of 27 cases); furthermore, 21 patients received subsequent CD22 CAR-T cells (six cases withheld from the second cell therapy) and the overall survival (OS) and event-free survival (EFS) rates at 18 months were 88.5% and 67.5%, respectively.
To investigate the long-term efficacy of combination treatment with CD19 and CD22
CAR-T cells in these patients with relapsed B-ALL after allogeneic hematopoietic cell transplantation (allo-HCT), the 5-year survival outcome was followed up.
MATERIALS AND METHODS
Except for the 27 patients previously reported, an additional three cases relapsed only with bone marrow minimal residual disease (MRD), who were not eligible for the trial but were administered CD19 and CD22 CAR-T cells following the same protocol in the same time period, were included in this follow-up study. CAR-T cells were transfected by lentiviral vectors encoding second-generation CARs composed of CD3ζ and 4-1BB, the interval time between two cell infusions was 1.5–6.5 months. The
Figure 1: The probabilities of overall survival and event-free survival, analysed by the Kaplan–Meier method.
OS (A) and EFS (B) in 24 patients who received both CD19 and CD22 CAR-T cells. OS (C) and EFS (D) in all 30 intention-to-treat patients including six cases without undertaking the second CD22 CAR-T. CAR: chimeric antigen receptor; EFS: event-free survival; OS: overall survival.
first cell infusion was performed between December 2017–October 2019, and the cut-off date for the last follow-up was as 31st December 2023.
RESULTS
Three MRD relapsed cases turned MRD negative after first CD19 CAR-T. Among 24 patients who finished both CD19 and CD22 CAR-T therapy, with a median follow-up of 64.4 (95% CI: 50.6–68.6) months, 12 patients relapsed and the late relapse (more than 2 years after first CD19 CAR-T cell infusion) occurred in three cases (25%). Of relapsed patients, six died of disease progression and six were still alive after treatment with other therapies (four received second alloHCT). More encouragingly, the other 12 patients remained in sustained remission since first CD19 CAR-T (n=11) or second CD22 CAR-T (n=1, who was with extramedullary disease and achieved partial remission after CD19 CAR-T). Kaplan–Meier survival analysis showed that the 3-year OS and EFS rates were 79% (95% CI: 57–91) and 54% (95% CI: 33–71), respectively, while the 5-year OS and EFS rates were 75% (95% CI: 53–88)and 50% (95% CI: 29–68), respectively (Figure 1).
In all 30 intention-to-treat patients, including six cases without receiving second CD22 CAR-T cells, the OS and EFS rates were 70% (95% CI: 50–83) and 50% (95% CI: 31–66) at 3 years, and 67%(95% CI: 47–80) and 46% (95% CI: 28–63) at 5 years, respectively.
After each cell infusion, CR/partial remission patients with incomplete blood cell count on Day 30 had cell count recovery in 1.3–6.7 months; 13 severe infections (Grade 3 or greater) beyond 1 month and within 2 years were observed in 11 patients, presenting with 11 lung infections (Grade 3, n=10; Grade 4, n=1) and two skin varicella-zoster virus infections (Grade 3). Most infections occurred within 1 year (76.9%; 10/13).
CONCLUSION
In patients with relapsed B-ALL postHCT, who previously had an extremely poor prognosis, the authors’ combination treatment of CD19 and CD22 CAR-T cells tremendously improved long-term survival with a 5-year OS of 75% and EFS of 50%.
References
1. Spyridonidis A et al. Outcomes and prognostic factors of adults with acute lymphoblastic leukemia who relapse after allogeneic hematopoietic cell transplantation. An analysis on behalf of the Acute Leukemia Working Party of EBMT. Leukemia. 2012;26(6):1211-7.
2. Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449-59.
3. Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-48.
4. Liu S et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. 2021;96(6):671-9.
Congress Interviews
Three leading experts, including the 2024 European Hematology Association (EHA) President, discuss their career and latest advancements in the field of haematology. Read on to learn about the latest initiatives in EHA, as well as the newest innovations and challenges in the field.
Antonio Almeida Head of Haematology at Hospital da Luz and Founding Dean of Católica Medical School, Lisbon, Portugal; President of the European Haematology Association (EHA)
The Mentorships
Mixer is really a new initiative to bring more senior haematologists in contact with younger haematologists
In recent years, the European Hematology Association (EHA) has launched several exciting initiatives. For example, the YoungEHA Mentorship Mixer at EHA 2024 is designed to connect early-career haematologists with experienced mentors. What are the key goals you hope to achieve with this initiative, and how do you see it shaping the career of young haematologists?
I think the most important thing to say in respect to this is that EHA is a members’ association, and as such, it's crucial that we cater to our members’ needs. This initiative of having a mentorship programme really came from young haematologists who felt that there are big discrepancies and big inequalities in various countries in Europe, in terms of how mentorship is given. Especially for those who want to go into research, who want to have a career as a physician scientist, we felt it was particularly important to have a mentor, an international mentor that could advise them on how to take their careers, and how to take their research projects. The Mentorships Mixer is really a new initiative to bring more senior
haematologists in contact with younger haematologists, in hopes to be establishing more long-term relationships that will shape their careers. This will enable them to direct their research and career paths into whatever they want to do in the future. I know that the focus is very much on young EHA members, but it also has a big focus on more mature EHA members, getting them involved in the EHA as much as possible, and helping with the careers of future haematologists.
Q2
Secondly, the EHAEuropean Molecular Biology Laboratory (EMBL)/ European Bioinformatics Institute (EBI) Computational Biology Training program aims to equip researchers with essential computational skills. Can you elaborate on the importance of computational biology in modern haematology research, and how this collaboration will enhance research capabilities in the field?
I think one thing that we must recognise is that computational biologists are a rare breed, and they're particularly important. In research, we all think very much of laboratory research and clinical research collection, and
generation, of data. However, data analysis, in silico analysis, is so critical, and it has can have such a huge impact, especially nowadays with all the new tools that are powered by artificial intelligence and other algorithms. So I think it is essential that researchers do have competences in computational biology, and that they do acquire all these new knowledges that can be had. This partnership with the EMBL/ EBI has been critical. They have the know-how, they know what there is to know in computational biology, and they have huge expertise to be able to teach this to researchers. This has been very important and very well received by the community. Once again, like the Mentorship Mixer initiative, this came very much out of the need from our researchers to know more about computational biology, and to gain proficiency in it, and I think this will also have a very big impact on what haematology research can do, and on the whole community. So I think it's a very good example of how, through partnerships with experts, we can really serve the community, and help everybody come up to speed with what they need.
Q3Finally touching on the EHA-American Society of Hematology (ASH) Translational Research Training in Haematology (TRTH) programme bridges the gap between laboratory research and clinical application. Could you discuss the significance of this collaboration with ASH, and how the TRTH programme is expected to impact the translation of scientific discoveries into patient care improvements?
Once again, I think this is a great example of collaboration between experts societies. The ASH has had a very large programme, for a long time, in translational research. We joined forces with them, really, so that our trainees in translational research could have a vision of what research is like on both sides of the Atlantic. This has been very, very productive. The whole idea is that the participants in this programme come out with a research project, which then
can submit for funding from large funding agencies. This has been amazing in terms of the people who we've helped in their careers, in terms of how projects really have come up to scratch and produced great results, also in terms of how the EHA and ASH have come together with common goals; to promote clinician scientists, to promote researchers in haematology, and to really change careers and produce world class research. This is very much a mentorship programme. It does benefit from expertise from both sides of the Atlantic. So, I think it has produced a state-of-the-art community of scientists who are able to take things into the future, and hopefully will continue to do so. It's a very exciting programme that has really been the birthplace of many other mentorship programmes that we have.
There is a huge part of imagination, of thinking, of collaboration that happens face to face
Q4We have now passed the 4-year anniversary since the World Health Organization (WHO) declared COVID-19 a global pandemic. Reflecting back on this time, how do you feel the COVID-19 pandemic impacted the field of haematology?
It is undeniable that COVID-19 had a huge impact on patients. Most of our patients have some source or other of immune suppression, and so they suffered heavily from COVID-19. Even after the vaccines, many of our patients still have COVID-19; this was particularly tragic. It's also quite poignant that this has led to a lot of research into COVID-19, into immune suppression, and how we can protect our patients from immune suppression, how we change our gears to serve the patient community, and to serve the needs at the time. I think one thing that's been particularly striking is how much value we've all started giving to network and contact, to actually meeting people. When we look at the EHA Congress, we now have a record
number of registered attendees (over 15,000), which really brings home the message that, yes, online meetings are fine; yes, we can do a lot of production when we're far apart; however, there is a huge part of imagination, of thinking, of collaboration that happens face to face, and that is just human nature. All the enthusiasm around belonging to the congress and being part of the community has come back in, and that has had a huge impact on the haematology community. I think it's not so much the travelling, but the recognising how important these connections are, and how important the networks are, and how we really need to foster them. All the numbers of congresses, all the results that have come out of collaborations, have really pointed towards that. So I'm very happy that we've been able to contribute not only to increasing the research network, but to increasing the actual clinical and professional network as a result of COVID-19.
Q5 How do you manage the dual responsibilities of being Congress President and an active researcher, and how do you see your role evolving in the future with respect to this?
I must say that within the EHA, we have fantastic support of all the committee members in a very competent office that actually enables things to happen, and allows us to focus on the essential, and not on the nitty gritty. I'm very fortunate with that. Being President of a professional society, especially one that dedicates itself to the area that we work in, is really part of everyday life. So, my leadership role is very much to ensure that the whole community, be it medical school, be it work, be it the association, benefits from the actions that we do, and that we actually cater for them in those needs. Hopefully, my role in the future will really be to facilitate this more and more by bringing about more research grants, more networking opportunities, and increasing the educational footprint that EHA has in haematology in Europe.
Q6 What key pieces of advice would you offer to young researchers aspiring to build a career in haematology?
The piece of advice that was most striking and helpful for me was to be focused, and yet flexible. I know that sounds a little bit contradictory, but I think in life and in career, we all know where the centres of excellence are. We all know that we want to work with fantastic people, we want to have good mentors, and all that is very, very important. However, what you forget is that life sometimes throws opportunities at us that are unexpected; while we're busy focusing on our area
of interest, opportunities and other areas of interest come along that really can mould our lives and our careers. That was very much my case. I started off as a red cell specialist, with a huge interest in red cells, ended up by doing a PhD in something totally unrelated, inherited GPI deficiency, and then moved to Portugal from the UK where I dedicated myself to myeloid neoplasms. This sort of flexibility, I think, is so enriching within haematology, which is such a vast field. For me, to have this flexibility has really been a game changer, and really enabled me to advance my career without necessarily being stuck in areas that might not have progressed along. So, be curious, and be interested in all of haematology. It's so rich, and it's so valuable to be able to do everything
that you know; you don't really need to be limited to one field of haematology. Just be open to what comes your way.
Q7
What do you find to be the most rewarding aspects of your work as a haematologist?
I think the most rewarding aspects is how fast haematology moves; and how we can really see changing practices in how we treat diseases, how we help patients, and how quickly research comes into clinical practice. I think that speed of translation, that speed of research, is really, really exciting in haematology, and it goes all the way from having exciting research to seeing it being put into practice, and seeing the patients benefit from it. So I
think the translational aspect of haematology is definitely the most exciting part of it all.
Q8
From your professional experience, what are some of the most significant challenges faced by haematologists in the clinical setting today?
Well, I think there are two big challenges. One is the enormity of information, and the enormity of new options that come, which really makes being up to date a challenge, but also makes treatment choices challenging. I think, as we look to the future, we certainly need better guidance from experts and from panels, but we also need to be able to digest information, to be able to focus on what the patient has, and what the patient's needs are. The second important challenge is that the patient populations change; we have more and more informed patients. This is not challenging in a negative way, but in redefining our role as physicians, we end up by not having necessarily that traditional ‘we know it all’ ‘God’ role, but very much having a role of accompanying patients in their disease, in their lives, and helping them overcome the challenges of the disease. To take that step back and be the physician who accompanies patients, and does not dictate what the patient should be doing, is a change in mindset. That is very challenging, and very important nowadays.
Elizabeth Macintyre
President of the Biomedical Alliance, Europe; and Past President of the European Hematology Association (EHA)
As the Head of the Hematology Laboratory at Necker–Enfants Malades Hospital for over two decades, what were some of the major challenges and achievements you have experienced?
Since starting in 1992, I've witnessed significant advancements in diagnostics. My background includes clinical and laboratory haematology from my time in the UK, but I transitioned to focusing on diagnostics. Upon my arrival, I was asked about my plans, and I responded, "I'm going to establish molecular diagnostics." Initially, there was scepticism about whether I had the courage to take on such a task. However, today, it's unimaginable to diagnose
Examining pathway abnormalities is crucial because targeting individual genetic abnormalities in T-ALL is ineffective
haematological malignancies without a molecular approach.
We successfully set up molecular diagnostics within the hospital diagnostic lab, benefiting immensely from a close collaboration with the research lab. This synergy was crucial; one could not have advanced without the other. In France, we are fortunate to have mechanisms that allow us to conduct innovative diagnostic work within our hospital budget, which has made a substantial difference.
Q2
How has the research at the Hematology Laboratory at Necker–Enfants Malades Hospital evolved over the years?
Our research at the Hematology Laboratory primarily focused on lymphoid malignancies. Early in my career, I worked in immunogenetics and was at the forefront of minimal measurable residual disease (MRD) detection. I pioneered the quantification of clonal immunoglobulin and T cell receptor gene rearrangements
for MRD detection in lymphoid malignancies. Although I am an adult haematologist, working in a paediatric hospital connected to an adult hospital has been beneficial. An adult haematologist initially brought me in, but a paediatrician from another hospital asked if I could implement MRD for paediatric acute lymphoblastic leukaemia. This led to the parallel development of oncogenetics and immunogenetics in our research.
Over the past 30 years, we've combined these approaches to identify specific characteristics of cancers that can be targeted for therapeutic purposes, which has been a fruitful area of research. We've focused on immature lymphoid malignancies at the intersection of adult and paediatric leukaemia, with each area enriching the other. We have concentrated on T cell acute lymphoblastic leukaemia (T-ALL), which involves blood and bone marrow. However, some closely related cancers, known
as T-lymphoblastic lymphoma, remain in the tissue. Despite their biological and genetic similarities, understanding why some metastasise to the bone marrow while others do not is a critical area of research.
France tends to prefer local approaches, but for rare cancers, centralisation is essential. We have gradually become a reference centre for molecular and cellular analysis of these disorders, recognising the importance of integrating cellular and molecular diagnostics. Our research success has depended on, in addition to exceptional scientists, two smart decisions made early on, in the 1990s: properly banking samples with the budget to conserve them and setting up a comprehensive database. These decisions were not great scientific foresight but rather sensible planning, enabling the scientific discoveries that followed.
Q3Your current research focuses on immature T lymphoid leukaemia and lymphomas. Could you elaborate on the importance of this research and any recent breakthroughs your team has achieved?
Because we're having this discussion during the European Hematology Congress in Madrid, I'd like to highlight two notable oral presentations from our group. Although I no longer lead the frontline research, I consider myself more of a mentor in the unit. I lead the research unit alongside Vahid Asnafi, who was once my PhD student and is now my superior in the hospital structure. He handles the hard science while I focus on public relations, making us very complementary.
At this year’s European Hematology Association (EHA) Congress, Guillaume Andrieu presented our team's research on patients with T-ALL and NOTCH1 pathway mutations,
The focus has been on the costs of therapeutics and medical devices, but diagnostics are becoming more expensive
which are associated with a favourable prognosis. Our goal is to leverage this positive prognostic subgroup to aid those with poorer outcomes. Examining pathway abnormalities is crucial because targeting individual genetic abnormalities in T-ALL is ineffective due to their vast genetic diversity. Unlike chronic myeloid leukaemia, which has a single targetable gene (BCR-ABL), T-ALL requires a pathway-based approach for therapeutic targeting. Andrieu's presentation included findings on how the NOTCH1 pathway led to investigations into ferroptosis and metabolic disruption.
Another significant presentation at EHA, was given by Aurore Touzart, MD/PhD in our team. Aurore worked with Matthew Simonin, a paediatrician who did his PhD in our group, to develop an updated genetic classifier that allows one to stratify individual patients into what kind of treatment they should have, initially in T-ALL. A typical classifier will focus on reducing treatment for patients at low risk and targeting novel therapies for patients at high risk. Such approaches are increasingly being integrated into functional drug screening approaches whereby clinicians can decide which patient needs which treatment at an individual patient level. This year, Aurore presented an extension of this approach to T lymphoblastic lymphoma.
Q4 What motivated you to take on the role of president-elect at the Biomedical Alliance in Europe, and how has your experience with the EHA prepared you for your role at the Biomedical Alliance?
I have worked with EHA for a very long time. I joined the European Affairs Department at EHA, and
I currently chair that activity. The EHA asked me to be their representative on the Biomedical Alliance in Europe. I may have been elected to the board of the Biomedical Alliance because I speak my mind, I speak English as my mother tongue, and I am also a woman, so I tick a few minority boxes. Sometimes we say, ‘See One, Do One, Teach One’, but I would say, ‘See One, Do One, Teach One, Organise or Regulate One’. We all know that health is a member state issue, but especially since COVID-19, there is increasing realisation that there are some things we can do better together, at European level, in health. Not working together on such aspects does not bear considering, especially with the current nationalist trends in Europe, born of fear of change, in my opinion. People are frightened of change, in general and in their professional practice. We can do so much together by working with the European Parliament and the European Commission, because the people who are writing the rules for Europe need to work with experts in the field. I firmly believe that for those of us that want the European health agenda
to be the space we deserve and desire, we have to work for it. That is probably why I got so involved in Biomedical Alliance, because there are transversal issues that have enormous potential if we get them right, but dreadful alternatives if we get them wrong.
Q5Can you discuss any collaborative projects or partnerships that the Biomedical Alliance in Europe is currently pursuing, and how these collaborations are expected to impact healthcare research and policy?
At the Biomedical Alliance in Europe, collaboration means working together on broad, crosscutting issues rather than focusing on specific areas like cancer or ageing, which are handled by specialised scientific societies. Our primary interactions are with policymakers, lawmakers, and politicians. There are three main groups of policy advocates: industry, patients, and healthcare professionals.
The industry has strong advocates with close ties to governments,
and patients have become effective advocates who are listened to by politicians because they are voters. The medical profession has long enjoyed a significant degree of trust, but this has been challenged in recent years. I believe it is essential for healthcare professionals to advocate as effectively as industry and patient representatives. Specialists often communicate only within their fields, making it difficult to convey their needs to lawmakers and politicians. If regulations don't translate into benefits and/or are not feasible/ reasonable for healthcare professionals, everyone suffers.
Our collaborations aim to facilitate clear communication in a multistakeholder format. The European Commission recognises increasingly that excessive regulation can be counterproductive. While regulations are necessary, they need to be streamlined, pragmatic, and comprehensible to all stakeholders to accommodate 27 different cultures under the same rules. Bureaucratic and regulatory zeal must be balanced with clarity and efficiency.
Medical oncologists manage solid cancer chemotherapy, driven by pathologists' diagnoses
Moderation is key, even within a discipline. Whenever we start to write guidelines, we encounter variations in treatment approaches. Collaborative projects and partnerships at the Biomedical Alliance are crucial for creating a harmonious European health space. I often engage with non-healthcare professionals, to represent academic perspectives. We aspire to optimise multistakeholder communication and ensuing confidence in order to build a vibrant healthcare industry in Europe, providing equitable healthcare to all.
While we don't set drug prices, medical specialists and educators must inform future and trainee doctors about the economic impacts of their actions. Using public funds wisely to prevent and treat diseases effectively, with minimal side effects and optimal costs, is vital. This starts with accurate diagnostics. Historically, the focus has been on the costs of therapeutics and medical devices, but diagnostics are becoming more expensive and crucial for targeted therapies, thus attracting interest from the for-profit sector and requiring regulatory surveillance.
A notable example is the Theranos disaster, where an inappropriate test benefitted from massive capital investment and aggressive marketing, out of all proportion to its diagnostic performance. We need regulations to ensure patient safety but without stifling innovation in the diagnostics sector. The Biomedical Alliance is committed to balancing innovation and safety in healthcare.
Q6
If we invest in diagnostics, for example, could there be potential in preventing diseases?
At the European health policy level, significant investments have been made in cancer with the ‘Beating Cancer’ plan. There have also been remarkable advancements in rare diseases through the European rare disease networks. However, many people suffer from lifestyle disorders, and there's still a lot we can do in this area. Recently, I read that the four major industries endangering lives are alcohol, tobacco, excessive food consumption, and fossil fuels. As one example from haematological cancers, individuals with clonal haematopoietic disorders are at higher risk for cardiovascular diseases, and evidence suggests that obesity worsens outcomes for those with haematological cancers. Screening for clonal haematopoiesis could allow for early education of these at-risk populations.
While preventative measures are crucial, we might not see screenings like mammograms for blood cancers. Many leukaemias are detected incidentally. Haematology is innovative but often associated with expensive therapies for rare patient subgroups. Fortunately, the Biomedical Alliance and the EHA maintain strong relationships with European Commission regulators, policy officers, and lawmakers, via the European Medicines Agency (EMA) for pharmaceuticals. Both hold joint sessions to facilitate communication and collaboration and are recognised for their commitment to collective, constructive actions.
Q7
Having attended EHA this year, were there any sessions or themes that you were particularly interested in, and why?
It’s a difficult choice, since there was a plethora on offer. I attended a session on addressing excessively costly therapies. The panel included government authorities, pharmaceutical companies, patient representatives, and a representative from a lowerincome European country. The room was full, with people standing, and having lively discussions. Five years ago, such sessions were attended mostly by EHA's European affairs department and patient representatives. While scientific innovation is vital, it's equally important to foster communication among stakeholders to maximise resources. Even the wealthiest countries cannot afford CAR-T cell therapies for everyone. As indications for CAR-T cell therapy extend to solid tumours, and perhaps soon to autoimmune disorders, the difficulties in being able to afford to offer these therapies to all patients
will become apparent even in the highest-income countries. We must find ways to improve public quality of life within budgets that are unlikely to exceed 10–14% of gross domestic product.
Q8
As a leader in the field of haematology, what advice would you give to young researchers and clinicians who are just starting their careers in this rapidly evolving area of medicine?
Choose haematology because it is a fantastic speciality, offering both diagnostic and therapeutic opportunities. Medical oncologists manage solid cancer chemotherapy, driven by pathologists' diagnoses. In haematology, you can both diagnose and deliver therapy. Additionally, trust your intuition. If you feel passionate about something honourable, you'll likely achieve honourable outcomes.
Q9
How have you seen the field change in your career? Are the challenges faced by young haematologists today the same that you faced?
The challenges remain largely the same. Society now emphasises perfect work–life balance, but my balance has always involved hard work, apparently not to the detriment of family wellbeing. My husband is also an academic haematologist, and we had our children relatively late. They consider that their parents work too hard for little financial recognition. They have seen us, mostly, work tirelessly because we are passionate about our work and believe in what we do. If we earned massive salaries and came home early with energy for leisure, we might have been great role models for relaxation. However, our children have inherited a strong sense of contributing to society, influenced partly by their grandfather who remained a contributor to society in an evolving fashion until he was more than 100 years old. He taught us that we receive through contributing.
Q1Your current research at Radboud university medical center focuses on hereditary and acquired thrombophilia, and women's issues in thrombosis and haemostasis. What motivated you to focus on these specific areas, and what are some of the most exciting advancements you've seen in this field recently?
Netherlands
Professor of Medicine and Head of the Department of Internal Medicine, Radboud University Medical Center, Nijmegen, the
I sort of coincidentally got into the field of clinical thrombosis and trials, looking at how to diagnose, prevent, and treat it
Let me start with what drives me. I sort of coincidentally got into the field of clinical thrombosis and trials, looking at how to diagnose, prevent, and treat it. Everything you study gets more interesting because you start to know more, and then you get to know what you don't know. So, that sort of happened naturally. However, as a clinician, as a doctor, I was very struck by the fact that there was hardly any high-level evidence for younger women, women with
hormone-related thrombosis, or pregnancy-related thrombosis. So, at some point, I decided to try and create that evidence and think of how we can advance the field so that we can provide better evidence-based care to this relatively large group. That is how it all started. It’s quite challenging because it was often thought that it was very difficult to randomise pregnant women, for instance, like we did in the Highlow study. Or because, for women with a history of venous thrombosis or pulmonary embolism, we have no idea what dose of low molecular weight heparin to prescribe. So, we just decided to study and start doing it. It took 10 years for a reason. The drive for me, mainly, is the fact that it's quite annoying that you can't base your clinical advice to patients on evidence.
Q2
As an EHA Presidential Session Speaker, you are set to present the following talk at EHA 2024, ‘Conducting multinational RCTs in pregnancy to improve maternal health in thrombosis: it CAN be done’. What are the key challenges and considerations in conducting such complex trials, and can you highlight any findings from your recent work?
I think that the challenge is, although very prevalent in my practice, in order to collect data on many women, you need collaborations, and very often you need multinational collaborations. That is one barrier, because for collaborations you need colleagues who are willing to participate. I think the biggest challenge, really, is the money. So, we build academic thrombosis networks in which a lot of colleagues have become friends, and we share the same passion. For the Highlow study, for instance, I received starting money from a pharmaceutical company, which was really great. I failed to get national money from the Netherlands for some reason; I mean, that happens too. However, my French colleagues, who have a very good academic thrombosis network, were very successful. They acquired money for the France part. Then, a couple of years later, Irish colleagues got money for the Ireland part. So, at the end of the day, if you look at the amount of funding, it was still very small for such a big trial, but it was successful; although it took 10 years. I think if we had more money, we could get it done more quickly. Money is the main issue. Very often it is thought that ethical problems arise, but the trials I am doing are with agents, like low molecular weight heparin, that have been registered and are being used already in clinical practice,
also in pregnant women. We know it's safe, but it's without evidence on optimal dosing. In fact, we have shown that women are very willing to participate in these trials and be randomised between one dose or the other. So, I think the main challenge is money.
Q3
You have been a principal investigator in several practice-changing trials such as ALIFE, ALIFE2, and Highlow. Could you share some key findings from these trials and how they have impacted clinical practice in the management of thrombosis?
In the Highlow study, we randomised women with a history of venous thromboembolism who became pregnant between a standard, prophylactic, low dose, that is derived from other indications like medical inpatients or prolonged prophylaxis after hip or knee surgery, or a higher dose that we hypothesised
would be more effective to prevent recurrence in pregnancy or the postpartum period. We randomised more than 1,100 women in eight countries, and we found that the primary outcome of a recurrence over that entire study period was not significantly different, which was against our hypothesis. There was a 30% risk reduction, but it wasn't significant, unfortunately. I think the most striking feature was the fact that we found that there was no numerical difference between the doses antepartum, but there was a statistically significant difference in the postpartum period. However, my practice has changed in that we used to provide a very high dose antepartum, and we now provide a low prophylactic dose during pregnancy, which facilitates neuraxial anaesthesia and all those things during delivery, and then escalate to an intermediate dose for the 6 weeks postpartum.
The ALIFE2 study is in a bit of a different field as it's about thrombophilia and recurrent miscarriage. I've been interested in recurrent miscarriage because it is associated with antiphospholipid syndrome, which is an established relationship, but also in inherited thrombophilia, which is present in about 15% of the general population. These women, for many years, have been globally treated with low molecular weight heparin with the aim to improve pregnancy outcomes, and this has been a debate for a long time. Back in 2003 was the first ALIFE study, where we looked at unexplained inherited miscarriage. We randomised women to low molecular weight heparin, to low molecular weight heparin and aspirin, or to placebo. We didn't find any effect, but we did find a signal in inherited thrombophilia, and that was the basis for ALIFE2, where we showed that in women with recurrent miscarriage and established inherited thrombophilia, low molecular weight heparin does not improve live birth. So, we should stop testing these women, we should stop prescribing low molecular weight heparin, and we should put all that money into research on how to improve outcomes in recurrent miscarriage. We should take it out of the haematology or thrombosis and haemostasis field. I think that impact should be huge, so I'm really an advocate for evidence-based medicine in maternal health.
If there's no evidence, you should just go and get it. Make it yourself. Find your colleagues. Don't be shy. Don't be scared. Not everything I've done has been a success, but the successes are the ones that I try to cherish and celebrate.
Q4Given your extensive involvement with the International Society of Thrombosis and Haemostasis (ISTH) and the INVENT-VTE network, how do you leverage these positions to drive advancements in thrombosis and haemostasis research, and what initiatives are you most excited about currently?
I'm an elected council member for ISTH, and that provides me with a large network, and also a podium to try and inspire people. I'm always open to mentoring people or providing my experiences, and I really hope to motivate younger people to not be turned off if it takes a bit longer to find success. Not everything you do will succeed, but if you get a couple of successes, then you can really make an impact. That is what ISTH does for me. INVENT-VTE is an international network of academic, national venous thromboembolism networks. It is a very powerful network to find colleagues in other countries, even the ones you don't know yet, and to facilitate multinational trials. It doesn't come with a lot of money, but there is some funding to, let's say, acquire another country to participate in your research by providing some money for legal issues, the startup fees, and so on. The network also provides standard procedures, so to speak, for academic trials. For example, adjudication committee platforms and a core set of outcome data, so that you don't have to rethink everything yourself,
If there's no evidence, you should just go and get it. Make it yourself.
but can take the best bits out of it. That is what INVENT-VTE does in close association with ISTH.
Q5
You have received numerous accolades, including the Ham-Wasserman Lecture award and the Hijmans van den Bergh penning, in recognition of your contributions to internal medicine. How do these recognitions influence your work, and what has been your proudest achievement to date?
So, the Ham-Wasserman Lecture award was provided by the American Society of Hematology (ASH) in 2016 for “pioneering work in inherited thrombophilia.” That was an enormous honour, and is still an enormous honour, but I'm not sure if it impacts your work. It is more a recognition of what you have done. Of course, it is an outside confirmation that what drives you is actually being
seen, and impacts people. So, I'm very proud of that. The Hijmans van den Bergh penning was not about thrombosis, but it was really about my contribution to internal medicine in the broadest sense. I'm very proud of that, because I'm the first woman, if you can believe it, that has been recognised in the 60-something years that it has existed. I'm proud that I can inspire young people by being a visible woman, by being both a clinician and an investigator, and some of the, let's say, ‘softer’ parts, like having a family, and still having time to work out or enjoy life a little bit in the broader sense. Does it really impact your work? No, but it does impact the way you are being seen.
Q6What key pieces of advice do you have for the next generation of aspiring haematologists wishing to pursue a successful career in this field?
Find a mentor. You can be very fond of your supervisor, and that is an extremely important person, but it is also nice to have a mentor outside of this. It can even be someone outside the field of medicine, who can talk to you about what drives you, what keeps you going, what is a drainer of energy, and what is giving you energy. To be quite honest, I sort of intuitively follow that path. Not all the time, of course, but we're always in this rat race, and it's really helpful to have some time to sit back and go back to the core, and ask the big questions. I think it's also important to provide some advice to mentors, because it's been such a thrill to work with younger people; to see some people who have stayed in the field and now are doing the same to young people today. I think that is fantastic.
Interviews
Two experts in haematology discuss their careers thus far, and outline their motivations for research. Conversations are centred around current topics: advancements in gene therapy, non-factor replacement products, and incorporating digital approaches to treat bleeding disorders.
Featuring: Niamh O’Connell and Pier Mannuccio Mannucci
Niamh O’Connell
National Haemophilia Director and Consultant Haematologist, National Coagulation Centre, St James’s Hospital, Dublin, Ireland; Clinical Professor, Department of Haematology, Trinity College, Dublin, Ireland
Q1 What motivated you to follow a career in haematology, and what continues to drive you today?
When I was a medical student in the small university city of Cork, in Ireland, there was one female consultant, and she was a haematologist. She was a wonderful clinician, who was very kind to her patients, and students, as well as other staff, and on some level I think this inspired me to think about haematology.
looking after them throughout their treatment. I think most haematologists enjoy this aspect; it is very satisfying.
Those were the main things that convinced me haematology was for me, and I have never regretted it for a minute.
Q2
In Ireland, patients are also partners in the co-design of our services
When I went on to do my House Officer training, one of the rotations I chose was haematology, and here I met another wonderful clinician. He was so thoughtful, and it was very interesting shadowing him, as he dealt with all sorts of blood abnormalities. The other thing he did was introduce me to the laboratory; our job in the UK and Ireland involves both clinical and laboratory haematology. There is a beautiful circularity here: seeing the patient with acute symptoms in an emergency department, reviewing the results in the laboratory, making a diagnosis, before going back to the person with a treatment plan, and
You are currently on the Executive Committee for the European Association for Haemophilia and Allied Disorders (EAHAD). How important are organisations like this in creating a European network, and a uniform message on the standard of care for people with rare disorders?
In a small European country like Ireland, there are relatively few haematologists and even fewer who are specifically trained and working in haemostasis. My network is essentially outside of my country and the community in haemostasis is global; we have both European communities and international organisations, which are critical for managing rare bleeding and thrombotic disorders. To give an example, in Ireland, we diagnose 4–6 cases per year of a very rare bleeding disorder called acquired haemophilia. If
I am not communicating with other colleagues all around the world, I am never going to be able to advance the care of those patients. It is just essential that we have these networks across the world. The world today is so much more connected for sharing knowledge and information, and discussing challenging cases. I cannot emphasise enough how important this network is, and EAHAD as an organisation provides the framework for that network in Europe.
Q3
How is EAHAD helping to provide comprehensive care for bleeding disorders, and what are the barriers to achieving this?
Unfortunately, there is a large global inequity with bleeding disorders, and at least threequarters of patients with bleeding disorders do not receive adequate care, including access to testing or treatment, and organised care. Organisation, in particular, is integral to delivering high-quality care.
There are also differences in healthcare models. Even across Europe, we see a lot of differences. It is important to understand what is done differently, and also where our similarities lie. One of the things I find reassuring is that whenever we are presented with a case, the scenarios are often the same; our patients are not different, aside from small cultural differences. The differences tend to be in how care is organised in a country, as well as how accessible this is, and this can be a challenge.
EAHAD helps us standardise the key principles of care, basing this on the known ways to organise care, and deliver better outcomes for people and families with bleeding disorders. This allows countries, like Ireland, to measure their services against the European principles. EAHAD is moving towards an accreditation model, providing formal inspections to verify that institutions are delivering the best possible care. Knowing that the centres where they are undergoing treatment are
registered with an organisation like EAHAD provides reassurance to patients that they are receiving the highest possible standard of care.
Another barrier to overcome is funding and resources to improve services for haematological disorders; these conditions are relatively rare and it can be difficult to convince the healthcare payers to prioritise this over other more common conditions. An audit by an external evaluator from a respected organisation such as EAHAD can highlight the aspects of care which need improvement, and this is often more effective in persuading funders to provide the necessary resources.
Q4 How have you incorporated a digital approach to your practice, and how can we expect to see bleeding disorders digitalised in the coming years?
This is something that the Haemophilia Service has been taking very seriously; we have established a Global Standards-1
It is just essential that we have these networks across the world
barcode-based track and trace system in Ireland for all vials of factor since 2004; a national electronic health record for haemophilia since 2006, updated in 2017; and a patient portal which was rolled out in 2023. We believe that patients should have access to their information, and we are trying to promote equal access to treatment in the 38 hospitals across Ireland, wherever in the country you live.
In addition to barcoding, we have a national cold chain delivery system for patients receiving clotting factor treatment at home and for delivery to hospitals. We have made sure that every hospital with an emergency department has several doses of all the clotting factors, to support acute treatment in peripheral hospitals.
We have kept momentum to a paperless record, with electronic prescribing in use throughout our hospital. Significant training has taken place to ensure that nurses and clinicians are entering information in electronic systems in the correct way, to facilitate analysis of this data to support quality improvement in care in the future.
Q5 What can other establishments learn from the approach taken at St James’s Hospital, the leading comprehensive care centre for adults with bleeding disorders in Ireland?
In some establishments, probably more so in the past, if the name of a consultant was over a patients’
bed, this was the only Consultant looking after them. This is not how we work in our National Haemophilia Service; there are five Consultants and we work together as a team, sharing all of our information. The value in this is that the patient receives a consensus view, particularly if you have a bleeding disorder which is rare, and/or challenging. This might be the consensus from the five of us, or we may have discussed this with colleagues in Europe and beyond.
In our treatment, we try to always involve the patient in discussions, providing benefits and downsides to potential treatment pathways. We make them aware that we are all thinking about their case, and we are all trying to come up with solutions. At the end of the day, the patient is always the decision maker, but it is very much a collaborative and shared decisionmaking process.
In Ireland, patients are also partners in the co-design of our services. The National Haemophilia service works hand-in-hand with the patient organisation on access to services, types of services provided, access to new therapies, and procurement of treatment. We are greatly assisted by having a statutory National Haemophilia Council and this body oversees our provision of service and is a vital communication channel to the national health service for us.
One of the most enjoyable parts of our work in recent years is our partnership with the National
Service for Haemophilia in Jordan. This has been really interesting; in some aspects I am reminded of services in Ireland several years ago, and we hope that other countries can benefit from our learnings, so that they too can incrementally improve, but at a faster pace.
Q6You are involved in clinical trials. Could you outline some of the exciting novel therapies for bleeding disorders which are currently emerging, and are expected in the near future?
It has been an incredible 10 years in haemophilia care. I don’t think any of us anticipated how many new treatments we were going to have. This is really exciting, as the new treatments have far less of a burden for administration, and they provide much better protection. There is a completely different future available now for patients transferring to us from paediatric centres, at around the age of 16 years. I meet these kids in the clinic, and it is just wonderful to think they now have a much better quality of life ahead of them.
One of the exciting treatments that I think is making a big difference is gene therapy, especially for factor IX deficiency. Clinical trials have shown this provides very protective treatment with a one-off infusion. There will soon be a menu of different licenced subcutaneous treatments for preventing bleeding. Also, coming up very soon is an extended half-life factor VIII treatment; delivered once weekly, this can provide really high protective levels for a large part of the week.
All of these options provide people with the ability to fulfil their own goals and dreams, living more of a normal life, whatever that might be for them, without having to contend with regular bleeding or joint disease, for example. None of these treatments are curative, but they will allow people to put their condition to the back of their mind, and do all of the other important things they want to, with the reassurance of a comprehensive care team as a safety net.
Q7What advice would you give to a young haematologist, looking to establish a career in the field?
I think having good mentors and advisors is always useful; if you can seek out someone in the field you are interested in as a trusted advisor, it is always a good idea to discuss your plans with somebody. You’ve got to follow your gut and your passions, too.
I think clinical research is important for patients and treaters. Clinical research, from sponsored and investigationled research to basic science research, will help to improve the quality of the care, but it also keeps us fresh and interested, and connected to a network of colleagues in Europe.
I come back from meetings energised, with all sorts of ideas. I would say go to the meetings, meet your colleagues; these are going to be the same people with you from the start of your career, in the middle, and at the finish. You might as well get out there and meet them!
Pier Mannuccio Mannucci
Emeritus Professor, Internal Medicine, University of Milan; Former Scientific Director, Research Hospital Ca’ Granda Foundation, Milan; Former Chairman and Professor of Internal Medicine, Department of Medicine and Medical Specialties, Maggiore University Hospital, Milan; Former Director, Angelo Bianchi Bonomi Hemophilia and Thrombosis Centre, Milan, Italy
Where does your passion for haemostasis and thrombosis stem from?
I think the beginning was when I graduated in the 1960s; my thesis was on red cell enzymes within haematology, but it had nothing to do with haemostasis and thrombosis. At that time, coagulation was not a topic of great interest, scientifically and also practically, because very little could be done. I thought that my knowledge of the enzyme biology could help me to tackle this field, which seemed to be interesting, because it was not widely covered at the time. Therapeutically, there was nothing then compared to what you see now in the field of thrombosis and bleeding disorders, so it was a challenging field. That is why I got interested, because I thought it was a field which was going to expand.
Q2Some of your work involved developing new drugs for the treatment of patients with haemophilia and von Willebrand disease. How have you seen the landscape change for patients living with these conditions over the course of your life?
Nowadays, the life expectancy of these patients has become similar to that of their peers
The mecca of blood coagulation was Oxford, UK, at the time, and I wanted to go there as a postgraduate student. I met an English colleague at a congress, and decided to spend 3 or 4 months with him at St Thomas’ Hospital, London, UK. This helped my application to Oxford, and gave me some good basic knowledge before I started. I was very lucky because, when I was at Oxford, Judith Pool was spending a sabbatical there. She was the person who discovered cryo-precipitation as a form of replacement therapy in haemophilia A and von Willebrand disease. She very patiently taught me to prepare cryoprecipitate, I learnt a lot from her, and she was very kind and gentle; a fantastic lady.
I began my career as an internist, with a special interest in haematology, at the University of Cagliari, Sardinia, Italy. I saw patients with haemophilia, and was interested by the gloomy problem that very little could be done for them. I observed a shift from the use of cryoprecipitate, to more concentrated plasmaderived coagulation factors, made available commercially in the 1970s. This really was the weapon that dramatically changed the situation, because they were quicker to prepare, and could be infused intravenously, rather than using a drip.
When I first got involved in the field, the life expectancy of patients with bleeding disorders was no longer than 30 years or so. Nowadays, the life expectancy of these patients has become similar to that of their peers, close to 80 years. What has happened in the last 20 years in the field of haemophilia is really a miracle.
In addition, an inhibitor of fibrinolysis (tranexamic acid) was developed around 60 years ago, and I have been really impressed with how this drug has progressed, beyond being solely an antidote for bleeding. This medication has proven very helpful in trauma patients, and is saving countless lives, as well
as continuing to prove useful for indications such as massive postpartum bleeding.
Q3
How important are organisations like the European Hematology Association (EHA), and American Society of Hematology (ASH), for sharing knowledge and advancing the treatment of bleeding disorders?
They are very important, because they are a forum for experts in the field. Haemophilia is only one part of haematology. EHA and ASH are both well known, but it was necessary to build something else in Europe. Together with two colleagues, Christopher Ludlam and Brian Colvin, we founded the European Association for Haemophilia and Allied Disorders (EAHAD). These organisations are important for clinicians, but also for patients. I would like to emphasise the importance of the
World Federation of Hemophilia (WFH), and how this congress encourages patient participation.
Q4
Over the course of your career, you have served on Editorial Boards, collaborated on many articles, and delivered countless talks. What do you consider to be your most memorable achievements in affecting change?
From my point of view, my most important contribution has probably been finding a treatment for haemophilia that is not based on blood products and blood derivatives. In the past, it was very difficult for patients with mild haemophilia and von Willebrand disease; there was a shortage of products, and they often went long periods with a lack of treatment. In 1977, myself and colleagues were the first to report the clinical use of
a pharmacological method for increasing factor VIII and von Willebrand factor.
What I am also particularly proud of in my career has been serving as editor, and on the Editorial Boards of important journals. I remember when I was given the option to be president of a society, or the Editor-in-Chief, I chose the second option. The work of a president is important, not only in terms of politics and also from the perspective of organisational science, but I very much like the idea of seeing papers produced by others. In this way, you stay up to date with the most important advancements in the field, and are able to judge, with the help of peer-review, provide authors with feedback, and make the best possible decisions. I think it is our duty as members of a scientific community to do this work, which was not remunerated at that time.
I have learnt so much from seeing what other reviewers suggest, and I think it is important to consider different views apart from our own.
Q5 Are there any novel therapies you are excited to see translated into practice in the near future, specifically related to side effects of haemophilia therapy, or rare coagulation disorders?
Speaking about disparities in life expectancy, in high-income countries the situation is very different to that in low- and middle- income nations. In the latter, there are several unmet needs which must be addressed. The development of a nonfactor product, which mimics the activity of factor VIII and can be given subcutaneously, has brought about a great amount of improvement in terms of quality of life. Instead of infusing patients every other day, treatments are now administered every fortnight, or even monthly, and I expect to see further improvements to this, to further ameliorate patient quality of life.
Emerging treatments for both haemophilia A and B are promising, and of course, there is now gene therapy. Only gene therapy can fulfil, in principle, a cure for these, and many other blood-related diseases. Talking about gene replacement therapy, this is not yet suitable nor licensed for use in children, because their liver is continuously changing. The issue here is that, to treat an inherited disorder, we must target children, not only adults; so, this is a problem to be studied further. I am sure that the true prophylaxis will be gene therapy, or gene editing, but this is not yet available.
Another area to focus on could be the variance in response to gene therapy for both the haemophilias. Currently, you can be met with either no response, or equally, an excessive response. The issue here is a lack of consistency, and there is room for improvement.
Only gene therapy can fulfil, in principle, a cure for these, and many other blood-related diseases
Q6
Speaking to a younger haematologist, what are some helpful points for them to incorporate into their work, allowing them to improve their practice/research ?
To a young person who wants to become a haematologist, I would say that, for me, this is still a fascinating field. The reason I like it is because of the alignment between what you see in the laboratory and in the clinic. You can predict this relationship to some extent, and that is not common across all medical specialties. I would say, train in both, and observe the close correspondence between these aspects.
T cells have protein receptors, that bind to protein fragments, known as antigens, on the surface of pathogenic invading cells. If recognised by the T cell as abnormal, the T cell will secrete toxic chemicals that degrade the target cell.
Mechanism of CAR-T:1
A sample of blood cells is taken from the patient, and T cells are separated out in a process known as apheresis
In the lab, the T cells are then genetically engineered to be able to produce CAR. These are proteins that are complimentary and responsive to the antigen on targeted tumour cells.
After a few weeks, the cells are multiplied, and the CAR-T cells are then thawed and infused into the patient.
Unfortunately, cancer cells can evade detection, so CAR-T therapy has been developed as an immunotherapy approach, boosting the patient’s immune system to better kill the cancer cells.
Recent Innovations in CAR-T Case Study:2
In recent years, CAR-T therapy has been promising in treating leukaemia, namely R/R B-ALL.
In May 2022, in a collaboration between GOSH for Children and UCL, a 13-yearold was the first person in the world to receive base-edited CAR-T cells for the treatment of resistant leukaemia.
She was diagnosed with T-ALL in 2021 and received a bone marrow transplant and chemotherapy, but the disease persisted.
Within 4 weeks of treatment, her leukaemia was undetectable.
Benefits and Potential Side
Benefits:
Targeted action: the therapy specifically targets and kills cancerous cells, minimising damage to healthy cells.
Personalised treatment: CAR-T therapy is tailored to the individual, enhancing its effectiveness.
Abbreviations:
AML: acute myeloid leukaemia; B-ALL: B-cell acute lymphoblastic leukaemia; BAFFR: B cell activating factor receptor; CAR: chimeric antigen receptors; CLL: chronic lymphocytic leukaemia; CRS: cytokine release syndrome; GOSH: Great Ormond Street Hospital; R/R: relapsed or refractory; T-ALL: T cell acute lymphoblastic leukaemia; UCL: University College London.
References
1. Leukemia & Lymphoma Society. Chimeric antigen receptor (CAR) T cell therapy. Available at: https:// www.lls.org/treatment/types-treatment/immunotherapy/chimeric-antigen-receptor-car-t-cell-therapy. Last accessed: 30 May 2024.
2. University College London (UCL). World-first use of base-edited CAR-T cells to treat resistant leukaemia. Available at: https://www.ucl.ac.uk/child-health/news/2022/dec/world-first-use-baseedited-car-t-cells-treat-resistant-leukaemia. Last accessed: 30 May 2024.
3. Mayo Clinic. Exploring receptor T cell rheumatologic Last accessed:
4. Memorial Sloan is first of its kind. myeloid-leukemia-is-first-of-its-kind.
Innovations
CAR-T Cell Therapy
Selected Clinical Trials: Steps:
been Study at the Mayo Clinic to assess the therapeutic efficacy of BAFFR CAR-T cells in BAFFR-expressing B cell haematological malignancies, such as large B cell, mantle cell, and follicular lymphoma CLL and B-ALL.3
Phase I clinical trial at Memorial Sloan Kettering Cancer Center, New York, USA, led by Jae Park, a haematologistoncologist, this trial is assessing CAR-T research for the treatment of AML.4
Study at Mayo Clinic testing IC19/1563, a CD19-targeted CAR-T therapy for R/R B cell malignancies.5
Researcher
Trial
Comparison Group
Effectivity
Side Effects6
Potential for long-term remission: some patients achieve long-term remission, reducing the likelihood of relapse.
Side Effects:
Patients might experience fever, chills, nausea, and difficulty breathing due to an allergic reaction to CAR-T cells.
CAR T-cells can cause neurotoxicity, leading to headaches, altered consciousness, confusion, speech changes, and seizures.
Test Person Safety
Analysis
Reduced need for conventional therapies such as chemotherapy and radiotherapy, improving patient’s quality of life.
Symptoms of CRS like fever, low blood pressure, and difficulty breathing can occur as the immune system releases large amounts of cytokines.
Exploring the Role of B-cell activating factor receptor (BAFFR)-based chimeric antigen (CAR-T) in BAFFR-expressing B-cell hematologic malignancies and autoimmune disorders. Available at: https://www.mayo.edu/research/clinical-trials/cls-20513089. 30 May 2024.
Sloan Kettering Cancer Center. New CAR-T cell T cell clinical trial for acute myeloid leukemia kind. Available at: https://www.mskcc.org/news/new-car-cell-clinical-trial-for-acutemyeloid-leukemia-is-first-of-its-kind. Last accessed: 30 May 2024.
Rapid breakdown of cancer cells can elevate uric acid levels in the blood, potentially overloading the kidneys.
5. Mayo Clinic. CD19-Directed CAR-T cell therapy for the treatment of relapsed/refractory b cell malignancies. Available at: https://www.mayo.edu/research/clinical-trials/cls-20512716. Last accessed: 30 May 2024.
6. Blood Cancer UK. What is CAR-T therapy? Available at: https://bloodcancer.org.uk/understandingblood-cancer/treatment/what-is-car-t-therapy/. Last accessed: 30 May 2024.
• Blood consists of many types of cells, such as white blood cells, red blood cells, and platelets.
• When a blood vessel breaks, platelets will localise to the site of injury and trigger a coagulation cascade. During this, an enzyme, thrombin, cleaves fibrinogen into fibrin monomers, which then polymerise together to form the blood clot.
Red blood cell
Platelet
• Platelet disorders refer to either the surplus, deficiency, or dysfunction of platelets. Symptoms can include excessive bruising, prolonged bleeding, fatigue, chest pain, and vomiting.
Examples of Drugs: FDA Approval Timeline4-5
Types of Platelet Disorders1
Bernard-Soulier disease: characterised by dysfunction of the glycoprotein Ib-IX-V complex, leading to increased bleeding tendency.
Glanzmann’s platelet disorder dysfunction of resulting in impaired increased bleeding
Innovative Therapies2,3
TPO-RA stimulate the production of platelets in the bone marrow and are used to treat thrombocytopenia, including ITP and myelodysplastic syndromes.
Antifibrinolytic agents, such as tranexamic acid, help reduce bleeding by stabilising blood clots and preventing their breakdown.
Lowe syndrome: a rare X-linked genetic disorder characterised by congenital cataracts, intellectual disability, and kidney abnormalities, which can include thrombocytopenia, caused by mutations in the OCRL gene.
Platelet release group of disorders storage or release to impaired platelet bleeding tendency.
Immunosuppressive therapy may be used in cases of immune-mediated platelet disorders like ITP; for which medications that suppress the immune system, such as corticosteroids, immunoglobulins, or immunosuppressants, may be prescribed to reduce platelet destruction. Anticoagulants agents blood associated hyperactivity thrombotic
Tranexamic acid (antifibrinolytic agent)
Ticolopidine (antiplatelet agent)
Clopidiogrel (antiplatelet agent)
Abciximab (antiplatelet agent)
Disorders
thrombasthenia: an inherited disorder caused by a deficiency or of the glycoprotein IIb/IIIa receptor, impaired platelet aggregation and bleeding tendency.
Hermansky Pudlak syndrome: characterised by oculocutaneous albinism, and other systemic manifestations, caused by defects in the biogenesis of platelet granules and lysosome-related organelles.
cell Cell without receptors Healthy platelet
Hermansky Pudlak syndrome platelet
Jacobsen syndrome: a rare chromosomal disorder characterised by multiple physical and developmental abnormalities, including heart defects, intellectual disability, and thrombocytopenia, caused by a deletion in the long arm of chromosome 11.
Jacobsen syndrome chromosome 11
release and storage pool defects: a disorders characterised by abnormal release of platelet granules, leading platelet function and increased tendency.
Healthy platelet Defective platelet
TAR syndrome: a rare congenital disorder characterised by thrombocytopenia, which is low platelet count, and bilateral absence or hypoplasia of the radius bone in the forearm.
Healthy platelet count Low platelet count
TTP: characterised by microvascular thrombosis, resulting in thrombocytopenia, haemolytic anaemia, and organ damage, often caused by a deficiency of the ADAMTS13 enzyme.
Eptifibatide (anti-coagulant agent)
Anticoagulants or antiplatelet agents are used to prevent blood clots in conditions associated with platelet hyperactivity or increased thrombotic risk.
Tirofiban (antiplatelet agent)
Romiplostim (TPO-RA)
Platelet transfusions may be necessary to increase the platelet count quickly in cases of severe thrombocytopenia or active bleeding. However, transfusions are typically reserved for emergencies or when other treatments are ineffective due to potential risks and complications associated with transfusions.
1. Great Ormond Street Hospital. Platelet disorders. 2018. Available at: https://www.gosh.nhs.uk/conditions-andtreatments/conditions-we-treat/platelet-disorders/. Last accessed: 30 May 2024.
2. National Institutes of Health (NIH). Treatment for blood disorders. 2022. Available at: https://www.nhlbi.nih.gov/ health/blood-bone-marrow-treatments. Last accessed: 30 May 2024.
3. Patient. Thrombocytopenia and platelet function disorders. 2020. Available at: https://patient.info/doctor/ thrombocytopenia-and-platelet-function-disorders. Last accessed: 30 May 2024.
4. Johnson SM et al. New and off-label uses of tranexamic acid. AACN Adv Crit Care. 2021;32(3):237-42.
5. Barry S. Coller. Historical perspective and future directions in platelet research. J Thomb Haemost. 2011;(Suppl 1):374-95.
Extensive Left Sided Venous Thrombi and Bilateral Pulmonary Emboli in the Context of May-Thurner Syndrome for a Patient Presenting with Acute Flank Pain: A Case Report
Editor's Pick
My Editor’s Pick for this year’s edition of EMJ Hematology is a compelling case report by Scadding et al. that highlights the importance of recognising May-Thurner syndrome, a congenital condition caused by left common iliac vein compression, in cases of extensive left-sided thrombus formation. This report emphasises the need for high clinical suspicion and discusses conservative management options, providing valuable insights for clinicians when thrombolysis is contraindicated.
Emanuele Angelucci Chair, Hematology and Cellular Therapy Unit, Ospedale Policlinico San Martino; Transplant Program Director, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Genova, Italy
Authors: *Sasha
Scadding,1 Deven Ramoutar1
1. Royal Free Hampstead NHS Trust, Royal Free London NHS Foundation Trust, UK *Correspondence to sasha.scadding1@nhs.net
Disclosure: The authors have declared no conflicts of interest.
Received: 20.05.24
Accepted: 10.06.24
Keywords: Anticoagulation, deep vein thrombosis, May-Thurner syndrome, pulmonary embolism, thrombolysis.
May-Thurner syndrome is an underappreciated differential in the context of deep venous thrombosis. It is a symptomatic congenital abnormality resulting from compression of the left common iliac vein, sometimes also referred to as iliac vein compression syndrome in the literature. This case reports the presentation of a female with acute flank pain to the emergency department following a recent laparotomy for a complicated tubo-ovarian abscess. She was subsequently diagnosed with a sub-acute thrombus extending from the left common femoral vein into the left inferior vena cava, as well as new bilateral pulmonary emboli. As the patient had relative contraindications to thrombolysis, a shared decision was made with the vascular team to opt for conservative management only. In this instance, this included long-term oral anticoagulant therapy and compression stockings. This case asserts that clinicians should maintain a high index of suspicion for May-Thurner syndrome in the context of extensive left-sided thrombus formation.
Key Points
1. Clinicians should maintain a high index of suspicion for May-Thurner syndrome and consider it in the diagnostic work-up of at-risk patient groups.
2. Shared decision-making with patients is integral in deciding whether to deviate from gold-standard treatment where relative contraindications/cautions are present.
3. The therapeutic management of chronic venous thrombi resulting from May-Thurner syndrome is not well defined in current literature.
BACKGROUND
May-Thurner syndrome (MTS) is defined as symptomatic compression of the left common iliac vein by the overlying right common iliac artery against the lumbar vertebrae.1 The compression of this specific element of the left-sided venous system was first observed to be a precipitating factor in deep venous thrombosis (DVT) formation by Virchow in 1851, who identified a link between this anatomical variant and left-sided lower limb clot formation.2 However, it was May and Thurner’s prominent 1957 study that exposed the underlying pathophysiology.3 Using 430 unselected cadavers, they revealed that intraluminal fibrous bands (termed ‘spurs’) tended to form within the left iliac vein if it was compressed, a result of chronic irritation of the venous endothelium by arterial pulsation.3 In addition, the results of this study also drew evidence for the prevalence of this abnormality. Although a small sample size was used, compression plus ‘spur’ formation was present in approximately 22% of the cadavers used.3 This has been corroborated by more recent retrospective radiological studies of patients with confirmed left-sided DVT. For example, one 2004 study, using spiral CT venography, found that 61% of patients with confirmed left-sided DVT had evidence of left iliac vein compression.4 Despite these studies, the overall prevalence of this anatomical variant remains challenging to ascertain as it is thought to be clinically silent in most individuals.5
The compression of the left iliac venous system can only be termed MTS in those who are symptomatic, with a currently
estimated prevalence of 14–32% in the general population.2 Many patients may present with signs consistent with venous hypertension, including left lower limb swelling or claudication. However, a significant proportion only present once they have developed signs consistent with left lower limb thrombosis or a potentially life-threatening pulmonary embolus. A significant sex difference has been noted amongst these symptomatic patients, with one 2018 systematic review estimating the female-to-male ratio at 2:1.6 Notably, this review also found that females were more likely to have a pulmonary embolus on presentation.6 Overall, the demographic most at risk of MTS has been identified as females aged 20–40 years.7
CASE PRESENTATION
A female in her 40s presented to the emergency department with a 6-hour history of paraesthesia and unilateral leftsided calf swelling. They subsequently developed severe left-sided flank pain 2-hours after the onset of swelling. The pain was severe enough to warrant multiple doses of IV morphine. Their significant past medical history included multiple recent admissions with left-sided tubo-ovarian abscess, culminating in an emergency laparotomy, with a 1-day intensive therapy unit admission post-surgery. Since discharge, they acknowledged a significant decline in her mobility levels. The patient was also known to be a heavy smoker.
On initial examination, the patient had mild hypotension (91/60) and low-grade pyrexia (37.7 °C) but was otherwise
haemodynamically stable. On palpation of the abdomen, there was left-sided renal angle tenderness without evidence of peritonism. The left calf was also visibly swollen, with pitting oedema to mid-shin, but was non-tender on palpation. The rest of the examination was unremarkable.
Laboratory studies requested on admission revealed a leucocytosis (18.7) with neutrophilia (12.7). Urine dipstick was negative. Initial coagulation studies, including D-dimer, had haemolysed and were sent for a repeat. An interim CT arterial portography considering her complex surgical history was requested and subsequently completed. This revealed a ‘highly suspicious acute/subacute DVT on a background of MTS with a tongue of thrombus extending from left common femoral vein into lower inferior vena cava’. There was also a ‘highly suspicious right lower lobe pulmonary embolism (PE)’. (Figure 1, Figure 2) D-dimer was also reported shortly after at 3,969 and other coagulation studies were unremarkable. The patient was subsequently admitted under the medical team and treatment dose anticoagulation was initiated with tinzaparin.
A dedicated CT pulmonary angiogram was requested following admission. This revealed ‘segmental and subsegmental PEs affecting the right lower lobe with evidence of wedge infarction in the right lower lobe’. (Figure 3) ‘Subsegmental PEs affecting the lower lobe’ were also identified. The patient was discussed with the vascular on-call registrar, who advised that there was no sufficient indication for urgent catheter-directed thrombolysis as an element of chronicity had been identified. Furthermore, their recent abdominal surgery was also a relative contraindication to any thrombolytic procedure. A thrombophilia screen was also sent at this point and was subsequently reported several days later as unremarkable. On discharge, follow-up was arranged in vascular and haemophilia clinics. On outpatient vascular review 5 days post-discharge, a shared decision was made with the patient to opt for conservative management only. This entailed continuation of anticoagulation, albeit with a switch made to rivaroxaban,
and orthotics assessment for an aboveknee stocking. They were also advised on smoking cessation and signposted to relevant resources.
DISCUSSION
Current literature suggests that MTS as a contributing factor to DVT formation is significantly underdiagnosed, particularly given the estimated population prevalence of iliac vein compression, as pointed to by cadaveric and radiographic studies.7,8 This is likely secondary to a combination of factors. Firstly, Doppler ultrasound is currently the most common modality used in emergency departments to diagnose acute DVT. However, it is often unable to adequately visualise the deep iliac vessels and therefore identify compression of the left iliac vein.9 Due to this, the gold standard for MTS diagnosis is venography with intravascular ultrasound, which has both a high sensitivity and specificity but is not necessarily a practical investigation in the acute setting.2 Secondly, for a patient, if a DVT is diagnosed and a transient risk factor is identified, further investigations are often not considered to identify any pre-existing vulnerabilities in the patient’s anatomy. This is significant as the population group most affected, younger females, are in a key demographic for additional risk factors such as the combined oral contraceptive pill and pregnancy. Imaging to identify MTS could therefore allow for further risk stratification, as part of the diagnostic work-up, once DVT is confirmed.
For patients recognised to have MTS without thrombotic complications, early management strategies may include compression stockings and systematic anticoagulation. However, this is not considered sufficient long-term due to failure to address the underlying anatomical precipitant.10 The mainstay of interventional therapy for MTS therefore consists of iliofemoral stent insertion.11 However, clinical outcomes, including stent failure or longterm patency, have been shown to be less favourable for patients with thrombotic sequelae of MTS versus those without.12 Therefore, there is a growing body of
1: Axial section from the CT abdomen pelvis with contrast demonstrating significant distension and filling defect in the left common iliac vein.
2: Coronal section from the CT abdomen pelvis with contrast demonstrating tongue of thrombus at the inferior part of the inferior vena cava.
Figure
Figure
research that suggests catheter-directed thrombolysis is preferable for patients with both MTS and DVT formation, with further long-term anticoagulation therapy postprocedure.13 However, in this specific case, management decisions were complicated by an element of chronicity identified by CT imaging, for which thrombolysis is not as useful.14 This was potentially linked to the recent removal of a tubo-ovarian abscess that was likely contributing to further external compression of the left pelvic veins. Furthermore, the patient’s recent open laparotomy within the previous month was also a relative contraindication. Therefore, the case also highlights the importance of shared decision-making between the multidisciplinary team and the patient. For this patient, once the above complicating factors were outlined to her, she opted for conservative management with systematic anticoagulation alone. Furthermore, regarding the duration of systematic anticoagulation for patients with MTS, there is no standardised
consensus. This is irrespective of whether interventional therapy has been pursued or not, with one small case series reporting variability in therapy from 6 months to lifelong.15 There is also no significant evidence base determining the choice of oral anticoagulation used.2
CONCLUSION
There are a growing number of case reports describing extensive thromboses with MTS acknowledged as a contributing factor.16-18 This report serves as an additional account to strengthen awareness of this anatomical syndrome and its consideration in the diagnostic work-up of select patient groups. Furthermore, this case also highlights the importance of shared decision-making with the patient when management options are not well defined, such as in the case of a sub-acute thrombus.
Figure 3: Coronal section from the CT pulmonary angiogram demonstrating a right lower lobe pulmonary embolism with evidence of wedge infarction.
References
1. Cockett FB, Thomas ML. The iliac compression syndrome. Br J Surg. 1965;52(10): 816-21.
2. Poyyamoli S et al. May-Thurner syndrome. Cardiovasc Diagn Ther. 2021;11(5):1104-11.
3. May R, Thurner J. The cause of the predominantly sinistral occurrence of thrombosis of the pelvis veins. Angiology. 1957;8(5):419-27.
4. Chung JW et al. Acute iliofemoral deep vein thrombosis: evaluation of underlying anatomic abnormalities by spiral CT venography. J Vasc Interv Radiol. 2004;15:249-56.
5. Kibbe MR et al. Iliac vein compression in an asymptomatic patient population. J Vasc Surg. 2004;39(5):937-43.
6. Kaltenmeier CT et el. Systematic review of May-Thurner syndrome with an emphasis on gender differences. J
Vasc Surg. 2018;6(3):399-407
7. Mousa AY, Abdu-Rahma AF. MayThurner syndrome: update and review. Ann Vasc Surg. 2014;27(7):984-95.
8. Harbin MM, Lutsey PL. May-Thurner syndrome: history of understanding and need for defining population prevalence. J Thromb Haemost. 2020;18(3):534-42
9. Patel NH et al. Endovascular management of acute extensive iliofemoral deep venous thrombosis caused by May-Thurner syndrome. J Vasc Interv Radiol. 2000;11:1297-302.
10. Hulsberg PC et al. Minimally invasive treatments for venous compression syndromes. Cardiovasc Diagn Ther. 2016;6(6):585-92.
12. Jiang L et al. Clinical outcomes at 3-years after stenting for thrombotic and non-thrombotic iliac vein compression syndrome patients. Clin Appl Throm Hemost. 2024;30. DOI:10.1177/10760296231220053.
13. Liu Q et al. Current status and prospect of stent placement for MayThurner syndrome. Curr Med Sci. 2021;41(6):1178-86.
14. Liu X et al. Safety and efficacy of pharmacomechanical thrombolysis for acute and subacute deep vein thrombosis patients with relative contraindications. Medicine. 2018;DOI:10.1097/ MD.0000000000013013.
15. Montes MC et al. Endovascular and medical therapy of May-Thuner syndrome: case series and scoping literature. J Med Vasc. 2021;46(2):809.
Spinal Plasmacytoma Transformed Into Solitary Sacral Amyloidoma: A Case Report
Authors: N. Neupane,1 *H. Kharel,1 M. Ammad-Ud-Din,2 S. Jamshed3
1. Department of Internal Medicine, Rochester General Hospital, New York, USA
2. Department of Hematology and Oncology, Moffitt Cancer Center, Tampa, Florida, USA
3. Department of Hematology and Oncology, Lipson Cancer Center, Rochester General Hospital, New York, USA
*Correspondence to Himal.Kharel@rochesterregional.org
Disclosure: Jamshed reports financial support from Jansen. All other authors report no conflicts of interest.
Acknowledgements: Informed consent was obtained from the patient.
Amyloidoma is a rare complication of plasmacytoma, that can involve the spine and present with compressive neurological symptoms. It is usually a diagnosis of exclusion, and is difficult to differentiate from other plasma cell disorders on imaging. Definite diagnosis requires a tissue biopsy. The treatment requires a multidisciplinary approach, with input from haematology, neurology, neurosurgery, and radiology for the optimum course of action, depending on the patient’s comorbidities and performance status. The authors hereby present a case of solitary sacral amyloidoma in a 52-year-old African American female. Only three other cases of solitary sacral amyloidoma have been reported in the literature.
Key Points
1. Plasmacytoma of the spine can present with residual amyloidoma after treatment.
2. The diagnosis of amyloidoma requires biopsy to exclude the mimics.
3. The treatment of spinal amyloidoma is challenging due to its close vicinity to the spinal cord, but early neurosurgical intervention has been shown to improve outcomes.
INTRODUCTION
Amyloidosis results from a misconfiguration of normal protein into β-plated linear sheets, leading to the formation and deposition of insoluble fibrils.1 These fibrils can sometimes form a localised tumour-like mass called amyloidoma at specific organ sites, even in the absence of systemic amyloidosis.2 Spinal amyloidoma can present with symptoms of nerve compression and radiculopathy, depending upon the site of involvement.3 Amyloidoma of the bone can sometimes be difficult to differentiate from other pathologies that commonly cause lytic lesions, like multiple myeloma (MM), plasmacytoma, and metastatic diseases.4 Therefore, the definitive diagnosis requires a tissue biopsy and Congo red staining, which produces an apple-green birefringence under polarised light.
PATIENT INFORMATION
A 52-year-old African American female with a past medical history of IgG lambda MM and sacral plasmacytoma presented with worsening back pain, bowel and bladder incontinence, bilateral lower extremity weakness, and saddle anaesthesia
concerning for cauda equina syndrome. She had been diagnosed with IgG lambda MM with paraprotein level of 0.2 g/dL 10 months previously, and had received radiation and bortezomib, lenalidomide, and dexamethasone, with subsequent normalisation of the light chain ratio, and complete disappearance of paraprotein. She had declined autologous stem cell transplant. The MRI of lumbosacral spine carried out at the time of plasmacytoma diagnosis showed an expansile and enhancing soft tissue mass overlying the right sacral ala, while the biopsy at that time showed presence of plasma cell neoplasm without any Congo red staining.
CLINICAL FINDINGS AND DIAGNOSTIC ASSESSMENT
On presentation, initial lab work revealed normal levels of haemoglobin, creatinine, calcium, and serum protein. MRI of the lumbar spine revealed an irregular expansile enhancing soft tissue mass along the right hemisacrum from the S1 to S4–S5 level, encompassing the right neural foramina of S2, S3, and S4 (Figure 1). These findings were identical to the CT scan finding carried out 2 months previously.
Figure 1: Coronal T1 showing large inferior T1 mass in right sacrum crossing midline.
Furthermore, serum immunoglobulin, serum protein electrophoresis, urine protein electrophoresis, and free light chain ratio were also normal (Table 1). A PET scan was carried out 1 month prior to the presentation, and did not show FDG uptake or evidence of metastasis in the area. With these findings, the suspicion of MM, plasmacytoma recurrence, and malignancies were low, leaving a suspicion for a sacral amyloidoma (SaA) as the diagnosis of exclusion. CTguided biopsy was performed, which
revealed a proteinaceous material showing apple-green birefringence under polarised light following Congo red stain, without evidence of plasma cell neoplasm, consistent with SaA (Figure 2). This was unlike the biopsy which occurred 10 months prior, at the time of initial diagnosis, which showed sheets of mature-appearing plasma cells. Furthermore, a colonoscopy with biopsy and cardiac MRI were also done, which ruled out systemic amyloidosis.
Table 1: Pertinent laboratory findings.
Figure 2: Biopsy showing proteinaceous material with no evidence of neoplastic plasma cells.
THERAPEUTIC INTERVENTION
The patient was started on dexamethasone, which helped to improve the back pain and incontinence within days. Hospital stay was further complicated by methicillin-sensitive staphylococcal bacteraemia, acute anaemia (haemoglobin <7 g/dL), and sacral fracture. The patient was managed conservatively, considering she had improvement of neurological symptoms with steroids; the absence of a fat pad between the spinal cord; and the large size of the mass, which placed her at a high risk of surgical and long-term neurological complications. In addition, bacteraemia made her a highrisk surgical candidate. She received dexamethasone 40 mg daily for 4 days. She was discharged with a spinal brace for outpatient follow-up.
FOLLOW-UP AND CLINICAL OUTCOMES
A repeat CT scan 2 months later revealed the mass was stable, and MM remained in remission without biochemical progression.
DISCUSSION
Amyloidoma is an aggregate of misfolded protein localised in an organ. It commonly involves the bladder, larynx, tonsils, skin, and lungs.5 Solitary spinal amyloidoma (SSA) is rare, with sacral spinal amyloidoma being exceptionally uncommon. In a review of literature, there were a total of 38 reported cases of SSA, out of which 18 were thoracic, 13 were cervical, four were lumbar, and only three were sacral.3,4,6-9 Like plasmocytomas, amyloidomas are usually found in the elderly, with 74% of cases being diagnosed between 60–80 years of age.3 The patient, however, had an usually early presentation at the age of 52, which may be due to her history of MM and sacral plasmacytoma, and may represent disease continuum. The exact mechanism or factors leading to transformation of the spinal lesion from plasmacytoma to amyloidoma is unknown, and needs further research. As amyloidoma is an expansile mass, the symptoms are generally progressive and are related to the site and neurological involvement.3-9 The most common presenting symptoms in patients with SSA, as expected, are either pain or neurologic compromise, with decreased
muscular strength (74.29%), and axial back pain (65.70%) reported. However, only 17% of cases present with acute symptoms.3 The patient had chronic back pain, likely due to the evolving mass, but presented with cauda equina syndrome when the mass encroached the spinal cord.
For every patient with cauda equina syndrome, initial imaging starts with an emergent MRI/CT myelography of the spine.10 An SSA generally appears as a hypointense lesion on T1- and T2-weighted images.3 CT imaging generally reveals lytic bone lesions with cortical destruction, and possibly pathological fractures, further raising diagnostic challenges, as the findings mimic MM and other primary neoplasms, like plasmacytoma, chondrosarcoma, or secondary metastatic lesions.11 A biopsy is required to distinguish between SSA and other mentioned differentials. An apple-green birefringence when stained with Congo red stain visualised under polarised light, without evidence of plasma cell neoplasm, is classically seen.3-5 Moreover, it should be noted that excluding systemic involvement is of utmost importance in such cases, to rule out systemic amyloidosis.
2. Pasternak S et al. Soft tissue amyloidoma of the extremities: report of a case and review of the literature. Am J Dermatopathol. 2007;29(2):152-5.
3. Pinheiro JP et al. Management and outcome of solitary spinal amyloidoma - a systematic literature review. World Neurosurg. 2020;140:325-31.
4. Klenke FM et al. Multiple myelomaassociated amyloidoma of the sacrum: case report and review of the literature. Global Spine J. 2014;4(2):109-14.
The treatment of the SSA is challenging, due to its close vicinity to the spinal cord, but early neurosurgical intervention has shown to improve outcomes. Based on the review by Pinherio et al.,3 out of 29 patients who underwent surgical resection for a spinal amyloidoma, 24 patients (82.76%) had postoperative neurological improvement. Out of the three cases of SaA, two underwent surgery, and both showed neurological improvement. Another case of SaA reported by Griffin et al.9 had no neurological symptoms, and therefore did not undergo surgical intervention. Based on few reports, surgery seems to be a reasonable approach in a symptomatic patient like this one. However, in this case, the authors elected for conservative management due to high surgical risk, and improvements in neurological symptoms with steroids.
CONCLUSION
Although cases of distant amyloidoma arising secondary to plasmacytoma have been reported, as per the authors’ knowledge, this is the first case report of plasmacytoma of the spine with residual amyloidoma after treatment. In cases of SaA presenting as cauda equina syndrome, it is imperative to initiate steroid therapy, and seek a neurosurgery consult for consideration of emergent decompression surgery.
5. Mahmood S et al. Natural history and outcomes in localised immunoglobulin light-chain amyloidosis: a long-term observational study. Lancet Haematol. 2015;2(6):e241-50.
6. Brawanski N et al. Differentiated plasma cell myeloma presenting as a solitary spinal amyloidoma: a case report, possible pitfall and review to the literature. Clin Neurol Neurosurg. 2015;137:1-4.
7. Bruninx G et al. [Isolated idiopathic amyloid tumor of the sacrum. An important differential diagnosis]. J Radiol. 2001;82(4):495-7. (in French).
8. Giorgi PD et al. Spinal cord compression in dialysis-related upper cervical amyloidoma -
a case report. Spinal Cord Ser Cases. 2021;7(1):40.
9. Griffin M et al. Amyloid tumor of the sacrum. A case report. Acta Cytol. 1995;39(3):503-6.
10. Bischoff RJ et al. A comparison of computed tomography-myelography, magnetic resonance imaging, and myelography in the diagnosis of herniated nucleus pulposus and spinal stenosis. J Spinal Disord. 1993;6(4):289-95.
11. Haridas A et al. Primary isolated amyloidoma of the lumbar spine causing neurological compromise: case report and literature review. Neurosurgery. 2005;57(1):E196.
Filgrastim Used for Infection Prophylaxis for Moderate Neutropenia Related to Primary Myelodysplasia Prior to Elective Surgery: A Case Report
Authors: Sonya Mannala,1 *Emmett Harrison1
1. College of Medicine, University of Saskatchewan, Saskatoon, Canada *Correspondence to eph909@mail.usask.ca
Received: 27.12.23
Accepted: 10.06.24
Disclosure: The authors have declared no conflicts of interest.
Myelodysplastic syndromes are a group of disorders that affect the bone marrow, subsequently affecting the growth and relative abundance of blood-forming cells in the circulating volume. Myelodysplastic syndromes often do not cause early signs or symptoms, and can be found during routine blood tests. Granulocyte colony-stimulating factors (G-CSF) have been used in the treatment of myelodysplastic syndromes with neutropenia. Filgrastim, a G-CSF, helps increase the number of circulating neutrophils. Therefore, it has been proven to reduce patient vulnerability to infections in instances such as chemotherapy-induced neutropenia. This case report describes a 66-year-old male who presented for a pre-operative assessment before an elective left total hip arthroplasty. Routine bloodwork showed a low neutrophil count, and the surgery was cancelled due to concerns about the patient’s risk of infection. Further testing included a bone marrow aspirate and core biopsy that showed mild megaloblastic erythropoiesis and a relative increase in the proportion of myeloblasts and promyelocytes. The patient was given a working diagnosis of early myelodysplasia, and a trial of a low-dose G-CSF was started. The neutrophil count was monitored at 6–72 hours. After 72 hours of administration of filgrastim, the patient’s blood neutrophil levels had improved outside the range of neutropenia. After clearance for surgery, the patient had a successful hip arthroplasty with no postoperative infection reported. No neutropenia was noted post-surgery. This case highlights the potential of filgrastim to be used as prophylaxis before an elective surgery to improve moderate neutropenia related to primary myelodysplasia.
Key Points
1. Myelodysplastic syndromes cause blood dyscrasias, such as neutropenia, leaving patients vulnerable to infection during elective surgeries.
2. Granulocyte colony-stimulating factors treat myelodysplastic syndromes by increasing the number of white blood cells in the bone marrow.
3. Filgrastim, a granulocyte colony-stimulating factor, can be used as prophylaxis before elective surgeries to improve moderate neutropenia related to primary myelodysplasia.
BACKGROUND
Myelodysplastic syndromes (MDS) are a group of blood disorders that occur due to ineffective haematopoiesis that results in abnormal blood-forming cells in the bone marrow. Neutropenia, or having a lower-than-normal level of neutrophils, has been associated with an increased risk of infection in myelodysplastic syndromes.1,2 Granulocyte colony-stimulating factors (G-CSF) help the bone marrow increase the number of white blood cells and are used to treat MDS for neutropenic complications.3-7 Filgrastim, a GCS-F, has been used in the treatment of neutropenia resulting from many causes. The side effects of this medication include alopecia, bone and joint pain, and gastrointestinal issues. As a prophylactic means for moderate neutropenia, filgrastim has not been thoroughly studied to determine its feasibility in improving neutropenia.
CASE PRESENTATION
A 66-year-old male presented to his family physician for a routine preoperative evaluation for an elective left total hip arthroplasty. He was otherwise asymptomatic and did not report a history of recurrent infections. His past medical history was remarkable for hyperlipidaemia and prostate cancer with radical prostatectomy (Table 1).
His surgery was cancelled due to the orthopaedic surgeon's concern that the risk of infection potentially outweighed the benefit of surgery, and the patient was
referred to Haematology. Haematology reviewed the patient and completed a left posterior iliac crest bone marrow aspirate and core biopsy. The biopsy showed normocellular marrow with trilineage haematopoiesis showing mild megaloblastic erythropoiesis and left shifted granulopoiesis. The associated molecular and cytogenetic studies on the bone marrow aspirate were unremarkable. The working diagnosis from Haematology was early myelodysplasia.
The patient was determined to get the elective hip arthroplasty due to a change in functioning, including requiring a walking aid, and inability to do activities that he enjoyed, such as golf. The haematologist discussed a trial of low-dose G-CSF 300 µg subcutaneously for infection prophylaxis for elective hip arthroplasty, including risks and benefits, and the need to trend the neutrophil count at 6–72 hours to assess its effectiveness. The patient consented to a trial of this medication for its off-label use.
DISCUSSION
A low-dose G-CSF has been shown to improve neutropenia associated with MDS. In one randomised controlled Phase III trial, one such G-CSF, filgrastim, was shown to be safe and effective in increasing blood neutrophil levels for patients with congenital, cyclic, and idiopathic neutropenia.3 Furthermore, in that trial, patients reported an improved quality of life due to a reduced frequency of fevers, inflammation, and infections. In addition, at a conventional dose of 3 mg/kg/d of a
granulocyte-macrophage colony-stimulating factor, the incidence of severe infections was reduced, and moderate neutropenia due to MDS was improved.8
A novel approach was taken in this patient by trailing a low-dose G-CSF subcutaneously for neutropenia based on the working diagnosis of early myelodysplasia. In this case, for the patient, there was a perceived risk of infection due to the elective arthroplasty and therefore, the surgery was cancelled. The patient still sought to continue with the procedure, after consideration of the benefits and risks, to improve his functioning. Therefore, a trial of a low-dose G-CSF was perceived to improve the patient’s neutrophils levels. Unfortunately, the G-CSF was not covered by the Saskatchewan Exceptional Drug Formulary without a formal haemeoncological diagnosis, and the medication had to be covered out of pocket.
The neutrophil values along with other values from the Complete Blood Count Differential and Morphology taken at 6, 24, and 72 hours are provided in Table 1 After 6 hours on filgrastim, the patient’s neutrophil levels improved significantly
from 0.70x109/L to 6.6x109/L. This increase in absolute neutrophil count meant that the patient was outside the range of even mild neutropenia.9 At 24 hours, the blood neutrophil count had further increased to 7.3x109/L, resulting in an above normal neutrophil count. At the final bloodwork draw, taken at 72 hours after administration of filgrastim, blood neutrophils levels decreased to 2.0x109/L. After this trial of filgrastim, the patient was cleared to undergo the elective procedure, and the surgery proceeded successfully. Neutrophil values along with other selected Complete Blood Count Differential and Morphology values 1 week prior to his surgery and post-surgery are presented in Table 1 Twenty four hours post-surgery, the neutrophil values were outside the range of neutropenia, and the patient showed no signs of infection. This trend suggests a potential role for filgrastim to treat moderate neutropenia. Further studies are warranted to determine if a low-dose G-CSF should be considered in a prophylactic regimen to treat moderate neutropenia and reduce infection risk prior to surgery.
Table
CONCLUSION
Neutropenia, because of myelodysplastic syndromes, presents a potential risk of infection following surgery. Filgrastim, a G-CSF, is a special protein that stimulates the bone marrow to produce neutrophils to fight infection. It has been used to reduce febrile neutropenia, infection rates, and hospitalisation in patients receiving myelosuppressive chemotherapy.8 This report details the case of a patient with moderate neutropenia prior to undergoing elective arthroplasty. Due to the neutropenia, the surgery was cancelled. With a working diagnosis of early myelodysplasia, a low dose G-CSF was considered as a potential method to
References
1. Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes: a review. JAMA. 2022;328(9):872–80.
2. Diamantopoulos PT et al. Real world data on the prognostic significance of monocytopenia in myelodysplastic syndrome. Sci Rep. 2022;12(1):17914.
3. Stopka T et al. G-CSF plus azacitidine versus azacitidine alone for patients with high-risk myelodysplastic syndrome: academic, open label, randomized trial. Blood Cancer J. 2022;12(7):105.
improve neutrophil levels. After a discussion of risks and benefits, a trial of filgrastim was agreed upon. After 72 hours, the patient’s blood neutrophils increased outside the range of neutropenia. Once cleared for surgery, the patient had a successful elective hip arthroplasty. No post-operative infection was reported, and no neutropenia was noted. This presents a potential application of filgrastim or another low-dose G-CSF as prophylaxis to treat moderate neutropenia prior to elective surgery.
CONSENT STATEMENT
Written consent for individual health information was obtained for this publication.
4. Shibata N et al. Real-world data analysis of perioperative chemotherapy patterns, G-CSF use, and FN status in patients with early breast cancer. Breast Cancer Res Treat. 2023;201(2):265–73.
5. Mouri A et al. Clinical significance of primary prophylactic pegylatedgranulocyte-colony stimulating factor after the administration of ramucirumab plus docetaxel in patients with previously treated nonsmall cell lung cancer. Thorac Cancer. 2019;10(4):1005–8.
6. Laali E et al. Appropriateness of using granulocyte colony-stimulating factor (G-CSF) for primary prophylaxis of
febrile neutropenia in solid tumors. J Oncol Pharm Pract. 2020;26(2):428–33.
7. Aslam S et al. Risk of chemotherapyinduced febrile neutropenia in intermediate-risk regimens: clinical and economic outcomes of granulocyte colony-stimulating factor prophylaxis. J Manag Care Spec Pharm. 2023;29(2):128-38.
8. Dale DC et al. A systematic literature review of the efficacy, effectiveness, and safety of filgrastim. Support Care Cancer. 2018;26(1):7-20.
9. Mithoowani S et al. Neutropenia. CMA J. 2022;194(49):E1689.
Introducing the Concept of Patient Blood Management and Haemovigilance in Government Sector Hospitals of Karachi, Pakistan
Background and Aims: While patient blood management (PBM) and haemovigilance are different, they are both significant following evidence-based clinical transfusion. PBM is defined as the timely application of evidence-based medical and surgical concepts designed to maintain haemoglobin concentration, optimise haemostasis, and minimise blood loss in an effort to improve patient outcomes. The main aim is to manage patients such that transfusion is only used when the benefits outweigh the risks. Haemovigilance is the set of surveillance procedures covering the entire blood transfusion chain, to minimise the risk of any transfusion-related event to the donor as well as the patient.
In this study, the authors audited, critiqued, and measured their own performance regarding the establishment of PBM and haemovigilance at government sector hospitals of the Regional Blood Centre Karachi, Sindh, Pakistan.
Materials and Methods: A retrospective analysis of the 3.5 years since the establishment of Regional Blood Centre (RBC) Karachi and its associated hospital-based blood banks was conducted. Data were taken from Blood Bank Management Information System software and analysed on SPSS version 23 (IBM, Armonk, New York, USA) . The authors calculated the frequency of transfusion reactions and donor-related adverse events, and also calculated cross match to infusion (CT) ratio and transfusion index.
Results: Initially, the cross match to transfusion ratio was 15:1, but with time and management, at the end of the third year, the team achieved a CT ratio of 1.7:1.0 for different hospitals. Transfusion index was also calculated to be 0.2 at the start of 2020 and 0.8 at the end of 2022. Similarly, no transfusion reaction was reported for the first 1.5 years since RBC’s establishment, and in the later 2 years, 59 reactions were reported. No whole blood was issued, a maximum surgical blood ordering schedule was initiated, and a restrictive transfusion strategy was applied.
Conclusion: While there is still progress to be made, the authors were able to reduce wastage and subsequently suggest the idea of a restrictive transfusion strategy, so that the right blood component is given to the right patient at the right time.
Key Points
1. The main aim of this study was to understand the implementation and importance of haemovigiliance and patient blood management.
2. This is a retrospective analysis of the overall impact of Regional Blood Centre on patient blood management and hemovigilance practices over 3.5 years.
3. The study has provided the basis for developing and implementing patient blood management and haemovigilance programmes that are tailored to the specific needs of government hospitals in Karachi. The findings also highlight areas for improvement, trends in practices, and the impact of teamwork on blood transfusion execution.
INTRODUCTION
One-fourth of the global population is anaemic, and as the population is gradually increasing day by day, so does the number of anaemia cases. Anaemia causes an increased risk of morbidity and mortality, particularly in reproductive-aged females and children less than 5 years old. The insufficient and inappropriate treatment of anaemia worldwide has also increased social and economic expenditure, reducing technical productivity as output is lesser as compared to the input on anaemic patients.1 As far as hospitalisations are concerned, the risk and severity of anaemia are increased due to several causes such as iatrogenic blood loss.2
Blood transfusion, when not used appropriately, is a dangerous and unsafe procedure.3 However, the appropriate use of transfusion can be safe, effective, and save millions of lives. Unfortunately, in the author’s region, the usage of blood products is a major concern that has not been extensively acknowledged yet. This includes the liberal use of blood products, mismanagement of blood products inventory, the safety of blood products with regards to transfusiontransmitted infections, alloimmunisation, and the lack of health care professional knowledge regarding transfusion.
PBM is a patient-centred, systematic, evidence-based approach to improve patient
outcomes by managing and preserving a patient's own blood, while promoting patient safety and empowerment.4 It involves the timely, multidisciplinary application of evidence-based medical and surgical concepts aimed at diagnosing and appropriately treating anaemia. It also includes the management of coagulapathic bleeding, minimising surgical and iatrogenic blood loss, and supporting the patient while appropriate treatment is required. To summarise, the concept of PBM is to reject the standard code of belief and approach of blood product transfusion in every case.5
The term haemovigilance was extracted from the words hema (blood) and vigilance (surveillance).6 It is a procedure that covers the whole transfusion chain, from collection of blood and its components to monitoring recipients. The objective of haemovigilance is the continuous development of the transfusion chain through corrective and preventive actions to improve patient as well as donor safety, and minimise wastage. Ideally, hemovigilance systems cover the whole transfusion chain and analysis of adverse events and reactions related to blood donation and transfusion.7
Transfusion decisions for acutely ill patients presenting with multiple co-morbidities may become complex and require a more in-depth understanding to minimise risks of transfusion and to identify, treat, and report reactions for health and safety at both the
individual and population levels.8 In this study, the authors tried to critique/ audit their own performance over the past 3.5 years since the establishment of the RBC. Objective and subjective parameters were utilised to recognise their achievements in the government sector hospitals of Karachi, and to overview their shortcomings in order to rectify them.
MATERIALS AND METHODS
In this retrospective analysis, the authors included all the donors as well as the patients they have treated over the past 3.5 years from January 2020–July 2023. A total of 81,066 donors were accepted at our centres at Sindh Government Qatar Hospital, Sindh Government Lyari General Hospital, and Dr. Ruth K.M. Pfau Civil Hospital Karachi, Pakistan. During this time, 186,767 blood products were issued, including packed red cells, platelets, fresh frozen plasma, cryoprecipitate, and pedi blood bags. Packed red blood cells accounted for 80,888 of products issues. The team calculated the cross match to transfusion (CT) ratio yearly, transfusion index yearly, transfusion reactions, and donor-related adverse events. Subjectively, they reviewed the maintenance of blood product temperature through data loggers, the maximum surgical blood ordering schedule, and the overall impact of their centre over the three government sector hospitals. All the data were analysed on SPSS version 23.
The formula used for calculating CT ratio was: number of units cross matched/ number of units transfused. Ratio of 2.0 or below was considered significant.9 The formula used for calculating transfusion index was: number of units transfused/ number of units cross matched. A value of 0.5 or more was indicative of significant blood usage.9
Formation of hospital transfusion committees was done at each hospital, chaired by the medical superintendent and constituting of a consultant haematologist, medicine and surgery faculty member, gynaecology and paediatrics faculty member, emergency department member, anaesthesia department member, and administrative
department member, with anyone else added as needed. The meeting was called once every quarter or as required by the chairperson.
RESULTS
A total of 81,066 donors were recruited in Royal Blood Centre Karachi and its associated hospital based blood banks from January 2020–July 2023. The majority of them were males. Out of these donors, the team had 154 donors with donor-related adverse events (0.18%). Out of the donors who fainted, for most it was their first time donating and it was due to vasovagal shock. All these donors were well managed by the blood transfusion officer (physician) and none of the donors had any serious adverse event. The cause of adverse events is shown in Figure 1. The team received transfusion reaction reports from 59 patients, which is not significant as per the product issuance. Regretfully, they were unable to obtain any transfusion reaction reports prior to June 2021 due to inadequate training in this area. Most of the reactions reported were allergic, followed by febrile non-hemolytic transfusion reactions, as represented in Figure 2.
They calculated CT ratio per year, as shown in Figure 3. In 2020, the ratio was 15:1, and in 2023 a ratio of 1.7:1.0 was reported. The transfusion index was 0.2 at the beginning of the period, followed by 0.8 in the middle of 2023. The subjective analysis of this study will be conferred in detail in the discussion.
DISCUSSION
Through this article, the authors aimed to critique and audit their performance with respect to PBM and haemovigilance. They were not able to find much data regarding these issues within their region; however, the World Health Organization (WHO) has recognised PBM as an urgent area of intervention due to excessive wastage and irrational use of blood and blood products.10 Ajay Gandhi et al.,11 published an article in India in 2021 regarding the current practices and feasibility in India regarding
PBM. They established an interdisciplinary expert group from different fields of both private and government sector hospitals. They developed a questionnaire, consensus statements, and manuscript development. They used evidence and references from previous literature and worked on perioperative diagnosis and management of anaemia. Cases of high-risk bleeders were differentiated from low-risk bleeders, and a consensus on a multidisciplinary team approach in all such settings was made. These findings are contrary to this study because the authors don’t have any such guidelines or literature; however, on a personal level, the team tried to introduce the concept of a maximum surgical blood ordering schedule. This was challenging due to a lack of knowledge among the clinical practitioners. The maximum surgical blood ordering schedule identifies the number of units typically required by 80–90% of the patients undergoing a specific surgical procedure where transfusion is likely.
This assists the clinician in ordering the appropriate number of units of blood for their patient, and was the author’s first step in lowering the CT ratio from 15:1 to 1.7:1. Waheed et al.12 published an article in 2022 regarding the CT ratio of a private sector hospital. It was found to be 1.0:1.0 ratio using retrospective data, which contradicts this
study’s findings of 2021 due to the authors working in government sector hospital. The huge amount of workflow at these hospitals means patients land usually in very critical condition with no transfusion medicine team in their departments. The authors conducted continuous medical education and awareness sessions, distributed pamphlets regarding the significance of blood and blood products, and posted posters in all the wards regarding perioperative anaemia management, the restrictive transfusion strategy, and managing anaemia without transfusion. Baig et al.13 in 2020 published an article from Lahore regarding CT ratio, which included government sector hospitals, and the CT ratio was found to be 3.3:1.0.
An article from Iran by Alavi-Moghaddam et al.14 reported a CT ratio between 1.4–1.1 in 2014. It was also conducted in a government sector largest hospital of Iran; however, the findings are quite different from this study’s as the country’s transfusion services are quite well developed. The most significant thing Pakistan needs is the addition of transfusion medicine as a physician subspeciality. This will help physicians, and those associated with the ordering of blood products, to understand the significance, dangers, and proper utilisation of blood and its products, and implement the PBM programme more effectively.
Figure 3: Cross match to infusion ratio annually.
Haemovigilance is also a concept that is not extensively discussed by transfusion authorities. The authors aimed to establish it at their local level in order to start a chain of haemovigilance provincially. As their blood bank is associated with three of the largest government hospitals of Karachi, it was quite challenging to maintain this cycle. Samukange et al.15 conducted research across 10 African countries and established, through grading and scoring, that haemovigilance in those countries was suboptimal. The authors of the current study also concluded that the haemovigilance system is underdeveloped; however, over the period of 3.5 years, the team made significant changes to the transfusion setup. Apart from managing the CT ratio and transfusion index, they also introduced a system of blood transport porters, 72 hours rule for cross match, 30 minutes and 4 hours rule of transfusion, maintenance of blood components temperature through transport containers (electronic) and data loggers, as well as doing regular quality control of blood products including the blood cultures. The team also didn’t issue a single whole blood after starting the transfusion service; whereas, the above-mentioned study reported the use of whole blood in many African countries. Demirağ and Hintistan16 published an article in 2020 regarding haemovigilance in nurses. They reported that nurses who had training in haemovigilance had higher frequency of performing better safety procedures than those who didn’t. The authors also recognised this difference since starting the transfusion services compared to now. For the initial 1.5 years, they were unable to get any of the transfusion reaction reported. With continuous training, education, distribution of transfusion reaction forms, and information on how to identify and report these reactions, the team managed to record 59 transfusion reactions over the next 2 years. Though it is still underreported, they took it as achievement, as there was no concept of these reactions before RBC’s establishment. The human resource team was very well trained regarding all these concepts, so 154 donor-related adverse events were reported. The majority of the donors were donating for the first time and had vasovagal shock. Along with a well-trained phlebotomist,
the team also had blood transfusion officer (doctors) trained for managing these donors well. The introduction of doctors in the blood collection department was exclusively done by RBC in these government hospitals. The formation of active hospital transfusion committees and regular meetings helped to increase the confidence of the team through discussing and solving issues together. In Asia, Japan has a very good centralised national haemovigilance centre, and they published reports on national level.17 The national blood policy of India has also taken significant steps and established haemovigilance policy on a national level.18
The Pakistan government’s Safe Blood Transfusion Programme took a step making the national transfusion guidelines (2014–2020) and included haemovigilance to be a part of it.19 Their aim was to: “Develop a comprehensive and nationwide hemovigilance system to monitor, evaluate, and improve clinical transfusion practice.”19 This study was in order to evaluate the author’s own performance, as well as to make the authorities acknowledge the need for patient blood management and haemovigilance. Meybohm et al.20 published an article representing the bundle approach in patient blood management instead of individualised. It would be a feasible approach to manage patients and can be acquired on national/provincial level. The WHO has recommended a draft for developing an efficient haemovigilance system in developing countries and following in could be useful.21
CONCLUSION
Patient blood management and haemovigilance are two important indicators in clinical transfusion practice. The data from operating these two systems effectively can be used as a quality indicator for blood transfusion safety, efficacy, and significant utilisation, as well as aid towards evidence-based transfusion medicine. Though the author’s setup lacks many aspects of these, they achieved a remarkable difference over 3.5 years to make transfusion safer and better in government sector hospitals.
References
1. Kassebaum NJ et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood. 2014;123(5):61524.
2. Hayden SJ et al. Anemia in critical illness: insights into etiology, consequences, and management. Am J Respir Crit Care Med 2012;185(10):1049-57.
3. Jones JM et al. Slowing decline in blood collection and transfusion in the United States - 2017. Transfusion. 2020;60(Suppl 2):S1-9.
4. Shander A et al. A global definition of patient blood management. Anesth Analg. 2022;135(3):476-88.
5. Association for the Advancement of Blood & Biotherapies (AABB). Standards for a patient blood management program (2023) 4th edition, Bethseda: American AABB.
6. De Vries RRP et al. Haemovigilance: an effective tool for improving transfusion practice. Vox Sang 2011;100(1):60-7.
7. International Haemovigilance Network (IHN). Haemovigilance. Available at: https://ihn-org.com/about/ haemovigilance. Last accessed: 1 June 2024
8. Halford B et al. Hospital medicine providers’ transfusion knowledge: a survey study. Transfus Med Rev. 2021;35(2):140-5
9. Association for the Advancement of Blood & Biotherapies (AABB), Cohn C et al., Technical Manual (2023) 21st Edition, Bethseda: AABB.
10. World Health Organization (WHO). The urgent need to implement patient blood management: policy brief. 2021. Available at: https://www.who.int/ publications/i/item/9789240035744. Last accessed: 1 June 2024.
11. Gandhi A et al. Patient blood management in India-review of current practices and feasibility of applying appropriate standard of care guidelines. A position paper by an interdisciplinary expert group. J Anaesthesiol Clin Pharmacol. 2021;37(1):3-13.
12. Samra Waheed et al. Blood ordering and transfusion practices: an insight toward better utility of blood products. Cureus. 2022;14(2):e22075.
13. Baig MI et al. Wastage of blood units at tertiary care hospitals of Lahore. Cureus. 2020;12(5):e8040.
14. Alavi-Moghaddam M et al. Blood transfusion practice before and after implementation of type and screen protocol in emergency department of a university affiliated hospital in Iran. Emerg Med Int. 2014;2014:316463.
15. Samukange WT et al. Implementation and performance of haemovigilance systems in 10 sub-saharan African countries is sub-optimal. BMC Health Serv Res. 2021;21(1):1258.
16. Demirağ H, Hintistan S. Knowing and use situations of hemovigilance system in the scope of blood transfusion safety of nurses: rural example. Bezmialem Science. 2020;8(4):388-97.
17. Japanese Red Cross Society. Blood Services 2019. 2019. Available at: https://www.jrc.or.jp/english/pdf/ BloodServices2019_WEB.pdf. Last accessed: 1 June 2024.
18. National AIDS Control Organisation (NACO). National Blood Policy. 2003. Available at: https://www.naco.gov. in/sites/default/files/National%20 Blood%20Policy_0.pdf. Last accessed: 1 June 2024.
19. Waheed U et al. Haemovigilance as a quality indicator in transfusion medicine: Pakistan’s perspective. Annals of PIMS. 2020;16(1):46-51.
20. Meybohm P et al. Patient blood management bundles to facilitate implementation. Transfus Med Rev. 2017;31(1):62-71.
21. World Health Organization (WHO). A guide to establishing a national haemovigilance system. 2016. Available at: https://www.who.int/publications/i/ item/9789241549844. Last accessed: 1 June 2024.
22. World Health Organization (WHO). The urgent need to implement patient blood management: policy brief. 2021. Available at: https://www.who.int/ publications/i/item/9789240035744. Last accessed: 1 June 2024.
Diffuse Large B Cell Lymphoma of Spleen: An Important Differential of a Nodular Splenomegaly: A Case Report
Authors: VPS Punia,1 *Aditya Chakravorty,1 Naman Bansal,1 AK Mandal,2 Shaavi Mittal,1 Akash Bharti1
1. Department of General Medicine, School of Medical Sciences & Research, Sharda University, Greater Noida, India
2. Department of Pathology, School of Medical Sciences & Research, Sharda University, Greater Noida, India
*Correspondence to adityajune.17@gmail.com
Disclosure: The authors declare no conflicts of interest. Informed consent was taken from the patient on whom the case report had been prepared.
Acknowledgements An idea regarding the preparation of this case report was generated initially. Multiple authors were involved in the active management of this case and also provided valuable input while conceiving the article. All the ideas and images were put together, and the final case report was prepared. All the images of various resected specimens, including spleen, jejunum, colon, and omentum, were incorporated in the case report. All authors read and approved the final case report.
Received: 19.04.24
Accepted: 27.06.24
Keywords: Diffuse large B cell lymphoma (DLBCL), nodular spleen, splenic malignancy.
Diffuse large B cell lymphoma (DLBCL) is the most common histological subtype of nonHodgkin’s lymphoma. However, splenic DLBCL is a relatively uncommon form of nonHodgkin’s lymphoma.1 In this case report, the authors present a 38-year-old male who was admitted to the hospital with a complaint of abdominal distension, left-sided abdominal pain, loss of weight, and loss of appetite for 2 months. The basic workup of this patient was suggestive of microcytic anaemia with a raised total lymphocyte count, platelet count, and erythrocyte sedimentation rate, while a nodular spleen with altered splenic echotexture was revealed on ultrasonography. Splenic malignancy was suspected and contrast-enhanced CT of the abdomen was planned, which gave an impression of an extra splenic mass lesion causing impingement on the spleen with continuity to bowel loops and thickening of the fascia, raising the possibility of a gastrointestinal stromal tumour; however, the presence of large conglomerated necrosed lymph nodes in the abdominal cavity pointed the diagnosis towards a splenic lymphoma. The splenectomy specimen had multiple nodular deposits and immunohistochemistry studies finally provided a clear-cut diagnosis of DLBCL–mucosaassociated lymphoid tissue lymphoma.
Key Points
1. Splenic diffuse large B cell lymphoma is an extremely rare malignancy and has been identified in less than 1% of patient with non-Hodgkin’s lymphoma.
2. It is important for the physician to consider splenic lymphoma as a differential diagnosis whenever a patient presents with nodular splenomegaly.
3. Diffuse large B cell lymphoma of the spleen is potentially curable with R-CHOP chemotherapy and splenectomy if detected in early stages of the disease.
INTRODUCTION
B cell non-Hodgkin lymphomas (NHL) are characteristically divided into aggressive and indolent tumours, out of which diffuse large B cell lymphoma (DLBCL) remains the most common and aggressively behaving NHL. DLBCL involves lymphoid tissue commonly, but non-lymphoid tissues are affected in up to 40% of patients. Due to functional heterogeneity and omnipresent distribution of lymphatic tissue, DLBCL can occur in bone marrow, central nervous system, gastrointestinal tract, liver, skin, and thyroid gland.1 However, splenic DLBCL is extremely rare and has been identified in less than 1% of patients with NHL.
Nodular splenomegaly can have many differentials including haemangioma, lymphangioma, gastrointestinal stromal tumour, abscess, infarct, metastatic deposits, and hamartoma. Ultrasonography and contrast enhanced CT (CECT) abdomen has an important role in the establishment of a specific diagnosis. DLBCL is potentially curable and responsive to combination chemotherapy with 6 cycles of R-CHOP (Rituximab, Cyclophosphamide, Hydroxydaunorubicin, Vincristine, Prednisone) every 3 weeks.
Here, the authors present a case of splenic DLBCL–mucosa-associated lymphoid tissue (MALT) lymphoma in a 38-year-old male who visited the hospital with a massive nodular spleen.
CASE REPORT
A 38-year-old male came to the emergency department with complaints of loss of appetite and loss of weight for 2 months, as well as abdominal distension, abdomen pain, and vomiting for 15 days. The patient had 3–4 episodes of vomiting per day for 15 days along with passage of dark stools. However, there was no complaint of fever, cough, breathlessness, epigastric pain, bloating, or night sweats.
On examination, the patient had pallor and dry skin. On abdominal palpation, a nodular and firm spleen approximately 11 cm from the left costal margin was detected. He had no palpable cervical, axillary, or inguinal lymph nodes. The complete blood count of this patient was suggestive of microcytic hypochromic anaemia (Hb: 6.8 mg/dL) and mean corpuscular volume, mean corpuscular haemoglobin, and mean corpuscular haemoglobin concentration values were well below the normal range. He had a raised total lymphocyte count (12.6 cmm), platelet count (493 cmm), and erythrocyte sedimentation rate (ESR; 100 mm/hr); whereas, liver functions, kidney functions, and serum electrolytes were within normal limits. Urine routine/microscopy didn’t reveal any abnormality, while Tridot, Rk39, and rapid malarial antigen tests were also negative for this patient. However, his lactate dehydrogenase (LDH) (569 U/L) and prothrombin time–international normalised ratio (18.1/1.44) were significantly elevated.
The patient also underwent bone marrow biopsy, which revealed marked erythroid hyperplasia with dimorphic maturation and eosinophilia. Ultrasonography of the whole
abdomen was done and was indicative of hepatosplenomegaly, cholelithiasis, altered splenic echotexture, and moderate ascites. The patient was finally subjected to CECT whole abdomen for a more conclusive diagnosis and imaging findings were suggestive of an extra splenic mass lesion in the left hypochondriac region causing impingement on the spleen on the medial aspect with continuity to bowel loops and thickening of fascias (possibility of gastrointestinal stromal tumour). Large conglomerated lymph nodes in the abdominal cavity with necrosis were seen. Other findings on CT revealed mild hepatomegaly, mild ascites, and cholelithiasis. The patient was immediately transferred from medicine to the surgery department as exploratory laparotomy with splenectomy was planned by the head surgeon.
Intraoperative findings post laparotomy included a grossly enlarged spleen with adhesion to the omentum and large bowel. The spleen was studded with multiple nodular deposits along with some deposits on the omentum. Due to extension of the mass to the jejunum and left colon, resection of the jejunum was done along with left hemicolectomy, omentectomy, and splenectomy. Apart from the abovementioned procedures, feeding jejunostomy and Hartmann’s procedure were also done.
All four samples of spleen, omentum, jejunum, and colon were sent for histopathology (Figure 1,2,3). Immunohistochemistry of these tumour cells was positive for CD20 and CD45 and negative for CD15, CD30, CK, and Vimentin.
A final impression of MALT lymphomaDLBCL type was made through this histopathological study.
Figure 1: Gross specimen of spleen with parenchymal damage.
2: Microscopy of spleen: tumour cells contain large vesicular to hyperchromatic nuclei.
Figure
Figure 3: Omentum with multiple nodules consisting of tumour cells.
DISCUSSION
Of all lymphomas, 90% belong to B cell origin. DLBCL is the most common subtype of non-Hodgkin’s lymphoma. However, splenic DLBCL is a very rare type of NHL and has been detected in only <1% of patients with DLBCL. Multiple extra-nodal involvement of DLBCL indicates adverse clinical outcomes and is influenced by several oncogenic mutations as well as tumour microenvironment alterations.2
Primary splenic lymphoma (PSL) was first described by Dasgupta et al.3 as lymphoma involving the spleen and splenic hilar lymph nodes with no evidence of relapse within the first 6 months post-splenectomy. Kehoe et al 4 defined PSL as NHL arising in the spleen or restricted to the spleen and its local lymph nodes. Kraemer et al.5 approved PSL in patients with splenomegaly, cytopenia of at least two haematological cell lineages, and absence of peripheral lymphadenopathy.
DLBCL is frequently associated with infections (hepatitis C virus,6,7 hepatitis B,8 HIV, and Epstein–Barr virus), autoimmune diseases (Sjogren’s syndrome, systemic lupus erythematosus, rheumatoid arthritis) and immunosuppressed conditions. However, this patient had a negative hepatitis B, hepatitis C, and HIV status, and had no evidence of consumption of immunosuppressant drugs or any autoimmune disease. The patient with splenic lymphoma presents with symptoms of low-grade fever, abdominal distension, abdominal pain, loss of weight, loss of appetite, and night sweats.9,10 Common laboratory findings observed in patients with PSL include raised erythrocyte sedimentation rate, LDH, serum β-2 microglobulin, and cytopenia of at least two haematopoietic cell lineages. This patient had a raised erythrocyte sedimentation rate and LDH along with microcytic hypochromic anaemia, elevated TLC, and platelet count.
CECT abdomen is the preferred investigation for accurate staging of indolent lymphomas; whereas, fludeoxyglucose F18 PET is essential for diagnosing aggressive lymphomas such as DLBCL.11 Sometimes, imaging findings can be easily confused with a splenic
abscess owing to the feature of liquefactive necrosis.12 The common differential diagnosis of nodular splenomegaly includes haemangioma, lymphangioma, hamartoma, abscess, metastatic deposits, or infarcts.13,14 Splenic lymphomas are usually of B cell origin, and other subtypes of NHL should be considered apart from DLBCL.
Follicular lymphoma is the second leading subtype of NHL, which is characterised by small lymphocytes with cleaved nuclei and a variable population of large cells with vesicular chromatin and prominent nucleoli. Immunochemistry for follicular lymphoma is positive for Bcl2 and CD10. Follicular lymphoma may transform into diffuse large B cell lymphoma in the spleen.15 The other common NHL include marginal zone lymphomas, mantle cell lymphoma, lymphoplasmacytic lymphoma, and Burkitt’s lymphoma (the rarest NHL and most common in childhood). All NHLs are staged according to Ann Arbor staging, which focuses on the number of tumour sites (nodal and extra-nodal) and the presence or absence of B symptoms. Splenic lymphomas are categorised into three stages, Stage 1: tumours confined within the spleen only; Stage 2: involvement of the spleen along with splenic hilar lymph nodes; and Stage 3: involvement of extra splenic lymph nodes or liver.16 The abovementioned case fits into Category 3 of splenic lymphoma. DLBCL has a higher incidence among Caucasians and men aged 60 years and above.17 The median age for diagnosis of DLBCL is 64 years. Patients with splenic lymphoma do not have any evidence of lymphadenopathy and present with a normal peripheral blood smear. The tumour consists of predominantly large neoplastic cells with ample cytoplasm, vesicular chromatin, and prominent nucleoli. These cells express B cell antigens CD19, CD20 and CD79A. CD10 and BCL6 expression is suggestive of germinal centre origin whereas MUM 1 expression corresponds to non-germinal centre. Combination chemotherapy with R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone) is the standard and a potential curative therapy for DLBCL.18,19 Early stages of this disease respond to either full dose chemotherapy of R-CHOP every 3 weeks for 6 cycles
or abbreviated chemotherapy for 3–4 cycles followed by involving field radiotherapy. For advanced disease DLBCL, a full course of chemotherapy is the only available option. For chemorefractory malignancies, chimeric antigen receptor T cells are a potential curative therapy. Epigenetic mechanisms such as DNA hypermethylation or histone deacetylation normally silence gene expression. Mutations of histonemodifying genes, especially EZH2, is commonly observed in germinal centre-derived lymphomas such as follicular lymphoma and DLBCL. EZH2 is essential to the formation of germinal centre in the secondary lymphoid tissue (lymph nodes and spleen). Recently, an orally active inhibitor of EZH2, tazemetostat, has received regulatory approval for patients carrying the EZH2 mutation.20 Splenectomy with multidrug
References
1. Taibi S et al. Diffuse large B-cell lymphoma revealed by splenic abscess: a case report. Cureus. 2021;13(10):e18771.
2. Shen R et al. Influence of oncogenic mutations and tumor microenvironment alterations on extranodal invasion in diffuse large B-cell lymphoma. Clin Transl Med. 2020;10(7):e221.
3. Dasgupta T et al. Primary malignant neoplasms of the spleen. Surg Gynecol Obstet. 1965;120:947-60.
4. Kehoe J, Straus DJ. Primary lymphoma of the spleen-clinical features and outcome after splenectomy. Cancer. 1988;62:1433-8.
5. Kattepur AK et al. Primary splenic lymphoma: a case report. Indian J Surg Oncol. 2013;4(3):287-90.
6. Shimono J et al. Clinicopathological features of HCV-positive splenic diffuse large B cell lymphoma. Ann Hematol. 2019;98(5):1197-207.
7. Yu SC, Lin CW. Early-stage splenic diffuse large B-cell lymphoma is highly associated with hepatitis C virus infection. Kaohsiung J Med Sci. 2013;29(3):150-6.
8. Deng L et al. Hepatitis B virusassociated diffuse large B-cell lymphoma: unique clinical features,
chemotherapy offers a good survival rate among patients with splenic DLBCLMALT lymphoma.21
CONCLUSION
Splenic DLBCL-MALT lymphoma is a pretty rare malignancy. It becomes very important for the physician to consider splenic lymphoma as a differential diagnosis. CECT abdomen can help distinguish between a splenic abscess and a malignant lesion because they look similar. Splenic DLBCL is potentially curable if R-CHOP chemotherapy is initiated in early stages of the disease. Early diagnosis with CECT abdomen, splenic biopsy, and immunochemistry can be of great help in initiating improving the survival rate of splenic diffuse large B cell lymphomas.
poor outcome, and hepatitis B surface antigen-driven origin. Oncotarget. 2015;6(28):25061-73.
9. Ingle SB, Ingle CRH. Primary splenic lymphoma: current diagnostic trends. World J Clin Cases. 2016;4(12):385-9.
10. Pirzada S et al. Primary splenic diffuse large B-cell lymphoma: an atypical presentation. Cureus. 2023;15(6):e40793.
11. Iioka F et al. A unique subtype of diffuse large B-cell lymphoma primarily involving the bone marrow, spleen, and liver, defined by fluorodeoxyglucosepositron emission tomography combined with computed tomography. Leuk Lymphoma. 2016;57(11):2593602.
12. Wadsworth PA et al. Primary splenic diffuse large B-cell lymphoma presenting as a splenic abscess. EJHaem. 2023;4(1):226-31.
13. Jain A et al. Primary splenic lymphoma presenting as splenic abscess: a rare entity. Annals Pathol Lab Med. 2016;3(4):C207-11.
14. Singh M et al. Primary Splenic Hodgkin’s Lymphoma: a case report. Res Rev J Med Health Sci. 2014;3(Suppl 2):38-41.
15. Makis W et al. Follicular lymphoma transforming into diffuse large B-cell lymphoma in spleen: simultaneous
appearance of both on 18F-FDG PET/CT and histology. Clin Imaging. 2017;43:88-92.
16. Baiomy TA et al. Primary splenic nonHodgkin lymphoma of diffuse large B cell type; a case report and review of the literature. Human Pathology: Case Reports. 2020;22:200459.
17. Lokesh KN et al. Diffuse large B-cell lymphoma in elderly: experience from a tertiary care oncology center in South India. South Asian J Cancer. 2017;6(2):72-4.
18. Olszewski AJ et al. Improved survival with rituximab-based chemoimmunotherapy in older patients with extranodal diffuse large B-cell lymphoma. Leuk Res. 2014;38(8):86673.
19. Miyazaki K. Treatment of diffuse large B-Cell lymphoma. J Clin Exp Hematop. 2016;56(2):79-88.
20. Chung C. A promising future for precision epigenetic therapy for follicular and diffuse large B-cell lymphoma? Blood Lymphat Cancer. 2022;12:99-106.
21. Yonghao O et al. Comparison of survival outcomes of different treatment modalities for patients with primary splenic diffuse large B cell lymphoma. Ann Hematol. 2023;102(7):1857-65.
The Impact of ‘Pre-conception’ on Conception: An Inadvertent Form of Infertility
Author: *Jeff Lipton1
1. Princess Margaret Cancer Centre, Toronto, Canada *Correspondence to jeff.lipton@uhn.ca
Disclosure: The authors have declared no conflicts of interest. Consent to describe this case was obtained from the patient.
Infertility post stem cell allograft is a common event, with the frequency uncertain because the wish for pregnancy is not commonly discussed. However, it must be remembered that pregnancy requires a functioning female and male reproductive system, and what would at first seem to be the reason for infertility is not always the case.
Key Points
1. When investigating infertility, especially after a stem cell allograft, both partners need to be assessed.
2. Infertility in females is not necessarily a consequence of stem cell transplantation.
3. Thorough gynaecological follow-up is essential in females post-stem cell allograft, not just for fertility issues, but for gynaecological problems.
INTRODUCTION
Stem cell allografting can be associated with many chronic issues that affect the quality of life of survivors.1,2 Younger individuals, both males and females, may have potential problems with issues of fertility.3-9 Some of these problems may actually precede the allograft and can be related to underlying disease, pre-allograft therapy, and other unrelated comorbidities. In an ideal world, the thought of fertility in survivors should
be considered and discussed by the transplant team, but sometimes events are already in progress because of ongoing therapy or because delaying the transplant may be risky to the patient. From a patient perspective, a focus on surviving the problem for which the transplant is being performed may very well overshadow any other considerations. For others, pregnancy was not wanted, they are beyond the age of pregnancy, or they or their partner are on permanent birth control. Fertility
preservation in females who undergo intensive chemotherapy, and in particular stem cell allografting, is a major concern for many younger individuals. The allograft can affect fertility at many points: disruption of the endocrine system; destruction of the ovaries; disruption of gynaecological structures, especially by chronic graftversus-host disease (GvHD); and ongoing therapeutics with potentially mutagenic or teratogenic medications, to name the major players. ‘Advanced’ age at time of transplant (>25 years), transplant around the time of menarche, early menopausal symptoms from previous therapy, and the use of total body, abdominal/pelvic, or craniospinal irradiation are contributors. Needless to say, these issues are also relevant to males, a discussion beyond the scope of this article.10
CASE
This 26-year-old female presented with a nosebleed and petechiae while 16 weeks pregnant. She was found to be pancytopenic and a diagnosis of severe aplastic anaemia was made. No other risk factors were noted other than the pregnancy. Previous history was notable for an ectopic pregnancy 2 years earlier, treated with termination, with unilateral oophorectomy and a dilation and curettage. In both cases, the partner was different from the one post-transplant.
Her sister was found to be 6/6 human leukocyte antigens-identical. Options were discussed, and she elected to proceed with a bone marrow transplant after termination of her pregnancy. Conditioning was cyclophosphamide 50 mg/kg daily for 4 days and total body irradiation 300 cGy, as well as GvHD prophylaxis with cyclosporine for 12 months and methotrexate for 4 days. The course was complicated by Grade 2 acute GvHD of skin and gut, requiring steroids that subsequently resulted in avascular necrosis of both hips, requiring replacement. No chronic GvHD was observed. Birth control as hormone replacement was used for the first year and then stopped. Spotty menstrual periods resumed at about 2 years and then normalised.
She had the same partner from before her pregnancy and ongoing. She had a strong desire for pregnancy post-transplant and they continued with unprotected intercourse that was unsuccessful in this regard. Referral to fertility clinics was done but it was concluded that she was infertile due to the transplant on a background of oophorectomy for previous ectopic pregnancy. Endocrine work-up, which included thyroid stimulating hormone, follicle stimulating hormone, luteinizing hormone, and estradiol levels, was not helpful. Anti-Müllerian hormone testing was not available at that point in time. Ultrasounds at different cycle time points were not done. Fertility stimulation was not attempted by her gynaecologist.
After 15 years with the same partner, they parted ways and, shortly after, she became sexually active with a new partner. At the age of 39 years, she presented with fatigue and lower abdominal discomfort. Work-up, elevated beta-human chorionic gonadotrophin, and ultrasound confirmed that she was pregnant. Unfortunately, she was subsequently found to have a blighted ovum and the pregnancy was terminated at 10 weeks.
Subsequent attempts at pregnancy and repeat fertility assessment were unsuccessful. Six years later, she became menopausal.
DISCUSSION
Although pre-transplant fertility preservation, if desired, is the ideal, it is not always possible because of the severity of the pre-existing disease and need for prompt therapy, successful egg or embryo procurement, and the availability of this option and cost issues, let alone the patient or physician even thinking about this at a time when the transplant is needed. Previous treatment may already have rendered the female infertile. Ovarian suppression during the peri-transplant period may not be an option, although this may also help in reducing vaginal bleeding in the thrombocytopenic period.
Preventative options for all side effects post-transplant should be part of the complete process.11-13 If time and safety permit, a complete evaluation of the patient, males as well as females, should be done, and discussions of options for fertility preservation should be discussed; if possible, oocyte or embryo preservation should be attempted if requested.11-15 Options for males are also essential.16 These include semen storage, and even testicular biopsy and sperm isolation. If possible without risking transplant outcome, alternate conditioning protocols that reduce exposure to radiation can be considered.17
Often neglected, good gynaecological care in the peri- and post-transplant period is essential.18 Gynaecological chronic GvHD can result in significant structural issues as well.
With normal couples, appropriate infertility work-up includes assessment of both the male and female.19-20 This is no less true in the post-allograft situation, even if a transplant-related cause is most likely. In this case, ‘pre-conceived’ notions of infertility by the patient, transplant team,
References
1. Calvo C et al. Haematopoietic stem cell transplantation in adolescents and young adults with acute lymphoblastic leukaemia: special considerations and challenges. Front Pediatr. 2022;9:796426.
2. Granat LM et al. Late complications after allogeneic hematopoietic cell transplant: What primary care physicians can do. Cleve Clin J Med. 2023;90(8):499-508.
3. Alexandroni H et al. Fertility preservation from the point of view of hematopoietic cell transplant specialists-a worldwide-web-based survey analysis. Bone Marrow Transplant. 2019;54(11):1747-55.
4. Bourlon C et al. Outcomes and challenges of reproductive health in hematopoietic stem cell transplantation survivors. Biol Blood Marrow Transplant. 2020;26(11):212731.
5. Forgeard N et al. Sexuality- and fertility-related issues in women after allogeneic hematopoietic stem cell transplantation. Transplant Cell Ther. 2021;27(5):432.e1-6.
and even consulting gynaecologists, due to a transplant, became dominant, and at no time was the original partner ever assessed for possible aetiology of the infertility. Infertility is not guaranteed with a stem cell allograft, especially if the female is younger, has a reduced intensity regimen without radiation, and minimal chronic GvHD. The same is true for sterility in the male. ‘Oops’ pregnancies can occur, and if not wanted, ongoing contraception, even in the case of minimally or absent menstrual function or ‘negative’ semen analyses, should be considered. Going back to a complete basic work-up for the infertile couple is essential because sometimes the cause is not what it at first seems.
Finally, restoration of fertility may be possible.21,22 Hormonal correction, surgical correction of gynaecological structures or in vitro fertilisation, use of stored eggs, sperm or embryos, use of donor sperm or eggs, surrogacy, and even organ (uterus) or mesenchymal stem cell transplant are, or may become, options. The message here is that appropriate referral to experts in this area should be available.
6. File B et al. The effect of hematopoietic stem cell transplantation on fertility and strategies for improvement. Bone Marrow Transplant. 2022;57(11):164956.
7. Appiah LC et al. Reproductive late effects after hematopoietic stem cell transplant in pediatric, adolescent, and young adult cancer survivors. Pediatr Blood Cancer. 2023;70(Suppl 5):e30551.
8. Lee YJ et al. Risk factors of menopause after allogeneic hematopoietic cell transplantation in premenopausal adult women. Eur J Haematol. 2023;111(3):449-57.
9. Gazzo I et al. Pregnancy complications after allogeneic hematopoietic stem cells transplantation: focus on the placenta. Placenta. 2023;132:27-31.
10. Pallotti F et al. The impact of male factors and their correct and early diagnosis in the infertile couple's pathway: 2021 perspectives. J Endocrinol Invest. 2022;45(10):180722.
11. Murphy J et al. A practical guide to gynecologic and reproductive health in women undergoing hematopoietic
12. Loren AW, Senapati S. Fertility preservation in patients with hematologic malignancies and recipients of hematopoietic cell transplants. Blood. 2019;134(9):74660.
13. Rotz SJ et al. International recommendations for screening and preventative practices for long-term survivors of transplantation and cellular therapy: a 2023 update. Transplant Cell Ther. 2024;30(4):34985.
14. Mulder RL et al. Fertility preservation for female patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2021;22(2):e4556.
15. Brodigan K et al. Safety of surgical fertility preservation procedures in children prior to hematopoietic stem cell transplant. Transplant Cell Ther. 2021;27(8):696.e1-4.
16. Tran KTD et al. Male fertility preservation and restoration strategies for patients undergoing gonadotoxic therapies. Biol Reprod. 2022;107(2):382-405.
17. Bender JD et al. Reduced-intensity conditioning mitigates risk for primary ovarian insufficiency but does not decrease risk for infertility in pediatric and young adult survivors of hematopoietic stem cell transplantation. Transplant Cell Ther. 2023;29(2):130.e1-8.
18. Milroy CL, Jones KP. Gynecologic care in hematopoietic stem cell transplant patients: a review. Obstet Gynecol Surv. 2010;65(10):668-79.
19. Garolla A et al. Practical clinical and diagnostic pathway for the investigation of the infertile couple. Front Endocrinol (Lausanne). 2021;11:591837.
20. Lavafian A et al. Investigation of the female infertility risk associated with anti-cancer therapy. Clin Transl Oncol. 2023;25(7):1893-905.
21. Rizano A et al. Exploring the future potential of mesenchymal stem/stromal cells and their derivatives to support assisted reproductive technology for female infertility applications. Hum Cell. 2023;36(5):1604-19.
22. Tsui EL et al. Restoring ovarian fertility and hormone function: recent advancements, ongoing efforts and future applications. J Endocr Soc. 2024;8(6):bvae073.