

Home»Post-MarketSurveillance(PMS)inMedicalDevices:USA&EU
Post-MarketSurveillance(PMS)inMedicalDevices:USA&EU
May72025

Asaregulatoryleader,yourresponsibilitydoesn’tendwithmarketapproval,itbeginsthereThis sentimentechoesacrossboardroomsandregulatorystrategymeetingsaspost-marketsurveillance (PMS)becomesacriticalpillaroflifecyclemanagementinmedicaldevicesInthetimeswherereal-world performance,patientsafety,andglobalcomplianceareunderthespotlight,PMSisastrategic advantage
Accordingtoglobalregulatoryexperts,theabilitytopredict,monitor,andactonpost-marketdataisfast becomingabenchmarkforexcellenceinmedicaldeviceoversightWhiletheUSFDAandtheEuropean Union(EU)bothprioritizePMStheirframeworksreflecttwodistinctphilosophies:theFDAsreactiveyet data-drivenmodelversustheEU’sproactivelifecycle-integratedsystemunderMDR2017/745
Forcompanieswithglobalaspirations,understandingthesenuancesin2025isessentialtosafeguarding brandreputationachievecomplianceandmaintainmarketaccess
UnderstandingPost-Market Surveillance�PMS�
Post-MarketSurveillancereferstotheongoingandsystematiccollection,analysis,andinterpretationof datarelatedtoamedicaldeviceafteritisplacedonthemarketIthelpsinidentifyingrisksevaluating benefit–riskratios,andsupportingregulatoryactionssuchassafetycommunications,recalls,orlabeling updatesPMSisalsoafundamentalinputintothemanufacturersCorrectiveandPreventiveAction (CAPA)andQualityManagementSystem(QMS)processes
USA�PMSRequirementsUnder FDA Regulations
IntheUnitedStates,PMSactivitiesareprimarilygovernedby21CFRPart803,whichcoversMedicalDevice Reporting(MDR)
KeyStakeholdersandResponsibilities
Manufacturers&Importers:Mustreportdevice-relateddeathsseriousinjuries,andcertain malfunctions
UserFacilities(eg,hospitals):RequiredtoreportdeathstotheFDAandseriousinjuriesto manufacturers(ortotheFDAifthemanufacturerisunknown)
Distributors:Notrequiredtoreportroutinelybutmustmaintainrecordsofcomplaintsandprovide themuponrequest
TimelinesforMDRSubmission
30calendardaysforstandardadverseeventreports
5workdaysforeventsrequiringremedialactiontopreventanunreasonableriskofsubstantial harm
10workdaysforuserfacilitiestoreporttotheFDAormanufacturersdependingonthenatureofthe event
ElectronicMedicalDeviceReporting(eMDR)
RecentPosts
FDALaunchesGenAIToolElsafor ScientificReviews
UnderstandingtheDrug RegistrationProcessinMexico
NewINVIMAPlanAimsto StreamlineRegulatoryProcesses inColombia
ImpactofInternationalRegulatory HarmonizationInitiativeson PharmaceuticalDevelopment
PharmacovigilanceAudits: EnsuringDrugSafety
PressRelease
DDReg-DeliveringRegulatory Excellencesince2009 ContactUs
Fieldsmarkedwithan*are required
Name*
Email*
Message*
IhavereadthePrivacy Policy*

VITALIC®-RIMSSoftware OnePlatform|Multiple Solutions BookForDemo
Since2015allMDRsubmissionsmustbemadeelectronicallyviatheFDAsElectronicSubmissions
Gateway(ESG)Manufacturerscanuse
eSubmittersoftware(forMedWatch3500AXMLfiles),or
AS2GatewayusingHL7ICSR-compliantXMLformats
MAUDEDatabase
SubmittedMDRsaremadepubliclyavailablethroughtheManufacturerandUserFacilityDevice Experience(MAUDE)databaseenhancingtransparencyandenablingstakeholderstomonitorsafety profiles
RecentDevelopments(2023–2025)
WhilenomajoramendmentstoPart803haveoccurred,theFDAhasreleased TechnicalupdatestotheeMDRvalidationprocess, Guidanceencouragingintegrationofreal-worldevidence(RWE)and EnhancementsalignedwithIMDRFharmonizationgoals
ManufacturersareadvisedtotracktheCDRHeMDRenhancementsscheduleforthelatestupdates
EU�PMSUnder Regulation�EU�2017/745 �MDR�
IntheEUPMSisgovernedbyChapterVIIandAnnexIIIofMedicalDeviceRegulation(MDR)2017/745The approachisproactiveandlifecycle-based,tightlyintegratedintoamanufacturer’sQMS
PMSPlanandSystem(Articles83–84)
ManufacturersmustestablishandmaintainaPMSsystem,documentedthroughaPMSPlanthatoutlines: Datasources(eg,complaints,literature,userfeedback)
Analyticalmethodologies,
Riskassessmentcriteria,and
Correctiveactionprotocols
PMSReportingDocumentation
PMSReport:RequiredforClassIdevicesandkeptinternally.
PSUR(PeriodicSafetyUpdateReport):
MandatoryforClassIIa,IIb,andIIIdevices
SubmittedannuallyforClassIIIandClassIIbimplantables;everytwoyearsforClassIIaandother IIbdevices
ReviewedbyNotifiedBodiesforClassIIbandIII
VigilanceReporting(Article87)
Seriousincidentsandfieldsafetycorrectiveactions(FSCAs)mustbereported
Timelines
2daysforseriouspublichealththreats
10daysfordeathorseriousdeterioration
15daysforallotherseriousincidents
TrendReporting(Article88)
Uniquely,EUMDRmandatesreportingofstatisticallysignificantincreasesinnon-seriousincidentsor sideeffectsthatmayimpactthedevice’sbenefit–riskprofileManufacturersmust DefinetrendthresholdsinthePMSPlan
Regularlyanalyzedataand
Reportfindingsifthresholdsareexceeded
EUDAMEDPlatform
TheEuropeanDatabaseonMedicalDevices(EUDAMED)isacentralizedITsystemfor: Deviceregistration
Clinicalinvestigations, Vigilancereporting
PMSactivities
Asof2025severalmodules(vigilancemarketsurveillanceandclinicalinvestigations)areoperational butfullmandatoryuseisstillpendingaEuropeanCommissionnotice
NotifiedBodiesRole
ForClassIIbandIIIdevices,NotifiedBodiesreviewPSURsanduploadevaluationsintoEUDAMEDThisadds alayerofexternaloversightabsentintheUSsystem
USA vs EUPMS�A ComparativeSnapshot
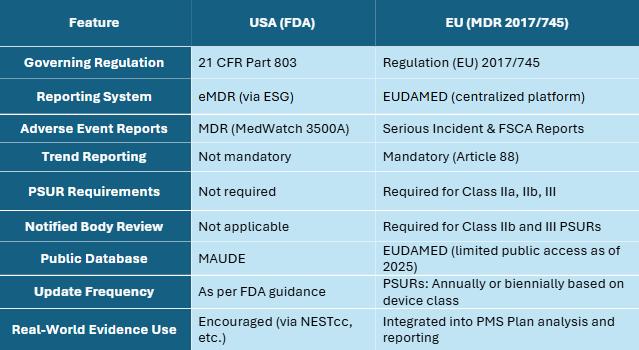
Conclusion
Post-marketsurveillanceismorethanaregulatoryrequirement itisacornerstoneofensuringongoing patientsafety,effectiveriskmanagementandproductperformance
IntheUS theFDA’sapproachemphasizesreactiveadverseeventreportingthroughMDRand publictransparencyviaMAUDE
IntheEU,MDRhasintroducedacomprehensiveandproactivesystem,featuringperiodicreporting trendmonitoringandoversightbyNotifiedBodies
Manufacturersoperatinggloballymustinvestin:
RobustQMSandCAPAsystems, Integratedregulatoryintelligenceand Digitaltoolstohandlediversesubmissionformatsanddatabases(eg ESGEUDAMED)
As2025unfoldsandregulatorscontinuerefiningtheirPMSframeworks,stayinginformedagileand alignedwithreal-worldevidence-drivensafetystrategieswillbecriticaltosuccess
About DDReg
DDRegisattheforefrontofsupportingmedicaldevicemanufacturersinnavigatingcomplexregulatory landscapeslikePMSWithoveradecadeofexperienceinregulatorystrategy,DDRegpartnerswith companiestoensuretheynotonlycomplywithevolvingregulationsbutalsoleveragepost-marketdata toenhancesafety,streamlinereporting,andprotectpatientwell-beingOurexpertiseinbothUSFDAand EUregulationsallowsustoguideyouthroughtheintricaciesofPMS,offeringtailoredsolutionsthatdrive globalcomplianceandoperationalsuccess
Readmorefromushere:UnderstandingSoftwareasaMedicalDevice(SaMD)


