
KEY DIFFERENCES BETWEEN THE
ANDIVDRINTHEEU WWW.MAVENPROFSERV.COM
THE
MDR

Compa r ison of the key diff er ences between the MDR a nd
IVDR in the EU
In Vitro Diagnostic Regulation (IVDR) and its Key Points
Medical Device Regulation (MDR) and its Key Points
T he Key Diff er ences between In Vitr o Dia gnostic Regulation (IVDR) and Medical Device Regulation (MDR)
Conclution and References
In the fast-paced world of medical devices, staying updated on regulatory changes is crucial for manufacturers, healthcare professionals, and patients alike. In the European Union (EU), two significant regulations have been introduced to enhance the safety and effectiveness of medical devices – the In Vitro Diagnostic Regulation (IVDR) and the Medical Device Regulation (MDR).
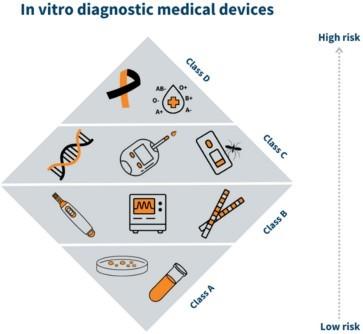
The In Vitro Diagnostic Regulation (IVDR), Regulation (EU) 2017/746, was adopted by the European Parliament and the Council in 2017 and came into effect on 25 May 2017. It replaced the previous In Vitro Diagnostic Directive (IVDD) 98/79/EC and introduced a more stringent framework for the approval and marketing of in vitro diagnostic medical devices (IVDs). This regulation specifically pertains to in vitro diagnostic medical devices distributed within the European Union.
1.Scope and Definition
2.Risk Classification
3.Conformity Assessment
4.Unique Device Identification (UDI)
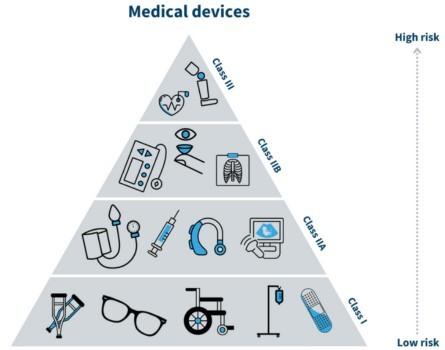
The Medical Device Regulation (MDR), Regulation (EU) 2017/745, was also adopted in 2017 and came into force on May 26, 2021. It replaces the Medical Device Directive (MDD) 93/42/EEC and aims to strengthen the regulatory framework for medical devices, ensuring a higher level of safety and efficacy.
1.Scope and Definition
2.Risk Classification
3.Conformity Assessment
4.Unique Device Identification (UDI)
Medical Devices for human use manufactured or sold into the European Union In vitro diagnostic medical devices for human use manufactured or sold into the European Union.
ARTICLES
123 articles, applied to all human medical devices
RISK CLASSIFICATION
113 articles, focuses solely on in vitro diagnostic devices
Clinical Evaluation report based on evaluation of clinical evidence or a clinical investigation Performance evaluation and performance studies to justify intended patient outcome
Class I (Low risk)
Class Is (Sterile)
Class Im (Measurable)
Class Ir (reusable)
Class A (Low risk)
Class B (Medium)
RISK CLASSIFICATION Class IIa (Low to Medium risk)
IIb (Medium to High risk)
III (High risk)
C (Medium to High)
POST-MARKET DATA
Post-Market Clinical Follow-Up
Post-Market Performance (Surveillance and Vigilance)
CLINICAL EVIDENCE
Focused mainly on safety and clinical performance in accordance to the intended purpose of the device
Focused mainly on performance of device and improved patient outcomes
POST MARKET
SURVEILLANCE REPORT (PMSR)
Required for Class I manufacturers – updated when necessary)
Required for Class A and Class B manufacturers –updated when necessary)
PERIODIC SAFETY UPDATE REPORT (PSUR)
Required for Class IIa, IIb and III manufacturers –updated at least annually
Required for Class C and D manufacturers –updated at least annually
23 applicable clauses as per Regulation (EU) 2017/745
20 applicable clauses as per Regulation (EU) 2017/746
Navigating the regulatory landscape of medical devices in the European Union demands a thorough understanding of both the In Vitro Diagnostic Regulation (IVDR) and the Medical Device Regulation (MDR). While these regulations share commonalities, such as risk-based classification and conformity assessment, differences in scope, implementation dates, and the level of notified body involvement necessitate careful consideration from manufacturers.
REGULATION (EU) 2017/745 OF THE EUROPEAN
PARLIAMENTAND OF THE COUNCIL of 5 April 2017 on medical devices, Regulation (EC) No amending Directive 2001/83/ EC, 178/2002and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
REGULATION (EU) 2017/746 OF THE EUROPEAN
PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on
in vitro diagnostic medical devices and repealing
https://mavenprofserv.com/comparison-and-highlighting-ofthe-key-differences-between-the-mdr-and-ivdr-in-the-eu/