All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IBI will be published in Winter 2025.
ISSN No.International Biopharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2025 Senglobal ltd.
Volume 8 Issue 3 – Autumn 2025
www.international-biopharma.com
04 Foreword TALKING POINT
06 An Expertise and Dedication to Advancing Cell Therapy Manufacturing: An Interview with Camille Bachelet of CELLforCURE by SEQENS
Founded in 2010, CELLforCURE was created to pioneer manufacturing in the emerging field of cell & gene therapy. Camille Bachelet of CELLforCURE discusses how the company’s recent acquisition by SEQENS well-positions them to accelerate their development as a CDMO dedicated to ATMPs.
10 The Future of Single-Cell Applications: Discussing the Importance of Bringing Single Cell Function into Focus with the Envisia Platform from Lightcast
Drug development ultimately comes down to how cells behave, whether an antibody blocks a pathway or enhances it, whether a T cell kills its target and whether a therapy persists long enough to matter in a patient. Jonathan Didier of Lightcast explains how singlecell functional analysis makes the difference.
12 The Future of Pharmaceutical Neutral Borosilicate Glass Tubes and Glass Containers
Founded in 1992, Changzhou Four Star Glass Company is changing the face of pharmaceutical glass manufacturing to a more environmentally friendly method.
TODAY’S BIOTECHNOLOGY
14 Empowering Flow Chemistry: Transforming Manufacturing Through Integrated Flow Technologies at Asymchem
Pharmaceutical manufacturing is entering a new era of transformation. Globally, regulatory expectations are rising and the demand for safer, cleaner and more efficient manufacturing practices is becoming increasingly urgent. Linglong Yi of Asymchem evaluates how flow chemistry is emerging as a key solution.
16 The Whole Picture: Total-Body PET and the Future of Biopharma
Advanced PET technology can deliver full-body scans in minutes, opening exciting new avenues for research and discovery. Dr. Juliana Maynard and Dr. Ian Wilson of Medicines Discovery Catapult explore how the UK’s National PET Imaging Platform can expand its potential impact through global data sharing and continued innovation.
20 Enabling Oral Delivery of TPDs: The CDMO Path to Oncology’s Next Frontier
No major scientific breakthrough comes without its challenges. This is certainly true of targeted protein degraders (TPDs), which have rapidly become one of the most dynamic areas within oral solid dose drug development. Dr. Rebecca Coutts of PCI explores how collaboration can create a bridge between laboratory innovation and real-world access to treatments for cancer and other chronic diseases.
REGULATORY AND COMPLIANCE
24 Breaking the Mould: MHRA’s Draft Paves the Way for Individualised mRNA Therapies
Scientists have long understood that cancers are as individual as the patients in which they develop and proliferate. We still classify cancers in different groups depending on tissue origin, morphology
and other factors, but genomic studies have revealed that on a cellular and molecular level, cancer cells can be very individual within a group. Steven Watt of A&M STABTEST evaluates whether individualised mRNA cancer vaccines could be the answer to the treatment of previously untreatable or unresponsive cancers.
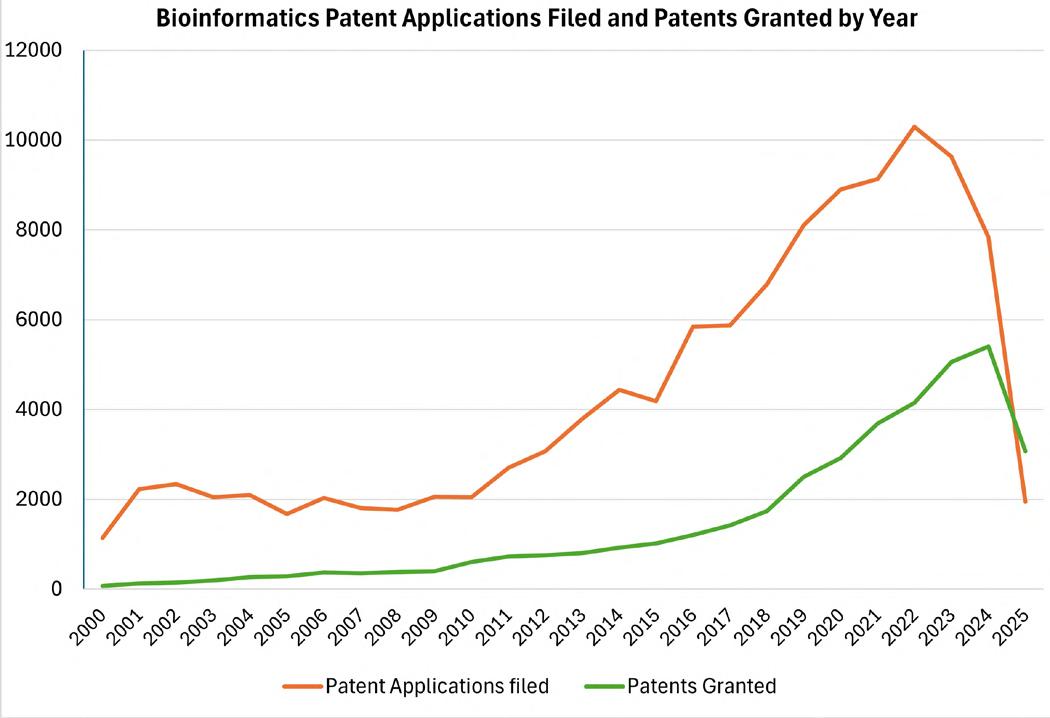
26 Building IP Value in Bioinformatics
Bioinformatics sits at the convergence of computational methods, life sciences, diagnostics and data analytics. Advances in computational power and efficiencies and the increased capabilities of AI methods mean are driving forward at pace. Dr. Janine Swarbick, Dr. Sofie McPherson, Ms Roxna Kapadia and Dr. Claire Green of HGF explore how combining robust patent portfolios with well-guarded trade secrets can allow companies to protect and extract value from their bioinformatics innovations.
RESEARCH / INNOVATION / DEVELOPMENT
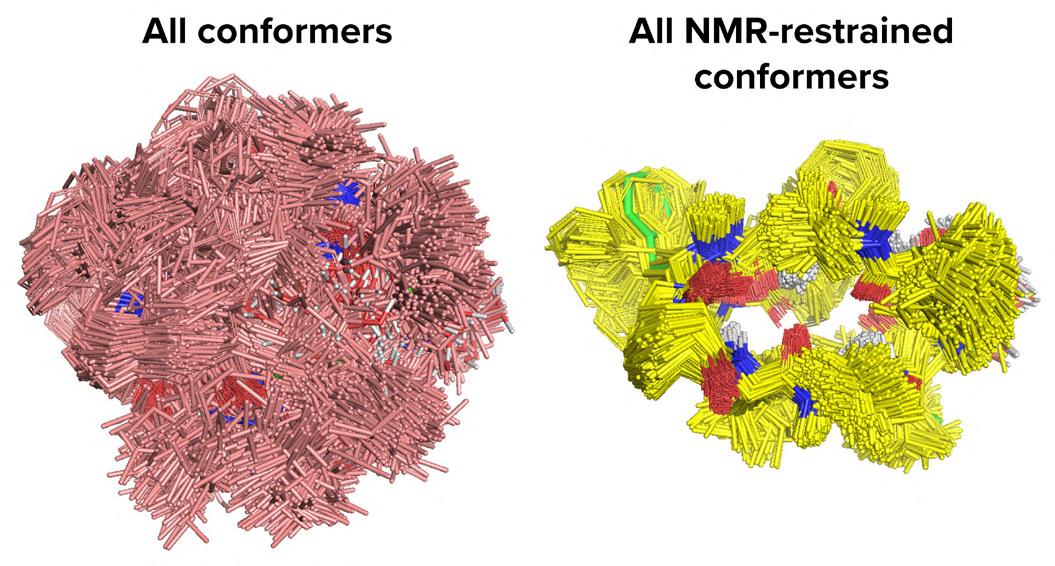
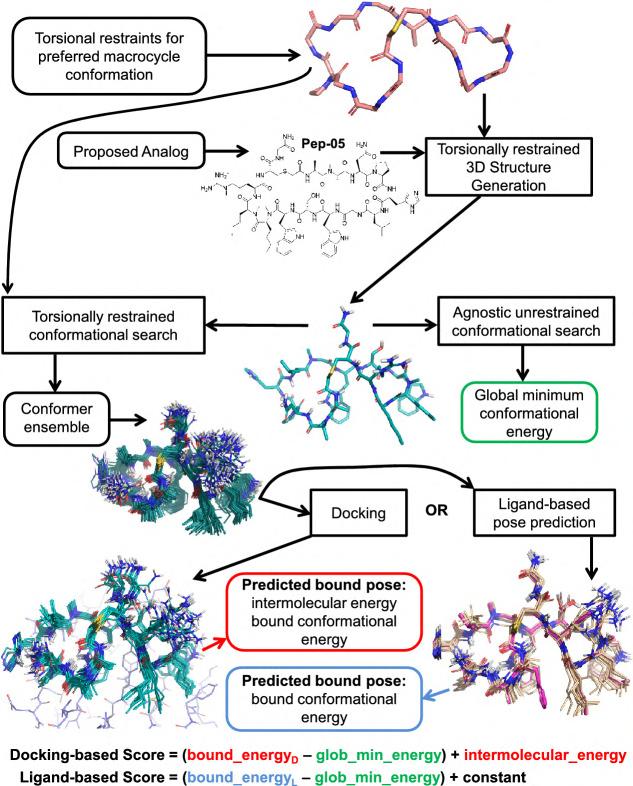
30 Beyond Small Molecules: How advances in 3D Modelling Are Opening New Frontiers in Macrocyclic Drug Discovery
While traditional small molecules excel at targeting buried active sites, they often struggle with flatter protein surface interactions.
Matthew Segall and Himani Tandon of Optibrium explore how advanced 3D modelling capabilities enable more accurate prediction of macrocyclic conformations, providing practical considerations for effectively working with these promising therapeutic agents.
34 Comparing Organoids and Patient-derived Xenograft Models for the Development of Antibody-drug Conjugates
Antibody-drug conjugates (ADCs) are one of the most rapidly expanding forms of oncology treatment and therapeutic successes have led to an unprecedented expansion in the number of ADCs in development. Benjamin Wilkin of Crown Bioscience argues that by taking a strategic approach, organoid and PDX models can be utilised in a complementary way to maximise their strengths and mitigate their limitations.
TECHNOLOGY
44 Lipid Nanoparticles (LNPs) for Nucleic Acid Delivery
Lipid nanoparticles (LNPs) are one of the most prominent tools for nucleic acid delivery. Their versatility allows the encapsulation of different cargos to address multiple disease models. Xavier Warnet of Tebubio explains how, with the advent of targeted delivery, the field of therapeutic applications is broadening.
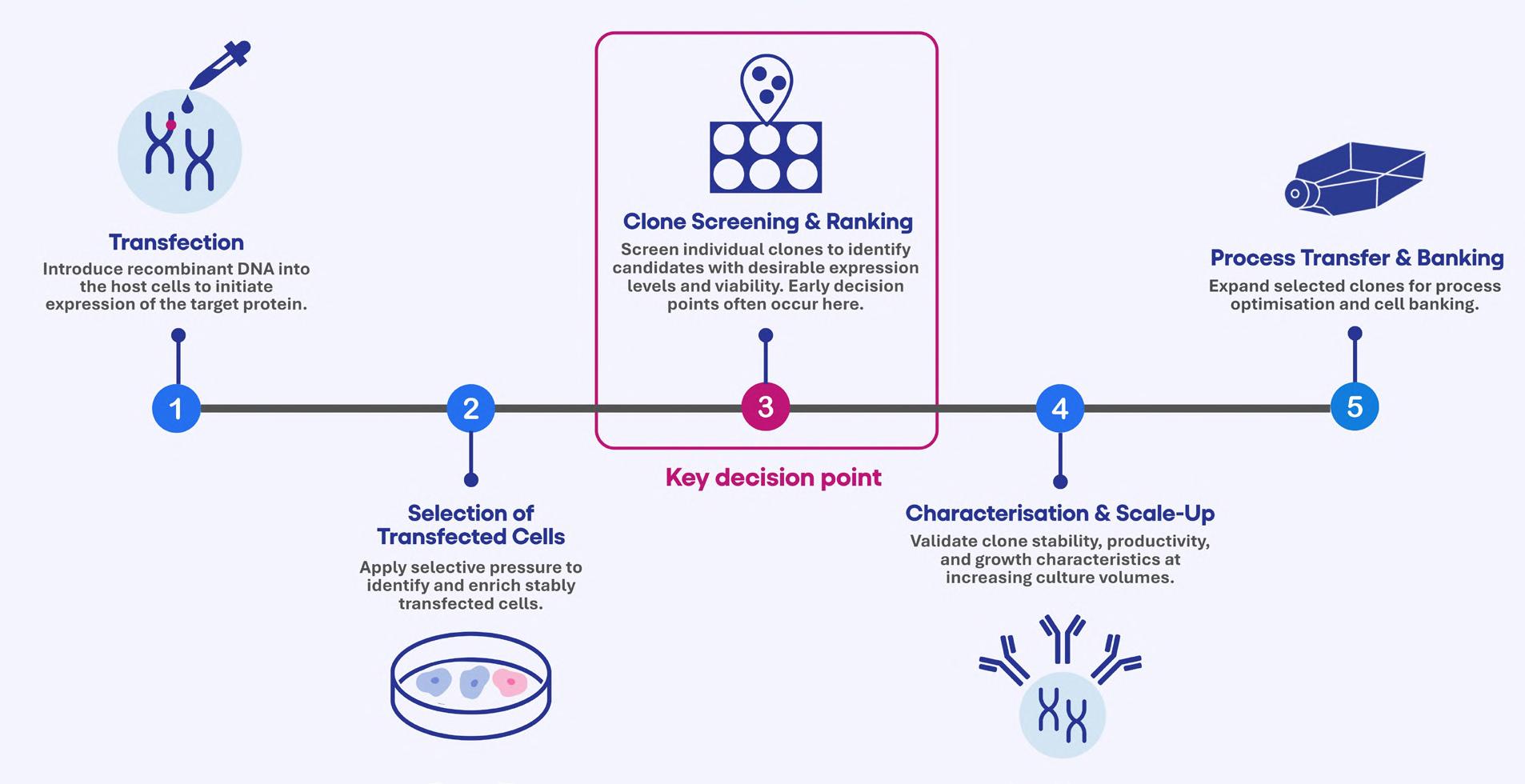
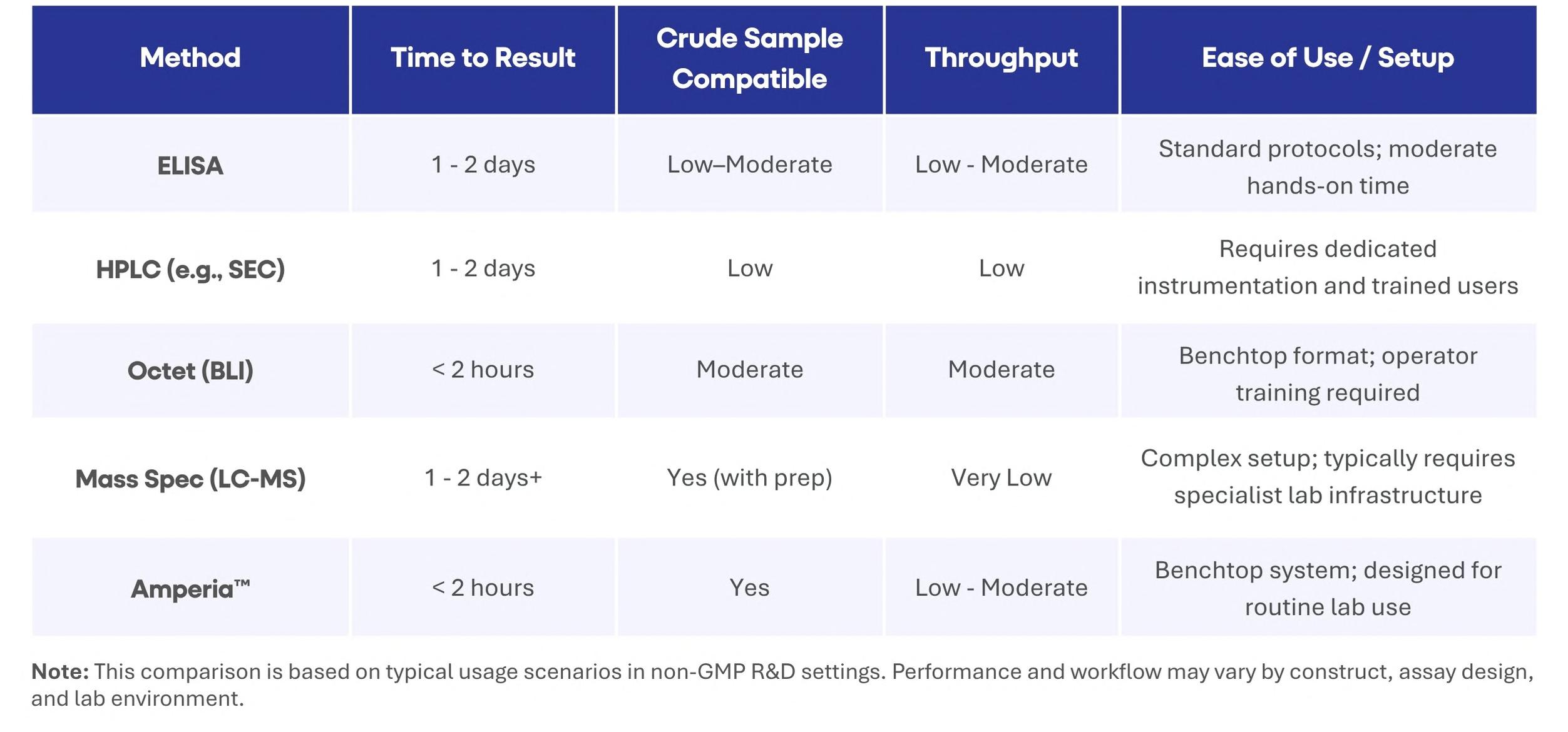
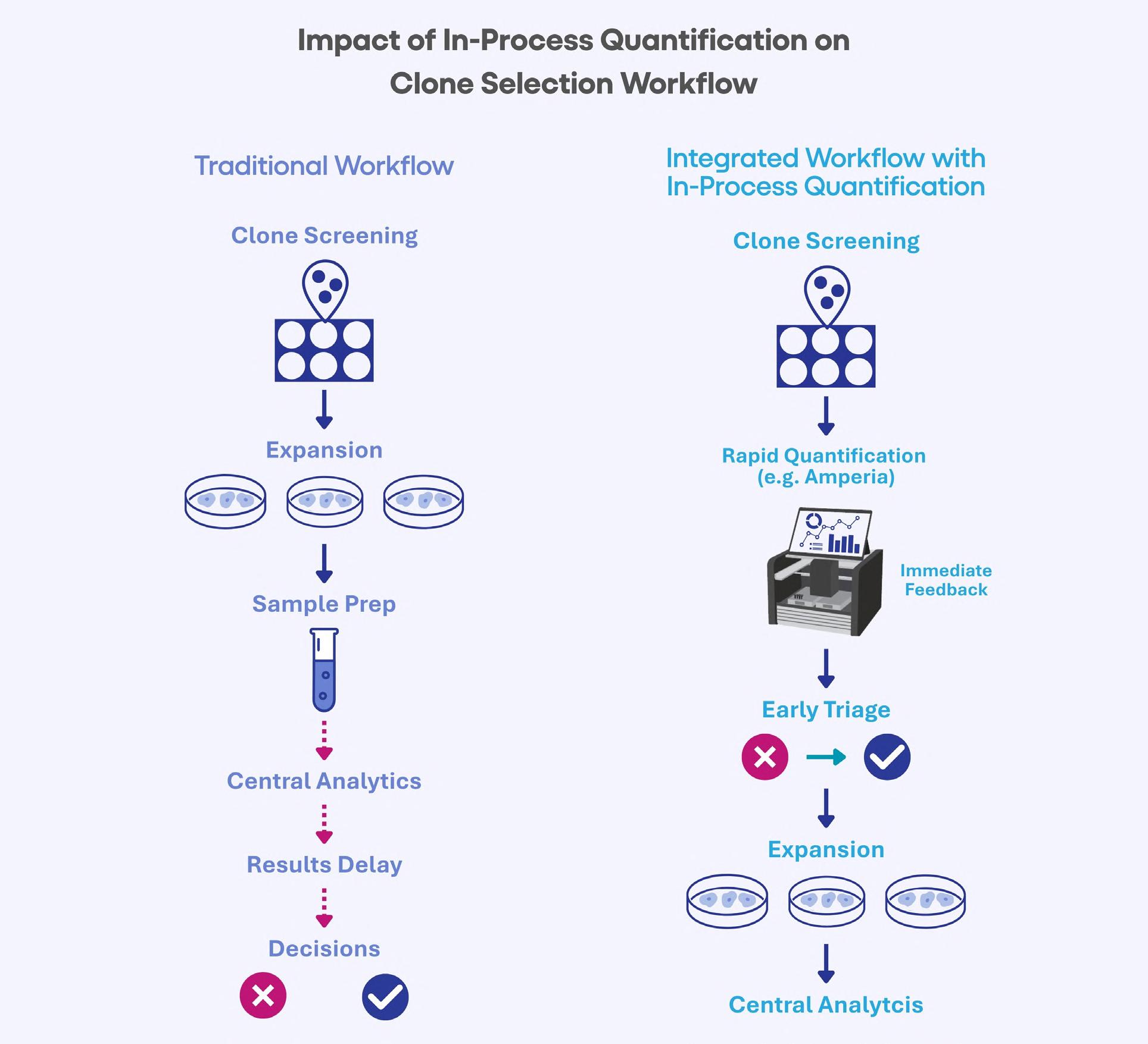
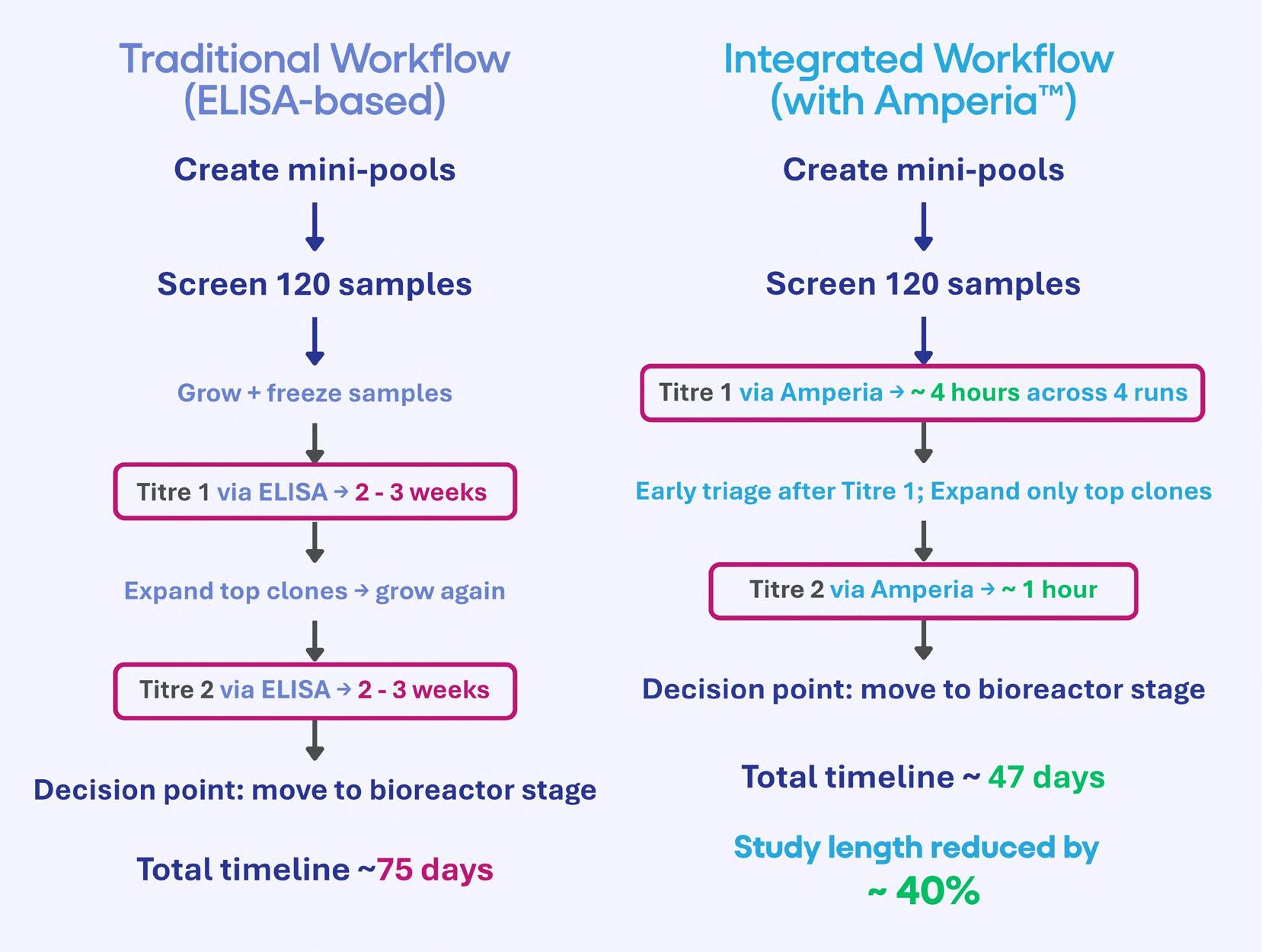
48 Accelerating Cell Line Selection with Integrated Analytical Strategies
Cell line development is a critical determinant of success in biologics manufacturing. Yet, early-stage clone selection remains one of the least analytically supported stages of development. Professor Alan Dickson and Dr. Paolo Romele of Abselion argue that the ability to make confident and data-driven decisions earlier in development will be key, not just to decreasing timelines, but also to improving the quality and manufacturability of molecules entering the pipeline.
MANUFACTURING AND PROCESSING
54 Leveraging Strategic Partnerships to Enhance Flexibility in Facility Design and Bioprocesses
Flexibility is an operational imperative in today’s fast-evolving biopharmaceutical industry. Soyeon Ahn, Daeryun Park, Hyoseok
Kim and Joomyung Lee of Samsung Biologics argue that flexibility in biomanufacturing is the product of ongoing discipline, engaging deeply with clients to gather diverse operational experiences, capturing those experiences as structured data and applying that data systematically to facility design and process execution.
LOGISTICS & SUPPLYCHAIN
66 Turning the Temperature Up on Cold Chain Logistics
As biopharma therapies become more sensitive, costly and complex, traditional compliance frameworks are no longer enough. Real-time visibility and IoT innovation are redefining what’s possible, and necessary, in temperature-controlled logistics. Alex Guillen of Tive explores the future of cold chain logistics, arguing that it’s about staying in control, ensuring life-saving medicines arrive safely, consistently and with total confidence.
CELL AND GENE THERAPY SUBSECTION
68 Developing Endotoxin Limits, Risk Assessment and In-process Testing for CGT Products
Cell & Gene Therapy (CGT) products face a unique challenge in the required pyrogen testing for injectable wares. Timothy Francis of FUJIFILM argues that the overall benefit of bringing endotoxin testing in-house, as opposed to using third-party testing, is that the QC control program can have the tools to take a proactive approach over a reactionary approach to the endotoxin in the product samples.
APPLICATION NOTE
38 Benefits of Mass Spectrometry from Process Development to GMP Release of Biomolecules – A Comprehensive CDMO Perspective from Richter BioLogics
In biopharmaceutical production, a thorough understanding of analytical processes and target molecules is essential for ensuring patient safety, as well as maintaining consistent and reliable product quality. Dr. Maja Erdmann, Daniel Goetz, Dr. Daniela Stummer and Dr. Ingo Goldbeck of Richter BioLogics assess how mass spectrometry presents a high-resolution, sensitive and extremely flexible and diverse technique, gaining increasing influence in the biopharmaceutical industry, particularly within the CDMO market.
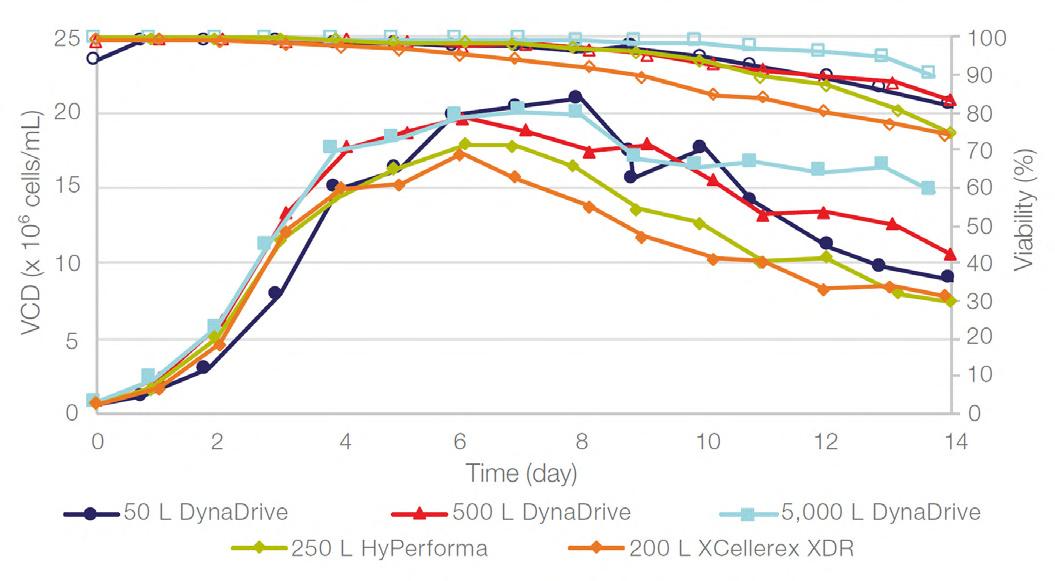
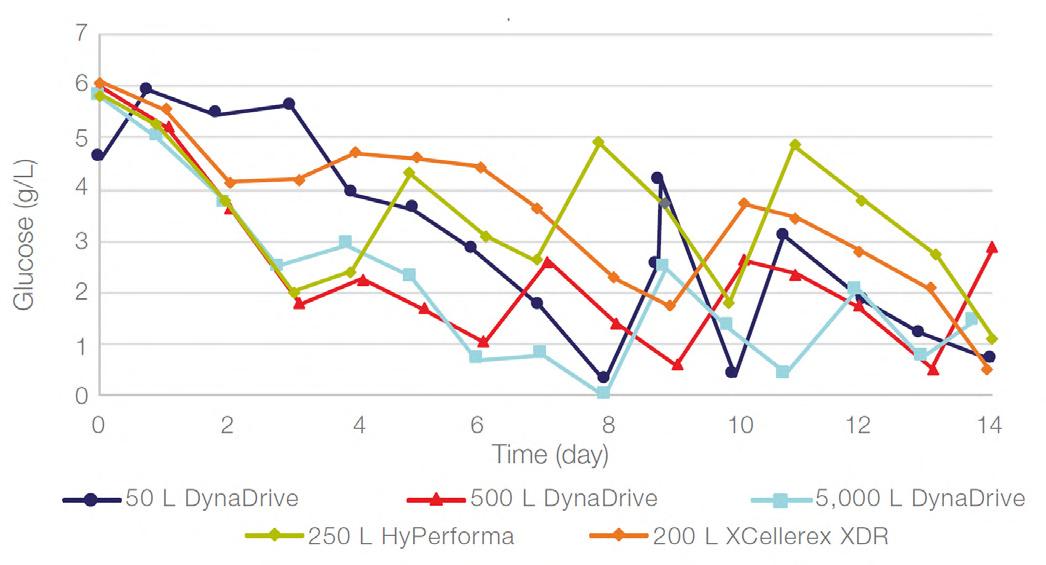
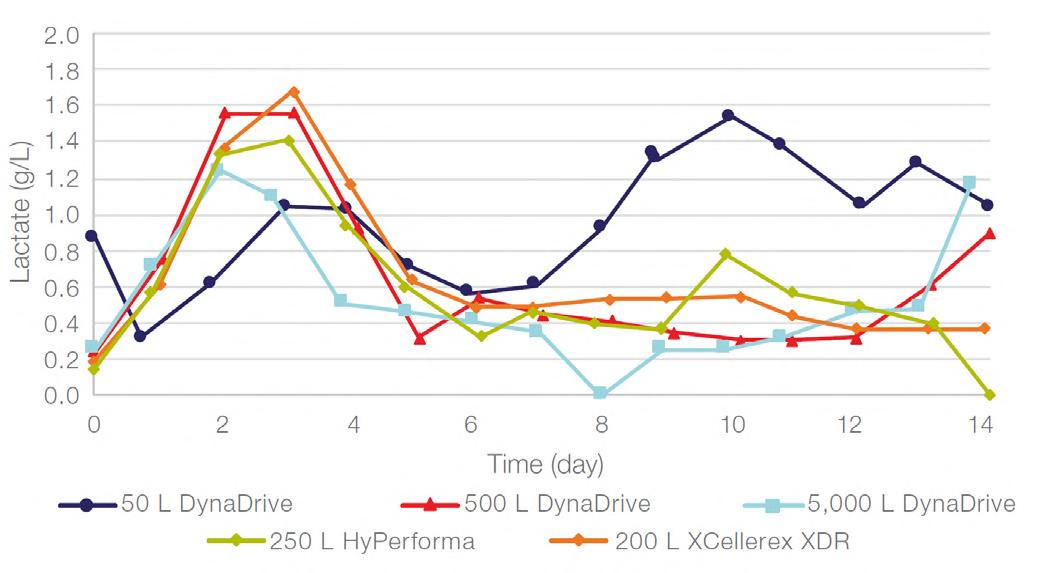
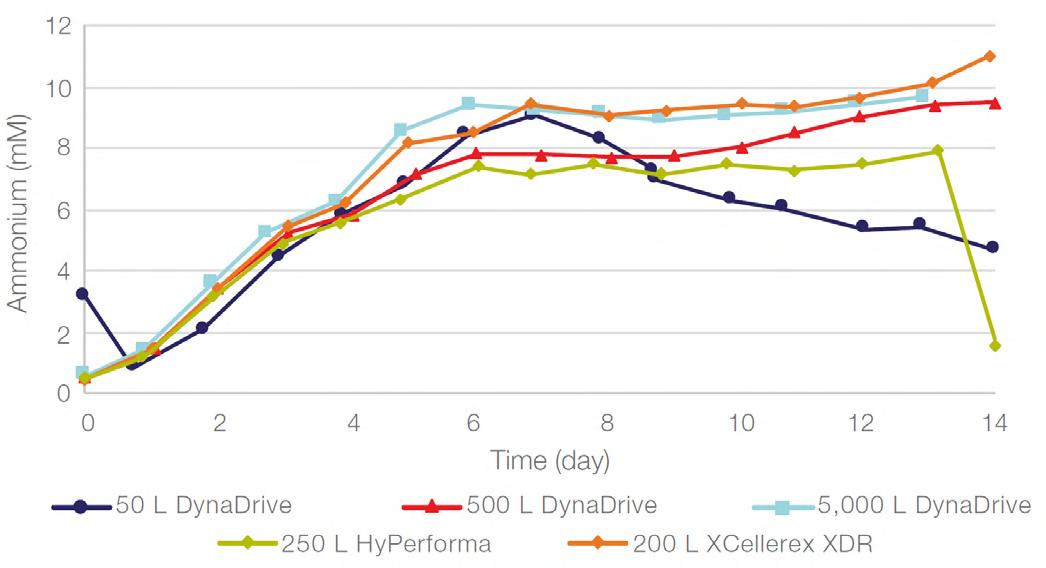
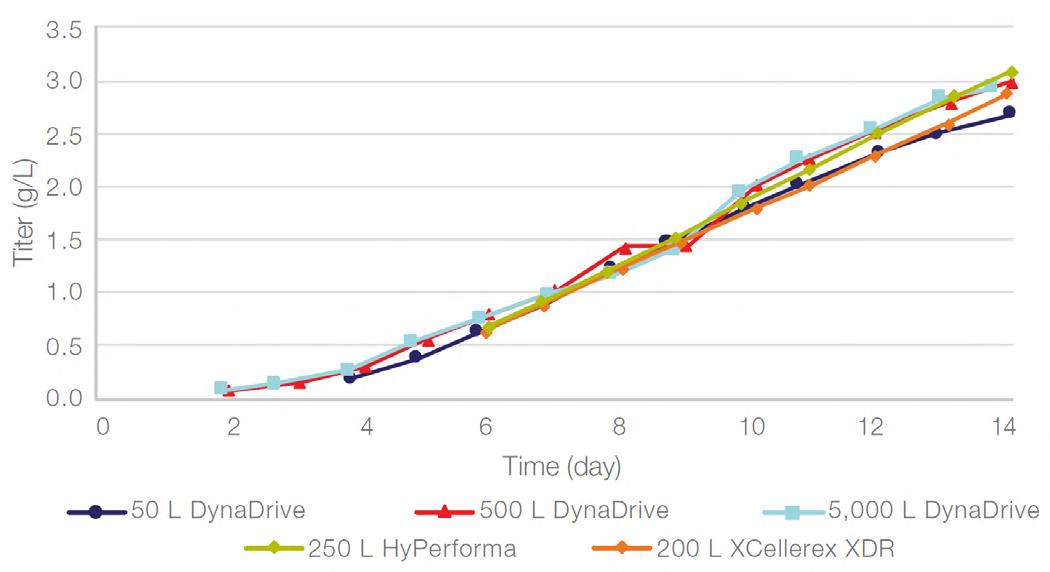
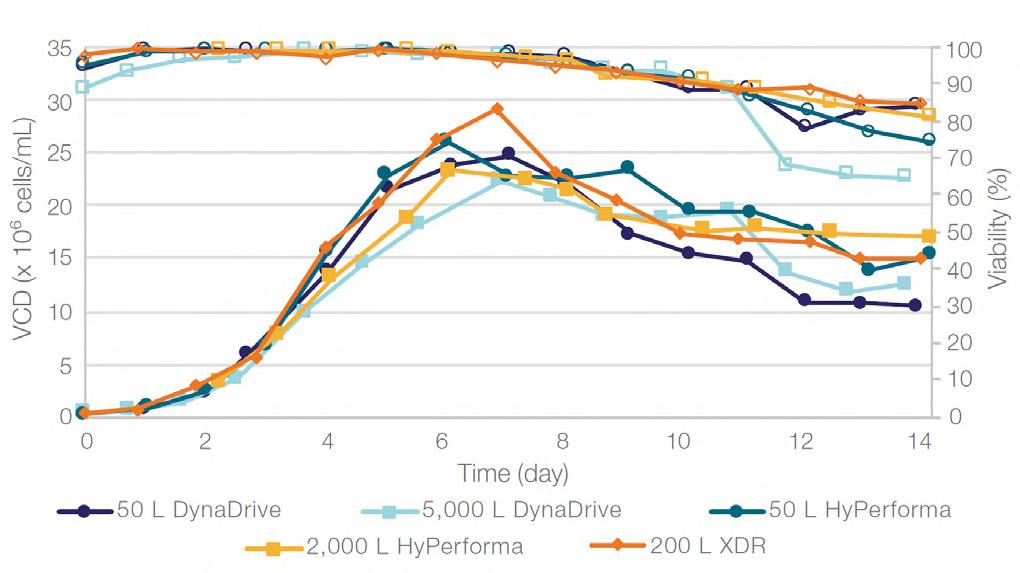
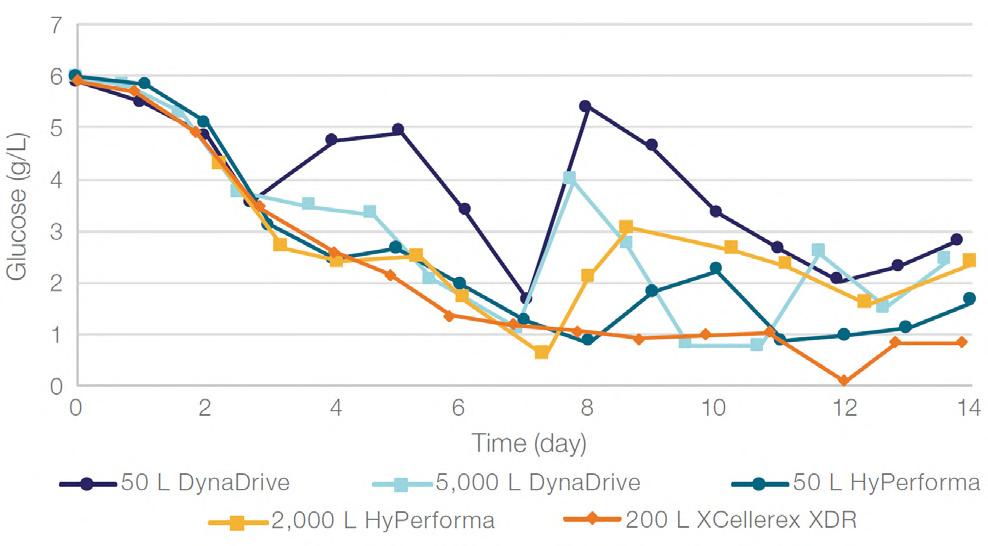
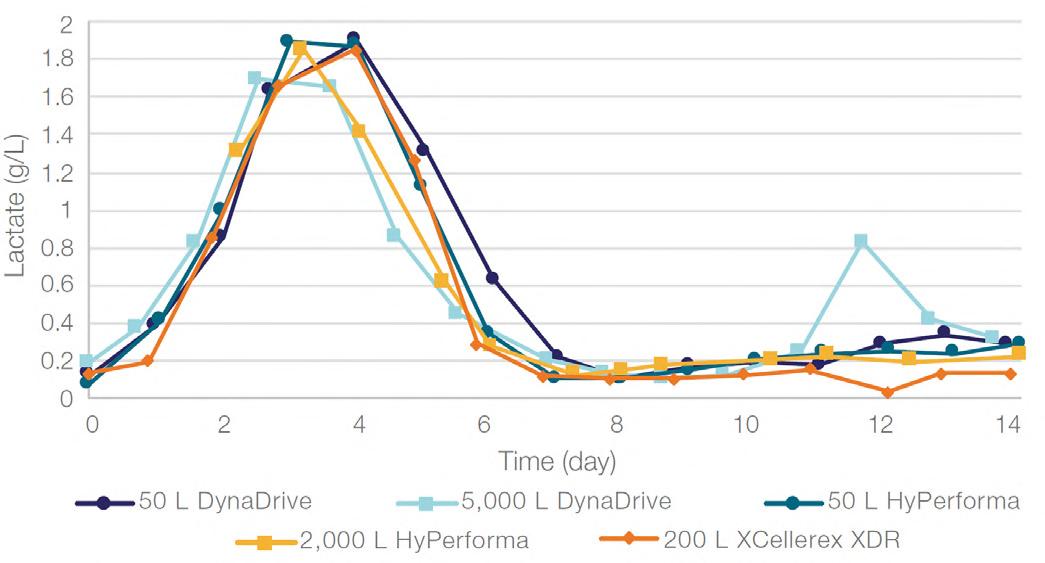
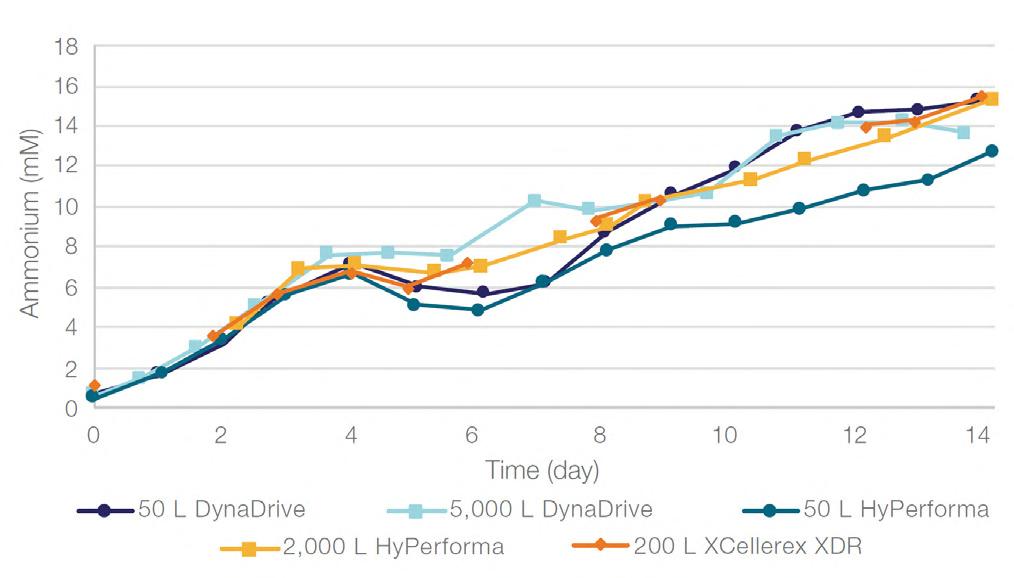
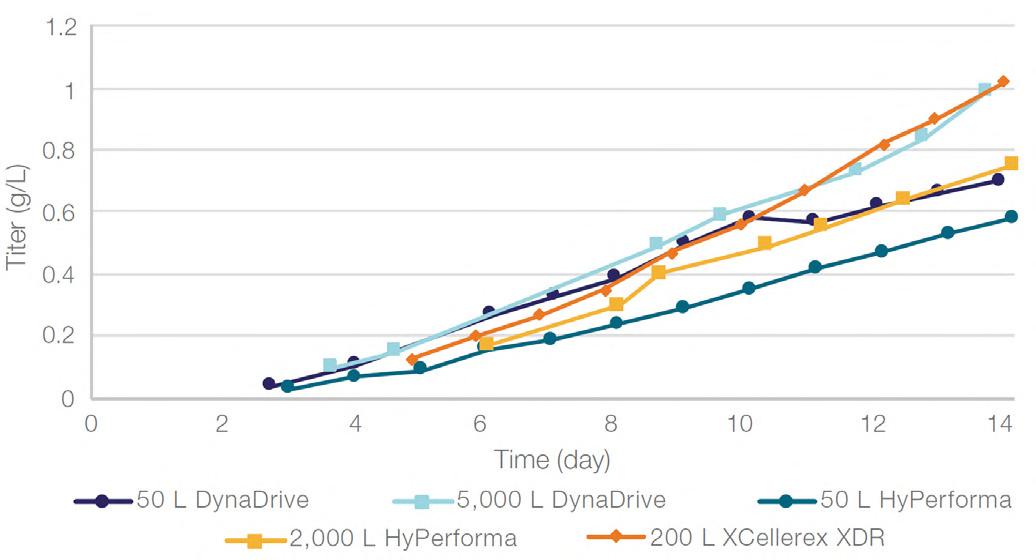
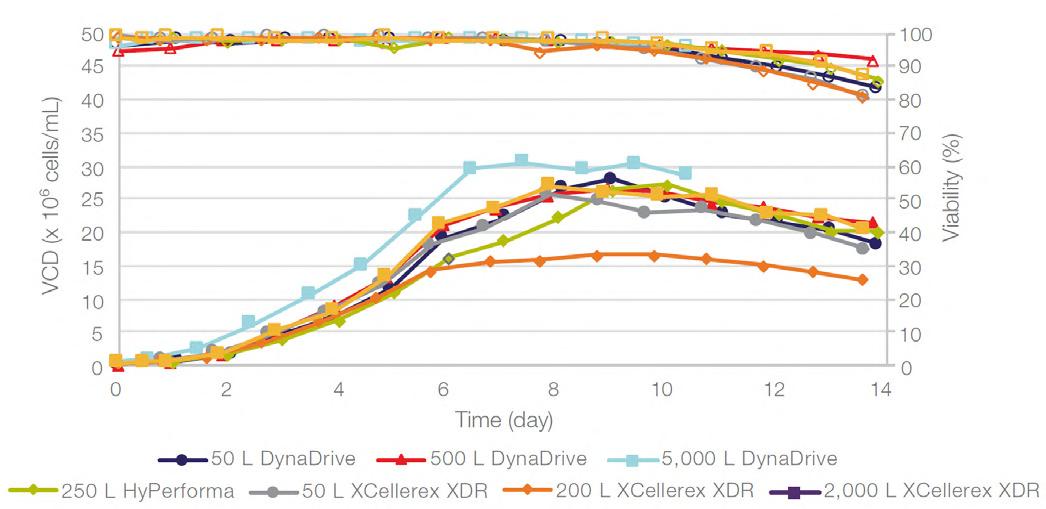
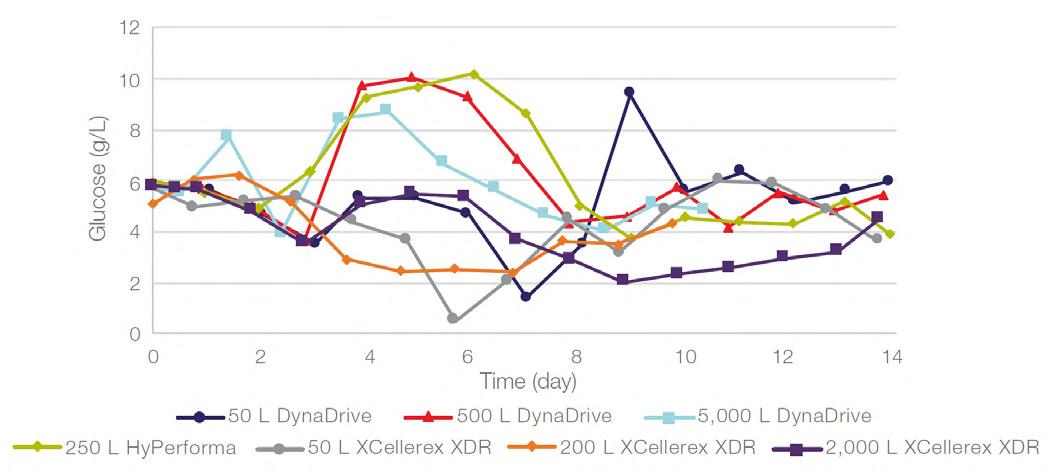
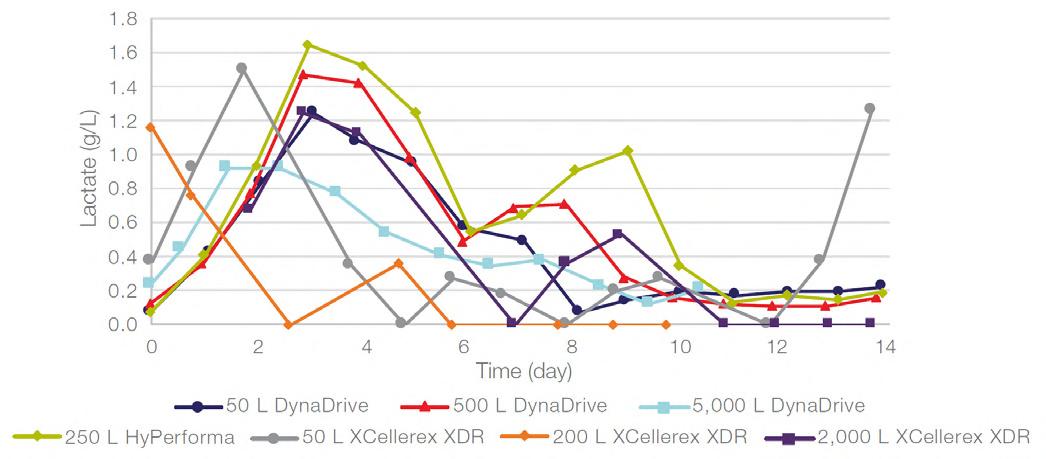
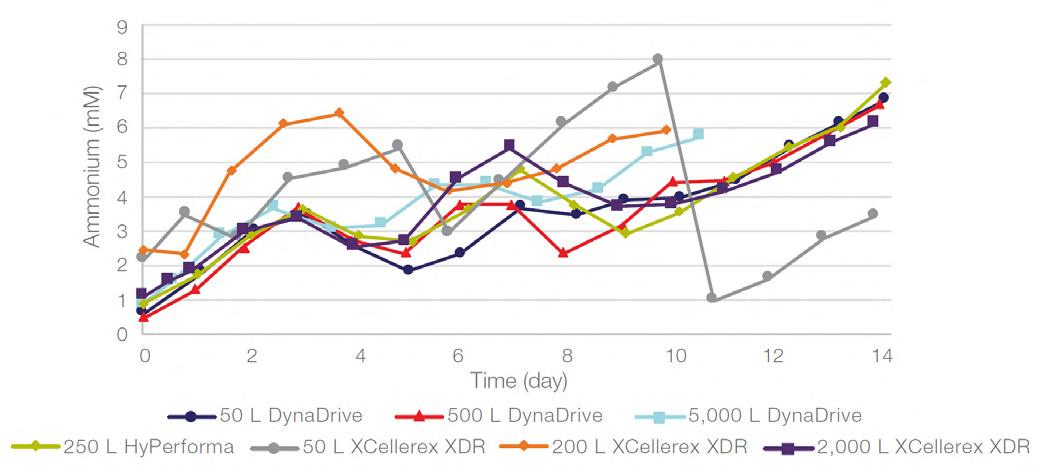
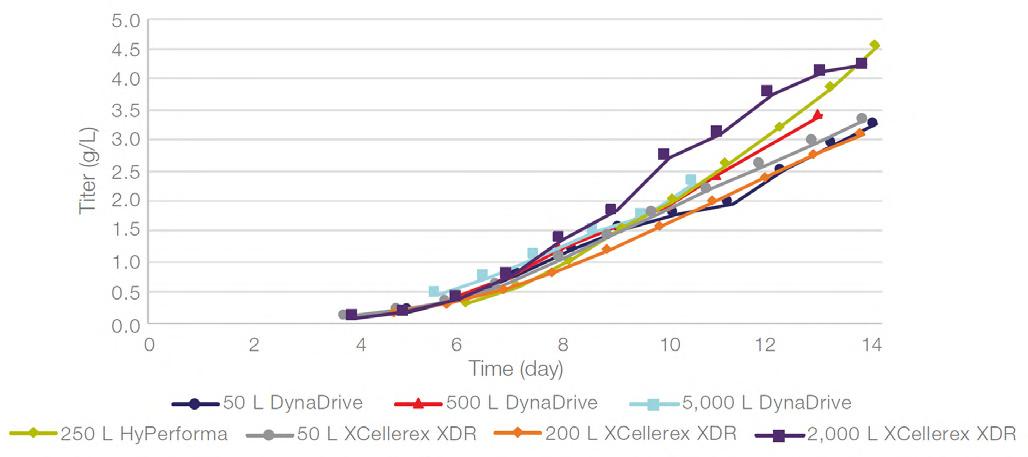
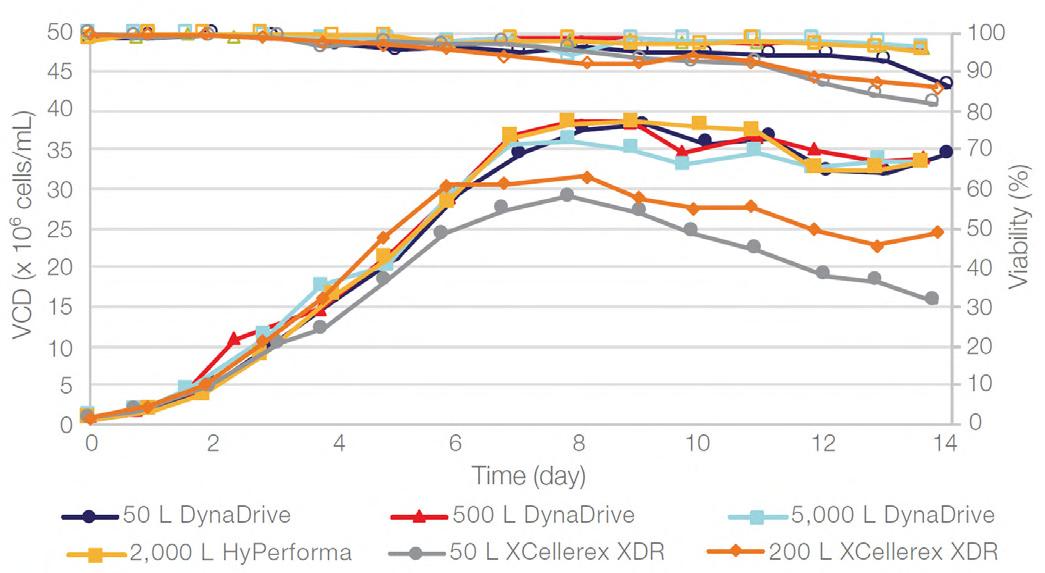
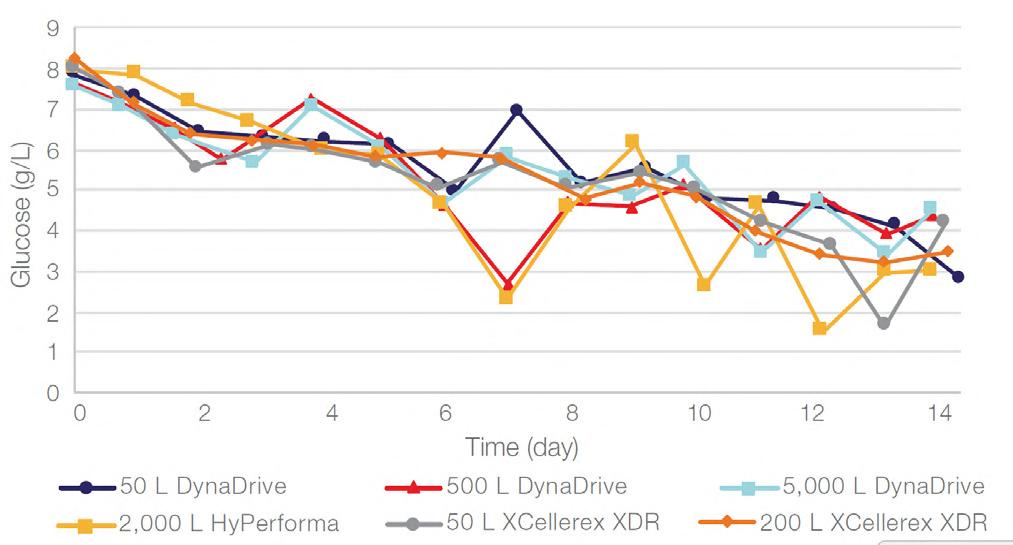
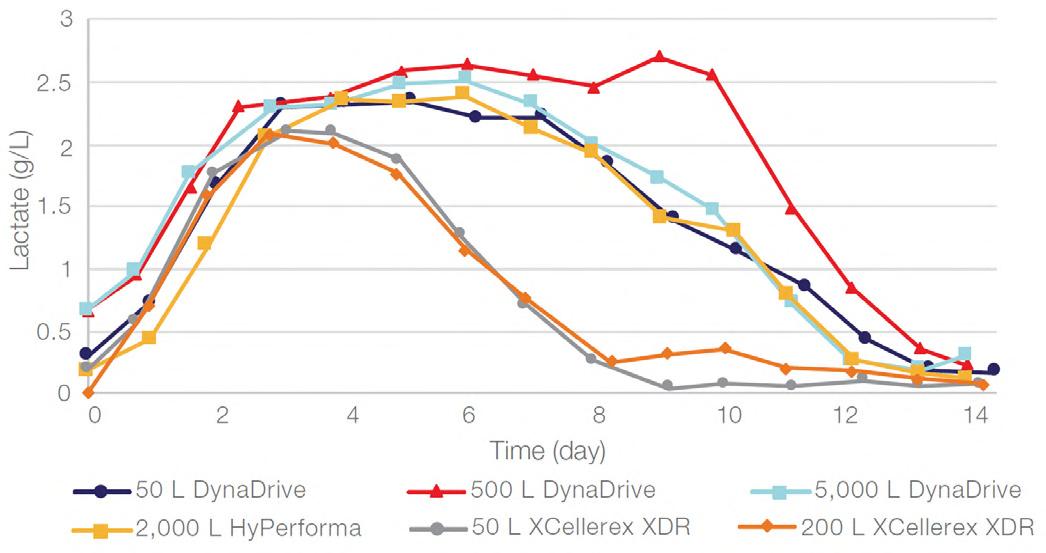
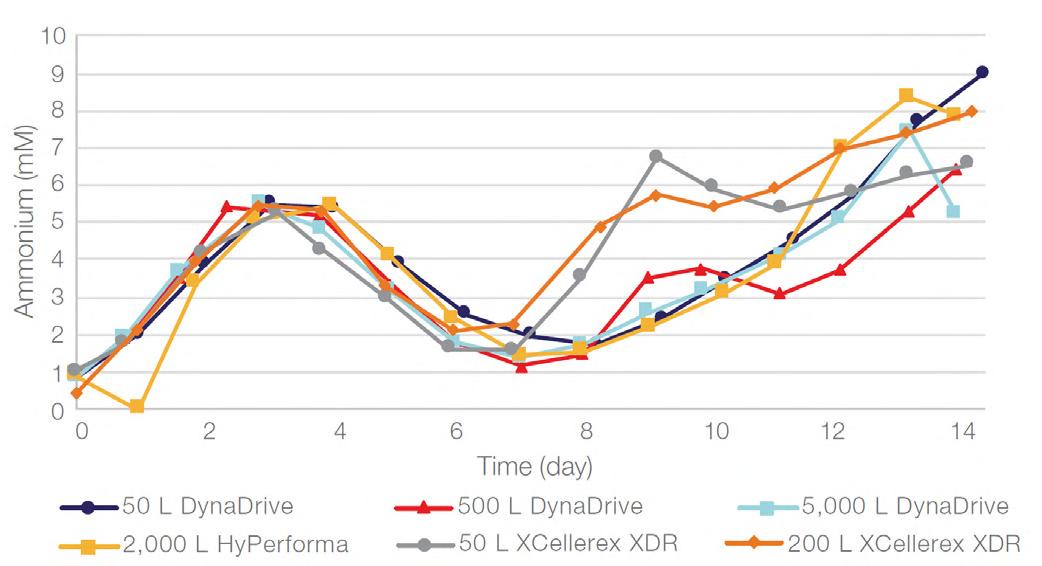
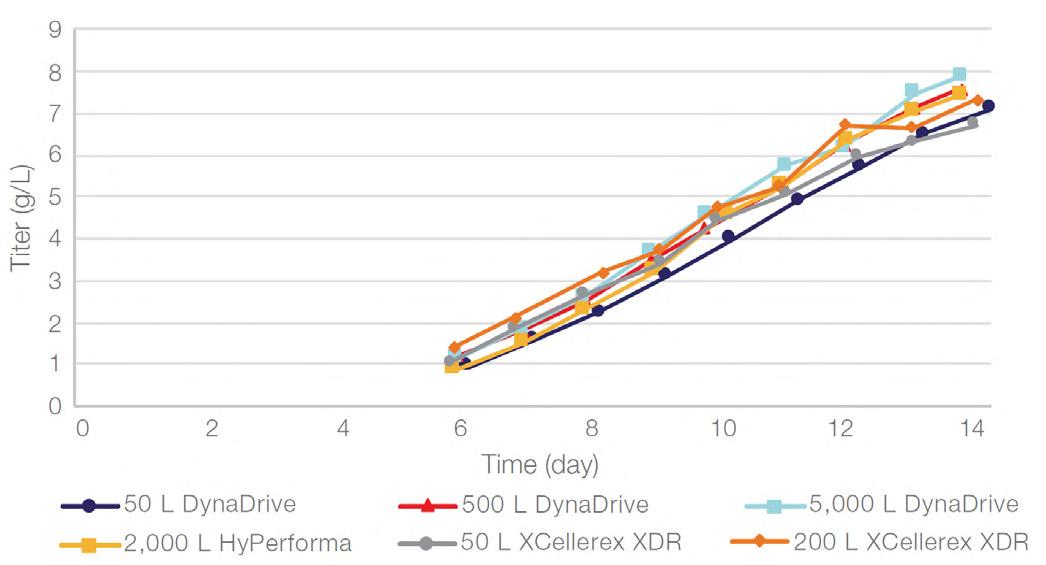
58 Scale-up Evaluation of the DynaDrive S.U.B.s
Commercialisation of a drug is a monumental milestone that hinges on years of lifecycle management. As a product enters commercialisation, product sponsors are positioned to derive demand from a fluid market. Qingwei Luo, Ben Madsen, Jeff Hou and Matt Zustiak of Thermo Fisher present how the design, scale and operations of the 5,000 L DynaDrive S.U.B. can be key in the commercialisation of a human therapeutic.
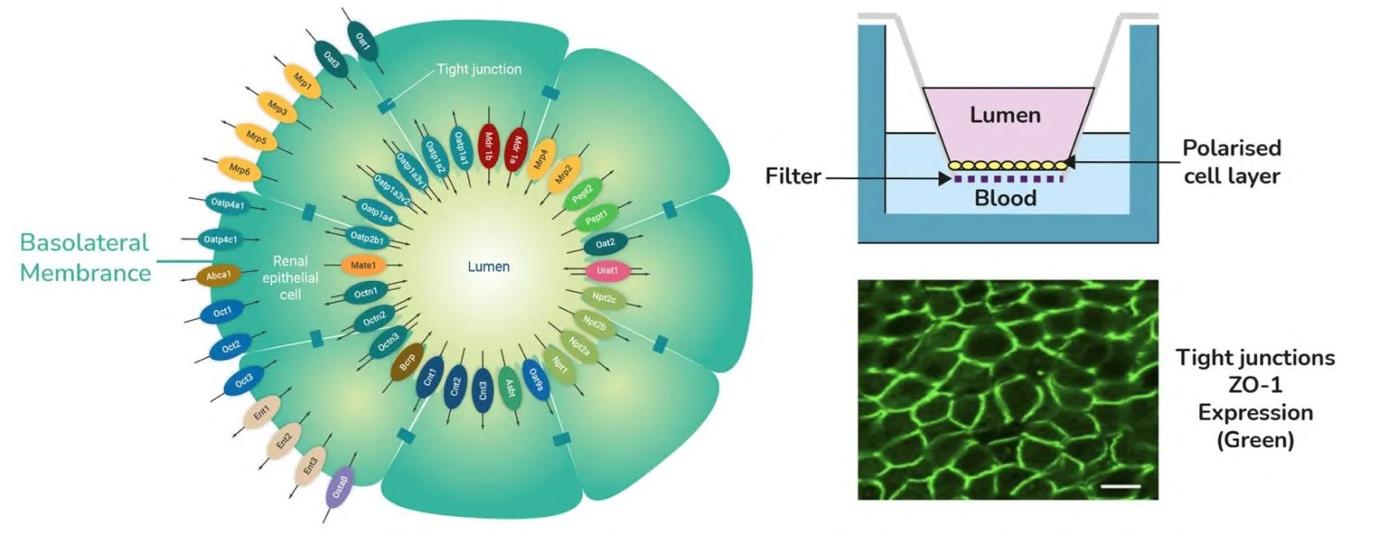
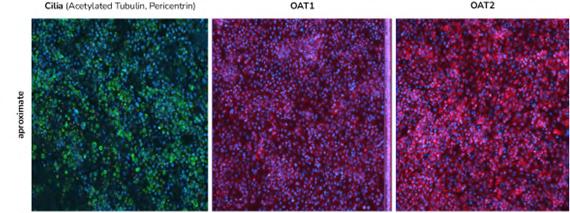
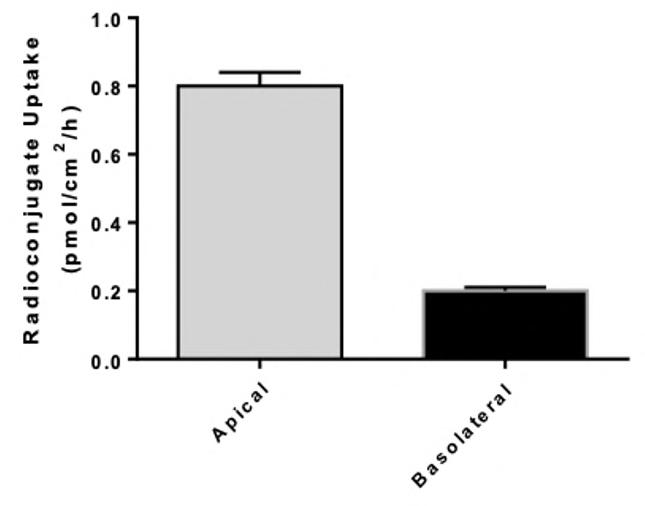
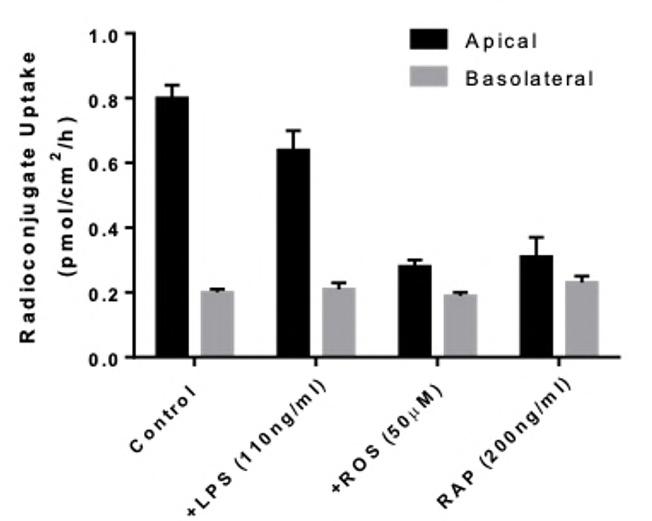
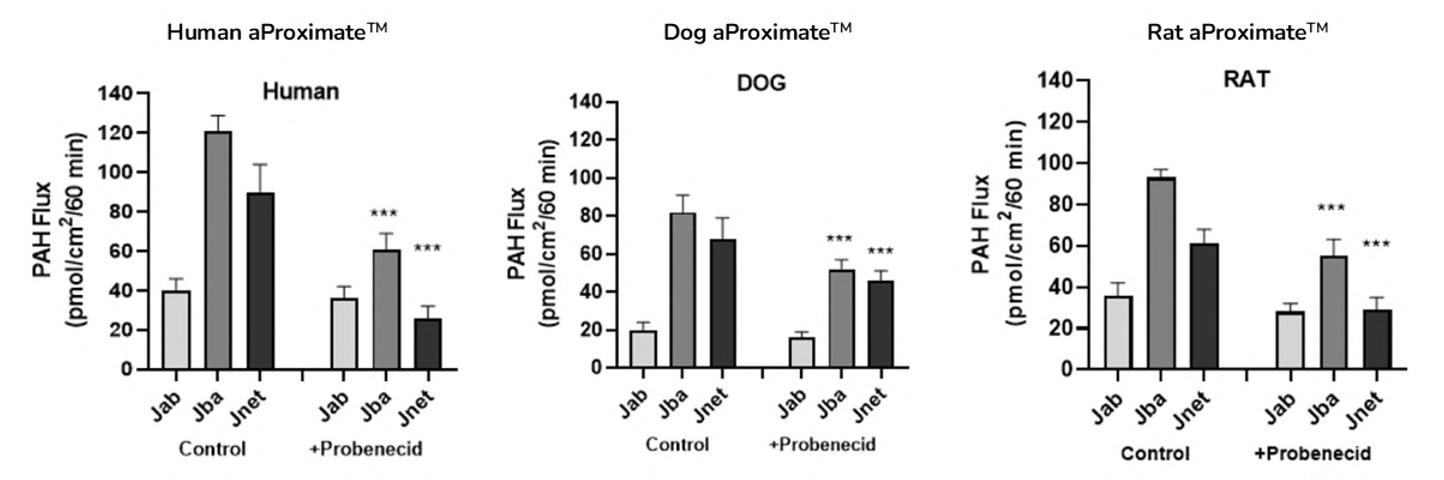
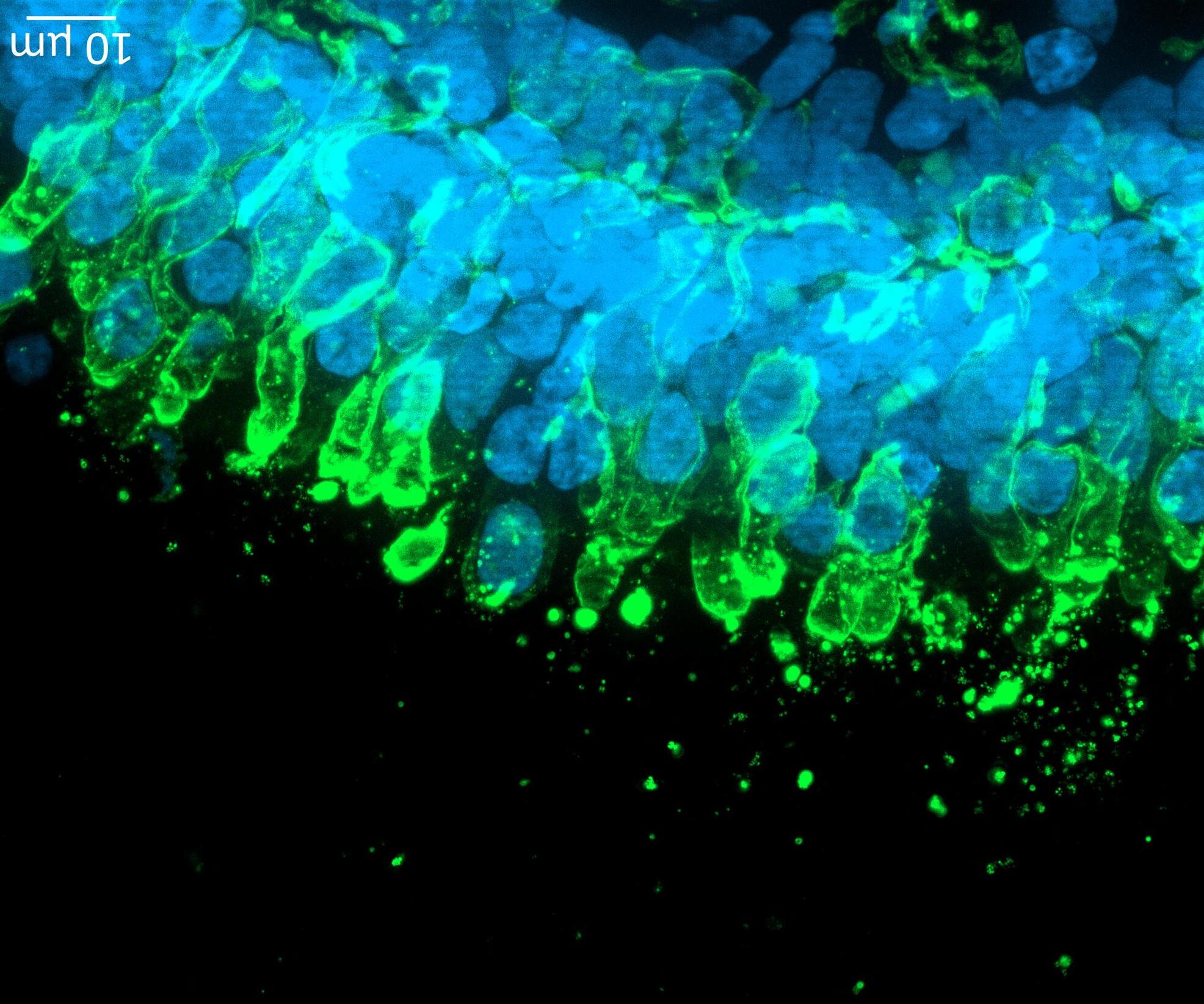
72 Use of the aProximateTM Proximal Tubule Cell Model for the Evaluation of Safety and Renal Accumulation of Radioconjugates and Large Molecules
The aProximateTM Proximal Tubule Cell (PTC) model offers a cutting-edge platform for the evaluation of drug-induced nephrotoxicity, radioconjugate retention and the accumulation of large molecules in the proximal tubule. Colin Brown of NewCells outlines the advantages of using the aProximateTM PTC model in predicting and assessing renal safety in the early stages of drug development.
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
Foreword
The global biopharmaceutical industry is navigating a pivotal moment. Recent U.S. announcements of 100% tariffs on imported patented pharmaceuticals are poised to reshape supply chains, accelerate onshoring and challenge companies to adapt quickly. While the full impact remains uncertain, the need for innovation, strategic partnerships and regulatory agility has never been clearer. This issue of International Biopharmaceutical Industry showcases how the sector is responding with resilience and forwardthinking solutions.
We open with cell and gene therapy, where Camille Bachelet of CELLforCURE by SEQENS discusses how their integration with SEQENS strengthens their position as a CDMO for advanced therapy medicinal products. Jonathan Didier of Lightcast highlights the power of single-cell functional analysis via the Envisia platform, demonstrating how deep cellular insight drives drug development. Complementary articles address endotoxin control and early-stage cell line selection, showing how analytical strategies are accelerating biologics development.
In manufacturing and biotechnology, Linglong Yi of Asymchem explores how integrated flow technologies are transforming pharmaceutical production with efficiency and sustainability in mind. Soyeon Ahn, Daeryun Park, Hyoseok Kim and Joomyung Lee of Samsung Biologics emphasise the critical role of flexibility in facility design and bioprocess execution, while Qingwei Luo, Ben Madsen, Jeff Hou and Matt Zustiak of Thermo Fisher highlight innovations in large-scale bioreactors that enhance operational efficiency and regulatory compliance.
Research and innovation pieces delve into emerging modalities and modelling approaches. Optibrium explains how advanced 3D modelling is opening new frontiers in macrocyclic drug discovery, and Benjamin Wilkin of Crown Bioscience discusses the complementary use of organoid and patient-derived xenograft models for antibody-drug conjugates. Xavier Warnet of Tebubio examines lipid nanoparticles as versatile delivery vehicles for nucleic acids, and Colin Brown of NewCells presents a
Welcome to our autumn edition of IBI. I have really enjoyed my time settling into the editorial team at Senglobal and getting to know many of our excellent contributors. In this edition, we have a brilliant Cell and Gene Therapy subsection starting on page 68. This features a number of great articles, including an excellent piece by Timothy Francis of FUJIFILM titled “Developing Endotoxin Limits, Risk Assessment and In-process Testing for CGT Products” to kick us off. Elsewhere, starting on page 16, Dr. Juliana Maynard and Dr. Ian Wilson of Medicines Discovery Catapult highlight the
• Cellia K. Habita, President & CEO, Arianne Corporation
• Deborah A. Komlos, Senior Medical & Regulatory Writer, Clarivate Analytics
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in Ethics
• Hermann Schulz, MD, Founder, PresseKontext
proximal tubule cell model for early assessment of renal safety in biologics.
Regulatory and compliance perspectives underscore the intersection of innovation and oversight. Steven Watt of A&M STABTEST reviews MHRA guidance on individualised mRNA cancer vaccines, while Dr. Janine Swarbick, Dr. Sofie McPherson, Ms Roxna Kapadia and Dr. Claire Green at HGF examine strategies to protect and extract value from bioinformatics innovations through patents and trade secrets. These insights reflect the dual pressures of scientific advancement and regulatory rigour.
Finally, in logistics and supply chain, Alex Guillen of Tive highlights the critical importance of real-time visibility and IoT-enabled cold chain management, ensuring that complex therapies reach patients safely. Dr. Maja Erdmann, Daniel Goetz, Dr. Daniela Stummer and Dr. Ingo Goldbeck of Richter BioLogics demonstrate how mass spectrometry is becoming a cornerstone analytical tool in CDMO operations, reinforcing quality, safety and process understanding across the industry.
Together, these contributions provide a panoramic view of an industry under pressure, but also brimming with opportunity. Even as trade policy introduces fresh uncertainty, the articles in this issue reaffirm a central truth: biopharma’s future will be built not only on molecules and modalities, but also on resilience, collaboration and the ability to turn disruption into progress.
Dr. Steven A. Watt, CBDO (Chief Business Development Officer) at A&M STABTEST GmbH
exciting potential that lies ahead with the UK’s National PET Imaging Platform technology in their piece titled “The Whole Picture: Total-Body PET and the Future of Biopharma.” As always, thank you to our brilliant contributors.
As the season changes and orange leaves start to fall from the trees, before we know it the end of the year will be upon us. With that in mind, please keep submitting your white papers to me and we can finish 2025 on a high with our winter edition!
• Rafael Antunes, Vice President Business Development, Aurisco Pharmaceutical Europe
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steven A. Watt, CBDO (Chief Business Development Officer) at A&M STABTEST GmbH
Your Partner for Inhalative Drug Testing
The complexity of Inhaled therapies demand analytical services at the highest standards to meet the demands of quality, precision and reproducibility over the complete product lifecycle.
Having a trusted partner makes all the difference.
With over 20 years of experience in inhaled drug testing, A&M STABTEST turns analytical challenges into confidence, providing precise, reproducible analytical solutions that ensure your development timelines are met, and products are released on time. We provide specialized analytical solutions for inhalable drug substances and products, supporting our customers throughout every stage, turning development projects into high value market -ready products, while ensuring compliance with cGMP and ICH guidelines.
Why choose us as partner for your success:
• Decades of experience with different formulations and device technologies (DPI, MDI, nebulizer and nasal spray)
• Analysis of inhaled ATMPs (mRNA and viral vectors)
• Dedicated laboratory with controlled humidity and temperature environment Partner with us for analytical solutions that overcome today’s challenges and meet tomorrow’s standards.
For more information, contact Dr. Steven A. Watt or scan the QR-Code below.
An Expertise and Dedication to Advancing Cell Therapy Manufacturing:
An Interview with Camille Bachelet of CELLforCURE by SEQENS
Can you please start by providing a brief overview of CELLforCURE’s history and how the company has developed since being founded?
Founded in 2010 by LFB, a French government-owned pharmaceutical company, CELLforCURE was created to pioneer manufacturing in the emerging field of cell & gene therapy. Designed as one of the first specialised CDMOs in Europe, CELLforCURE built a GMP facility tailored to ATMP (Advanced Therapy Medicinal Products) production.
In 2015, CELLforCURE reached a major milestone when it received GMP authorisation from the ANSM, allowing it to manufacture ATMPs for both clinical and commercial use in Europe. This recognition positioned CELLforCURE as a trusted manufacturing partner in the growing cell therapy ecosystem.
In 2016, CELLforCURE manufactured clinical vials of an allogeneic CAR-T cell therapy, marking one of the first GMP productions of its kind in Europe.
In 2019, Novartis acquired CELLforCURE and, from 2020 to 2023, CELLforCURE produced the first approved autologous CAR-T therapy.
In 2023, following a strategic decision made by Novatis, SEQENS acquired CELLforCURE, marking a new chapter. Now under SEQENS, CELLforCURE continues to deliver end-to-end CDMO services in ATMP manufacturing from early clinical phases to commercial supply, leveraging more than a decade of operational and regulatory expertise.
Today, CELLforCURE by SEQENS remains one of the key CDMOs in Europe dedicated to the manufacturing of innovative cell and gene therapies.
Thinking back to when the company started in 2010, what have been the key areas of growth in the past 15 years for CELLforCURE?
We began as one of the first European GMP facilities entirely dedicated to the production of advanced therapy medicinal products (ATMPs). Over the past 15 years, we’ve expanded our activities from clinical-stage manufacturing to full commercial production, with a strong emphasis on reliability, scalability and compliance.
A key area of growth has been the diversification of our technological platforms. Initially focused on a limited set of cell types, we now support a wide range of autologous and allogeneic therapies, including CAR-T cells, NK cells, MSCs and other innovative products. This diversification has enabled us
to meet the evolving needs of our clients in both oncology and regenerative medicine.
We have also continuously strengthened our quality systems and regulatory expertise to align with global standards, supporting the transition of our clients’ products from early-stage development through to market approval. Finally, one of the most important drivers of our growth has been our people. We’ve built a multidisciplinary team with deep scientific, regulatory and operational knowledge to navigate the complexity of advanced therapies and deliver high-quality products to patients.
What would you say are the key attributes of CELLforCURE’s work?
CELLforCURE stands out through its comprehensive expertise and dedication to advancing cell therapy manufacturing. One of the key attributes of our work is our real-life, hands-on experience in both clinical and commercial manufacturing. We have developed deep knowledge across a wide variety of cell types, enabling us to deliver reliable and high-quality manufacturing services tailored to the specific needs of each client.
Quality and regulatory compliance are foundational to everything we do. We maintain rigorous standards in quality assurance to ensure that all products comply with the stringent requirements of clinical trials and regulatory agencies. This focus guarantees the safety and efficacy of therapies manufactured in our facilities.
Another essential attribute is our collaborative partnership approach. We work closely with biotech companies, from startups to more established innovators, acting as a trusted partner to translate groundbreaking research into scalable and compliant manufacturing processes. This partnership mindset
helps accelerate the development and industrialisation of novel therapies.
Lastly, CELLforCURE’s manufacturing capabilities are highly scalable and flexible. We support clients throughout the entire product lifecycle, from early clinical development phases to commercial-scale production. This adaptability allows us to meet evolving client needs while ensuring seamless transitions between development stages.
Together, these attributes position CELLforCURE as a reliable and forward-thinking CDMO in the cell therapy space.
Please explain the experience that CELLforCURE gained from being part of NOVARTIS and how this allowed the company to develop.
Being part of a global pharmaceutical leader has been a transformative experience for CELLforCURE, allowing the company to accelerate its development both technically and organisationally. In this context, the site benefited from significant investments, upgrading its infrastructure and bringing it in line with global CAR-T manufacturing standards. This has significantly enhanced our capabilities in terms of capacity, compliance and operational excellence.
As part of a global pharmaceutical leader, CELLforCURE benefited from direct exposure to the industrialisation of complex cell and gene therapies. We gained hands-on experience in large-scale GMP manufacturing of CAR-T therapies, including processes designed for both clinical and commercial supply. This was made possible thanks to the integration into Novartis’ global network, the implementation of best practices and collaboration with cross-functional international teams.
In addition, Novartis’ decision to entrust CELLforCURE with the production of advanced therapies validated the expertise of our teams and reinforced our positioning as a centre of excellence in Europe. The site now operates with six flexible GMP production suites, covering over 10,000 m² and is fully equipped to serve biotech, academic and pharmaceutical partners.
Ultimately, this experience helped us mature as a CDMO, ready to support the next generation of cell and gene therapy developers from early development to market launch.
Now that you have been acquired by SEQENS, please explain how you anticipate this aiding the further development of CELLforCURE’s future.
Following its acquisition by SEQENS, CELLforCURE is well-positioned to accelerate its development as a CDMO dedicated to ATMPs.
As a leading CDMO group with extensive experience, SEQENS brings significant added value to CELLforCURE, from commercial
insight to robust client project execution. This support is particularly strategic as CELLforCURE restarts its activities in the advanced therapies field.
SEQENS also provides a full range of shared services essential for the smooth and efficient operation of any site within the Group, including IT, HSE, legal, procurement and human resources.
Finally, as the foundation of SEQENS’ Cell & Gene Therapy Business Unit, CELLforCURE works closely with SEQENS teams to identify synergies and define future service offerings for customers in the ATMP field.
How has your own expertise and that of your colleagues aided the development of CELLforCURE by SEQENS’s work?
As an Innovation and Partnership Manager at CELLforCURE by SEQENS, my role is to bridge the latest cutting-edge technologies with our manufacturing expertise. Holding a PhD in immunology and having provided scientific support at Miltenyi Biotec, I bring a strong scientific foundation that allows me to understand the biological complexities and technical challenges of cell therapy development.
This expertise helps me evaluate emerging technologies critically and identify those with the highest potential to enhance manufacturing efficiency and product quality. At CELLforCURE by SEQENS, I collaborate closely with multidisciplinary teams including development, manufacturing, quality control and quality assurance. Such cross-functional collaboration ensures smooth integration of innovations while respecting regulatory requirements. Our combined knowledge guarantees that every step, from cell sourcing and engineering to final product release, meets stringent quality and reproducibility standards.
By uniting scientific insight with manufacturing know-how and strong teamwork, we continuously foster innovation at CELLforCURE by SEQENS. This approach improves process efficiency, shortens development timelines and supports the delivery of therapies worldwide.
This shared commitment and collaboration are key drivers at CELLforCURE by SEQENS and are essential in the rapidly evolving cell therapy landscape.
CELLforCURE by SEQENS oversees projects from start to finish. Please tell us a bit about how that process works and why overseeing the entire thing helps with efficiency.
What sets CELLforCURE by SEQENS apart is our ability to support products, not only in clinical stages, but all the way through to commercial manufacturing. Our site is one of the few in Europe with experience in the commercial supply of ATMPs. This makes us a strategic partner for companies looking to optimise the transition from clinical to commercial stages.
At CELLforCURE by SEQENS, we offer end-to-end support for ATMP manufacturing, from technology transfer and process development to GMP manufacturing, quality control and final product release. This integrated approach allows us to act as a true partner throughout clients’ product lifecycle.
We start by working closely with our clients to ensure a smooth and robust technology transfer. From there, our teams collaborate on process optimisation and scale-up, ensuring that the manufacturing process is not only compliant but also tailored to the specific needs of each project. Our in-house quality and regulatory teams are involved from day one, helping to anticipate challenges.
By managing the full value chain under one roof, we reduce the risk of delays, avoid communication silos and ensure continuity across project phases. This seamless coordination enhances both speed and reliability, two key factors in the development and commercialisation of cell and gene therapies. It also gives our clients a single point of contact, which simplifies decision-making and increases transparency.
A key USP of CELLforCURE by SEQENS is the quality control facility, boasting 800 square meters of quality control area and 100 square meters of banking area. Please briefly explain your facilities and what makes them unique in the field.
A key strength of CELLforCURE by SEQENS lies in our state-ofthe-art quality control facility, where approximately 90% of analyses are performed in-house. This extensive infrastructure enables us to conduct a wide range of critical tests, including sterility, identity, potency, stability, environmental monitoring, raw materials testing, in-process quality control, as well as final product quality control and batch release.
What makes our facility unique is the high level of integration and control, allowing seamless project development and ensuring fast, reliable delivery of products, thereby reinforcing our position as a trusted comprehensive solution provider in the cell and gene therapy field.
It was recently announced that CELLforCURE by SEQENS have formed a partnership with Galapagos to support the manufacturing for clinical development of their CAR T-cell therapy for upcoming trials. Please explain a bit about this new project and how CELLforCURE by SEQENS’s experience makes it a good fit.
CELLforCURE by SEQENS has been chosen by Galapagos to manufacture CAR T-cell therapy candidates for upcoming clinical trials, expanding the decentralised manufacturing network in Paris and the broader France region.
The selection of CELLforCURE by SEQENS as a manufacturing partner is based on its proven expertise in GMP-compliant, commercial-scale cell therapy production. As a recognised leader in France, CELLforCURE by SEQENS has worked closely
with French hospitals for over five years, actively contributing to the establishment of the first local CAR-T manufacturing pathways in the European Union.
Located strategically in Les Ulis, near major treatment centres in Paris and the wider France area, CELLforCURE by SEQENS enables Galapagos to realise its ambition of a decentralised manufacturing network that brings treatments closer to patients, allowing faster and more efficient access to care.
What is next to come for CELLforCURE by SEQENS?
CELLforCURE by SEQENS’s top priority is, and will remain, to serve its clients by relieving them of the complex burden of ATMP manufacturing from development through to commercialisation. With large GMP facilities in Europe and a proven track record in commercial production, our mission is to provide reliable, scalable and compliant manufacturing solutions that allow our clients to focus on what matters most, delivering therapies to patients.
In parallel, CELLforCURE by SEQENS is committed to supporting emerging biotech companies by fostering partnerships with a range of stakeholders. The goal is to facilitate and accelerate the development of advanced therapy projects.
Camille Bachelet (Scientific Innovation and Partnership Manager) holds a PhD in Immunology. She completed her doctoral research at Imagine Institute, focusing on the immune system of children with lymphoma. She then spent four years at Miltenyi Biotec as Scientific and Technical Support for France, supporting researchers and clinicians in the development and implementation of advanced cell and immunotherapy technologies.
Camille Bachelet
The Future of Single-Cell Applications:
Discussing the Importance of Bringing Single Cell Function into Focus with the Envisia Platform from Lightcast
1. Can you please start by providing a brief overview of Lightcast’s history and how the company has developed since being founded?
The foundational technology of Lightcast is actually spun out from Base4, an early stage Cambridge-based company that was initially working with optical-electrowetting (oEWOD) technology for DNA sequencing. However, it became clear there was potential for cell-based assays, including singlecell applications, which at that point, focused largely on static molecular profiles.
And so Lightcast was founded in 2019 to further develop a version of oEWOD that enables precise manipulation, tracking and functional characterisation of cells at singlecell resolution. In late 2023, we launched the Luminary Early Access Program. In May 2024, we then opened the Boston Innovation Centre to support the expansion of that program and in May 2025, we revealed the Envisia platform to the world at PEGs Boston.
2. Lightcast take a function-focused approach to their work. Please explain what this means and how it differs from other offerings.
In many drug discovery programmes, functional analysis is performed relatively late in the pipeline and often after early candidate pools have already been filtered using molecular or binding data alone. The problem is that those screens are inherently indirect; a strong binder or a promising transcriptomic profile doesn’t always translate into the cell behaviour that drives therapeutic effect. By the time researchers get around to functional assays, the most potent clones, for example that rare antibody that blocks and recruits or the T cell with true serial killing capacity, may already have been discarded.
Lightcast is aiming to shift this dynamic. Envisia enables functional readouts like secretion, binding, cytotoxicity at the single-cell level, and does so in a multiplexed, sequential and traceable way. That means researchers can generate a direct functional fingerprint for each cell while still recovering it for sequencing and re-expression or outgrowth. In practice, it ensures that high-performing candidates aren’t lost in the noise and that functional data drives decision-making from the very start.
3. Please explain more about the technology that is on offer from Lightcast.
Previewed earlier this year, Envisia integrates oEWOD, droplet microfluidics and machine learning (ML) into a benchtop system that makes functional single-cell analysis practical for any lab.
Core Technology
Droplet-based compartmentalisation: using a gentle stepemulsification, cells are encapsulated in picolitre droplets, preventing crosstalk and ensuring accurate readouts.
oEWOD manipulation: light-driven control allows droplets to be moved dynamically, merged as required to build assays and workflows, and then dispensed off-platform in real time based on user-defined criteria. This enables highly flexible workflows that adapt to evolving research needs.
ML filtering: after the initial droplet generation, real-time algorithms overcome inefficiencies in random droplet loading. In typical systems, only around 27% of droplets contain the desired contents, compared with 95% occupancy achieved by Lightcast using this ML-based filtering.
Traceability: each droplet, its contents, interactions and readouts are tracked, from load to dispense. This enables
researchers to link functional performance on-platform to off-platform analysis, such as sequencing.
The result is a system that not only captures the functional diversity of single cells but also lets researchers rapidly recover the exact ones they want to take forward.
4. How do you see Envisia impacting drug development?
Envisia creates a fully traceable environment in which individual cells can be isolated, paired with reagents or other cells, assayed and ultimately recovered. For drug developers, this means the ability to rapidly screen immune cells, antibodyproducing cells, or engineered cell therapies for the functions that matter most, not just binding, but killing, persistence and serial activity. And we can run these interactions sequentially, building increasingly complex functional workflows. We can then take hits off-platform for further analysis, such as sequencing and re-expression, or perhaps transcriptomic analysis, thus generating a more comprehensive view of each hit. This should lead to better candidate selection, reduced attrition and faster progression from discovery into development.
5. Why is single-cell functional analysis critical to the drug development process?
Drug development ultimately comes down to how cells behave, whether an antibody blocks a pathway or enhances it, whether a T cell actually kills its target and whether a therapy persists long enough to matter in a patient. Yet, across the industry, bulk assays are still the default way of measuring these effects. They give you an average signal across millions of cells, which can be useful, but you risk missing the top performing candidate.
That’s where single-cell functional analysis makes the difference. Instead of averaging, it lets researchers see exactly which individual cells are driving the activity and in what way. You can find that subset of T cells with serial killing capacity or the antibody-producing cells that not only bind but trigger the right downstream effect. This is especially critical in areas like antibody discovery, ADCs and cell therapies, fields where function is the ultimate predictor of whether a treatment will succeed in patients. By moving beyond bulk assays and looking directly at function one cell at a time, researchers gain a level of clarity and precision that simply hasn’t been possible before.
6. Please can you provide a brief insight into what the drug development market currently looks like and why the work being done by Lightcast is so important?
The industry is under immense pressure; development timelines are long, costs are high, and success rates are lower than anyone would like. At the same time, new therapies like cell and gene therapies show huge promise, but they require a
Talking Point
new type of toolkit to really uncover how they behave. Again, that’s where we see Lightcast’s value. By helping researchers measure what cells do, not just what they are, we’re giving them a way to make better decisions earlier. That can save time, money and ultimately help patients faster.
7. Please can you explain what the key markets being targeted by Lightcast are and why these areas have been identified?
Right now, our main focus is on antibody discovery, ADCs and cell therapies. In these markets functional analysis is now seen as essential. An antibody might bind beautifully, but if it doesn’t trigger the right response, it won’t make it as a drug. Cell therapies are even more complex, as you need to know if a T cell can persist, kill and sometimes even kill again. Those are exactly the kinds of questions Envisia was built to answer.
8. How are Lightcast able to work collaboratively with other companies to accelerate drug discovery research?
We see collaboration as a natural part of what we do. Our technology slots into existing workflows, so researchers can generate functional data on Envisia and then combine it with sequencing, imaging or re-expression. We’ve also set up early-access programmes that allow us to work closely with partners to explore new applications. It’s mutually beneficial; we learn from their biology and they get to push the limits of our platform. We are currently working with a number of big pharma companies who, for obvious reasons, we cannot disclose. But we also have instruments with several key academic sites in both Europe and the US.
9. Please provide us with a brief insight into what’s to come next for Lightcast.
As the Autumn conference season gets up and running, we’ll be out providing updates. We’re already working on expanding the menu of assays researchers can run, so they can explore everything from antibody binding to cell killing on the same platform. We’re also continuing to build out partnerships with pharma and biotech groups worldwide. We firmly believe that Envisia has the potential to become the de facto standard for functional single-cell biology!
Jonathan Didier, PhD, is the Field Application Team Leader at Lightcast. He previously worked for Berkeley Lights, Inc. He received his PhD from Carnegie Mellon University and did postdoctoral work at Harvard University's Wyss Institute, focusing on clinical applications of droplet microfluidics.
Jonathan Didier
Setting new standards in ATMP production reliability
GENEX is a groundbreaking machine concept designed specifically for small and medium batch production. This innovative system redefines the future of pharmaceutical production. With GENEX, Bausch+Ströbel has developed an innovative approach that significantly reduces the risk of contamination and sets new safety standards in aseptic processing. The fully automated GENEX filling and packaging system utilizes robot technology, automating size changes and environmental monitoring while ensuring full compliance with GMP regulations. GENEX is the key to tomorrow’s pharmaceutical production, combining the highest safety standards and product quality.
GENEX Features
Today's Biotechnology
Empowering Flow Chemistry
Transforming Manufacturing Through Integrated Flow Technologies at Asymchem
Leading the Shift to Greener Processes
Pharmaceutical manufacturing is entering a new era of transformation. Globally, regulatory expectations are rising and the demand for safer, cleaner and more efficient manufacturing practices is becoming increasingly urgent. Sustainability is no longer optional; it is now a driving force that is pushing the industry to rethink long-established approaches. Flow chemistry is emerging as a key solution, but its full impact depends on more than just technology. Meaningful progress happens when process expertise, equipment design and engineering integration come together. At Asymchem, they bring these capabilities under one roof. With deep expertise and a proven track record in flow chemistry, they help clients improve efficiency, reduce environmental impact and scale the production of RSMs, intermediates and APIs with confidence.
Scope and Capabilities
Asymchem was among the first CDMOs to implement continuous manufacturing for pharmaceuticals and fine chemicals, beginning in 2008. Since then, they have delivered more than 500 flow projects over kilo-scale and enabled technology transfer projects at scales up to 10,000 MT. With nearly two decades of experience and fully integrated capabilities, Asymchem is equipped to address some of the most complex challenges in pharmaceutical manufacturing. They have established capabilities to perform virtually all reactions in flow mode, encompassing liquid-liquid, gas–liquid, solid-liquid, gas-liquid-solid and other multiphase reactions, with demonstrated excellence in hydrogenation, nitration and lithiation, etc. These technologies enable the precise, scalable and efficient production of pharmaceutical intermediates and APIs.
At Asymchem, innovation is core to how they solve complex manufacturing challenges. To address the difficulties of continuous solid-liquid reactions, they developed and built their own proprietary flow reactor, using metformin synthesis as the test platform throughout its design and optimisation. This breakthrough led to multiple patents for continuous metformin production and resulted in China’s first API continuous manufacturing verification under the ICH Q13 guidelines. By leading this effort, Asymchem is helping to drive broader adoption of continuous manufacturing across the industry.
Pioneering in Green Innovations
Sustainability is a cornerstone of Asymchem’s philosophy. Flow processes inherently minimise solvent and energy use, reduce waste and enhance safety. This commitment was recognised when Asymchem became the first recipient of the ACS CMO Excellence in Green Chemistry Award in 2022. The award was granted for the development of a custom photochemical flow reactor designed to perform a challenging [2+2] cycloaddition. This innovation replaced a six-step batch synthesis with a single-step continuous
process, reduced waste by 72 per cent and enabled production at the 250-kg scale. The success of this project established a new benchmark for green chemistry within the industry and reflects Asymchem’s continued investment in developing sustainable and high-performance manufacturing technologies.
In-House Equipment Fabrication
In modern flow chemistry, process innovation must be supported by advanced engineering capabilities. While many CDMOs rely on third-party equipment with long lead times and limited flexibility, Asymchem has established a fully integrated system that combines proprietary design, precision machining and comprehensive equipment lifecycle support. This system is anchored by a dedicated engineering team and a 10,000 m² manufacturing centre. The facility is equipped with advanced technologies, including metal laser 3D printers, five-axis CNC machining centres and robust simulation and modelling tools. Through this in-house fabrication capability, Asymchem delivers client-centric, end-to-end solutions that address process, regulatory and scale-up requirements. Customised flow reactors can move from initial design to delivery in as little as one to two weeks, enabling seamless integration and significantly accelerating project timelines.
Global Expansion: The Sandwich Site
Asymchem’s integrated approach to flow chemistry is exemplified by their UK site in Sandwich. The facility features CE-certified proprietary flow equipment, including plug flow reactors, heterogeneous hydrogenation systems, biocatalysis platforms and Grignard capabilities. This installation has created a flexible, fully integrated environment for rapid process development and scale-up, representing a key step in Asymchem’s broader expansion into Western markets. With these capabilities in place, their Sandwich site serves as an innovation hub for flow chemistry. Client projects can be rapidly developed and tested in a controlled laboratory setting, allowing for efficient process optimisation. Once a process meets performance and quality requirements, they provide tailored equipment solutions ready for direct deployment at the client’s site.
Linglong Yi
Linglong Yi, Chief Engineer of CFCT, has built his career around the development, technology transfer and commercial-scale production of small-molecule drugs. He is particularly recognised for his extensive expertise in continuous manufacturing, having led multiple projects from concept to successful commercialisation. As the Chief Engineer of Asymchem's Center of Continuous Flow Technology (CFCT), he oversees the development, validation and implementation of flow continuous technologies across the organisation and in external collaborations.
TrueQuant pan-RNA-Seq
Key Benefits
• FFPE samples
• Liquid biopsies / EVs from only 25 µl of plasma
• Degraded RNA / Low RIN-value
• Ultra low sample Input: 10 pg total RNA – single cell
• Fast, single tube protocol
• Gel free
• UDIs included
• Sequencing on any Illumina® NGS instrument
• Bioinformatics optional
Today's Biotechnology
The Whole Picture: Total-Body PET and the Future of Biopharma
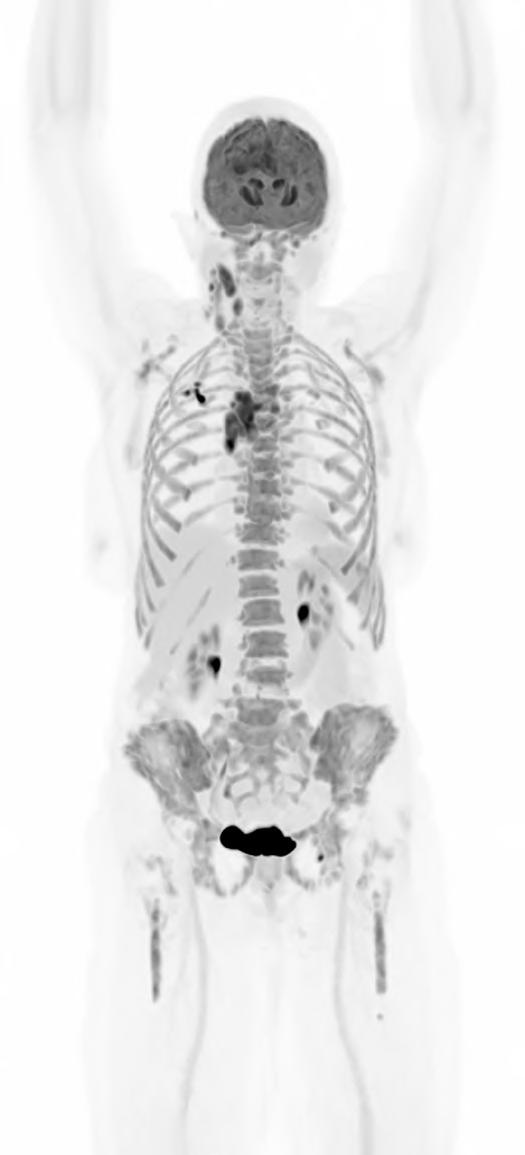
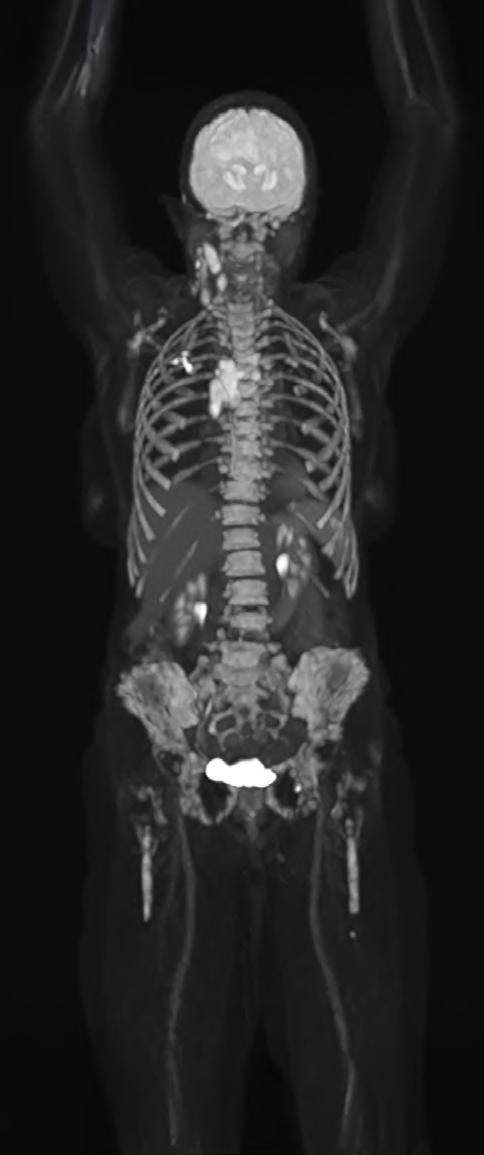
What if a clinical scan could transform biopharmaceutical innovation and patient care, streamlining drug development, clinical trials, and complex disease diagnosis? Total-body Positron Emission Tomography (PET) imaging is turning that vision into reality, offering a full-body, real-time view of human physiology.
Total-body PET is redefining what is possible in molecular imaging and biopharmaceutical development. This advanced PET technology can deliver full-body scans in minutes, with a sensitivity of up to 40x greater than conventional standard field PET scanners1 and potential for significantly lower radiation exposure. This opens exciting new avenues for research and discovery. It enables clinicians to study disease in real time across the whole body, revolutionising how we diagnose, stage and treat complex conditions like cancer, cardiovascular disease and neurodegenerative disorders. For patients, this means faster scans and earlier interventions, with the opportunity for more personalised treatments. For research and industry, it unlocks a powerful new platform for evaluating novel therapies, tracking their behaviour and accelerating their path to market.
But realising the full potential of total-body PET requires more than cutting-edge scanners. It demands a robust infrastructure, one that includes a reliable supply of radiotracers and an integrated framework for data sharing and collaboration. In the UK, the National PET Imaging Platform (NPIP) is building exactly that. What it reveals is not just a clearer image of the body, but a blueprint for the future of biopharmaceutical development.
FROM CONCEPT TO CLINIC – THE EVOLUTION OF TOTAL-BODY PET
PET is a type of imaging technology that helps clinicians and researchers visualise cellular and molecular processes inside the body. Prototype PET scanners emerged in the 1950s, gained traction in nuclear medicine during the 1970s and were later adopted clinically for disease diagnosis, staging and monitoring.
PET scanning involves the injection of radiotracers into the body. Radiotracers are made up of two parts: a radionuclide (a compound or molecule that emits radiation), combined with a molecule or drug that guides the radionuclide to a specific area of the body to be studied or targeted for treatment. As the radionuclide decays, it will emit gamma rays inside the body, which are detected by the PET scanner. Through image processing, the detected gamma rays are used to build a detailed picture of where the radiotracer has accumulated. This picture helps clinicians and researchers to diagnose diseases and make decisions about treatments.
Continued technological advances in hardware and software have led to a new generation of PET scanners with far greater sensitivity and resolution. While conventional PET relies on multiple bed positions and generating multiple images to acquire
a whole picture of the body, total-body PET has a field of view wide enough to take an image from head to toe in a single scan, and a detector capable of 40x higher sensitivity.¹ This means the whole body can be imaged faster, at higher resolution and in real-time. It also means lower doses of radiotracer are required to generate the same amount of detail (Table 1).1,3
Feature
Conventional PET Total-Body PET
Large Axial Field of View (LAFOV) 15–20cm >100cm*
Sensitivity Moderate Ultra-high (up to 40x higher for whole-body images)
Average Total-Body scan time** 10–30 min 30 seconds–3 minutes
Average radiation dose for a full-body scan** ~370 MBq ~9.25 MBq
Dynamic imaging potential (continuous capture of data over time)
Patient throughput
Tracers can be followed for ~3 half-lives, limiting dynamic imaging
Lower
Tracers can be followed for 5–6 additional half-lives, enabling whole-body, real-time imaging
Potential for twice as many clinical scans per day
*Scanners in the NPIP network have a LAFOV of between 106–194cm. **Based on (18F)-FDG scan procedures.
Table 1. Differences between conventional PET scanners and total-body PET scanners.1,4,5,6
Importantly, total-body PET does not replace conventional PET imaging. Conventional PET remains indispensable in clinical practice, especially when imaging needs are localised to a specific area of the body. But for complex diseases that span multiple organ systems or in cases where lower doses of radiation are required, total-body PET offers a transformative new lens:
• In oncology, total-body PET enables earlier detection of metastases and monitoring of tumours throughout the body, as well as increasing the potential for radiotheranostics (radiopharmaceuticals that can both detect and treat disease).7,8
• In cardiology, it helps us to understand how heart disease affects, or is affected by, other organ systems, supporting approaches to systems-based or preventative cardiovascular medicines.9
• In neurology, it allows the brain to be imaged at the same time as the rest of the body, which is critical for our understanding of complex neurodegenerative diseases.10
• In infectious disease and immunology, it offers a powerful tool for visualising infection and immune responses throughout the body, opening new avenues for treatment.11
• Finally, the higher sensitivity of total-body PET reduces the amount of radiotracer required for a high-resolution image, enabling paediatric studies in children and other patient populations where higher levels could be unsafe.12
In short, total-body PET is paving the way for a future where a systems-level understanding of human physiology may be possible. This will enable new insights into complex disease mechanisms and therapeutic targets where whole-body context is essential, or where study was previously difficult.
TRANSFORMING DRUG DISCOVERY
In addition to enabling more comprehensive study of complex disease mechanisms and therapeutic targets, total-body PET imaging is also set to change the way we assess and validate those targets and progress novel therapeutics through clinical trials. It does this by offering a more complete, accurate and efficient way to study their behaviour and role in the human body.
Total-body PET Provides Whole-body Systems Insight
One of the most powerful capabilities of total-body PET is the potential for whole-body pharmacokinetics (PK) and pharmacodynamics (PD) analysis. Through capturing real-time data on how a radionuclide-tagged drug accumulates and clears across the entire body or by using another radiotracer to measure its effect, researchers can gain a better understanding of its behaviour far earlier and with more confidence than with traditional imaging or blood sampling. This is especially valuable for drugs with systemic effects on the body, like cell therapies, RNA therapies and biologics.
Total-body PET Delivers Longitudinal Safety Data
The heightened sensitivity of total-body PET also opens the door to the use of a wider range of imaging agents or radionuclides and, therefore, longer-term studies in the human body. Conventional PET typically relies on short-lived radionuclides such as Fluorine-18 (¹⁸F) and Carbon-11 (¹¹C), which have half-lives of approximately 109 minutes and 20 minutes, respectively. These radionuclides decay quickly in the body, which is important for limiting the radiation dose to the patient, but limits their utility for imaging over longer time scales. In contrast, the ultra-high sensitivity of total-body PET allows for the use of longer-lived radionuclides like Zirconium89 (⁸⁹Zr). ⁸⁹Zr has a half-life of approximately 3.3 days,13 so when linked to an active molecule, total-body PET can measure its distribution and behaviour across the whole body up to 30 days post-injection.14
This is particularly useful for studying therapies like biologics. Biologics are slow circulating, so data captured over days or even weeks in the body can provide critical safety and efficacy insights to support their progression through clinical trials.14
Total-body PET Empowers Translational Medicine Earlier
When it comes to the study of complex biological therapies in early trials, total-body PET is strategically placed. Its ultra-high sensitivity offers a powerful alternative to tissue models and provides the basis for earlier first-in-human studies.15 Due to the lower dose of radiotracer required for visualisation, researchers can conduct microdosing studies in humans using concentrations small enough to minimise the risk of adverse events. It also enables the study of much earlier-stage drug effects or low-dose responses that conventional methods are not sensitive enough to detect. This offers a transformative new pathway for the study of novel biopharmaceuticals, helping us to identify promising candidates sooner and rule out those that
Today's Biotechnology
are ineffective earlier in clinical trials with greater precision, using real human data.
Total-body PET Improves the Efficiency of Clinical Trial Recruitment
Further down the drug development pipeline, total-body PET may also enable more targeted clinical trial recruitment. Increasingly, biomarkers (biological markers of disease) are used during clinical trial recruitment to stratify subjects, i.e. divide patients into groups based on their biological profile, and therefore their predicted response to the treatment is being studied. Total-body PET allows even more precise biomarker screening during patient recruitment, meaning that patients who are likely to demonstrate therapeutic efficacy can be selected more efficiently, increasing the likelihood of regulatory success.
Total-body PET Enables Better Trial Design
Total-body PET imaging supports more effective and adaptive clinical trial design by enabling real-time, whole-body assessment of drug distribution and response. This allows researchers to make more informed adjustments to factors like dosing during the trial, potentially improving outcomes and reducing adverse effects.
Moreover, the data provided by total-body PET has the potential to enhance the development of healthcare ‘digital twins’. Digital twins are ‘virtual patients’ resembling actual patients, created by combining available genetic and clinical data. This new concept in healthcare allows clinicians and researchers to simulate disease progression and predict treatment outcomes based on mathematical models of patients with the same phenotype.16
While still an emerging concept, digital twins represent a dramatic leap forward in biopharmaceutical development. Digital twins could offer predictive modelling capabilities that would enable researchers to optimise the design of clinical trials. They would allow for the simulation of a wide range of patient scenarios, dosing strategies and treatment schedules, far beyond what would be feasible to study in human participants. Ultimately, the result is more precise dosing and scheduling data, improving the clinical benefit of the drug when it reaches the market.
By combining total-body PET data with multi-omics and other clinical datasets, researchers will be able to build more robust and reliable digital twin models that will strengthen the foundations of the concept and support its transition from experimental research to mainstream practice.
BARRIERS TO ADOPTION
However, as with many technologies, the widespread integration of total-body PET into today’s drug development pipelines will face several hurdles. It requires investment in infrastructure, both in the systems and facilities where they will be installed.6 Central to this is the need for an infrastructure capable of managing the huge data volumes generated by whole-body dynamic imaging, spanning storage, processing and analysis.17 Integration into existing clinical workflows presents additional challenges, as protocols must be adapted and staff trained, not only in operating the new systems, but also in interpreting complex multi-organ data and conducting new studies using emerging radionuclides.
Today's Biotechnology
Radionuclide supply presents another critical barrier in some countries. At present, in the UK, the production of many of the radionuclides that are valuable for total-body PET, such as Zirconium-89 and Copper-64, is limited. This challenge is compounded by a lack of sovereign manufacturing capacity, leading to overreliance on unreliable production and importation of radionuclides made outside the UK in ageing nuclear reactors. To combat this, there is an ongoing need and support research and innovation funding, policy change and new routes to production.
Even with the technology integrated, unlocking its full potential will require transforming how we use and share its data.
NPIP: A NATIONAL PLATFORM FOR COLLABORATION AND DISCOVERY
At the heart of this transformation in the UK is the National PET Imaging Platform (NPIP), a £32 million government-funded initiative delivered by Medicines Discovery Catapult, the Medical Research Council and Innovate UK, that is deploying advanced total-body PET scanners across the country.18 Launched in 2023 through investment from the UK Research and Innovation’s Infrastructure Fund,19 NPIP is delivering more than just a technological upgrade. Crucially, the platform is providing standardised protocols and a national infrastructure for collaboration and data sharing that aims to foster innovation across trials and institutions. The platform is designed to accelerate drug discovery and redefine the future of what is possible in healthcare, providing unprecedented access to high-resolution, whole-body molecular imaging data.
Total-body PET in Crohn’s disease
A new research project is utilising the UK’s NPIP total-body scanner network to study intestinal fibrosis in Crohn’s disease for the first time, one of its most debilitating complications. The study aims to explore whole-body disease dynamics in Crohn’s and advance therapeutic development.20
All total-body PET systems in NPIP’s network have been harmonised with respect to camera functionality and adhere to protocols that guide best practice in image capture and data interpretation. This standardisation helps to ensure consistency in imaging data, supporting and building regulatory confidence in clinical trials, especially those that span multiple centres.
NPIP also aims to ensure that, where possible, clinical data is centralised and harmonised, empowering researchers to tackle complex biomedical challenges together. The NPIP network is developing a fully functional and accessible database, led by the UK’s Medical Research Council, for the scientific community to leverage.
The availability of large imaging datasets like this is critical to driving forward innovation in systems biology, biopharmaceutical development and patient care using total-body PET scanners. Through machine learning and artificial intelligence, we will be able to use stored data to develop novel algorithms that will be able to predict and diagnose disease earlier, as well as optimise clinical trials and suggest personalised treatment plans.21 This could lead to a world where
clinicians will be able to ‘see and treat’ patients for increasingly complex diseases in the same visit.
However, arguably the most critical factor in scaling the impact of total-body PET will be the continued collaboration across platforms like NPIP and the accessibility of imaging data from research and clinical sites around the world. Diverse datasets are essential for training artificial intelligence models to provide unbiased outputs in healthcare, which in turn enables more accurate diagnostics and personalised treatments across populations.22 With broader reach, more inclusive data and continued access to that data, the outputs of this technology will only become more precise and equitable.
CONCLUSION
Total-body PET is more than a sensitive imaging tool; it is a strategic asset with the potential to reshape the future of biopharmaceutical development. Enabling earlier, safer and more precise evaluation of therapies, it sets the stage for a future where drug discovery is more efficient, more inclusive and deeply data-driven. This means more confidence in emerging therapeutics from regulatory bodies and more effective therapies for patients reaching the market faster.
But the true transformation lies in how we deploy this technology, not in isolation but as part of a connected and collaborative ecosystem. The UK’s National PET Imaging Platform offers a blueprint, providing a benchmark for the integration of total-body PET technology into existing healthcare systems and opportunities to expand its potential impact through global data sharing and continued innovation.
REFERENCES
1. Cherry, S. R., Jones, T., Karp, J.S., et al. Total-Body PET: Maximizing Sensitivity to Create New Opportunities for Clinical Research and
2. Belcari, N., Bisogni, M.G. & Del Guerra, A. Positron emission tomography: its 65 years and beyond. Riv. Nuovo Cim. 46, 693–785 (2023).
3. Daube-Witherspoon, M.E., Pantel, A.R., Pryma, D.A., et al. Total-body PET: a new paradigm for molecular imaging. Br J Radiol. 95 (1140). (2022).
4. Slart, R.H.J.A., Tsoumpas, C., Glaudemans, A.W.J.M., et al. Long axial field of view PET scanners: a road map to implementation and new possibilities. Eur J Nucl Med Mol Imaging. 48, 4236-4245. (2021).
5. Bailey, D.L., Meikle, S.R., Calamante, F., et al. The Australian National Total-Body PET Facility – A Shared Resource and Risk Model for Implementing Total-Body PET. J Nucl Med. (2025).
6. Alberts, I. More, S. Knapp, K., et al. Is Long–Axial-Field-of-View PET/ CT Cost-Effective? An International Health–Economic Analysis. J Nucl Med. (2025).
7. Pantel, A.R., Viswanath, V., Daube-Witherspoon, M. E., et al. PennPET explorer: human imaging on a whole-body imager. J Nucl Med. 61: 144-151. (2020).
8. Herrmann, K. Schwaiger, M. Lewis, J.S., et al. Radiotheranostics: a roadmap for future development. The Lancet Oncology. 21 (3): e146-e156. (2020).
10. Chung, K.J., Abdelhafez, Y.G., Spencer, B.A., et al. Quantitative PET imaging and modeling of molecular blood-brain barrier permeability. Nat Commun. 16 (1): 3076. (2025).
11. Patel, N., Bergstrom, M., Murphy, P.S., et al. PET in the characterization of immune diseases and development of therapeutics. Oxford Open Immunology. 6 (1). (2025).
12. Djekidel, M., AlSadi, R., Abi Akl, M., et al. Total-body pediatric PET is ready for prime time. Eur J Nucl Med Mol Imaging. 49: 3624–3626. (2022). https://link.springer.com/article/10.1007/s00259-022-05873-y
13. Liu, Q., Wang, X., Yang, Y., et al. Current Perspectives on 89Zr-PET Imaging. Quant Imaging Med Surg. 12 (6): 3300–3313. (2022).
14. Pandya, D.N., Hantgan, R.R., Budzevich, M.M., et al. The Role of 89Zr-Immuno-PET in Navigating and Derisking the Development of Biopharmaceuticals. J Nucl Med. 61 (5): 665–670. (2020).
15. https://www.fda.gov/media/186092/download?attachment, visited on 23 Jul 2025.
16. Asghar, U.S., Chung, C. Application of digital twins for personalized oncology. Nat Rev Cancer (2025).
17. Mingels, C., Chung, K.J., Pantel, A.R., et al. Total-Body PET/CT: Challenges and Opportunities. Semin Nucl Med. (2024).
18. https://npip.org.uk/, visited on 25 Jul 2025.
19. https://www.ukri.org/what-we-do/creating-world-class-research-andinnovation-infrastructure/funded-infrastructure-projects/, visited on 23 Jul 2025.
20. https://www.accord.scot/about-accord/accord-news/grant-securedinvestigate-role-full-body-petct-imaging-track-fibrosis, visited on 31 Jul 2025.
21. Sundar, L.K.S., Gutschmayer, S., Maenle, M., et al. Extracting value from total-body PET/CT image data – the emerging role of artificial intelligence. Cancer Imaging. 24: 51. (2024).
22. Chinta, S.V., Wang, Z., Palikhe, A., et al. AI-driven healthcare: Fairness in AI healthcare: A survey. PLOS Digit Health. 4 (5): e0000864. (2025).
Dr. Juliana Maynard
Dr. Juliana Maynard, Head of Translational Imaging at Medicines Discovery Catapult and Director of Operations and Engagement for the National PET Imaging Platform, is an expert imaging scientist with over 20 years of experience in translational imaging and therapeutic precision medicine. Juliana developed, established and is now the Director of Operations and Engagement at the National PET Imaging Platform, where she is accountable for strategically building and growing collaborative opportunities to use the platform and its cutting-edge total-body PET imaging technology. She is also Head of Translational Imaging at Medicines Discovery Catapult.
Email: juliana.maynard@md.catapult.org.uk
Dr. Ian Wilson
Dr. Ian Wilson, PET Engagement & Delivery Manager for the National PET Imaging Platform has over 30 years of experience in the development and commercialisation of radiopharmaceutical therapies and CT, MR, ultrasound and optical imaging agents. Formerly CEO of ImaginAb Inc and Edinburgh Molecular Imaging, he is now responsible for the delivery of the National PET Imaging Platform.
Email: ian.wilson@md.catapult.org.uk
Today's Biotechnology
Enabling Oral Delivery of TPDs: The
CDMO Path to Oncology’s Next Frontier
No major scientific breakthrough comes without its challenges. This is certainly true of targeted protein degraders (TPDs), which have rapidly become one of the most dynamic areas within oral solid dose drug development.
The idea behind TPDs, encompassing both proteolysistargeting chimeras (PROTACs) and molecular glues, emerged in the early 2000s as a novel means of removing, rather than merely inhibiting, disease-causing proteins. The first PROTACs, described in 2001 by the Crews and Deshaies laboratories, used peptide linkers to connect a target ligand with an E3 ubiquitin ligase ligand, ultimately directing the target protein for degradation. While conceptually elegant, these early molecules were large, unstable and lacked permeability, making them unsuitable as therapies. Molecular glues had a precedent in drugs such as thalidomide, but their discovery was largely serendipitous and their mechanisms poorly defined.
For years, TPD research remained largely confined to academic settings, constrained by limited availability of E3 ligases, the absence of predictive models for ternary complex formation, synthetic difficulties and the prevailing view that molecules of such size could never be orally bioavailable. Over the last decade, however, advances in structural biology, computational modelling, small-molecule chemistry and clinical validation have shifted this perspective. The central challenge is no longer whether TPDs can function, but how to deliver these inherently bulky and poorly soluble molecules in a form that the body can effectively absorb.
The CDMO Advantage
TPD molecules are frequently described as “brick dust” compounds because of their large and complex structures, which translate into poor solubility in the gastrointestinal tract. Even when dissolution is achieved, their bulk also restricts permeability across intestinal membranes, meaning only a fraction of the administered dose reaches systemic circulation. The consequence is low bioavailability in patients, a frustrating paradox in which highly potent therapies for serious, often life-threatening diseases such as cancer face fundamental delivery hurdles.
In parallel, the past two decades have brought significant progress in the capabilities of contract development and manufacturing organisations (CDMOs), particularly in formulation and process science. The most established CDMOs have supported the development and manufacture of hundreds, if not thousands, of molecules over this period, building deep institutional knowledge of best practices in process development and large-scale manufacturing. Those with experience in handling highly potent compounds at occupational exposure band 5 (OEB5, ~10 ng/m³) bring an added advantage: the ability to
manage challenging molecules safely and effectively. While TPDs are not always categorised as highly potent, the expertise and infrastructure developed for OEB5 compounds provide a strong foundation for managing their complexity.
That said, even among larger CDMOs, not every organisation possesses the full suite of in-house capabilities needed to enhance the bioavailability of complex modalities. Advanced enabling technologies such as spray drying, hot-melt extrusion and nanomilling demand specialist expertise and dedicated facilities. The leading providers in this area are typically those focused on these cutting-edge platforms. Encouragingly, an emerging model within the pharmaceutical supply chain is addressing this gap, the rise of strategic partnerships designed to align CDMOs, technology specialists and drug developers in pursuit of optimal solutions.
Oncology is the industry’s growth engine and the proving ground for oral targeted modalities. Estimates vary by methodology, but most place the global oncology market at ~$225–$356 billion in 2024–2025, with forecasts clustering around $600–$900 billion by 2034 (≈11% CAGR).1,2,3 At the same time, small molecules still dominate US approvals. In 2024, 64% of CDER’s novel approvals were small molecules, broadly consistent with recent years, and the majority of these are administered orally. Industry summaries indicate that ~60% of FDA NME approvals are small molecules and ~80–90% of those are oral, underscoring the continued centrality of OSD in oncology pipelines.
Potency and complexity further raise the bar on development and manufacturing. Multiple analyses suggest ~45% of recent small-molecule NCEs qualify as highly potent (HPAPI),4,5 demanding OEB-4/5 controls and specialist containment and handling. For TPDs, this landscape is even more acute; market studies project rapid category growth as programmes mature, with published CAGRs ranging from ~21% to ~35% through 2035.6,7
Put together, oncology’s scale, the predominance of oral small molecules and the rise of HPAPIs make CDMOs with deep potency, formulation and tech-transfer expertise indispensable, particularly when moving TPDs from concept to robust, scalable OSD products at speed.
Strategic Partnerships
Introducing new technologies into a CDMO environment can be a lengthy, resource-intensive and costly endeavour, particularly when bridging technical skills gaps. This naturally raises a question: why not leverage complementary expertise across the industry and connect them within a collaborative network that spans the pharmaceutical supply chain?
Such an approach allows CDMOs to focus on their core strengths (such as process development, technology transfer,
Today's Biotechnology
scalable manufacturing, clinical and commercial packaging, and essential support functions like regulatory and analytical services) while simultaneously tapping into best-in-class enabling formulation technologies. The result is access to cutting-edge capability without the risks of protracted timelines or costly integration efforts. Yet forming partnerships is only the starting point; it is effective partnership management that ultimately determines whether a therapy reaches the clinic and patients in the fastest, most efficient and most cost-effective way possible.
Traditionally, a drug sponsor would present its substance to a CDMO, only to discover that an enabling technology was required elsewhere. The sponsor would then have to engage an upstream specialist, whose work was often disconnected from the CDMO that would eventually assume responsibility for the product. This fragmented process created inefficiencies, slowed progress, and most importantly, delayed patient access to critical therapies.
A CDMO’s real differentiator lies in its project management expertise, developed over decades of guiding complex molecules
through multiproduct facilities and across every stage of the development lifecycle. The solution, therefore, is to establish the CDMO as the project’s single point of contact (SPOC). In this role, the CDMO coordinates directly with its strategic partners, aligning on drug substance characteristics, product requirements and the most suitable enabling technologies. Detailed technical exchanges occur within a single, managed framework, ensuring seamless knowledge transfer and preventing costly omissions or delays in downstream development and manufacturing. In oncology, where speed to clinic literally affects patient outcomes, the SPOC model ensures that enabling technologies for oral targeted molecules, such as TPDs, are fully integrated and time-efficient, streamlining transitions from discovery to first-in-human trials.
Enabling Technologies
Adopting a platform-agnostic approach ensures that the client’s product dictates the choice of technology, rather than forcing a molecule into an ill-suited process. For “brick dust” TPDs in particular, several established enabling technologies have proven effective in enhancing bioavailability.
Spray drying converts TPDs into amorphous solid dispersions by dissolving the drug substance within a polymer matrix at the molecular level. By removing the crystalline structure that typically restricts dissolution, the process yields a high-energy amorphous form that dissolves more readily in gastrointestinal fluids. For TPDs, spray drying can significantly improve the apparent solubility of these large, hydrophobic molecules while offering scalability from early development through to commercial production.
Hot-melt extrusion (HME) achieves intimate molecular-level mixing between TPDs and pharmaceutical polymers without
Today's Biotechnology
the use of solvents. Through the application of controlled heat and shear, the drug substance is blended with carrier polymers to form a homogeneous solid solution or dispersion. As a continuous process, HME delivers excellent content uniformity while improving both solubility and permeability by disrupting crystalline structures and establishing more favourable thermodynamic conditions for dissolution.
Nanomilling reduces TPD particles down to the nanometre scale, vastly increasing the surface area available for dissolution. This mechanical size reduction can accelerate dissolution rates by orders of magnitude, in line with well-established dissolution principles. For TPDs, often hindered by both poor solubility and sluggish dissolution kinetics, nanomilling provides a straightforward means of enhancing bioavailability, especially when combined with stabilising surfactants and polymers to maintain long-term stability.
Together, these technologies offer complementary pathways to overcoming the solubility and permeability challenges inherent to TPDs. By applying the right approach, or even a combination of methods, developers can unlock the therapeutic potential of these complex molecules and advance them toward clinical and commercial success.
Looking Ahead
While today’s TPDs benefit from proven enabling technologies, one certainty remains: the pharmaceutical landscape will continue to evolve. Cancer biology continues to drive innovation in oral targeted therapies, particularly in addressing “undruggable” targets, protein classes that traditional small molecule inhibitors cannot modulate. TPDs, including PROTACs and molecular glues, offer a compelling route to degrade these proteins, a mechanism that is now increasingly viable as formulation science and CDMO-enabling technologies align.
As formulation data accumulates across the industry, enabling platforms will advance in step, further broadening the toolkit available to drug developers. The shift toward closer collaboration between CDMOs and specialist formulation partners comes at precisely the right moment. With the TPD market projected to expand at a CAGR of roughly 21% through
2035, outpacing even biologics, commercial momentum is now firmly aligned with their therapeutic promise.
For patients awaiting these therapies, such collaboration delivers more than operational efficiency; it creates the essential bridge between laboratory innovation and real-world access to treatments for cancer and other chronic diseases. By uniting deep manufacturing expertise with state-of-the-art formulation and processing technologies, the industry is now positioned to realise the therapeutic vision that has surrounded TPDs for over two decades. In this light, the long-standing “curse” of these complex molecules may at last be giving way to the prospect of meaningful, life-changing therapies.
Dr. Rebecca Coutts graduated from the University of Bath with a BSc in Pharmacy before completing a PhD in Pharmaceutics at Cardiff University. Over her 25-year career, Coutts has shown a passion for development services and currently holds the role of Senior Director, Pharmaceutical Development. Before this, she held roles including General Manager for PCI Pharma Services at its Tredegar site, Director of Development with specialist expertise in the development and processing of highly potent molecules, and Associate Director of Contained Manufacturing
pci.com
talkfuture@pci.com
Building
High potent solutions include:
• High potent pharmaceutical development, clinical trial and commercial manufacture
• Containment down to an OEL of 0.01µg/m3
• Xcelodose® microdosing technology and roller compaction
• Tablets, capsules, powders, liquids, suspensions, semi-solids and drug-in-capsule/vial
• Analytical laboratory and QP services
• Clinical and commercial packaging
Regulatory and Compliance
Breaking the Mould:
MHRA’s Draft Paves the Way for Individualised mRNA Therapies
Scientists have long understood that cancers are as individual as the patients in which they develop and proliferate. We still classify cancers in different groups depending on tissue origin, morphology and other factors, but genomic studies have revealed that on a cellular and molecular level, cancer cells can be very individual within a group. We also know from antibody-based therapies that the patient’s immune system can be used to fight cancer cells. Antibody-based therapies have been, and newly developed antibodies will continue to be, successful in the treatment of cancer. But the idea of directly activating the patient’s humoral immune response against tumour cells promises a lasting vaccine-like response. Individualising this immune response, tailored to unique (mutated) proteins produced by patients' own cancer cells, opens a new avenue of vaccine-like cancer therapies. Individualised mRNA cancer vaccines may be the answer to the treatment of previously untreatable or unresponsive cancers.
Unfortunately, individualised treatments are not something that drug developers, manufacturers and regulatory authorities are used to dealing with. The MHRA draft guideline on individualised mRNA cancer immunotherapies indicates that regulatory authorities are willing to discuss new ideas and QC-concepts required to industrialise the individualisation of mRNA therapies.
The publication of the MHRA’s draft regulatory guidance on individualised mRNA cancer immunotherapies marks more than just a technical milestone in regulatory science. It addresses the fundamental challenges to the way medicines have been conceptualised, manufactured and approved for over a century. Once, the industry relied on the predictability of mass-produced drugs designed, tested and delivered in homogeneous batches. Now, individualised drugs demand a regulatory framework for therapies that are, by definition, unique to every patient. Traditional drug development, however, is built upon scale. A single molecule, discovered and refined, undergoes years of preclinical and clinical testing across populations to establish safety and efficacy before being manufactured at scale. Regulatory oversight centres on batch integrity, ensuring that each vial, tablet or syringe from a given lot is chemically and functionally identical. Quality assurance is grounded in reproducibility over the complete lifecycle of the product. Although individualised therapies break with this paradigm, most elements of the production will still have to be reproducible and traceable to ensure that the quality in terms of identity, purity and quantity is designed into the product and, more crucially, that each single batch is received by the intended patient. A guideline should address these points.
The MHRA draft document challenges the fundamentals of traditional mass-produced medicines and accommodates
the concept of an individualised mRNA cancer vaccine, which is designed against each patient’s tumour molecular profile, integrating mutational data into a bespoke RNA construct. No two patients’ products are exactly the same, nor are they intended to be. Here, the very definition of a batch is inverted; the patient-specific product is the batch. Whereas the new Ph. Eur. chapters 5.36 mRNA vaccines for human use, 5.39 mRNA substances and 5.40 DNA templates, coming into effect on January 1st 2026, only conceptualise how mRNA vaccines are already designed and manufactured, they do not make reference to how such vaccines may be individualised. The MHRA takes the bold step of taking the specific regulatory requirements of individualised mRNA cancer vaccines head-on, rather than leaving innovators to navigate a regulatory vacuum. The guidance acknowledges that classical comparability, stability and potency testing models cannot simply be retrofitted. Instead, it gestures towards a more flexible, risk-based approach, focusing on the robustness of the platform technology, the reliability of bioinformatic pipelines and the control of manufacturing processes, rather than on product-byproduct characterisation.
Critics may rightly question whether regulatory science can keep pace with technological ambition. What happens when accelerated manufacturing timelines, sometimes mere days from biopsy to vaccine, collide with the need for meaningful quality oversight? The MHRA’s consultation process is crucial, but the eventual guidance must strike a balance, enabling innovation without eroding the safeguards that patients and clinicians rely on. Therefore, this guidance is hopefully conceived as a living document that should be amended when technology outpaces current thinking; no one can predict the future. This should especially be reflected when individualised mRNA therapies leave the vaccine space and move into other indications like infectious disease, rare disorders, or autoimmune conditions. This forward-thinking guidance document, positioning the MHRA as a regulatory pioneer, could attract developers and clinical trials to the UK, enhancing its scientific leadership in the field. Contrary to libertarian thinking, measured regulatory oversight can be the ground to grow innovation and foster entrepreneurial success, especially in the medical field. Clearly, the early implementation of the FDA as a strong agency drafting regulations with patient safety as their foundation and an open mind to science and innovation to foster the development of new lifesaving drugs, has been key to the success of the US-pharma landscape. The draft guideline for individualised mRNA cancer vaccines proposed by the MHRA has a very similar spirit, regulate but invite new thinking and innovation.
However, the UK needs to remain aware that it does not regulate in isolation. To be a leader in this field, the MHRA should align with the EMA and the FDA. Divergent frameworks risk creating regulatory silos, complicating multinational development and slowing patient benefit. The MHRA’s boldness,
Regulatory and Compliance
therefore, must be matched with diplomacy. Anyone developing individual cancer therapies is well informed to look at related guidelines proposed by other authorities, like the FDA and EMA. The FDA's draft guidance for Analytical Procedures for mRNA Vaccine Quality has been a first out there to propose sets of detailed analytical methods designed to evaluate the quality of mRNA vaccines throughout all production steps. EMA has followed with a very similar approach, having divided their quality expectations into three chapters (5.36 mRNA vaccines for human use, 5.39 mRNA substances and 5.40 DNA templates), reflective of the three main production steps required for mRNA manufacturing. Although these documents do not reference individualised mRNA therapies, they have a stronger CMC focus and fix a shared vocabulary and baseline for release testing, impurities, potency and critical process parameters. If regulation is a two-storied house, the MHRA draft lives upstairs (clinical and regulatory pathway), while the new EMA chapters and the FDA draft guideline form the foundation (CMC and quality standards). All of these documents have shared scientific and technical understanding of mRNA-based therapies and should be read together for a successful mRNA development program.
But the greatest challenge for the success of these potentially lifesaving drugs is not rooted in regulatory oversight. It lies in the restoration of trust in science, scientists and scientific and pharmaceutical institutions. Legislators need to understand the scientific process and that in science, there is no black and white, and that answers to questions may change over time as more data emerges and the knowledge base grows. Scientists need to realise that this is difficult to understand for people outside of the scientific community, who are increasingly told that things can just be either black or white. Patients, patient groups and legislators need to be informed about potential benefits and risks (side effects) in a clear and unbiased language, outlining what we know and don´t know. And this is where this MHRA guideline may also have a part to play, taking the individualised mRNA therapies out of the corner of the Advanced Therapy Medicinal Products and away from gene and cell therapies, where concepts are difficult to grasp and therefore daunting for patients and their families. Giving individualised mRNA therapies their own label separately from ATMPs will make it far easier to communicate to all stakeholders
what mRNA vaccines are. What does individualisation in this context mean? What diagnostics and information are required to achieve individualisation? And lastly, the potential benefits as well as risks or side effects of mRNA cancer vaccines.
None of this guarantees that personalised cancer vaccines will fulfil their promise. Science is still wrestling with immune escape, tumour heterogeneity and the sobering reality that not all predicted neoantigens (tumour-specific proteins) drive durable responses. But the regulatory and quality scaffolding is finally catching up. Of course, regulatory authorities can play it safe and wait until the first individualised products are brought to submission and then, based on what they have been presented, start drafting a regulatory framework. Or, they can communicate to developers that they understand that individualisation of therapies is very different to how standard medicines are produced, controlled, distributed and regulated; they see the challenges faced and this is how they want those to be addressed. The MHRA draft guideline signals to companies trying to make individualised mRNA cancer therapies a reality that if you are able to industrialise personalisation, your submission will be evaluated by an agency that has competence and a clear vision to enable a fair process.
Dr. Steven Watt
After studying molecular biology at Bielefeld University and graduating with a PhD in genetics and molecular biology in 2005, Dr. Steven Watt was granted a position as an assistant professor at the department of proteome and metabolome research at Bielefeld University, where he was in charge of a mass spectrometry service unit. In 2008, he changed to industry working, as an instructor and consultant for accurate-mass high-resolution mass spectrometry applications in pharma. In 2010, he joined A&M STABTEST Labor für Analytik und Stabilitätsprüfung to establish a Business Development unit. During his time at A&M, Steven also established the cell-based bioassay operations. He currently holds the position of Managing Director / CBDO.
Regulatory and Compliance
Building IP Value in Bioinformatics
Bioinformatics sits at the convergence of computational methods, life sciences, diagnostics and data analytics. Advances in computational power and efficiencies and the increased capabilities of AI methods mean developments in genomics, diagnostics, computational biology and AI-driven drug discovery are driving forward at pace. Patenting in this field shows ever increasing activity, indicative of the commercially valuable nature of innovation in this field. However, with these emerging and rapidly developing technologies come intellectual property complexity and challenges. This article considers key patenting trends and patent protection considerations in bioinformatics.
What is Bioinformatics?
Bioinformatics is the science of using computational tools and methods to analyse biological data, particularly large datasets such as genomic sequences or protein structures. In the context of pharmacy, bioinformatics plays a vital role in drug discovery, development and personalised medicine by helping researchers identify new drug targets, predict how drugs will interact with specific genes or proteins and tailor treatments based on individual genetic profiles. As pharmaceutical research becomes increasingly data-driven, bioinformatics serves as a key bridge between laboratory science and clinical application.
Patent Filing Trends
Since 2000, the number of patent filings in bioinformatics has held steady at around 2000, with an increase from around 2010, which led to around 10,000 patent filings in 2022 and 2023. Of those filings, the number of granted patents has increased at a growing rate since 2000, with around 5000 patents granted in 2024.
The data shows a decrease in filings after 2023, which is likely an artefact due to patents filed but not yet published (typically a patent is published 18 months after filing), so not contributing to the data. Also, the data shows a decrease in the number of grants after 2024 because the data was captured partway through 2025.
Looking at where bioinformatics innovations are taking place and where patent protection is being sought, the highest number of patent filings in bioinformatics are in the jurisdictions of China, the USA, the European Patent Office and Japan, with a significant number of “International” applications being filed with the World Intellectual Property Organisation, which are then entered into national/regional jurisdictions after 30 or 31 months from the initial filing date.
It is clear that bioinformatics is a rapidly developing field of increasing importance wherein patent applications are filed to obtain commercial protection for innovations. Next, we consider how patent applications filed in Europe may be examined and what to consider to ensure robust patent protection.
Patent Examination in Europe at the Convergence of Life Science and Software – Is It Technical?
To be patentable, an invention must be novel (not known anywhere publicly before the filing date of the patent application) and inventive (not obvious from what is already known). In bioinformatics (and other software-related innovation), the patent application will also be examined to determine whether the invention falls into a category of exclusion from patentability. There are two main exclusion categories to consider when drafting patent claims:
1. Under Article 52(2) of the European Patent Convention (EPC), “discoveries, mathematical methods, programs for computers, and presentations of information as such” are excluded from patentability as being non-technical. The “as such” is important because if the invention includes technical features solving a technical problem, the invention can be framed to avoid being excluded from patentability. Thus, if the invention is an algorithm that processes molecular data, this may be excluded from patentability as software per se. However, if the invention is framed as a method of taking molecular data as input from real world measurements, processing the molecular data using the algorithm and providing a technical output, such as an indication of the likelihood of a drug being suitable to treat a medical condition, the invention is more likely to avoid an objection under Article 52(2) EPC.
Regulatory and Compliance
2. Under Article 53(c) of the EPC, “methods for treatment of the human or animal body by surgery or therapy and diagnostic methods practised on the human or animal body” are excluded from patentability. A key to avoiding an objection under Article 53(c) EPC is to understand what constitutes a diagnostic/therapeutic method practised on the human body vs. in vitro methods.
To avoid a bioinformatics invention falling under one, or both, of the exclusion criteria, the invention should be tied to computing hardware; to an in vitro step or method; operating on real world measured or sensed data; or yielding a concrete technical result.
Once the invention is accepted as being technical (and not excluded), the examiner will consider whether the invention provides an inventive step. Only features contributing to the technical character of the invention are considered when assessing inventive step; purely mathematical or algorithmic steps are not considered for inventive step. Again, careful patent application drafting can help support the case for the inventiveness of the invention by tying the computational steps to technical effects.
Practical considerations to support technical character include:
• Explicitly reciting hardware, computing environments, sensor use, laboratory automation or data acquisition devices.
• Providing technical rationale for inventive features, such as reduced memory requirements, improved accuracy, use of real world data inputs and provision of clinically relevant diagnostic outputs.
• Detailing non-obvious algorithmic improvements and tying them to technical effects.
For example, inventions relating to “diagnosing a disease from genomic data” or “determining abnormally methylated regions via an automated pipeline” can get over the “technical” hurdles, provided they are tied to the use of technical means, computer systems or laboratory equipment, rather than being interpreted simply as a statistical inference.
Trade Secrets and Confidential Know How
Patents offer territorial protection but involve public disclosure when the patent application is published. A patent application must contain enough description and detail that someone else working in that technological space can recreate the invention.
In bioinformatics, the computational factors involved in innovations, such as algorithms, training data, parameterisation, software pipelines and computational models, allow for IP protection to be crafted around combinations of patenting and trade secrets, while retaining some valuable know-how secret within the business.
For a trade secret to exist and receive legal protection in Europe, it must meet three key requirements:
1. The information must be secret. The information should not be publicly available or easily obtainable through legitimate means.
2. The information must have commercial value because it is secret.
3. The person lawfully in control of the information must have taken reasonable steps under the circumstances to keep it secret.
While the patent must disclose enough detail to allow someone else to implement the invention, it is typically not necessary to provide full disclosure of features of the innovation which themselves provide a competitive advantage. For example, while a predictive model for drug discovery must be described in enough detail in a patent application to allow someone else to implement the model, the actual training data set used need not be disclosed (though the overall characteristics of the training data set may be required if that leads to the technical advantage the invention provides). Similarly, while a regression algorithm may be used, the specific weighting parameters in the model may not need to be included in the patent application. In this way, the know-how involving your time and effort in determining particularly effective parameters and trained models may be retained in the business as a trade secret and add value, particularly if those details are difficult to reverse engineer.
Therefore, often a hybrid approach can be an effective way to protect bioinformatics innovations, patenting the overall technical method (e.g. an algorithm implemented in a diagnostic pipeline), while retaining the detailed model weights or data curation pipeline as trade secrets.
Strategic Framework: Deciding Patent vs Trade Secret
The following table summarises some factors to consider whether patenting or retaining a trade secret can be an appropriate form of IP protection for your bioinformatics invention:
Factor Patent Strategy Trade Secret Strategy
Disclosure level Full public disclosure
Legal exclusivity
Technical disclosure risk
Territorial, 20 years
Confidential, not publicly revealed
Potentially indefinite, but only if kept secret
Opponents can learn design Rivals may reverse engineer or independently develop
Enforcement Infringement proceedings can be brought
Suited for Novel technical architecture, overall functionality of algorithms
Conclusion
Breach of confidentiality/ confidentiality agreements
Proprietary training data, model parameters, curated datasets
Patenting bioinformatics inventions in Europe presents both opportunities and challenges. Many innovations in this space should be considered from the point of view of relating to excluded subject matter and mixed-type inventions of technical and non-technical features. Careful consideration of the innovation and crafting a patent claim set with potential patenting objections in mind from the outset can help ensure as robust a patent application as possible.
Alongside patenting, trade secrets can provide a powerful tool well-suited to specific details, typically which provide a
Regulatory and Compliance
competitive edge and which are difficult to reverse engineer, such as proprietary data assets or algorithmic details giving particularly accurate or advantageous results.
By combining robust patent portfolios with well-guarded trade secrets, companies can protect and extract value from their bioinformatics innovation across diagnostics, drug discovery, precision medicine and beyond.
Dr. Janine Swarbrick is a chartered UK and European Patent Attorney, specialising in patent protection for physics and computing technologies, with a special interest in software in healthcare. She works with a range of clients developing digital health technologies, and she co-leads the firm's MedTech, Digital Health and Bioinformatics sector. Before entering the IP profession, Janine was an experimental physicist using microscopies, synchrotronbased spectroscopies and computer modelling to understand electronic structures of molecular systems.
Dr. Sofie McPherson
Dr. Sofie McPherson is a Chartered UK and European Patent Attorney. She specialises in MedTech, Digital Health and Bioinformatics. She has significant experience in connected medical ecosystems in a range of medical applications, including training, diagnostics and treatments. Sofie has a degree in Materials Engineering in Medicine and a PhD in Biomaterials with a focus on the development and characterisation of synthetic bone grafts. She has practical and academic experience in tissue engineering, electron microscopy and material characterisation systems.
Roxna Kapadia
Roxna Kapadia is a UK and European Patent Attorney specialising in bioinformatics with a strong foundation in life sciences, particularly in microbiology and immunology. With expertise spanning computational biology, genomics and intellectual property law, she bridges the critical gap between cutting-edge bioinformatics research and patent protection strategies. Roxna brings a unique perspective to bioinformatics patent prosecution and portfolio development, combining technical understanding of biological data analysis, algorithm development and computational methodologies with comprehensive knowledge of European patent law.
Dr. Claire Green
Dr. Claire Green is a trainee within the Life Sciences team at HGF and specialises in patent protection for bioinformatics, computational biology and AI inventions. Claire has significant experience drafting and prosecuting patent applications for AI and computer-implemented inventions. Claire has a first-class degree in neuroscience, an MSc in clinical statistics and a PhD in bioinformatics. She has published and co-authored several papers in the field of bioinformatics, computational biology, genetics and neuroimaging.
Document, search, and collaborate
Scientific data management with new add-on functionality to
make discoveries faster
Generate, QC, and analyze results
Register entities to secure your assets
Keep track of entities
Deep learning and AI
Connect instruments, data, and tools
Manage and analyze experimental data
Assay informatics
Plot and mine results
Beyond Small Molecules: How Advances in 3D Modelling Are Opening New Frontiers in Macrocyclic Drug Discovery
While traditional small molecules excel at targeting buried active sites, they often struggle with flatter protein surface interactions. Macrocyclic compounds offer a promising alternative, binding with better affinity and selectivity to address previously "undruggable" targets. However, their larger size and structural complexity present unique computational challenges. This article explores how advanced 3D modelling capabilities enable more accurate prediction of macrocyclic conformations, providing practical considerations for effectively working with these promising therapeutic agents.
Macrocycles are an exciting class of compounds to tackle previously undruggable targets. In this article, we’ll explore how advances in computational modelling of macrocycles are unlocking efficiency improvements in macrocycle drug discovery that were previously only available for small molecules.
Beyond the Traditional Small Molecule Therapeutic Space For decades, Lipinski's Rule of Five has provided guidelines for the characteristics of compounds that are more likely to yield oral bioavailability.1 These include a molecular weight threshold of 500 Da, which has somewhat limited the exploration of chemical space by disfavouring larger molecules.
While small molecules excel at binding to deep, well-defined pockets within proteins, they struggle with broader, flatter surfaces, such as those that characterise protein-protein interactions.2 These are attractive therapeutic targets as they are fundamental to cell signalling and signal transduction.
Macrocycles, which are defined by ring structures containing 12 or more heavy atoms, bridge the gap between small molecules and biologics. They can bind to flatter, surface-level protein surfaces with high specificity, and despite surpassing the 500 Da threshold, can achieve good oral bioavailability. As a result, the industry has seen greater interest and investment in macrocyclic drug discovery.
Early concerns about metabolic stability were addressed by demonstrating that the constrained, cyclic architecture provides sufficient protection against proteolytic degradation.3 Additionally, strategic incorporation of non-natural amino acids can further enhance stability whilst maintaining biological activity.
Recent macrocyclic peptides that have been approved by the FDA for therapeutic use include rezafugnin (Rezzayo®), which is used to treat candidemia and invasive candidiasis in adults, and Lurbinectedin (Zepzelca®), for the treatment of metastatic small cell lung cancer.
The Computational Challenge: Why Size and Flexibility Matter
This same size and flexibility that makes macrocycles a promising therapeutic agent, also makes them much more difficult to model using traditional computational methods. With a higher number of rotatable bonds, macrocycles can adopt a vast number of 3D conformations. This flexibility cannot be ignored as it impacts every aspect of a molecule’s drug-like characteristics, including on- and off-target potency, and ADMET and physicochemical properties.
Traditional methods that work well for small molecules in modelling their 3D structure and how they bind to a protein target, as well as accurately predicting drug-like properties, falter here as they can’t efficiently capture this complex conformational landscape.
As a result, the predictions are either much less accurate, too slow or require an unfeasibly high computational cost. For practical drug discovery, we need modelling that is both accurate and fast enough to integrate into design-make-test cycles.
The Three Pillars of Accurate 3D Macrocycle Modelling
Recent advances have transformed our ability to accurately predict macrocyclic behaviour, making it possible to routinely use 3D macrocycle modelling in hit-to-candidate workflows.
1. Efficient, High-quality and Comprehensive Conformational Sampling
High-quality conformational sampling aims to characterise the biologically relevant ensemble of low-energy 3D conformations, rather than just finding the single lowest energy structure. The bioactive conformation often differs from the global minimum, representing a higher-energy state that is stabilised when the molecule interacts with its target.
Traditionally, conformational ensembles have been generated using templates or pre-calculated torsional libraries, which limit the speed and generality of those methods. However, modern approaches use physically intuitive molecular movements to explore a molecule’s conformational space without relying on rigid templates or pre-set torsional libraries. This allows for a more direct and unconstrained exploration of the chemical landscape based purely on molecular energetics in a tractable time and computational cost.
Such advances are highly transformative for macrocycles, and today we can routinely achieve conformational search accuracy for macrocycles that rivals what we see with smaller and simpler non-macrocyclic ligands.5 Many medicinally relevant macrocycles with up to 21–23 rotatable bonds, once thought “too big to handle”, can be explored efficiently at timescales compatible with modern drug discovery pipelines. As a result,
Research / Innovation / Development
conformational sampling is no longer a limiting step, but a practical tool that can be integrated directly into early drug discovery pipelines where speed and reliability are critical.
2. Leveraging Biophysical Restraints
While unrestrained computational searches can be fruitful for very large and complex peptidic macrocycles, it is time-consuming and identifying a conformation close to the bound form becomes increasingly challenging with the increase in size. In these cases, integrating biophysical restraints from techniques such as nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, or cryogenic electron microscopy (Cryo-EM) can help focus the search on biologically relevant regions of conformational space. These biophysical restraints bridge the gap between purely theoretical sampling, narrowing down the vast available space and increasing the likelihood of finding the bioactive conformation efficiently.
3. Understanding Molecular Strain
Another critical component of 3D macrocycle modelling is the accurate estimation of the bound ligand strain. This refers to the energetic penalty a molecule pays when its atoms or bonds adopt geometries in the bound state that deviate from their preferred, lowest-energy arrangement in the unbound state. While small, rigid molecules typically experience minimal strain differences in bound versus unbound states, the story is very different for macrocycles due to their size and flexibility.6
In the case of large macrocycles, their intramolecular interactions and geometries can be as significant as their ligand-protein interactions. As a result, even seemingly small
molecular modifications can drastically change the overall backbone of the molecule, shifting the global energy minimum and introducing additional strain in the bound conformation.
Deriving accurate strain estimates directly from Protein Data Bank coordinates, however, remains challenging. Traditional methods for fitting ligand poses to electron densities determined by X-ray crystallography often rely on a single static conformation, which can exaggerate strain by forcing atoms into geometries that fail to reflect the molecule’s intrinsic flexibility. Novel real-space refinement approaches address this limitation by integrating force field calculations with experimental data. By considering ensembles of low-energy conformations compatible with the observed electron density, these methods provide more realistic estimates of strain.7 Thus, combining experimental structural data with advanced computational refinement enables the discovery and optimisation of macrocycles with realistic, low-strain conformations, supporting more efficient and reliable macrocyclic drug design.
How Does a Better Understanding of Macrocyclic Flexibility Improve Drug Discovery?
These advances unlock computational capabilities previously reserved for small molecules:
1. Precision Molecular Docking
Molecular docking predicts how a ligand would fit into the binding site of the target protein and provides a score that can be used to prioritise or rank compounds for further study. It’s a valuable method for virtual screening and lead optimisation, while providing detailed insights into molecular interactions.
For large, flexible macrocycles, successful docking depends critically on accurately sampling ligand conformations, as discussed above. Starting with a diverse, low-energy ensemble of ligand conformations increases the likelihood of finding a geometry that is complementary to the protein pocket during docking. Additionally, it has been shown that refining confirmational searches with NMR or X-ray crystallography data gives a more accurate starting point for docking experiments, generating the correct bound poses of new compounds and thereby better estimating ligand strain and protein-ligand intermolecular binding energy.
Combining improved conformational search with the intelligent use of prior structural knowledge and enhanced scoring functions achieves pose prediction accuracies rivalling those for small molecules. Understanding the binding modes of compounds guides the design of new analogues with better binding affinity, reducing the number of compounds that must be synthesised and tested.
Furthermore, combining conventional docking scores for protein-ligand interactions with a rigorous assessment of strain enables active compounds to be identified and inactive compounds rejected prior to synthesis and testing. Illustrative applications have shown that this can save up to 90% of compound synthesis and testing.8
Moreover, understanding whether activity is driven by binding interactions with the protein or molecular strain informs strategies for further optimisation.
Figure 1. Conformational sampling of macrocyclic peptide, Aureobasidin A. As the size and flexibility of a macrocycle increases, the number of accessible conformers grows exponentially.
Figure 2. NMR-derived restraints used to narrow down the conformational search space of Aureobasidin A. Such restraints help to increase the likelihood of obtaining a bioactive conformation within the pool.
Research / Innovation / Development
2. Enhanced Ligand-based Virtual Screening
3D ligand-based methods are based on the principle that molecules that adopt similar shapes and interactions in three dimensions are likely to show similar biological activities. Ligand-based methods are particularly valuable when a 3D structure is not available for the target protein, meaning that structure-based approaches such as docking cannot be applied. However, ligand-based methods can also complement structurebased virtual screening; the combination often provides better results than either individual approach.10
While the size, flexibility and complex conformational landscapes of macrocycles have traditionally made 3D similarity searches challenging, we can now effectively incorporate conformational ensembles and strain estimates to evaluate multiple accessible conformations. This dramatically increases the likelihood of identifying conformations that engage the protein effectively, even when screening chemically diverse molecules that can adopt alternative conformations to preserve key interactions.
The integration of experimental data further enhances these capabilities. Conformational restraints, derived by exploiting NMR data to identify low-energy solution ensembles of a lead compound, can be used to focus ligand-based molecular similarity optimisation. This approach has proven highly effective for predicting bound poses and prioritising lead-compound analogues.8
By moving beyond single, lowest-energy conformations to comprehensive ensemble-based approaches that consider shape, electrostatics and pharmacophoric features across multiple poses, these methods now identify active compounds that would have been missed by traditional single-conformation screening.
Summary
Macrocycles provide a promising solution to a key challenge in drug discovery, addressing protein targets with large, flat
Schematic diagram showing how NMR-restrained macrocyclic conformational preferences can be exploited for enhanced binding pose prediction, either through structure-enabled molecular docking or ligand-based similarity screening experiments. Taken from Jain et al. 2023 (CC BY 4).8
binding surfaces that traditional small molecules cannot effectively modulate. These ring structures offer the binding affinity and specificity of biologics, whilst maintaining the potential for oral bioavailability and cell permeability.
Computational modelling of macrocycles has evolved from an intractable problem to a practical reality through three
Figure 3.
Research / Innovation / Development
critical advances. Firstly, modern sampling methods can now accurately and efficiently explore the complex conformational landscapes of these flexible molecules. Secondly, accurate strain estimation allows us to better understand and predict the energetic costs of binding, preventing wasted synthesis efforts on molecules that appear promising but would require prohibitive conformational changes. Lastly, the ability to effectively integrate experimental data from NMR, X-ray and cryo-EM provides real-world constraints that make it possible to model even particularly large and complex macrocycles.
These capabilities now enable drug discovery teams to apply the full toolkit of ligand and structure-based computational methods to macrocycles, to prioritise optimal compounds for synthesis and testing. We’re now able to achieve similar accuracy as with predictions for small molecules, and importantly, with sufficient speed and efficiency to incorporate into a routine drug discovery workflow.
REFERENCES
1. Lipinski, C. A.et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 (1997).
2. Dougherty, P. G., Qian, Z. & Pei, D. Macrocycles as protein-protein interaction inhibitors. Biochem. J. 474, 1109–1125 (2017).
3. Khatri, B., Nuthakki, V. R. & Chatterjee, J. Strategies to enhance metabolic stabilities. Methods Mol. Biol. 2001, 17–40 (2019).
4. Du, Y. et al. FDA-approved drugs featuring macrocycles or mediumsized rings. Arch. Pharm. 358, e2400890 (2025).
5. Cleves A.E and Jain, A N. ForceGen 3D structure and conformer generation: from small lead-like molecules to macrocyclic drugs J Comput Aided Mol Des. 31(5), 4190439 (2017)
6. Jain, A. N. et al. A Distributional model of bound ligand conformational strain: from small molecules up to large peptidic macrocycles. J. Med. Chem. 66, 1955–1971 (2023).
7. Jain, A. N. et al. XGen: Real-space fitting of complex ligand conformational ensembles to X-ray electron density maps J Med Chem. 63(18), 10509-10528 (2020)
8. Jain, A. N. et al. Complex peptide macrocycle optimization: combining NMR restraints with conformational analysis to guide
structure-based and ligand-based design. J. Comput. Aided Mol. Des. 37, 11 (2023)
9. Cleves, A. E., Tandon, H. & Jain, A. N. Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands. J. Comput. Aided Mol. Des. 38, 33 (2024).
10. Cleves, A. E. & Jain, A. N. Structure- and ligand-based virtual screening on DUD-E+: performance dependence on approximations to the binding pocket. J. Chem. Inf. Model. 60, 4296–4310 (2020).
Matthew Segall
Matthew Segall, CEO, Optibrium, has an MSC in Computation from the University of Oxford and a PhD in Theoretical Physics from the University of Cambridge. He has led teams developing predictive models and intuitive decision-support and visualisation tools for drug discovery and has published over 40 peer-reviewed papers and book chapters. In 2009, he founded Optibrium, which develops ground-breaking AI software and services, that improve the efficiency and productivity of drug discovery.
Email: matt@optibrium.com
Himani Tandon
Himani Tandon, Principal Scientist, Optibrium, works in the research division at the Company, developing cutting-edge software solutions that support small-molecule and macrocycle design in drug discovery. Her work focuses on applying 3D structure-based and ligand-based design strategies for lead discovery and optimisation. Himani holds a PhD. in Computational Structural Biology and Bioinformatics from the Indian Institute of Science and completed her postdoctoral research at the MRC Laboratory of Molecular Biology.
Email: himani@optibrium.com
Comparing Organoids and Patient-derived Xenograft Models for the Development of Antibody-drug Conjugates
Antibody-drug conjugates (ADCs) are one of the most rapidly expanding forms of oncology treatment and therapeutic successes have led to an unprecedented expansion in the number of ADCs in development, with over 200 currently in clinical trials.
While ADCs represent an exciting new advance, the attrition rates for potential drug candidates remain particularly high at every stage of development. Additionally, progress has been far from linear, with several ADCs gaining approval only to be withdrawn and then reapproved. One of the reasons for this is the complexity of these therapeutics, both in terms of their multiple components and their complicated, multi-step mechanisms of action.
To overcome these challenges and maximise the potential of this promising new drug class, sophisticated preclinical testing platforms are essential. In recent years, organoids and patient-derived xenograft (PDX) models have emerged as the gold standards of ADC development. Each model type offers distinct advantages and limitations, which can be leveraged or mitigated by using the most appropriate models during different preclinical stages. Understanding how to effectively utilise these models together is foundational to the successful development of ADCs.
Utilising Organoids in ADC Development
Organoids are three-dimensional cellular structures that retain the form and function of tumour structures. Recent advances in organoid technology have been shown to support more complex, rigorous and informative validation of ADC performance ahead of larger, more expensive clinical trials. Organoids are particularly appropriate for:
Mechanism of Action Testing
ADCs operate through complex, multi-step processes involving multiple components, so the ability to isolate and analyse each aspect in isolation is essential to understanding why certain ADCs succeed. As organoids offer a controlled environment and a more complex and physiologically relevant system than two-dimensional cell cultures, they are well-positioned to support fundamental mechanisms of action studies. This enables teams to focus on individual processes, including binding, internalisation and payload release, to understand the subtle mechanisms within ADCs that determine their success or failure.
Large Panel Screening
During the early stages of ADC development, high-throughput capabilities are essential, allowing developers to assess dozens of different antigen, antibody and payload combinations so they can find and prioritise the most promising combinations. Advanced organoid culturing methods support increased
scalability, unlocking large panel screening at relatively low costs. Many different models can be tested simultaneously, so data can be generated rapidly. Therefore, organoid models can reduce timelines, widen the scope of preclinical testing and increase the chances of study success.
Bystander Killing Effect Visualisation
The bystander killing effect is a key feature of ADCs, as it allows released payloads to not only kill the tumour cells they specifically target, but also neighbouring tumour cells. As patientderived organoids preserve the heterogeneity of different cell populations found within tumours, they allow researchers to visualise, analyse and better exploit this important tactic. This has the potential to improve the efficiency of ADCs and extend their therapeutic effect so that tumours with heterogeneous antigen expression can be effectively treated.
Binding Studies
Multiple factors and processes play a role in the binding action between the antibodies within ADCs and the antigens they target within tumour cells. This process is both complex and central to improving the potential of ADCs in development. Organoids mimic tumour tissue architectures, so they can effectively recapitulate the extracellular matrix and important cell-cell interactions, helping research teams to better understand and optimise ADC penetration and delivery.
The Benefits of Using PDX Models During ADC Development
However, as organoids are usually organ-specific, they cannot fully reflect the systemic effects of diseases or treatments that occur within patients. For example, they can’t be used to model the role of systemic immune responses, which are involved in both ADC efficacy and toxicity. Additionally, these models are limited to on-site studies only, making them unsuitable for evaluating interactions between circulating drugs, healthy tissues and off-target binding sites. Using PDX models alongside organoids can help research teams overcome many of the limitations of organoid models by providing patient-relevant contexts that recapitulate the real behaviour of ADCs.
Specifically, PDX models play an important role within ADC development during:
Biodistribution Studies
Biodistribution studies, which assess how ADCs distribute through both tumours and the body, are vital to our understanding of ADC efficacy, safety and optimisation. PDX models are considered to be the most effective preclinical model to replicate intratumour heterogeneity and preserve intrinsic tumour architectures, which is why they are widely employed in pharmacokinetic and pharmacodynamic exploration. PDX models are therefore the most suitable model for investigating whether ADCs reach their target effectively or cause intolerable toxicity. In turn, this informs dosing strategies and the design of more effective and safer next-generation ADCs.
Research / Innovation / Development
Off-site Toxicity Studies
Off-site toxicity studies evaluate both off-site, on-target toxicity (which occurs when ADCs bind with the target antigen in healthy tissue) and off-site, off-target toxicity (where ADCs bind to healthy tissues that don’t demonstrate the target antigen). These studies are particularly important, as excessive toxicity and unfavourable risk-benefit profiles are a leading cause of ADC failure. This highlights the importance of clinically relevant models like PDXs.
Immunocompetent and Immunosuppressed Studies
Immunocompetent models retain a functional immune system, so developers can use them to investigate the ways ADCs interact with immune cells, unlocking studies that combine ADCs and immunotherapies. While immunosuppressed models are established by transplanting tissue into immunocompromised mice, teams can study the mechanisms of resistance seen in ADCs and investigate their cytotoxic effect on tumour cells directly.
Despite their strengths and numerous applications, PDX models are not without their limitations. For example, they incur
significantly higher costs than two-dimensional cell cultures or organoids. Additionally, they take longer to develop and are more labour-intensive, so they cannot be used for large-scale screening.
Maximising the Potential of Organoid and PDX Models During ADC Development
To maximise the potential and overcome the inherent drawbacks of different model types, complementary models should be employed at different stages of preclinical development. PDX and organoid models can be further enhanced with the application of sophisticated bioinformatic approaches.
Traditional biomarker approaches rely on measuring average target expression across entire tumours, which misses crucial spatial patterns that dictate whether ADCs can effectively reach their targets. Spatial biomarker analysis has recently emerged as a revolutionary new technology that fuses molecular biology and advanced imaging tools . Spatial profiling reveals the significant variation in ADC concentration and effectiveness caused by tumour heterogeneity. As a result, the unique characteristics of individual tumours are now being
Research / Innovation / Development
centred in oncology research, changing treatment design and the way patients who are most likely to benefit from treatment are identified.
Advances in co-expression analysis represent another exciting area of research in oncology. Co-expression patterns, where groups of genes are turned on or off together in the same cells or tissues, can indicate that these genes work together in biological pathways. Co-expression analysis identifies opportunities to exploit shared patterns in cancer cells via coordinated targeting. Avoiding co-expression patterns is also a fruitful tactic, as identifying co-expression seen across tumours and healthy cells can help developers avoid combinations that would cause intolerable toxicity.
AI is being called a game-changing technology and it is set to transform the future of precision medicine. While the full scope of its potential remains to be understood, drug developers are already adapting their approach to data collection with AI in mind. Teams are increasingly focused on capturing comprehensive information from every piece of tissue during development, so that this data can be fed into ever more powerful AI systems in the future to identify subtle biomarkers that cannot currently be isolated.
These advanced bioinformatics are also informing clinical trial patient selection, as advanced preclinical findings can be integrated with clinical biomarker strategies. This helps research teams identify patient populations that are most likely to respond to treatments where toxicity poses the lowest risk. By improving patient selection in this way, clinical trial success rates can be improved while regulatory approval is accelerated.
Conclusion
The complexity of ADCs has been described as a “double-edged sword” as it is key to both the challenge they pose to developers and the scope of their potential. As each ADC comprises four key components (target antigen, targeting antibody, chemical linker and cytotoxic payload) which can be optimised independently, there are countless opportunities for evaluation and optimisation. This is impossible without the application of advanced preclinical models, which allow researchers to
identify promising candidates and prioritise those that show the most efficacy without unacceptable toxicity.
By taking a strategic approach, organoid and PDX models can be utilised in a complementary way to maximise their strengths and mitigate their limitations. Organoids can be utilised for mechanistic characterisation and high-throughput screening to prioritise candidates, while PDX models offer a patientrelevant platform that bridges the gap with clinical settings. These models can be further optimised with sophisticated bioinformatics approaches to improve patient outcomes and reduce preclinical timelines.
Benjamin is a Product Marketing Manager at Crown Bioscience. His role spans from product management to product marketing with an emphasis on scientific content development. Benjamin's background specialises in in vitro advanced cell models, including organoids, organ-on-a-chip and bioprinting for preclinical research.
Benefits of Mass Spectrometry from Process Development to GMP Release of Biomolecules –A Comprehensive CDMO Perspective
In biopharmaceutical production, a thorough understanding of analytical processes and target molecules is essential for ensuring patient safety, as well as maintaining consistent and reliable product quality. Mass spectrometry (MS) represents a powerful toolbox to assess this knowledge and partially even compensates the need for a bundle of methods covering different analytical parameters due to its highly versatile applications. While mass spectrometry is often applied in the developmental phase, its use in biopharmaceutical routine testing is often hampered since requirements for specialised knowledge and GMP compliance are significant hurdles.
At Richter BioLogics, a leader in the microbial Contract Development and Manufacturing Organisation (CDMO) space, these hurdles were taken, opening the way to support development, production and release of biopharmaceutical products at multiple levels with an ESI-TOF (Electrospray Ionisation – Time Of Flight) based LC-MS (Liquid Chromatography-MS) system as a single device, beginning with characterisation of upstream process components up to GMP compliant release testing of large biomolecules.
The implemented ESI-TOF LC-MS system thus covers a wide range of applications over the whole product lifecycle, providing additional benefits to complete the analytical portfolio.
“Knowing your molecule” is a pivotal requirement in biopharmaceutical industries to ensure high product quality and patients’ safety. Understanding the properties and critical quality attributes (CQAs) of target molecules and their behaviour throughout the production processes allows decision about and setup of a suitable analytical panel for product characterisation, which provides the basis to deliver complex biomolecules with highest quality and sufficient yields by the most efficient processes in biopharmaceutical production. The need to characterise production processes and target molecules is defined in ICH Q141, stating that process and product understanding should be the first step in the analytical lifecycle. In biopharmaceutical production, molecules and processes are therefore characterised by a broad analytical portfolio from development to final release testing, covering analytical parameters such as content, potency, identity, purity, process and product-related impurities, which are generally addressed using various analytical equipment and techniques.
The analytical panel may vary throughout a product`s life cycle, depending on the complexity and aim of sample characterisation. Especially in the development phase, LC-MS (liquid chromatography mass spectrometry) is often applied
from Richter BioLogics
for purity evaluation and characterisation, since it allows direct insights into the molecular composition of formulated bulk, in-process samples and its impurity profiles, and thus represents a suitable tool to gain product and process knowledge in a timely and efficient manner.
One of the most striking benefits during this phase is the identification of the product as well as product-related variants and modifications on the molecular level at various process steps and under different process settings. Thus, LC-MS represents a valuable tool, not only to optimise processes, but also to gain knowledge on product quality characteristics and main impurities as well as degradation pathways, which can support the definition of critical quality attributes (CQAs) and the set-up of an analytical panel. Furthermore, LC-MS provides valuable information upon implementation and development of a wide panel of LC methods, opening the opportunity to identify peaks and to verify peak purity. The obtained qualitative data are beneficial to understand the product on a molecular level, identify CQAs and gain insight into process understanding.
Furthermore, process understanding can be improved using the LC-MS system as a semi-quantitative tool on a small molecule level at the very beginning of process optimisation, the upstream processing (USP), to improve product quality and yield.
Besides these scientific advantages, LC-MS analyses can be performed within short time frames and even compensate the need for a wide analytical panel for process and product characterisation. Due to this, LC-MS provides significant benefits for the improvement of time and costs during development and production.
Despite these advantages applied for development purposes, the impact of LC-MS-based characterisation in a GMP-related context is often limited due to a constrained compliance of equipment, software and methods. Moreover, most mass spectrometry devices require a specialised team with relevant expert knowledge to develop and validate the required LC-MS methods. Nevertheless, the capabilities that LC-MS offers in this field are no less valuable than in areas of development and can provide a versatile tool in fields of method validation, release testing, reference standard characterisation, as well as stability studies, as soon as the GMP-relevant aspects are fulfilled. In this context, the recently updated ICH harmonised guideline Q2(R2) for validation of analytical procedures sets the validation design for quantitative LC-MS methods, further promoting LC-MS in GMP applications and reducing efforts and, thereby, costs for GMP-compliant implementation.
The following chapters provide exemplary insights into the individual applications of LC-MS analyses as well as the GMP readiness process.
Application Note
Application of LC-MS in Upstream Bioprocess Development (Media Screening)
The upstream process (USP) at the very beginning of biotherapeutic production already sets the course for successful mid- and downstream processing, impacting yield, as well as quality of the final biopharmaceutical product. Thus, optimisation of the fermentation during USP, including, for example, feed media composition and feeding strategies for improved cell health and growth, as well as product titre and quality, has an essential impact on the final product.
At Richter BioLogics, the acquired LC-MS system has been established for quantitative monitoring of key nutrients and metabolites (e.g. amino acids and vitamins) in cell culture media and supernatants following the LC-MS system manufacturer’s application notes and amino acid/cell culture standard. While common USP optimisation strategies often rely on general aspects, such as cell growth curves, gas and carbon source profiles and product titre, the additional monitoring of specific components of cell culture supernatants, which remain uncertain in common approaches, can significantly speed up USP optimisation by elucidating critical key components.
Cell culture components are identified by their measured retention time and mass on the high-resolution ESI-TOF mass spectrometer in full scan mode and small molecule acquisition range (50–800 m/z). This allows, for example, for the quantification of isobaric components, such as Leucine and
Isoleucine (see Figure 1, a and b). Cell culture supernatants are diluted appropriately and the calibration curve is set up based on the manufacturer’s instructions. Independent of the calibration curve unit as amount or concentration (see Figure 1, c), the LC-MS software provides the relevant output (e.g. molar concentration of a component in the undiluted sample). Quantification of key components over time and by direct comparison between fermentation runs delivers valuable knowledge for USP optimisation, which is also graphically supported by the LC-MS software (see Figure 1, d).
As illustrated in Figure 1, LC-MS is a method which can selectively identify and quantify target cell media components in complex cell culture supernatants, revealing differences in their levels of consumption throughout fermentation processes.
Accordingly, LC-MS allows for a more target-oriented and streamlined development of early biotherapeutic processes, consequently leading to improved yield and quality of the final product in a time- and cost-efficient manner.
Application of LC-MS in Downstream Bioprocess Development
The benefits of LC-MS analyses to give direct insights into molecular details of samples and provide additional process knowledge were successfully applied at Richter BioLogics by identifying unknown peaks observed during downstream process monitoring. The respective species was observed in
quantification in two comparative fermentations (USP1 and USP2) performed with different cell culture media. a, Zoom of an extracted ion chromatogram (XIC) of isobaric amino acids Ile and Leu; b, Mass spectrum of Ile (protonated form in positive polarity and isotope peaks highlighted
c, Calibration curve of Ile; d, Trending plot bar chart of Ile responses for selected injections (calibration curve, samples of USP 1 and 2).
Figure 1: Media Screening examples for Isoleucine
in green);
Application Note
analytical UV-based RP-UPLC of in-process samples during the optimisation of several process parameters. Since the nature of this species and its impact on process-accompanying content determination by UV-based RP-UPLC was unknown, further characterisation was required. To this end, the UV-based RP-UPLC method was transferred to the ESI-TOF LC-MS system and samples of interest were analysed in intact mass modus (high range covering 400–7000 m/z). Deconvolution by the MaxEnt1 algorithm of mass spectra, averaged at peak maxima, revealed average molar masses of relevant species observed in analytical RP-UPLC. Based on these data, the relevant species was identified as a truncated isoform and its impact on content determination was assessed accordingly. The identification process of unknown peaks in RP-UPLC is illustrated in Figure 2.
LC-MS analysis provided the basis for a time- and cost-efficient manner by targeted and scientifically based development. Thus, this case illustrates the power and advantages of LC-MS at early stages of process development, which are equally suitable and transferable to other questions in process development.
Besides its application for identification purposes, LC-MS can also be applied as a valuable tool to sensitively quantify processrelated impurities. An LC-MS workflow for quantitation of residual albumin was developed at Richter BioLogics on the ESI-TOF LC-MS based on intact mass modus (high range covering 400–7000 m/z), covering a linear range of 2–50 ng albumin. Optimisation of the method involved the gradient, quality of reagents and consumables, detector settings and evaluation procedures based on total, as well as extracted ion chromatograms (TIC, XIC). The accuracy of the method was verified using different control samples.
The implemented method opens the possibility to cover residual albumin quantification during development, but also in release testing of respective APIs in a fast, sensitive and specific manner.
Requirements for the Operation of Mass Spectrometry in a GMP Environment
The power of LC-MS for analytical support during the developmental phase is based on its inherent analytical advantages to directly assess knowledge about product characteristics and product-related impurities, making it a versatile tool to report purity as well as identity data for a wide panel of samples and studies. These advantages are, in principle, equally applicable for product evaluation in a GMP-related context. Nonetheless, despite the versatile scientifically sound application options, usage of LC-MS methods in a GMP environment can be limited since application of instruments and techniques in a regulated environment requires defined prerequisites regarding equipment, software and methods.2,3 These prerequisites include aspects of instrument and software qualification, data integrity concepts, as well as training of operators and reflect a complex setup especially for LC-MS systems.
Analytical systems with GMP-relevant applications must fulfil several requirements. First of all, documented evidence must be in place verifying the suitability of the system for its intended purpose with regard to instrumental settings, IT infrastructure, data integrity and related documentation. These requirements are documented and verified during the qualification process, including phases of user requirements definition (URS), design (DQ), operational (OQ) and performance qualification (PQ).
Figure 2: Illustration of the impurity characterisation process. a, Zoom of UV-based chromatogram obtained by RP-UPLC; b, 3D Mass spectrum components observed in RP-UPLC; c, Averaged raw mass spectrum of main peak observed in RP-UPLC; d, deconvoluted mass spectrum of main peak observed in RP-UPLC
Furthermore, a detailed description of the system, the workflows, maintenance and qualification, and a review of analytical and metadata must be in place and QA (quality assurance) approved prior to GMP usage.
As the first step in extending Richter BioLogic’s analytical portfolio using mass spectrometry, we defined and documented the user requirements (URS) for the planned LC-MS system according to our internal SOPs. The following aspects were covered:
• Technical prerequisites
• Requirements regarding instrumental settings and ranges for all LC-MS modules
• Requirements related to the infrastructure
• Provider of the instrument
• Provision of documentation
• Quality system and quality management of the provider
• IT aspects
• Electronic records and signatures
• User profiles
• Data storage
Based on the above-mentioned URS documentation and prerequisites, an LC-MS ESI-TOF equipped with a software generally compliant with 21 CFR part 11 was chosen as a candidate and evaluated regarding compliance with the requirements in
the process of design qualification. All aspects of the URS were subsequently verified during operational and performance qualification either by the external provider or by internal assays.
Additionally, a working instruction was issued covering and describing the following aspects in a detailed manner:
• Description of the LC-MS system and its IT environment
• Description of analytical procedures
• General chapter
• Workflows describing intact mass as well as peptide (mass) mapping analysis
• Requirements for data reporting
• Requirements for data review, including data integrity aspects
• Administrative aspects
• Definition of policies
• Folder structure
• Audit trail settings
• User profiles and user privileges
• Data storage and back-up
• Aspects of qualification and maintenance
Based on the above-mentioned documentation, GMP compliance of the LC-MS system with regard to instrumental settings and ranges, as well as performance and review of analyses, data integrity and administration is ensured.
Application Note
Application of LC-MS in GMP-relevant Aspects
Successful verification and documentation of instrumental and software parameters, as well as available and clearly defined procedures for application of an LC-MS system, paves the way for a broad panel of LC-MS applications in GMP-related topics.
For example, the benefits of mass analyses are essential for the characterisation and release of reference standard material, since primary reference standards used in the QC environment of API (active pharmaceutical ingredient) production are recognised as presenting the highest metrological qualities with their property values accepted without reference to other standards (see Ph. Eur. 5.124). For their qualification (as well as batch comparison in the case of standard replacement), they are extensively characterised (see ICH Q72), which generally includes additional analyses such as determination of molecular weight, primary sequence, N-/C-termini, (post-translational) modifications and higher order structure (e.g. tertiary structure by disulphide bridging). As these parameters can be addressed by intact mass and peptide (mass) mapping workflows, LC-MS essentially contributes to primary reference standard qualification.
Moreover, OOX events such as out-of-specification, expectation or trend occasions have to be clearly documented and thoroughly investigated in a GMP environment.5,6 LC-MS can provide valuable information for the investigation process, complementing the insights gained by the concerned method and contributing to the final conclusion of the OOX procedure.
Furthermore, intact mass as well as peptide (mass) mapping analyses represent powerful techniques to identify and report CQAs for batch release analyses of biopharmaceutical products, covering purity and identity quality attributes, providing direct insights into product quality on a molecular level. Especially, simultaneous detection and quantitation of product-related impurities in formulated bulk samples enables LC-MS analyses to be a suitable tool for monitoring sample purity over time, providing a solid data set for evaluation of stability studies.
Summary
Mass spectrometry presents a high-resolution, sensitive and extremely flexible and diverse technique, gaining increasing influence in the biopharmaceutical industry, particularly within the CDMO market, where Richter BioLogics stands out as one of the leaders. Although its application in a GMP environment requires comprehensive activities such as system qualification and compliance in data integrity, the advantages of utilising this technique, once established under GMP, are striking; process development; production and release of biopharmaceutical products guided at multiple levels throughout the entire process and product life cycle; covering small to large biomolecules and various analytical parameters in a time-and cost-efficient manner. Beginning with upstream process optimisation up to GMP-compliant release testing of large biomolecules, mass spectrometry efficiently increases product quality and thus, patient safety.
REFERENCES
1. International Council for Harmonisation (ICH): ICH Q14 – Analytical procedure development, 12.11.2023, accessed 07.08.2025 from www.ema.europa.eu/en/documents/scientific-guideline/ich-q14-
2. International Council for Harmonisation (ICH): ICH Q7 – Good Manufacturing Practice for Active Pharmaceutical Ingredient, accessed 07.08.2025 from www.ema.europa.eu/en/documents/ scientific-guideline/ich-q-7-good-manufacturing-practice-activepharmaceutical-ingredients-step-5_en.pdf
4. European Pharmacopoeia, Chapter 5.12. Reference Standards. Ph. Eur. Suppl. 11.8. Strasbourg, France: Council of Europe; 2025.
5. EudraLex – Volume 4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Part I: Basic Requirements for Medicinal Products. Chapter 6: Quality Control. Brussels: European Commission; Revision March 2014, accessed 07.08.2025 from https://health.ec.europa.eu/document/download/ c74c8720-27bf-4252-808f-d65a206a90bb_en
6. Laboratory Management Guidance: Out of Expectation (OOE) and Out of Trend (OOT) results, ECA Analytical Quality Control Working group; 2015.
Dr. Maja Erdmann
Dr. Maja Erdmann is a biochemist and senior scientist in the LC department of Richter BioLogics, specialised in LC-MS analyses of biopharmaceutical products.
Daniel Goetz
Daniel Goetz is a pharmaceutical biotechnologist and scientist in the LC department of Richter BioLogics, focusing on LC-MS analyses of biopharmaceutical substances.
Dr. Daniela Stummer
Dr. Daniela Stummer is a chemist and senior scientist in Department “Separation Techniques” at Richter BioLogics, specialised in UV- and MS-based LC analyses of biopharmaceutical products.
Dr. Ingo Goldbeck
Dr. Ingo Goldbeck is a biochemist and Head of Department “Separation Techniques” at Richter BioLogicsGmbH.
Richter Biologics is your professional and experienced partner offering CDMO solutions from gene to product all from one source.
Richter Biologics: expert for late stage and commercial production.
RICHTER BIOLOGICS
Suhrenkamp 59, 22335 Hamburg, Germany
Phone: +49 40 55290-801
www.international-biopharma.com YOUR PRODUCT — OUR COMPETENCE AND DEDICATION FOR MORE THAN 35 YEARS!
BusinessDevelopment@richterbiologics.eu
Lipid Nanoparticles (LNPs) for Nucleic Acid Delivery
Lipid nanoparticles (LNPs) are one of the most prominent tools for nucleic acid delivery. Their versatility allows the encapsulation of different cargos (siRNA, miRNA, AONs, mRNA, DNA) to address multiple disease models (cancer, infections, neurodegenerative disorders, etc.). Herein, we summarise some of the main applications of LNPs and the latest developments in the encapsulation and delivery of different nucleic acids.
Lipid Nanoparticles
LNPs is a broad term encompassing different lipid-based structures. Herein, we will present a brief history of the evolution that led to the development of LNPs for COVID-19 vaccines.
Liposomes were one of the first nanoparticles used for drug delivery. They can be of different lamellarity and sizes (~ 50–250 nm for drug delivery) and can encapsulate both hydrophobic and hydrophilic molecules. Liposomes provide high loading capacity and biocompatibility. However, they remain fragile (sensitive to hydrolysis and oxidation), with a rapid circulation time due to their interactions with high- and low-density lipoproteins (HDL and LDL). To overcome these barriers, extensive work on the physicochemical properties of liposomes has been conducted over the years, which has revealed critical parameters regarding lipid-based delivery systems (Hald 2022):
• Cholesterol reduces the transfer of phospholipids to HDL, thereby increasing the residence time of liposomes in plasma, while also being critical to the structure of the LNP.
• LNPs with saturated fatty acyl chains are more stable in the blood than unsaturated ones; however, this stability must be balanced against the importance of unsaturated chains in fusogenicity.
• Particle size influences LNPs' stability, with smaller vesicles having longer half-life compared to larger ones.
• Net charge of LNPs influences their half-life, with negatively charged liposomes being less stable than neutral liposomes, whereas positively charged particles are more toxic and quickly removed from circulation.
As a result, liposomes have been successfully used in clinical trials, albeit for small drug delivery only (i.e. daunorubicin, doxorubicin). However, despite improvements, liposomes remain unstable in vivo as they bind to other serum components. To limit this phenomenon, liposomes can be coated with inert molecules by introducing PEGylated lipid into liposome preparations, creating “Stealth” liposomes for drug delivery. Yet, these systems are only suitable for the delivery of small drugs (Doxorubicin, DOXIL®).
First trials of nucleic acid encapsulation were performed using cationic lipids and new synthetic lipids, such as DOTMA
and DOTAP (Felgner, 1987). These are still being used in vitro along with DOPE (Lipofectin).
Despite their efficiency for entrapment of nucleic acids, these systems (and their derivatives) did not enter the market due to their toxicity, unspecific interactions with extracellular components and overall short half-time. This led to the development of ionisable lipids, a novel class of lipids which present an electrostatic charge depending on the pH (Mrksich 2024). Ionisable lipid nanoparticles (Hald 2022) combine ionisable lipids with a lesser proportion of structural lipids (DOPE or DSPC often) and PEGylated lipid, and provide the advancements mentioned above, including high cholesterol content (for stability, structure and ApoE-mediated endocytosis).
All these advancements, along with the optimisation of nucleic acid sequences (see below), supported the use of messenger ribonucleic acid (mRNA) for vaccine purposes, introduced in the 1990s (Martinon 1993) and further developed by companies such as CureVac, BioNTech and Moderna in the 2000s–2010s for vaccines against cancers. The COVID-19 pandemic accelerated the introduction of mRNA-based vaccines into the market.
Following these successes, the field of LNP-based delivery of nucleic acid has grown exponentially. The design and screening of efficient novel ionisable lipids is a key parameter to further develop nucleic acid delivery technology and improve endocytosis, one of the main limiting factors. Recently, design of experiment (DoE) approaches have also emerged as important tools for high-throughput optimisation of LNP formulations (Cui 2022), allowing researchers to screen for key parameters such as PEGylated lipid types and ratios, constituent ratios, or mixing parameters.
Due to their lipidic nature and high cholesterol content, LNPs tend to accumulate in the liver, limiting their use for extra-hepatic therapies. The introduction of SORT systems (Selective Organ Targeting) allowed scientists to target LNPs to the lung, spleen or liver, depending on lipid type (typically: cationic lipid for the lung, anionic lipid for the spleen and ionisable lipids for the liver). The SORT system was further developed to target nucleic acid to the kidneys (siRNA). The screening strategies mentioned above were also used to design formulations targeting endothelial cells and the pancreas. Other studies have demonstrated LNPs’ ability to transfect cardiac cells, even with low specificity.
Conjugation of peptides or antibodies onto LNPs appears to be a potent method for delivery of nucleic acid to specific tissues and organs, including the lungs (PECAM-1 Ab), T-cells (CD5-Ab), bone marrow (CD117-Ab), or the brain (T7 peptide).
Oligonucleotides
(ASO, siRNA, miRNA-targeting oligonucleotides)
Antisense oligonucleotides (ASO) are short oligonucleotides
(single- or double-stranded) that can bind to RNA in a targetspecific manner and modulate protein expression. They comprise a large variety of molecules encompassing small interfering RNA (siRNA), gapmer ASOs, splice switching ASOs (ssASOs), agomirs, and antagomirs, to name a few (Moumné 2022).
Depending on their structure, they can knockdown a particular transcript (gapmer or siRNA) either as pre-mRNA or mRNA, using different mechanisms (RNAseH for gapmer and RISC for siRNA, Moumné 2022). They can also modulate splicing (ssASOs) by binding to specific elements on the pre-mRNA, resulting in inclusion or exclusion of target exons.
All oligonucleotides benefit from chemical modifications which increase stability, specificity and circulation time while reducing their toxicity. These modifications include: the phosphate linkage (i.e. phosphorothioate, peptide nucleic acid (PNA)), the ribose moiety (Locked nucleic acid (LNA), 2’-O-methyl, 2’fluoro) and the addition and/or substitution with DNA stretches.
Alongside the traditional LNP systems described in section one, several lipid-based systems have been used and/or developed for oligonucleotide delivery:
• MEND system (Multifunctional envelope-type nano medicine): an LNP with an outer core functionalised with peptides.
• ssPalm system: ionisable lipids with a pH switch as well as biodegradability following reduction of S-S bonds in the cytoplasm (Akita 2013). A second generation of ssPALM used vitamin A and E as part of the hydrophobic backbone of the ionisable lipids.
• Solid lipid nanoparticles for the delivery of siRNA.
To date, only one siRNA-LNP has been approved by the FDA (Patisiran®, developed by Alnylam Pharmaceuticals in 2018). However, several oligonucleotides-based drugs using LNPs as a mode of delivery are in clinical trials: siRNA in neutral liposomes for cancer targeting, dual siRNAs for solid tumour treatment
(ALN-VSP02, Alnylam Pharmaceuticals), siRNA directed against the Heat Shock protein HSP47 for treatment of liver fibrosis/ cirrhosis (BMS-986263, Bristol Myers Squibb) or idiopathic pulmonary fibrosis (ND-LO2-s0201, Nitto Biopharma) and miRNA mimic for solid tumour treatment (INT-83, InteRNA).
Few miRNA-based therapeutics enter clinical trials as, unlike their siRNA counterpart, they usually modulate the expression of different genes/proteins, increasing the risk of toxicity and off-target effects. This can be bypassed by improving the specificity of delivery using targeted LNPs. Recent studies show that by carefully engineering lipid particles, it is possible to target a miR agonist in vivo to the skeletal muscle or an antagomir to microglia, both in vitro and in vivo (Badr 2024).
It should be noted that LNPs are not necessarily the most potent delivery method for oligonucleotides. Indeed, of the 12 FDA-approved ASOs, none use LNPs. Similarly, besides LNPs, several other platforms for siRNA delivery have been developed, for example, hexosamine conjugation for liver delivery (Gal-Nac).
mRNA
Messenger RNA (mRNA) is a transient single ribonucleic acid which acts as an intermediate between genes and proteins. With the introduction and development of in vitro translation (IVT), these molecules are now considered promising new drug candidates. Three types of mRNA are being investigated: non-replicating mRNA, self-amplifying RNA (saRNA) and Circular RNA (CircRNA). mRNA and saRNA share structural features: a 5’-cap structure, 5’- and 3’-UTRs enclosing the open reading frame, and a polyA tail (Omidi 2024). CircRNA does not contain a 5’-cap (replaced by an IRES) nor a polyA. Extensive research into the structure of mRNA has explored: modifying the capping structure to improve protein translation (Cap1, Trinucleotide cap analogues for co-transcriptional capping and their variations, Ishikawa 2009), optimising the UTRs to improve stability and proper translation of the mRNA, optimising the ORF (codon usage, RNA secondary structure) and optimising the length and structure of the polyA tail to achieve higher stability.
Technology
One of the main drawbacks of nucleic acids is that they activate Toll-Like Receptors (TLRs), leading to immune response. The introduction of modified nucleotides (pseudouridine, 1-methylpseudouridine, or 5-methylcytidine, Kariko 2021) has decreased the immunogenicity of mRNA by reducing TLR recognition.
mRNA-based therapeutics are a growing field with expanding applications, including:
• Viral vaccines:
• Herpes zoster (mRNA-1273 by GlaxoSmith-Kline, phase III; mRNA-1468 by Moderna, phase I/II)
• Cytomegalovirus (mRNA-1647 by Moderna, phase III;)
• Respiratory syncytial virus (mRNA-1345 by Moderna, approved; LNP CL-0137 by Sanofi, phase I/II)
• Influenza (mRNA-1010 by Moderna, phase III; mIRV by Pfizer, phase III)
• Zika virus (mRNA-1893 by Moderna, phase II)
• COVID-19 (Kostaive by Arcturus), first saRNA-based vaccine approved by European and Japanese authorities.
• Cancer vaccines:
• Melanoma (mRNA-4157 by MSD and Moderna, phase III; BNT111 by BioNTech, phase II)
• Pancreatic cancer (BNT122/Autogene Cevumeran by BioNTech)
• Non-small cell lung cancer (BNT116 by Moderna, phase II)
• Solid tumours (mRNA-4359 by Moderna, phase I/II).
• Protein replacements:
• OTC deficiency (ARCT-810-03 by Arcturus, phase II)
• Propionic acidaemia (mRNA-3927 by Moderna, phase I/II)
• Cystic fibrosis (MRT5005-101 by Translate Bio, phase I/II).
• Protein expressions:
• Heart failure (AZD8601 by AstraZeneca, phase II)
• Solid tumours (BNT142 by BioNTech, phase I/II).
• Immunotherapies:
• Solid tumours (BNT151 by BioNTech, phase I/II).
• Epigenetic modifiers:
• Cancers (OTX-2002 by Omega Therapeutics, phase I/II).
DNA
The use of DNA as a cargo is advantageous as it is less toxic than whole pathogens, allows prolonged expression compared to mRNA, can be easily manufactured and engineered, and is generally more thermostable than mRNA. Due to its versatility, plasmid DNA (pDNA) can be used to express a specific protein, peptide, toxin, or other oligonucleotides (i.e. siRNA).
Unlike the nucleic acid cargos mentioned above, pDNA does not require extensive chemical modifications. Toxicity can result from the presence of certain backbone elements (antibiotic resistance gene, or bacteria-derived sequences) or the encoded gene itself (CpG islets).
Vector design optimisation has led to the emergence of minicircle DNA, MIDGE DNA, doggybone DNA and linear DNA. Similarly, promoter optimisation and enhancers are required for optimal expression in a specific tissue or cell type.
Despite these advantages, there are several limitations inherent to the pDNA system: the inability to transfect DNA directly into the nucleus (it must target dividing cells), transfection efficiency, low risk of genome integration and cytotoxicity.
Two different transfection methods have proven successful for pDNA: physical methods (i.e. electroporation) and chemical methods (i.e. nanoparticles).
Of the two, electroporation is most commonly used and remains the most efficient method for DNA transfection as it bypasses the whole endocytic process. However, cytotoxicity can become an issue, especially in vivo, where it can be difficult to scale the method to larger species.
The development of new transfection strategies is therefore essential. Although research into COVID-19 vaccines prompted breakthroughs in LNPs, they mostly concerned RNA technologies (see above), and DNA encapsulation was overlooked.
However, strategies similar to those developed for RNA have been successfully applied for pDNA transfection in vitro and in vivo, including optimisation of ionisable lipids, lipid proportions and the use of ssPALM.
Recent studies have also confirmed the potential of pDNA-LNP as a vaccine and anti-cancer treatment (Chai 2025).
Several studies have also identified different parameters that could potentially be translated to RNA delivery. Co-delivery of pDNA with siRNA has facilitated the prolonged expression of the target in vivo by silencing transcription factors involved in the regulation of inflammatory response pathways. Similarly, incorporation of anti-inflammatory lipids (nitro-oleic acid) has reduced inflammation following pDNA-LNP administration, increasing the duration of expression. Combining cationic polymers and lipids allows researchers to efficiently encapsulate larger DNA molecules while maintaining prolonged expression. The recent development of dedicated platforms for pDNA-LNP designs enables the optimisation of both the liposome composition and pDNA properties (promoters/enhancers sequences).
Despite these advancements, pDNA-LNP-based delivery studies lag behind research into RNA-LNP. Further research will help expand the field of applications for pDNA-based therapeutics.
Perspectives
Although the studies mentioned above have increased the development of targeted LNPs to specific organs and tissues and improved endocytic properties, additional research is required for more precise targeting and better endosomal escape.
With the advent of targeted delivery, the field of therapeutic applications is broadening. Advances in molecular biology, for example, the development of new base- and prime-editing tools, are fuelling further progress in this field.
Several studies have already applied LNP to CRISPR/cas systems and base editors (i.e. Adenine and cytosine base editors), allowing the modification of genomic DNA in vitro and in vivo (from point mutations to insertions/deletions of different sizes). The applications are extensive (reviewed in Yuan 2025) and LNPs can be used to transfect: Cas9/sgRNA
Technology
using pDNA-LNP in tumours; Cas9/prime editors proteins carrying a sgRNA inside a LNP; and Cas9 mRNA and sgRNA in different LNPs or in a single LNP. The therapeutic potential of LNP-based drugs is great and, with ongoing research into increased tissue specificity and improvement in LNP formulations overall, the field is set to grow dramatically over the next couple of years.
REFERENCES
1. Hald Albertsen C, Kulkarni JA, Witzigmann D, Lind M, Petersson K, Simonsen JB. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv Drug Deliv Rev. 2022 Sep;188:114416. doi: 10.1016/j.addr.2022.114416.
2. Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987 Nov;84(21):7413-7. doi: 10.1073/pnas.84.21.7413.
3. Mrksich K, Padilla MS, Mitchell MJ. Breaking the final barrier: Evolution of cationic and ionizable lipid structure in lipid nanoparticles to escape the endosome. Adv Drug Deliv Rev. 2024 Nov;214:115446. doi: 10.1016/j.addr.2024.115446.
4. Martinon F, Krishnan S, Lenzen G, Magné R, Gomard E, Guillet JG, Lévy JP, Meulien P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993 Jul;23(7):1719-22. doi: 10.1002/eji.1830230749.
5. Cui L, Pereira S, Sonzini S, van Pelt S, Romanelli SM, Liang L, Ulkoski D, Krishnamurthy VR, Brannigan E, Brankin C, Desai AS. Development of a high-throughput platform for screening lipid nanoparticles for mRNA delivery. Nanoscale. 2022 Jan 27;14(4):1480-1491. doi: 10.1039/d1nr06858j.
6. Moumné L, Marie AC, Crouvezier N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics. 2022 Jan 22;14(2):260. doi: 10.3390/pharmaceutics14020260.
7. Badr A, Daily KP, Eltobgy M, Estfanous S, Tan MH, Chun-Tien Kuo J, Whitham O, Carafice C, Gupta G, Amer HM, Shamseldin MM, Yousif A, Deems NP, Fitzgerald J, Yan P, Webb A, Zhang X, Pietrzak M, Ghoneim HE, Dubey P, Barrientos RM, Lee RJ, Kokiko-Cochran ON, Amer AO. Microglia-targeted inhibition of miR-17 via mannose-coated lipid nanoparticles improves pathology and behavior in a mouse model of Alzheimer's disease. Brain Behav Immun. 2024 Jul;119:919-944. doi: 10.1016/j.bbi.2024.05.006.
8. Omidi Y, Pourseif MM, Ansari RA, Barar J. Design and development of mRNA and self-amplifying mRNA vaccine nanoformulations. Nanomedicine (Lond). 2024;19(30):2699-2725. doi: 10.1080/17435889.2024.2419815.
9. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009 Oct 8;461(7265):788-92. doi: 10.1038/nature08476
10. Karikó K. Modified uridines are the key to a successful message. Nat Rev Immunol. 2021 Oct;21(10):619. doi: 10.1038/s41577-02100608-w. PMID: 34580453.
11. Chai D, Wang J, Lim JM, Xie X, Yu X, Zhao D, Maza PAM, Wang Y, Cyril-Remirez D, Young KH, Li Y. Lipid nanoparticles deliver DNA-encoded biologics and induce potent protective immunity. Mol Cancer. 2025 Jan 13;24(1):12. doi: 10.1186/s12943-024-02211-8.i
12. Yuan Y, Li Y, Li G, Lei L, Huang X, Li M, Yao Y. Intelligent Design of Lipid Nanoparticles for Enhanced Gene Therapeutics. Mol Pharm. 2025 Mar 3;22(3):1142-1159. doi: 10.1021/acs.molpharmaceut.4c00925.
Xavier Warnet, PhD is a Project Manager at Tebubio, specialising in RNA therapeutics and advanced LNP technologies. Xavier focuses on optimising RNA delivery for oncology and infectious disease applications.
Xavier Warnet
Accelerating Cell Line Selection with Integrated Analytical Strategies
Cell line development is a critical determinant of success in biologics manufacturing. It lays the foundation for product yield, quality and long-term process stability. Yet, despite major advances in host engineering, vector systems and culture conditions, early-stage clone selection remains one of the least analytically supported stages of development.1
Therapeutic proteins, including monoclonal antibodies, enzymes and fusion proteins, are now central to modern treatment strategies for cancer, autoimmune conditions and genetic disorders. For each of these modalities, a well-characterised and scalable production cell line is essential. This makes cell line development not just a technical necessity, but a strategic priority for biopharmaceutical developers.
In practice, decisions about which clones to progress are often based on incomplete or delayed data, typically obtained only after expansion or purification. This not only introduces inefficiency but also increases the risk of overlooking high-performing candidates. As the pace of biologics development accelerates, the gap between biological complexity and analytical capability becomes harder to ignore. Speed and confidence in cell line selection can ultimately be the difference between a successful biologic and a stalled program. To address this, developers are increasingly turning to in-process analytical tools, such as rapid protein quantification platforms, to gain earlier insight and support more data-driven selection workflows.
This challenge is illustrated in Figure 1, which maps a typical cell line development process and highlights key decision points where data is often limited or delayed.
CHO Cell Line Development: Then and Now
Chinese hamster ovary (CHO) cells have long served as the preferred host for recombinant protein production due to their adaptability, capacity for human-like post-translational modifications and extensive regulatory track record. Over the last two decades, process yield and robustness have improved significantly through host engineering and upstream optimisation.2 Despite these advances, the fundamental architecture of clone screening, from transfection to selection of the top clone, remains largely unchanged.3
This persistence reflects the extensive research that has shaped our understanding of recombinant protein production in CHO cells. Over several decades, studies have uncovered the molecular and metabolic mechanisms that influence expression outcomes, including chromatin accessibility, nutrient signalling and stress adaptation.4,5 While molecular engineering and media optimisation have become increasingly sophisticated and streamlined, analytical methods at early screening stages have not kept pace. This disconnect continues to limit the ability to make timely and data-informed decisions during cell line development.
The need for more immediate and informative measurement tools is especially acute as developers pursue more complex formats. From bispecific antibodies to unstable or difficult-toexpress fusion constructs, early insight into expression performance is critical to avoid costly delays or missed candidates.
Figure 1: Cell line development workflow overview. High-level overview of the key stages in recombinant cell line development, highlighting where early analytical decisions are typically made.
From Practical to Pioneering
Raw materials, reagents, and custom solutions for therapeutics, vaccines, and diagnostics-supporting science from discovery to approval.
Table 1: Comparison of protein quantification methods for early clone evaluation. A side-by-side comparison of commonly used analytical tools, showing trade-offs in speed, sample compatibility and infrastructure needs. ELISA: Enzyme-Linked Immunosorbent Assay; HPLC: High-Performance Liquid Chromatography; SEC: Size Exclusion Chromatography; BLI: Biolayer Interferometry; LC-MS: Liquid Chromatography–Mass Spectrometry.
Gaps in Clone Selection and Evaluation
Today’s clone evaluation workflows still depend heavily on traditional protein assays such as enzyme-linked immunosorbent assay (ELISA), high-performance liquid chromatography (HPLC) and sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE). These methods are robust but slow, batch-based and often decoupled from the point of decision.6 They typically come into play only after significant expansion, requiring large amounts of product and delaying critical insights. As a result, early-stage agility is limited. The trade-offs between these commonly used analytical methods, including differences in speed, sample requirements and suitability for early-stage workflows, are shown in Table 1. These limitations help explain why developers often face challenges in generating timely and informative data to guide confident decision-making.
In the absence of fast and direct product measurements, developers frequently rely on proxy readouts such as cell growth or fluorescence. While these allow for higher screening throughput, they often fail to capture meaningful differences in cellular productivity, expression stability or stress response. Having worked extensively on CHO clone characterisation and bioreactor modelling, we have seen how subtle phenotypic differences at the screening stage can lead to significant downstream consequences. Yet tools capable of capturing these differences early in development, and at a scale suitable for exploratory workflows, remain limited. What is often missing is not a lack of metrics, but the ability to access meaningful data early enough to guide confident decisions.
Addressing this bottleneck does not require replacing existing assays. Instead, it calls for augmenting current workflows with tools that provide just enough information, just in time.
In-Process Quantification to Support Early Decisions
Bench-level protein quantification is gaining traction as a
practical solution to support earlier clone triage and more efficient resource allocation. Whether during screening, scale-down optimisation or early vector assessment, having access to concentration data within the same lab and the same day offers a meaningful advantage.7
While not intended to replace gold-standard analytics, platforms such as Abselion’s Amperia™ provide a pragmatic way to reduce uncertainty and close feedback loops earlier in the process. By delivering actionable data at the screening or early expansion stage, in-process quantification helps teams make better use of their time, incubator space and downstream capacity without disrupting existing workflows.
This approach complements established analytical strategies by enabling faster access to quantitative data during early development. While not a substitute for the detailed characterisation required later in the pipeline, early quantification supports more informed prioritisation and helps minimise delays during screening. The impact of this shift is illustrated in Figure 2, which contrasts traditional and integrated workflows to highlight how in-process quantification can streamline early decision-making.
Collaboration Highlight: University of Manchester Case Study Professor Alan Dickson’s group at the University of Manchester partnered with Abselion to evaluate whether benchtop quantification could provide practical support for early clone selection in a real-world setting. The study included samples from both the mini pool stage and individual clones, covering key points in the selection process where earlier data could support more informed decisions. The aim was to assess whether having access to protein concentration data at these stages could help prioritise candidates more effectively and reduce the risk of advancing underperformers.
Using crude culture supernatants, the team rapidly quantified antibody expression and observed clear ranking correlations
with gold-standard ELISA results obtained later in the workflow. In-process benchtop quantification supported earlier triage of low-producing clones and streamlined the transition to bioreactor testing, helping to make these steps more efficient and responsive.
The impact of this approach is illustrated in Figure 3, which compares conventional and integrated workflows. Incorporating Amperia into the process helped reduce assay turnaround times and enabled faster progression through the study overall.
Looking Forward: Connecting Data, Decisions and Development
As biologics pipelines grow in both complexity and urgency, there is increasing recognition that upstream workflows must evolve beyond incremental improvements. The ability to make confident and data-driven decisions earlier in development will be key, not just to decreasing timelines, but also to improving the quality and manufacturability of molecules entering the pipeline.
In this context, the role of in-process analytics is shifting. Rather than being seen as standalone tools, benchtop
quantification systems are becoming part of a more integrated strategy that connects clone selection, cell line optimisation and early process development within a tighter feedback loop.7
The goal has always been to help close the gap between academic insight and industrial application, whether through advanced cell line modelling, novel host engineering strategies, or now, more accessible analytical approaches. The ideal future is one in which every decision from clone to clinic is informed by timely and context-relevant data. Achieving this will require the integration of real-time analytics with data modelling, automation platforms and AI-driven decision tools. Just as importantly, it will require practical tools that empower researchers at the bench, not only in high-throughput labs, but across the diverse environments where biologics are designed, optimised and translated.
REFERENCES
1. Tejwani, V., Andersen, M. R., Nam, J. H., et al. High-throughput and automation advances for accelerating single-cell cloning, monoclonality and early phase clone screening steps in mammalian
Figure 2: Impact of in-process quantification on clone selection workflow. A visual comparison of traditional and integrated workflows. In-process quantification enables earlier decision-making, reduces reliance on late-stage assays and helps prioritise productive clones faster.
Figure 3: Case study: Integrated workflow enables faster clone selection. Workflow comparison of ELISA-based and Amperia-supported clone selection processes at the University of Manchester. Use of benchtop analytics reduced turnaround time and shortened total study duration by approximately 40%. cell line development for biologics production. Biotechnol. Prog. 37(6), e3208 (2021).
3. Clarke, H., Mayer-Bartschmid, A., Zheng, C., et al. Biotechnol. Prog. 40(4), e3449 (2024).
4. Torres, M., Hawke, E., Hoare, R., et al. Deciphering molecular drivers of lactate metabolic shift in mammalian cell cultures. Metab. Eng. 88, 25–39 (2025).
5. Meeson, K., Watson, J., Rosser, S., et al. Flux sampling suggests metabolic signatures of high antibody-producing CHO cells. Biotechnol. and Bioeng. 122(7), 1898–1913 (2025).
6. Wang, T., Chen, H., Zhao, Y., et al. Preliminary screening of CHO cell pools for recombinant protein expression: Integrated strategies and workflow considerations. Front. in Bioeng. Biotechnol. 10, 858478 (2022).
7. Shi, J., Ho, A., Snyder, C. E., et al. Accelerating biopharmaceutical cell line selection with label-free multimodal nonlinear optical microscopy and machine learning. Commun. Biol. 8, 157 (2025).
Prof Alan J Dickson
Alan J Dickson is Professor of Biotechnology at the University of Manchester’s Institute of Biotechnology and Director of the Centre of Excellence in Biopharmaceuticals. His research focuses on molecular and systems-level strategies, such as chromatin analysis, omics and mammalian cell engineering, to improve bioproduction of therapeutic proteins and viral vectors. He leads interdisciplinary collaborations across academia and industry to advance bioprocessing and biopharmaceutical development.
Email: alan.dickson@manchester.ac.uk
Dr. Paolo Romele
Paolo Romele is VP of Science at Abselion, where he leads R&D across biosensing technologies, consumables and assay development. He holds a PhD from the University of Brescia and has conducted advanced research at STMicroelectronics and the Max Planck Institute for Polymer Research. With deep expertise in electrochemical sensing and applied engineering, he focuses on creating robust, practical solutions for biomolecule measurement in life science and bioprocessing contexts, such as Amperia™.
Email: paolo.romele@abselion.com
Monodisperse 2kDa PEGs
Maximum Precision
Minimal Variation
Our monodisperse PEGs deliver consistent purity, reliable results, and total peace of mind.
Manufacturing & Processing
Leveraging Strategic Partnerships to Enhance Flexibility in Facility Design and Bioprocesses
Flexibility is an operational imperative in today’s fast-evolving biopharmaceutical industry. Contract development and manufacturing organisations (CDMOs), as integrated end-to-end service providers, need to pivot with agility by accommodating shifting product demands, modularising facilities, and continuously improving processes to be more versatile. Achieving this level of flexibility requires a strategic approach rooted in data, and that data is most effectively acquired through sustained, strategic partnerships with global pharmaceutical and biotechnology companies.
Flexibility for CDMOs can be defined as a data-driven, strategic operation. It develops incrementally as CDMOs diligently locate, analyse and apply data generated during projects for continuous improvement in operations. The data that underpins flexibility goes beyond information on batch yields or assay results. It includes project execution experience, product specifications, such as the size and configuration of bioreactors and the number of equipment and piping arrangements required for different molecules, and process know-how developed through years of hands-on problemsolving. This kind of knowledge is built in the day-to-day execution of collaborative projects between CDMOs and their biopharmaceutical partners.
Through these collaborations, CDMOs develop an understanding of numerous production requirements and challenges. Data derived from a diverse range of manufacturing processes is leveraged to continuously enhance process designs and strategies with every project. Over time, this body of data becomes the foundation for designing flexible facilities and implementing tailored bioprocesses, capabilities that enable timely adaptation to new client needs, emerging modalities and operational hurdles.
Facility Design: The Architectural Backbone of Flexibility
As process and product data accumulate, CDMOs can leverage these insights to address the most fundamental aspect of manufacturing flexibility, facility design. A CDMO facility is a dynamic architecture built to evolve. Electronic manufacturing batch records (eMBR) and manufacturing execution systems (MES) establish the digital backbone of such facilities, enabling standardised yet adjustable documentation, real-time process monitoring and traceability across batches.
Armed with detailed operational data, facility designers can determine the optimal target range, the bell curve of likely process types and scales, and design for the most probable scenarios while avoiding costly over-engineering for outliers. This data-driven facility design allows CDMOs to strike a balance between readiness and efficiency.
One tangible outcome of this balance is a reduction in product changeover times. For large-scale CDMOs that rely on fixed equipment rather than single-use systems, modifying installed infrastructure can be onerous. Designing a facility that either accommodates a wide range of processes or can be readily adapted, therefore, delivers efficiency and productivity.
At the facility level, this balance is achieved through layouts that integrate hybrid stainless steel and single-use systems to combine long-term capacity with agile changeover readiness. Modular cleanrooms can be reconfigured as needs evolve, while utility backup systems provide the capability to swap equipment or adjust layouts without halting operations. A bioprocess facility designed using data and built on experience creates an environment in which flexible upstream and downstream processes can operate.
Upstream Processing: Turning Experience into Flexible Practice
The upstream phase of biologics manufacturing, most notably cell culture, offers some of the richest opportunities to apply data gleaned from client partnerships. Every project adds to a growing library of recipes: process analytical technology (PAT), dual-feeding methods and centrifugation strategies. By collecting and analysing these variables in a structured way, CDMOs can identify what works best for particular process profiles and promptly retrieve that knowledge when a similar need arises.
1. Process Analytical Technology
The first mechanism reinforcing flexibility in upstream processes is the application of PAT, which drives integral cell culture systems, such as biocapacitance and Raman probe systems. In the latter case, Raman spectroscopy uses laser-based vibration energy to detect a molecule’s chemical composition, enabling qualitative and quantitative analysis of multiple components through a single probe. Unlike conventional methods that require offline sampling, PAT enables in situ real-time measurements inside the bioreactor.
This multivariate data analysis tool strengthens flexibility in several ways. By reducing the required sampling cadence, PAT enables the rapid detection of contamination risks and reduces the need for human intervention. Real-time data streams provide actionable insights into parameters, such as nutrient concentrations and cellular metabolism, which can then be fed into automated control systems to promptly adjust cell culture strategies.
2. Dual-feeding Methods
Another mechanism that reinforces upstream flexibility is dual feeding. By combining bolus and continuous feeding methods, CDMOs can accommodate a diverse category of biologics with different nutrient uptake profiles. Bolus feeding drives high-density growth phases, while continuous feeding
Advancing every day
Supplying what’s needed now and for what’s next
The biotechnology industry is poised to deliver amazing products and solutions that will dramatically change millions of lives for the better. Aldevron is ready.
As a globally trusted CDMO, our years pioneering ground-breaking solutions to advance biologic manufacturing for research use, as well as cGMP for clinical and commercial applications, has enriched us with a body of knowledge that is critical to support these next series of breakthroughs.
Contact us to advance your program.
ald.bio/contact-us
Manufacturing & Processing
maintains steady-state conditions for more sensitive cell cultures. Having both options readily available eliminates the need for process redesign when switching products. Implementing this approach effectively requires validating both feeding modes across cell lines, maintaining modular feeding hardware and customising feeding methods in real time.
Flexibility becomes tangible when dual-feeding methods are applied to large-scale manufacturing. For instance, in N-stage bioreactor operations with multiple harvest trains, the capability to route bioreactor output to any available train maximises scheduling flexibility and reduces bottlenecks. Similarly, media-hold vessels originally allocated to an N-1 stage perfusion setup can be reconfigured to supplement N-stage production runs when capacity constraints arise. In another case, large-stock media preparation can be retooled from vessel-to-vessel transfer into bag-based filtration, ensuring rapid turnaround for high-volume batches. Each case demonstrates how upstream operations built on dual-feeding methods create a portfolio of issue-response scenarios, enabling CDMOs to resolve client challenges quickly.
3. Centrifugation Strategies
The third mechanism that enhances flexibility in upstream processing is continuous centrifugation, achieved through the interchangeable use of bowls tailored to product specifications. The two main bowls are CSI and CFI. CSI, or self-cleaning bowls, eject solids periodically, while CFI, or continuous-discharge bowls, remove impurities continuously, an advantage for high-cell-density cultures. The capability of selecting between two bowl designs based on product characteristics optimises yield, reduces product loss and enables stable long-term operations.
These upstream mechanisms convert cumulative project experience and data into flexible operational strategies while also informing post-project technical reviews. After each stage of a project is complete, CDMOs should log process variables in centralised knowledge systems and apply data analytics to pinpoint the parameters most critical to yield and quality. When these conversion steps are carried out consistently and carefully, flexibility in upstream processing becomes not only a concept but also a tangible capability.
Downstream Processing: Enhancing Flexibility With Versatile Systems
1. Multi-train Systems
In manufacturing complex biologics, such as multispecific antibodies and antibody-drug conjugates (ADCs), downstream processing often determines whether a production timeline can stay on track. For these modalities, multiple downstream process (DSP) trains provide the flexibility to ensure stable, high-quality products and to conserve resources, especially resins.
A DSP train is an integrated purification line consisting of chromatography columns and skids, filtration systems, buffer preparation tanks and in-line analytical monitoring tools. With a single train, operational constraints can quickly pile up. When manufacturing bispecific or trispecific antibodies, the parental antibody purification may need to be run independently or,
in some cases, simultaneously to maintain product quality. A single-train setup limits these options, introducing scheduling conflicts and forcing compromises on hold times, product stability and throughput.
A multi-train configuration, on the other hand, allows CDMOs to develop purification strategies that account for product stability, batch size and regulatory timelines. If a product’s hold time is short or stability is low, one train can handle its purification immediately while another processes the parental antibody or a different molecule in parallel. This parallelism facilitates rapid adjustments to production timelines, whether to meet changing client demands, accommodate regulatory filing deadlines or recover lost time from upstream delays.
From a productivity standpoint, multiple trains minimise downtime during product changeovers or routine maintenance, helping CDMOs manage assets for full utilisation. With multi-train systems, CDMOs can also fine-tune project schedules to align with diverse client project milestones, an advantage that can be decisive in competitive outsourcing scenarios.
Despite these benefits, establishing multi-train systems presents some practical limitations and dilemmas. Installing an additional DSP train requires significant capital expenditure, not just for the purification line itself, but also for the corresponding upstream capacity, such as multiple harvest systems and additional bioreactor trains. Without sustained demand, these assets risk being underutilised, undermining the return on investment. Maintenance costs, depreciation and staffing requirements all compound these financial challenges.
For many CDMOs, especially those operating in uncertain market conditions or with variable order pipelines, a blanket multi-train implementation is not economically viable. As a result, these systems are installed selectively, targeting facilities where throughput needs, client demands and modality focus create a compelling business case.
2. Adaptable Purification Columns
While multi-train systems enhance capacity and scheduling flexibility, adaptable purification columns provide precision and scalability within individual purification processes. In downstream processing, this adaptability means less reliance on a single universal design and more on a facility’s capability to deploy columns of varying sizes and specifications to match each client process.
The choice of column size depends on upstream titre, resin type, resin load capacity and the stability of the product. If a client’s process yields a large volume of product with relatively low stability, a larger column is required to complete purification quickly, minimising hold times and preserving quality. Conversely, for smaller batches or more stable products, a smaller column may be deployed to optimise resin usage and processing efficiency.
This range of options is a key driver of success in complex molecule manufacturing. Multispecific antibodies, ADCs and other advanced biologics require different conjugation ratios, batch sizes and purification chemistries. Without the capability to match column size and configuration to these variables,
Manufacturing & Processing
CDMOs risk either overburdening processes, leading to longer purification times, or underutilising equipment.
The opposite of an adaptable setup is a rigid column inventory: fixed sizes, limited resin types and a one-size-fitsall approach. This rigidity can preclude accommodating a client process without significant reconfiguration or new capital investment, both of which slow down project initiation. From a flexibility perspective, investing in a wide range of purification column sizes and compatible hardware allows CDMOs to integrate client processes efficiently, without forcing either party to undertake costly modifications.
Adaptable purification capabilities are also directly tied to quality and stability outcomes. For complex molecules, small variations in purification conditions can affect the potency, safety and shelf life of products. By selecting the optimal column configuration for each run and closely monitoring critical parameters, such as chromatograms and process trends in real time, CDMOs can maintain product quality while adapting to varying process demands.
An adaptable purification column portfolio builds client trust. A CDMO equipped with adaptable columns can execute client processes with the right equipment, at the right scale, without unnecessary delays or compromise. In an industry where complex molecules are rapidly taking market share, such capabilities are a competitive imperative.
Implementing Flexible, Data-driven Operations On Digital Systems
eMBR and MES platforms drive operational flexibility across facility design, upstream and downstream bioprocessing. eMBRs replace paper-based records, ensuring real-time, standardised batch documentation while reducing transcription errors, delays and compliance risks. MES integration connects production floor systems, quality management and laboratory information management systems, creating a reliable digital infrastructure.
By collecting data in real time and automating workflow execution, these systems enable proactive process control, faster batch release and robust traceability. Upstream feeding strategies, PAT measurements and centrifugation data are integrated for responsive control, while downstream purification timelines are shortened because of automated adjustments. The outcome is a harmonised, agile environment where knowledge, experience and digital capabilities converge to enhance operational flexibility.
Flexibility As An Ongoing Discipline
Flexibility in biomanufacturing is not achieved through a single design choice or technology upgrade. It is the product of ongoing discipline, engaging deeply with clients to gather diverse operational experiences, capturing those experiences as structured data and applying that data systematically to facility design and process execution.
Strategic partnerships serve as the catalyst for this process, enabling CDMOs to look beyond their own operations and integrate best practices and insights from across the industry. In doing so, they enhance their flexibility and, in turn, strengthen their ability to achieve client satisfaction.
Flexibility is not only about being prepared for what comes next. It is also about establishing an infrastructure and culture in which CDMOs and their biopharmaceutical partners continue to thrive as one team, now and in the future.
Soyeon Ahn
Soyeon Ahn is a senior director of drug substance manufacturing and a principal scientist at Samsung Biologics. With more than 15 years of experience in the biopharmaceutical industry, she leads critical initiatives for the leading CDMO, including plant and equipment validation, technology transfer projects for global pharmaceutical clients and regulatory audit preparation for drug substance manufacturing.
Daeryun Park
Daeryun Park is a lead engineer at Samsung Biologics. He brings over 20 years of experience managing CGMP small molecule HPAPI manufacturing facilities and CAPEX projects at companies including Alpha Therapeutic Corp, Apotex Pharmachem and Genentech. Park is also a registered Professional Engineer in Chemical Engineering in California.
Hyoseok Kim
Hyoseok Kim is a director of upstream manufacturing and a lead engineer at Samsung Biologics. At a 240,000-litre facility, he oversees cell culture operations and manages facility safety. His core responsibilities include managing manufacturing issues, leading investigations, reviewing technology transfers and overseeing plant design, validation and inspection. With over a decade of experience in GMP operations, Kim ensures compliant production by effectively managing schedules and on-site operations.
Joomyung Lee
Joomyung Lee is a director of downstream manufacturing and a lead engineer at Samsung Biologics. At a 240,000-litre facility, he oversees purification operations, ensuring excellence in good manufacturing practices, managing technology transfers and leading regulatory audit readiness and process validation. With over a decade of experience, Lee directs numerous smalland large-scale productions, cultivating a culture of rigorous safety and quality standards.
Scale-up Evaluation of the DynaDrive S.U.B.s
Part 3: Pilot and Manufacturing Scale Comparison of DynaDrive S.U.B. Capabilities
Commercialisation of a drug is a monumental milestone that hinges on years of lifecycle management. As a product enters commercialisation, product sponsors are positioned to derive demand from a fluid market. Analysis of supply will inevitably question the cost of goods sold (COGS) and whether it is a process of scale-out or scale-up. In some cases, the decision must be made on how to scale up the process, which may require leaving the existing 2,000 L single-use bioreactor (S.U.B.) systems and moving the process into larger stainless steel vessels. Up until this point, there is a volumetric gap separating the single-use technology and stainless steel bioreactors. Existing 2,000 L S.U.B.s are supply chain limiting, while stainless steel systems can pose other challenges when demand remains fluid.
Modern process approaches have allowed increased product output, elevating titres past 10 g/L in some cases. These output achievements require increased production efficiency and/ or input, pushing many bioreactor systems outside of their operating capabilities. For example, as oxygen transfer rate (OTR) becomes a limiting factor, most traditional S.U.B.s rely primarily on increased sparging flow to increase oxygen mass transfer. Maintaining a dissolved oxygen (DO) target in high-demand cell culture can be difficult due to limitations in the amount of power that can be delivered through the existing drivetrain. Delivering the gas through a micro-sparger has become a strategy that is widely used to improve OTR in traditional S.U.B.s and typically requires a secondary sparger to facilitate the removal of dissolved CO₂ (measured as partial pressure of CO₂ or pCO₂). However, certain cell lines have been identified to be sensitive to the higher shear produced by micro-sparging, and thus, process scale-up should not depend on this method alone to ensure sufficient O₂ delivery or pCO₂ removal.
The next generation of S.U.B.s, such as the Thermo Scientific™ DynaDrive™ Single-Use Bioreactor (S.U.B.), which vastly improve mixing and mass transfer performances, can now enable scales of up to 5,000 L while remaining in the single-use format. Previous limits are easily addressed via the DynaDrive S.U.B. series, while the operating range is suitable to leverage an existing process using S.U.B.s and to allow for a smooth transfer to the new technology. The DynaDrive S.U.B.s are multifunction reactors with a range of applications, including intermediate production scale for preclinical and clinical materials, scalable seed train vessels with wide turndown ranges (up to 20:1) and perfusion processes at 50 L and 500 L scales, both for production and for use in large-scale intensified fed-batch applications. Additionally, the DynaDrive S.U.B.s include improved features over previous and alternative S.U.B. options as noted below:
• Superior enhanced drilled-hole sparger (DHS) helps provide repeatable and reliable performance.
• Flexible drive train with multiple impellers allows increased power input while offering reduced shear rates.
• Cuboid design contributes to better Thermo Scientific™ BioProcess Container (BPC) fit, increased mixing efficiency and allows for more productive use of facility footprint.
• Turndown capability (20:1) reduces facility requirements and increases flexibility in applications and scale-up.
• Minimal hold-up volume (<1%) post-harvest and drain.
• Each system is equipped with BPC-loading platforms, reducing handling and setup time, increasing safety and providing consistent BPC loading. BPC loading can be accomplished in less time than with other 50 L and 500 L S.U.B.s and in less than 45 minutes at the 5,000 L scale.
• Improved exhaust system for the 5000 L S.U.B., allowing for increased gas flow rates necessary for the increase in scale.
These major design changes have enabled a power-to-volume (P/V) ratio of up to 80 W/m3 in all sizes, T95 mixing times of less than 45 seconds, and kL a performance of at least 40 hr-1 at all scales (Tables 1–2).
Additionally, the DynaDrive S.U.B. allows for process scale-up and transfer from Thermo Scientific™ HyPerforma S.U.B.s, offering benefits of consistent BPC film, assurance of supply, robust quality controls and BPC integrity. End users can continue using previously qualified traditional and single-use probe options, as well as inlet and exhaust filters and other peripheral components integrated through high-strength porting and line sets.
Goal
The goal of this study was to evaluate the performance of the DynaDrive S.U.B., HyPerforma S.U.B. and Cytiva™ XCellerex™ XDR S.U.B. using four different cell lines (Table 3) with an XCellerex XDR S.U.B.–specific process developed in either the HyPerforma or XCellerex XDR systems for each cell line. These experiments were designed to demonstrate that these processes could be successfully transferred across different systems without impacting the process performance or product quality. A range of host cell platforms and processes were selected to demonstrate robustness and applicability across the diverse landscape in upstream bioprocessing.
Case Study 1: Vessel Evaluation Using ExpiCHO-S Cell Line in 14-day Fed-batch Run
Methods
Standard vial thaw, resuscitation and propagation procedures were used in initiating the cell train. Once cells were successfully thawed, they were expanded in a stepwise manner through a series of shake flasks and single-use vessels up to, and including, the N-2 stage. The N-1 stage for each seed train was performed in either a WAVE™ bioreactor or a stirred-tank seed bioreactor for the XCellerex XDR and HyPerforma S.U.B.s. For the N-1 step feeding the DynaDrive S.U.B. arm, the same vessel was used for both the N-1 and N stage. Specifically, a
10:1 turndown was used in the 50 L scale and a 20:1 turndown was used in the 500 L and 5,000 L scales at the N-1 stage. After a 3-day growth as N-1 culture, fresh production medium was added to the S.U.B. to reach the initial volume and seed density of the N-stage. Operating conditions used across all experimental arms are described in Table 4. Specifically, a daily bolus feed of 2X concentrated Gibco™ Efficient Feed™ C+ was added from day 3 to day 13, and glucose was added as needed. Daily samples were collected for cell counts, viability, dissolved gases, metabolites and titre. Titre samples were filtered and frozen starting on day 6 for ProA-UPLC analysis.
Results and Conclusions
Viable cell density (VCD) and viability for the cultures (Figure 1) show consistent growth profiles among the cultures with peak cell densities between 17.26 x 10⁶ and 21.25 x 10⁶ viable cells/ mL and viability >74% at harvest. Metabolite data collected offline indicated some fluctuations with glucose (Figure 2) and levels of metabolic byproducts, including lactate (Figure 3) and ammonium (Figure 4).
As observed in Figure 2, there were some fluctuations in glucose feeding, especially in the 50 L, 200 L and 250 L vessels as the glucose feed trigger was initiated on different days. However, lactate trends in Figure 3 remained consistent except for the 50 L DynaDrive S.U.B. vessel, where lactate accumulation increased from day 7 onwards. This was coupled with a drop in ammonium from 9.03 to 5.36 mM (Figure 4), while all other vessels followed a consistent upward trend.
Table 1. Comparison of Pilot-scale S.U.B. Capabilities
Table 2. Comparison of Manufacturing-scale S.U.B Capabilities
Table 3. Cell Lines Used to Evaluate Performance Across the Different Platforms
Figure 1. Viable cell density (VCD) and cell viability comparison of ExpiCHO-S (cell line 1) cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 2. Glucose profile for ExpiCHO-S (cell line 1) cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Temperature (ºC) Day 0–5: 37.0 Day 5–14: 34.0
pH control
headspace (sLPM)
DO cascade
Feeding strategy
Air supplemented with O2 through DHS (80 µm)
4.
Acid control: sparged CO2 Base control: 1 N NaOH
Air supplemented with O2 through the 20 µm sparger
Air supplemented with O2 through the 20 µm sparger
Air supplemented with O2 through DHS (233 µm)
Daily bolus of EfficientFeed C+ and glucose (as needed)
Volumetric productivity across the different platforms (Figure 5) measured 2.70–3.09 g/L on day 14 and showed a consistent trend from day 6 to 14 across all vessels. Product quality (PQ) samples were miniprepped and evaluated by qualified capillary gel electrophoresis (CGE) and size exclusion chromatography (SEC) methods. Assessment of the charge species across the different vessels showed some differences in acidic, main and basic species (Table 5). For example, the 250 L HyPerforma and 50 L/500 L/5,000 L DynaDrive S.U.B.s remained consistent, while the 200 L XCellerex XDR S.U.B. showed a slight shift towards the basic species. Assessment of product impurities via SEC showed consistency across all bioreactors above the 50 L bioreactor scale, while the 50 L showed a slightly higher monomer content due to lower molecular weight (LMW) aggregate species. Based on the productivity and quality results for the ExpiCHO-S cells, the data show strong consistency in productivity across all scales. A similar trend was also observed in the 250 L HyPerforma and the 500 L and 5,000 L DynaDrive S.U.B.s. While some minor PQ changes were observed at the 50 L DynaDrive and 200 L XCellerex XDR S.U.B.s, more work is required to determine a connection to a process or vessel change.
Case study 2: Vessel Evaluation Using Freedom CHO-S Cell Line in 14-day Fed-batch Run
Methods
The methods used for cell resuscitation, propagation and cell mass accumulation were outlined in case study 1. Changes in cell line, and thus process and operating conditions for each bioreactor, are described in Table 6. A 2X concentration of EfficientFeed C+ feed was added continuously from day 3 through to day 11, and glucose was supplemented as needed. Daily samples were collected for cell counts, viability, dissolved gases, metabolites and titre. Titre samples were filtered and frozen starting on day 6 for ProA-UPLC analysis or immunoturbidimetric analysis via Roche Cedex™ Bio HT Analyzer.
Results and Conclusions
The peak cell density for this cell line ranged from 22.42 x 10⁶ to 29.12 x 10⁶ viable cells/mL and viability at harvest ranged
Air through both DHSs. O₂ supplemented through micro DHS (233 µm)
Table
Basic Operating Parameters for Evaluation of ExpiCHO-S Cells in the S.U.B.s
Figure 3. Lactate profile for ExpiCHO-S (cell line 1) cell culture in the Hyperforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 4. Ammonium profile for ExpiCHO-S (cell line 1) cell culture in the Hyperforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 5. Titre results for ExpiCHO-S (cell line 1) cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels starting from day 2 to 14.
DO cascade
Feeding strategy
Table 5. ExpiCHO-S Cells (cell line 1) Product Quality From the Bioreactor Day 14 Sample
N2 and O2 through the DHS
EfficientFeed C+ supplement added on continuous drip from days 3–11 and glucose as needed
from 63.5 to 81.1% (Figure 6). The basic metabolites of glucose, lactate and ammonium showed good consistency in trends across the different scales, especially lactate and ammonium
Figure 6. VCD and viability comparison of the Gibco™ Freedom™ CHO-S™ cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process. 50 L DynaDrive and 50 L HyPerforma VCD and viability were measured by Roche Ceded HiRes™ Analyzer, while 2000 L XCellerex XDR, 2,000 L HyPerforma, and 5,000 L DynaDrive VCD and viability were measured by Beckman Coulter Vi-CELLTM XR.
(Figures 7–9). A comparison of productivity across all vessel types showed a range of 0.58–1.02 g/L on day 14 (Figure 10). The instrumentation used to measure productivity for all the 50 L runs was different compared to the ≥200 L runs, likely contributing to the observed productivity differences between the bioreactor scales.
Day 14 samples from the 200 L XCellerex XDR and 5,000 L DynaDrive vessels were miniprepped, and the product quality of charge variants and high and low molecular species was evaluated. It was clearly observed that potential critical quality attributes, such as charge variant species across the two samples, were less than 2% different, while SEC established a less than 0.5% difference in product impurities (Table 7). This consistency in product quality further highlights the opportunities and benefits in transferring existing processes based on Freedom CHO-S cells into the DynaDrive S.U.B. when searching for a larger vessel size.
Figure 7. Glucose profile for the Freedom CHO-S cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 8. Lactate profile for the Freedom CHO-S cell culture in the HyPerforma, DynaDrive,
Table 6. Operating Parameters for Freedom CHO-S Evaluation in Different S.U.B.s
Figure 9. Ammonium profile for the Freedom CHO-S cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 10. Titre results for the Freedom CHO-S cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process. 50 L DynaDrive and 50 L HyPerforma titres were measured by Cedex analyzer, while 200 L XCellerex XDR, 2,000 L HyPerforma, and 5,000 L DynaDrive titres were measured by ProA-UPLC.
Table 7. Freedom CHO-S Cell Line (cell line 2) Product Quality From Bioreactor Day 14 Sample
Feeding strategy Feed A and Feed B added as a bolus feed starting from day 3 and glucose as needed starting from day 6
8. Operating Parameters for CHO DG44 Cell Line Evaluation in the Pilot-scale S.U.B.s
Case study 3: Vessel Evaluation Using CHO DG44 Cell Line in 14-day Fed-batch Run
Methods
The methods used for cell resuscitation, propagation and cell mass accumulation were outlined in case study 1. Changes in cell line, process and operating conditions for each bioreactor are described in Table 8. Specifically, feed A and feed B were added as a bolus feed from day 3 to 14, and glucose was supplemented as needed. Daily samples were collected for cell counts, viability, dissolved gases, metabolites, and titre. Titre samples were filtered and frozen starting on day 6 for ProA-UPLC analysis.
Results and Conclusions
Case study 3 has the greatest distribution in vessel size and culture volume, covering the DynaDrive family of 50 L, 500 L and 5,000 L, and the XCellerex XDR family of 50 L, 200 L and 2,000 L S.U.B.s while also including the 50 L and 250 L HyPerforma vessels (Table 8). Peak VCD of the CHO DG44 cell
line shows similar trending across the different vessels with a range of 16.71 x 10⁶ to 30.50 x 10⁶ cells/mL (Figure 11). The 200 L XCellerex XDR culture had the lowest peak VCD of 17 x 10⁶ cells/mL and the 5,000 L DynaDrive culture had the highest VCD, peaking at 30 x 10⁶ cells/mL. The differences observed in growth are also reflected in the fluctuations observed in the various metabolites. While glucose was closely maintained post day 6 at 4.0 g/L and lactate remained below 1.65 g/L, there was significant metabolite fluctuation with this cell line even within the same bioreactor family (Figures 12–14).
Regardless of the different growth and metabolite levels from different vessel types and scales, the productivity only showed two separate trending patterns (Figure 15). For the cultures ≤200 L, a range of 3.11 to 3.33 g/L was observed, while cultures ≥250 L showed a range of 4.23 to 4.76 g/L.
To further elucidate the impact of process parameters on product quality, cIEF and SEC (Table 9) were performed on day 14 samples from all vessel types and volumes. Charge
Table
variant distribution was more consistent across cultures ≥250 L compared to cultures ≤200 L. Specifically, the charge variants from the 250 L HyPerforma, 500 L DynaDrive, 2,000 L XCellerex XDR and 5,000 L DynaDrive S.U.B.s were all within 6% of each other, whereas the DynaDrive 50 L S.U.B. had at least a 10% difference in acidic and main species when compared to XCellerex XDR 50 L and 200 L. In contrast to the charged species variability, product-related impurities, such as high
and low molecular weight species, were more consistent across all vessel types and volumes. Specifically, there were ≤6% of aggregates and <3% of fragments in samples for each bioreactor. Overall, although different growth and metabolites were observed among different cultures, the productivity and product quality data showed consistency with vessel volume regardless of the vessel type, suggesting the success of the process transfer from one type of vessel to another.
Methods
The methods used for cell resuscitation, propagation and cell mass accumulation are outlined in case study 1. The process and operating conditions for each bioreactor are described in Table 10. Specifically, feed A and feed B were added as a bolus feed from day 2 to 13, and glucose was supplemented as needed. Daily samples were collected for cell counts, viability, dissolved gases, metabolites and titre. Titre samples were filtered and frozen starting on day 6 for titre measurement.
Figure 11. VCD and viability comparison of the CHO DG44 cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Table 9. CHO DG44 Cell Line (cell line 3) Product Quality From Bioreactor Day 14 Sample
Figure 12. Glucose profile for the CHO DG44 cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 13. Lactate profile for the CHO DG44 cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 15. Titre results for the CHO DG44 cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 14. Ammonium profile for the CHO DG44 cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Case study 4: Vessel Evaluation Using CHO-M Cell Line in 14-day Fed-batch Run
Seeding density (x 106 cells/mL)
pH control
DO cascade O₂ through the DHS (80 µm)
Feeding strategy
Results and Conclusions
Acid control: sparged CO2 Base control: 1.5 M sodium carbonate
O₂ through the DHS (500 µm)
O₂ through the DHS (500 µm)
O₂ through the DHS (233 µm)
O₂ through the DHS (582 µm)
Feed A and feed B added on as a bolus feed from days 2–13; glucose was added as needed
O₂ through the DHS (457 µm)
*Agitation of the 2,000 L HyPerforma S.U.B. was set to the vendor-suggested maximum agitation of the vessel.
Table 10. Operating parameters for CHO-M cell line evaluation in different S.U.B.s
Results from case study 4 showed the peak VCD with a range of 29.08 x 10⁶ to 38.53 x 10⁶ cells/mL, while maintaining over 82% viability on the final day of the process (Figure 16). Glucose, lactate and ammonium profiles all followed their general trends and were consistent across vessels and scales (Figures 17–19). Specifically, lactate concentrations in the XCellerex XDR batches were below 1 g/L by day 7, while for both the HyPerforma and DynaDrive vessels, lactate reached below 1 g/L at later days. This change in lactate consumption was not observed to impact the titre range of 6.74 to 7.89 g/L (Figure 20), and the highest titre batch was the 5,000 L DynaDrive at 7.89 g/L. Using the CHO-M cell line, product quality from the 500 L and 5,000 L DynaDrive, 2,000 L HyPerforma and 50 L and 200 L XCellerex XDR bioreactors was evaluated (Table 11). For all scales and bioreactors evaluated, the CHO-M cells product
was purified using the same single-step chromatography purification method.
Some differences were observed across the charge variant species among the XCellerex XDR, HyPerforma and DynaDrive vessels. The differences in product impurities are less pronounced, as the aggregate and fragment content show a difference of 1.2% or less across all vessels studied. Overall, the CHO-M cell line showed consistent productivity and similar product quality when the process was transferred from XCellerex XDR and HyPerforma S.U.B.s to DynaDrive S.U.B.s.
Conclusion
Preliminary runs with DynaDrive S.U.B.s suggest that the process transfer from XCellerex XDR or HyPerforma S.U.B. to DynaDrive
Figure 16. VCD and viability comparison of the CHO-M cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 17. Glucose profile for the CHO-M cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 19. Ammonium profile for the CHO-M cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Figure 18. Lactate profile for the CHO-M cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process.
Application Note
Figure 20. Titre results for the CHO-M cell culture in the HyPerforma, DynaDrive, and XCellerex XDR vessels over a 14-day process
S.U.B. is achievable. While there is not a fit-for-all strategy for a vessel change or scale-up, the P/V scale-up strategy for agitation worked quite well for our studies. In the case studies presented in this paper, we were able to successfully transfer processes that had been developed for either the 2,000 L XCellerex XDR or HyPerforma bioreactor to the 5,000 L DynaDrive S.U.B.
Through the studies with four cell lines, it was clear that the process defines the product. With the processes for the CHO-S cell lines and the CHO-M cell line, there was enough robustness within the process to overcome the vessel change and scale-up. Conversely, with the processes for CHO DG44 cells, the changes in vessel type and volume impacted cell growth, metabolites and product quality. In cases like this, a risk-based approach
Table 11. CHO-M Cell Line Product Quality from Bioreactor Day 14 Sample
may go hand in hand with the process transfer or scale-up. Understanding the process robustness and vessel capabilities is necessary for a seamless transfer from one system to another.
Overall, the design, scale and operations of the 5,000 L DynaDrive S.U.B. can be key in the commercialisation of a human therapeutic. As such, the case studies in this paper serve as the basis for future process transfer from existing systems into the DynaDrive family of S.U.B.s.
Qingwei Luo
Qingwei Luo, PhD, Manager, Process Development, Thermo Fisher Scientific
Ben Madsen
Ben Madsen, Bioprocess Lab Manager, Research and Development, Thermo Fisher Scientific
Jeff Hou
Jeff Hou, PhD, Director, Scientific and Technical Affairs, Thermo Fisher Scientific
Matt Zustiak
Matt Zustiak, PhD, Director, BioCollaboration Centre, Thermo Fisher Scientific
Acknowledgment
We would like to thank Jordan Cobia, Paula Decaria, Hailey Mauro, Katelyn Parkinson, Andrew Sattler, and AFS for their contribution to the work.
Logistics & Supply Chain
Turning the Temperature Up on Cold Chain Logistics
As biopharma therapies become more sensitive, costly and complex, traditional compliance frameworks are no longer enough. Real-time visibility and IoT innovation are redefining what’s possible, and necessary, in temperaturecontrolled logistics.
Temperature control has always been a non-negotiable element in pharmaceutical logistics. But therapies have become more sensitive, routes are more complex and costs are sky-high, resulting in the biopharma industry losing around $35billion annually due to failures in temperature-controlled logistics. In this environment, compliance alone is no longer sufficient. Meeting the minimum standard does not guarantee the safety, efficacy, or timely delivery of life-saving treatments. And yet, many cold chain systems still rely on reactive monitoring and legacy infrastructure that can’t keep up with modern demands.
If the pharmaceutical industry wants to protect patients and products, it must reframe cold chain logistics as a strategic differentiator, not just a regulatory requirement. That starts with smarter, real-time visibility and IoT-driven resilience built for the realities of today’s pharma landscape.
Regulatory Compliance Isn’t Failing – But it is Falling Short Regulatory frameworks like FDA 21 CFR Part 11 and EU GDP have helped standardise critical components of pharmaceutical transport, from documentation to storage and handling. But they were never designed to address the speed, personalisation and volatility driving today’s drug development and delivery.
Consider the rise of cell and gene therapies (CGT). These are highly personalised, extremely time-sensitive treatments that can cost millions per dose and often require cryogenic conditions at -150°C or below. Delays, temperature excursions, or even minor deviations in humidity or shock can render a shipment useless, meaning a missed treatment window, a restart of the manufacturing process and in the worst cases, irreversible consequences for the patient.
These therapies and other cold chain-dependent products like biologics, mRNA vaccines, and oncology drugs are putting pressure on systems that were built for a different era. Compliance alone cannot keep up with the pace or complexity of these innovations.
Cold Chain Blind Spots Put Product and Patients at Risk
The financial and clinical stakes of a compromised shipment are staggering. Reports published in early 2025 show that 37% of companies still lack visibility into the real-time location or condition of their shipments, leaving them in the dark until it’s too late. Even when tracking is available, it’s often static, like barcode scans or delayed carrier updates. These provide snapshots, not continuous assurance.
This is especially dangerous for temperature-dependent, global pharmaceutical supply chains that must navigate customs, weather events, traffic delays and infrastructure inconsistencies across borders. Blind spots in these scenarios lead to lost product, regulatory fallout and even patient harm.
And the stakes are only getting higher. The global cold chain packaging market is forecast to grow from USD 29.1 billion in 2025 to USD 50.5 billion by 2035, reflecting the rising investment needed to safeguard sensitive shipments. Hospitals remain the central distribution channel, expected to account for more than 52% of total market share in 2025 due to their role as primary hubs for advanced diagnostics, drug administration and interventional procedures. This underscores both the economic and human importance of temperature-sensitive shipments, where even a single logistics failure can trigger public health crises, erode trust and result in multimillion-dollar losses. As the industry expands into emerging markets, the margin for error becomes even narrower.
Bridging the Gap Between Control and Confidence
Real-time visibility, powered by Internet of Things (IoT) technology, is redefining what temperature-controlled logistics can and should look like. IoT trackers can give logistics teams continuous access to five critical metrics, including:
1. Temperature and humidity
2. Shock and tilt events
3. Light exposure (to detect tampering)
4. GPS-based location
5. Route deviations and dwell time
And that’s just the start. This level of insight turns cold chain management from a guessing game into a controllable process. Teams can act on alerts the moment an excursion occurs, reroute shipments preemptively, or intervene with partners at key transit points. In many cases, these interventions preserve product integrity and prevent costly delays or waste.
Beyond individual shipments, historical data from IoT trackers enables systemic improvement. Trends can be analysed to optimise packaging, validate new shipping lanes, fine-tune SOPs and proactively inform audits or inspections. That means not only fewer errors, but better decisions over time.
Smart Shipments = Cold Chain Resilience
One of the most overlooked aspects of cold chain resilience is the psychological confidence it gives stakeholders. When a hospital pharmacist, QA leader or regulator can see real-time shipment data and a full audit trail, they gain peace of mind that a product has remained viable from origin to destination.
In the past, pharmaceutical companies would often discard a product if there was any doubt about its condition, even without conclusive evidence of a breach. That’s no longer necessary. IoT
devices offer an indisputable data record, allowing teams to make informed, defensible decisions and reduce unnecessary waste.
Real-world examples show just how powerful this level of visibility can be when lives and millions of dollars are at stake. Take the case of Optimize Courier, a global leader in time-critical and temperature-sensitive logistics. Prior to adopting real-time visibility tools, the company relied on passive data loggers that revealed issues only after shipments were delivered. By integrating real-time 5G GPS trackers into 97% of its high-value shipments, Optimize transformed its operations, making temperature excursions all but extinct.
The impact is both operational and financial. In one case, these alerts prevented the loss of a $500,000 pharmaceutical shipment by flagging that it had been mistakenly placed in a refrigerated cooler. Optimize immediately intervened, recovered the shipment and ensured it reached its destination on time and within the correct ambient range. Beyond saving product, this visibility has strengthened trust with customers who depend on Optimize for life-saving treatments, diseaseeradicating research and even organ transport.
From Validation to Continuous Assurance
Validation is another area where IoT is driving change. Traditional cold chain validation relies heavily on simulated conditions in climate chambers or small pilot runs. But these setups often fail to capture the true environmental variability of a live shipment.
By contrast, IoT trackers collect real-world data at scale. Companies can use this information to revise lane qualifications, update packaging specs and even adjust shelf-life calculations based on actual transit conditions. This continuous loop of data and optimisation allows validation to evolve from a one-time box-checking exercise to an ongoing assurance process.
Some progressive pharma companies are already leveraging virtual models of shipments that integrate IoT data with predictive analytics. This cutting-edge scenario modelling, otherwise known as “digital twins,” helps to stress-test conditions before products even leave the warehouse. This helps logistics teams move from reactive firefighting to proactive, data-driven decision-making.
The Future: Predictive, Sustainable and Collaborative
The ultimate promise of IoT-enabled cold chain systems lies in their predictive power. When combined with analytics, historical shipment data and even weather forecasts, companies can begin to anticipate risks before they occur. AI-powered route planning and exception alerts will increasingly guide decisionmaking, minimising delays and optimising outcomes.
Just as importantly, the same technologies advancing reliability are also opening doors to sustainability. Cold chain logistics is notoriously energy-intensive and companies are under growing pressure to reduce carbon emissions without compromising patient safety. Smarter route planning, reusable packaging and greener transport modes, enabled by visibility data, can help organisations strike the balance between environmental responsibility and operational excellence.
Logistics & Supply Chain
Collaboration will also be key. No single manufacturer, carrier, or technology provider can solve these challenges in isolation. The most resilient supply chains will be built on partnerships that share real-time data, harmonise processes across borders and adopt industry-wide standards for interoperability.
What Comes Next: Future-Proofing the Pharma Cold Chain
The pharmaceutical supply chain is at a tipping point. The industry has made massive strides in drug innovation, but logistics hasn’t always kept pace. Now, with the help of new innovations like IoT-enabled smart sensors, real-time tracking and integrated data platforms, cold chain leaders have the opportunity to modernise operations and mitigate the risks that compliance alone can’t cover.
The future of cold chain logistics is both transparent and intelligent. This is more than staying compliant; it’s about staying in control, ensuring life-saving medicines arrive safely, consistently and with total confidence. In a world where even the smallest temperature fluctuation can mean the difference between success and failure, that level of control isn’t just an operational upgrade. It’s a moral obligation.
Alex Guillen is an established executive with a proven record in global business and market development, with well-rounded experience in multicultural sales management and brand building. Guillen has extensive experience and expertise in cold chain as Global SME, Life Science and Pharma at Tive, Guillen leads sales and business development within the company's rapid-growth Life Science division. Previously, Guillen served as a Board Member and leader of Corporate Strategy at SWITRACE S.A, a developer of temperature and humidity data loggers compliant with the Pharma and Biotech industries. Guillen’s extensive experience also includes serving as Global Cold Chain Director of Fisher Clinical Services, CEO of Escort Cold Chain Solutions SA and Director for Commercial Operations for Novartis Vaccines.
Developing Endotoxin Limits, Risk Assessment and In-process Testing for CGT Products
Cell & Gene Therapy (CGT) products face a unique challenge in the required pyrogen testing for injectable wares. Since this test was written for conventional pharmaceuticals and medical devices, CGT products face special challenges in applying the endotoxin test to these goods.
Pyrogens are molecules that induce the human immune system to initiate a febrile response. Many of these are cytokines that are part of the immune system. These signalers are called endogenous pyrogens. However, molecules that originate from outside the body that initiate this pyrogen pathway are called exogenous pyrogens.
The most potent pyrogens are the lipopolysaccharide (LPS) molecule components of the outer membrane of gram-negative bacteria. It was known that drugs would frequently cause symptoms of septic shock even when sterilised. In 1942, the United States Pharmacopeia (USP) initiated USP <151> as the first pyrogen test requirement for parenteral and intrathecal injections.1
In 1972, it was discovered that the same LPS components that caused the human febrile response were responsible for the clotting of Atlantic Horseshoe Crab hemolymph in response to Gram-negative bacteria.2 This led to the 1980 publication of USP <85> Bacterial Endotoxin Test (BET), which allows for the usage of this extract (Limulus Amebocyte Lysate – LAL) to be used in place of the pyrogen test.3
In 2024, the USP published USP <86>, which allows for the recombinant protein found in LAL to be used to detect endotoxin in products. This allows for horseshoe-crab-free test reagents to be used for endotoxin testing.4
As we reach the realm of specific therapies that are personalised for each individual patient, lead times change the ability to perform the appropriate tests on the product. Although this is a much bigger issue in terms of sterility testing, endotoxin testing can still reach a time limit that can be pressing on the needs of the user.
Specifically, many users will have outsourced their QC tests, which may not be acceptable in terms of lead time for the needs of the therapeutic. To bring the testing in-house, an internal testing environment needs to be set up. However, this can be an issue as CGT samples are not simple and require various considerations for testing. Some of these considerations include determining the correct endotoxin limit for the product, overcoming interferences, non-traditional lot sizes and testing needs in non-conventional QC lab settings.
Determining the Endotoxin Limit of the Product
First, the acceptable endotoxin limit of the product needs to be determined. This is different from other forms of QC testing, such as sterility testing, that already have a predetermined set limit, or the limit is to show that the contaminant is not detected. Originally, the need for an endotoxin limit came about as the BET is more sensitive than the rabbit pyrogen test. In the original comparison of the methods, it was determined that most tests reported results consistently between the pyrogen and the endotoxin test. However, there were several cases where the lysate reagent reacted when no pyrogenic response was observed. This led to the need to standardise LAL reagent measurements and correlate them to pyrogenicity. This resulted in the adoption of a globally harmonised Reference Standard Endotoxin to provide calibration for each formulation of LAL reagent and the setting of a threshold pyrogenic dose (TPD) to correlate measurements in endotoxin units against the pyrogenicity of the product.6 In the years since, many harmonised standard levels for pharmaceuticals, excipients,
Subsection: Cell and Gene Therapy
water and accessory buffers have been set. However, for novel products, the manufacturer needs to consult the TPD in establishing the limits.
The TPD is set to 5 Endotoxin Units per kilogram of body mass per hour of injection or bolus dose (5 EU/kg/hr).3 Based on a 70 kg patient mass, this leads to a maximum bolus/hourly pyrogenic allotment per patient of 350 EU. Dividing this by the maximum bolus/hourly dose volume will result in the endotoxin limit for the product. For example, a dose of 10 mL would have a calculated endotoxin limit of 35 EU/mL. This provides the maximum endotoxin limit that should be used for the product. Care should be taken to examine what other products will be given with the product that could also add to the endotoxin given to the patient at this time. Although not a requirement, it is good practice to accept a more stringent endotoxin limit based on these considerations.
Mitigating Product Interference and Choosing the Correct Endotoxin Test Method
Once the endotoxin limit of the product is set, the determination of the endotoxin detection method to be used can begin. USP <85> outlines methods from simple endpoint qualitative techniques with a sensitivity of 0.25 EU/mL to quantitative techniques with a sensitivity down to 0.0005 EU/mL.
Choosing the correct test method will not be as simple as comparing the endotoxin limit of the product to the LOD/ LOQ of the test method. Cellular materials, proteins, pH, lipid complexes, and certain ions and detergents cause known interference with the BET method.
Dilution is the most reliable method to mitigate interference. The Maximum Valid Dilution (MVD) can be found by dividing the endotoxin limit of the reconstituted sample to be tested by the sensitivity of the product. For example, a product with an endotoxin limit of 10 EU/mL being tested with a test method with a lowest endotoxin sensitivity of 0.001 EU/mL would have an MVD of 10000. This value indicates how much the product can be diluted and still be applicably tested by the chosen BET test technique.7
Most product interferences can be mitigated by product dilution. However, this dilution factor may be significant. Finally, sensitive test reagents are valuable not just for testing to low endotoxin limits but also to accommodate testing highly diluted products to overcome test interference.
Although dilution will overcome most interference, there are several factors that may need additional treatment, which may require further consideration. First, proteins that exhibit enzymatic interference on the protein cascade found in the LAL reagent (whether naturally derived or recombinant reagent) will need to be inactivated, as they will cause interference at low concentrations. The easiest method is to perform heat treatment of the product at 70°C for approximately 15 minutes. However, if this causes the sample to congeal, other treatments, such as a digestion buffer or an endotoxin extraction resin, may be utilised. Second, chelating agents and alkali-earth cations can disrupt the binding of the LPS to the Factor C protein of the endotoxin detection cascade. Addition of an appropriate buffer may be required. Finally, the physical insoluble material of a
product with high cell material concentration can potentially interfere with the optical readers performing the test. A magneto-optical reader can potentially be used to overcome this interference when dilutions cannot be performed.8
Overcoming Time Constraints and Testing Needs in Non-conventional Lab Settings
The next consideration that manufacturers of CGT products will need to consider is how to bring the testing to their site. The BET is a relatively rapid QC test, usually not taking more than 1.5 hours for the result. However, this rapidity is lost if the manufacturer needs to outsource their endotoxin testing needs. Although consideration is greater for sterility testing, as USP <71> requires a 14-day turnaround, leading to sterility testing being done at risk in many cases, I recommend that endotoxin test results should always be known before administration. First, although not a sterility test, the endotoxin test is also a very sensitive indicator of Gram-negative contamination. Second, endotoxin test methods are available that can be performed simply and at the point of use to produce results within 15 minutes. For users of testing services for pyrogen and endotoxin services, I recommend that at least endotoxin monitoring equipment be brought in-house to confirm the products are endotoxin-free before administration. Additionally, robust endotoxin testing can be performed in-house with investment in equipment and training that will pay for itself many times over when the outsourced endotoxin testing can be eliminated.
For a CGT product manufacturer that currently outsources endotoxin testing but is looking to bring the test in-house, a phased implementation is recommended. This phased implementation could resemble the following. First, low-volume, point-of-use endotoxin equipment with FDA-licensed LAL reagent can be brought onsite to monitor the endotoxin content of water sources and the final products in conjunction with the outsourced testing. Second, when the final product testing is shown to match the outsourced testing and the team is sufficiently proficient in the test, new products can begin to be developed, in which the entire endotoxin validation is performed in-house. Third, as more products are tested in-house, higher volume equipment can be purchased with FDA CFR pt.11 compliant software to manage the test results. Finally, all products can be gradually brought to in-house testing as the team’s proficiency is complete.
Setting up a Testing Plan for Non-traditional Lot Sizes
Traditionally, endotoxin testing has been performed on at least three samples per manufacturing lot, taking from the beginning, middle and end of the run. However, the FDA and USP have encouraged users to take a risk-based approach to the correct sampling numbers and plan to be used, with emphasis on the need for in-process confirmatory tests.7,9
Additionally, traditionally the interference testing for the BET involves testing three lots of each product.
For highly customised CGT products that do not have the traditional manufacturing model of many identical products consisting of one lot, but rather many smaller “lots” that are uniquely manufactured and may not be replicated. For these considerations, one sample of each lot may be what is being
Subsection: Cell and Gene Therapy
tested to give both interference and endotoxin assurance. In these cases, the regulatory documents highlight the need for the user to demonstrate that the risk of this testing is mitigated by control-process testing that occurs upstream and over time to confirm the manufacturing environment is under control.
It is in these cases that low-cost, frequent, single gel clot tests may give invaluable insight spread over time and over the entire manufacturing process, supporting the final-product testing assurance. Additionally, the in-process testing will most likely have to overcome less product interference than the final product, as the simpler buffers and media will be more accommodating to the LAL reagent.
Conclusion
With the endotoxin test techniques found in USP <85> and <86>, a CGT manufacturer needs to approach their endotoxin needs not as a one-size-fits-all solution, but as several different techniques that can be adopted to give the overall endotoxin sterility assurance. The most information for the lowest cost will be had when low-resource, frequent tests can be performed along the entire production stream that complements the robust final product testing before final product release.
There are several challenges that CGT product manufacturers uniquely face in performing the endotoxin test. But these are not factors that are insurmountable. The overall benefit of bringing endotoxin testing in-house, as opposed to using third-party testing, is that the QC control program can have the tools to take a proactive approach over a reactionary approach to the endotoxin in the product samples. Interested manufacturers are encouraged to reach out, as the reagent manufacturer will be able to provide invaluable insight and validation support in the adoption of the method.
REFERENCES
1. United States Pharmacopeia NF. USP <151> Pyrogen Test, (2017). https://doi.org/10.31003/USPNF_M98900_01_01
2. Young, Neal, Levin, Jack, & Prendergast. An Invertebrate Coagulation
System Activated by Endotoxin: Evidence for Enzymatic Mediation. The Journal of Clinical Investigation 51, 1790-1797 (1972).
3. United States Pharmacopeia NF. USP <85> The Bacterial Endotoxins Test, (2018). https://doi.org/10.31003/USPNF_M98830_02_01
4. United States Pharmacopeia NF. USP <86> The Bacterial Endotoxins Test with Recombinant Reagents, (2025). https://doi.org/10.31003/ USPNF_M16015_02_01
5. Wachtel, Ruth, & Tsuji, Kiyoshi. Comparison of Limulus Amebocyte Lysates and Correlation with the United States Pharmacopeial Pyrogen Test. Applied and Environmental Microbiology June 1977, 1265-1269 (1977).
6. Weary, Marlys, Donohue, Georgiana, Pearson, Frederick, & Story, Kenneth. Relative Potencies of Four Reference SEndotoxin Standards as Measured by the Limulus Amoebocyte Lysate and USP Rabbit Pyrogen Tests. Applied and Environmental Microbiology. De. 1980, 1148-1151 (1980).
7. United States Pharmacopeia NF. USP <1085> Guidance on the Endotoxins Test, (2020). https://doi.org/10.31003/USPNF_ M2245_03_01
9. Food and Drug Administration. Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers, (2012). https://www.fda. gov/regulatory-information/search-fda-guidance-documents/guidanceindustry-pyrogen-and-endotoxins-testing-questions-and-answers
Timothy Francis
Timothy Francis is the Senior Technical Specialist for the Pyrogen Testing Products Division of FUJIFILM Biosciences. He holds a B.S. In Biology and an M.S. in Curriculum and Instruction in Science. He came into this role with five years of experience teaching the natural sciences at a college level. He is proficient at taking the complex, technical aspects of a topic and breaking them down into clear, understandable pieces that all connect back to the big picture. He draws upon this experience to provide professional technical support and training for the PYROSTAR™ line and to help you with your technical needs.
Use of the aProximateTM Proximal Tubule Cell Model for the Evaluation of Safety and Renal Accumulation of Radioconjugates and Large Molecules
The aProximateTM Proximal Tubule Cell (PTC) model offers a cutting-edge platform for the evaluation of drug-induced nephrotoxicity, radioconjugate retention and the accumulation of large molecules (ADCs, ASO, antibiotics, peptides, etc) in the proximal tubule. This white paper outlines the advantages of using the aProximateTM PTC model in predicting and assessing renal safety in the early stages of drug development, focusing on its application in the context of renal radioconjugates and large molecule dynamics.
Model Overview
The aProximateTM PTC model replicates the physiology and function of human proximal tubule cells. These cells are crucial for assessing kidney toxicity, drug-drug interactions
and the pharmacokinetics of new therapeutics, not only of traditional small molecules, but also of the new modalities of large molecules and particularly radioconjugates, which are prone to renal accumulation. The cells in the model maintain differentiation, expressing high levels of renal transporter, including Megalin and Cubilin, which mediate protein and large molecule uptake in the nephron (Figure 1.). This model has been validated against various parameters, including Trans Epithelial Electrical Resistance (TEER), uptake and flux of small and large molecules and specific biomarker expression of toxicity, ensuring its physiological relevance and accuracy in safety evaluations.
Safety Evaluation & Radioisotope Accumulation
The very specific, targeted and focused exposure of tumour cells to radioligand with minimal collateral damage represents a major breakthrough in cancer therapy. Unfortunately, this
Figure 1: (B) Immunofluorescence staining of aProximateTM proximal tubule cells for Tubulin, Pericentrin, OAT1 and OAT2.
Figure 1: (A) Schematic representation of the aProximateTM PTC cell model representing the high expression of all key transporters, the formation of an epithelium with tight junctions and polarity, and the use of TranswellsTM for cell culture to recreate a lumen and a blood compartment to assess renal transport directionality.
modality has the potential for severe off-target damage. The kidney is particularly at risk due to its potential accumulation of radioconjugates within the cortex of the kidney, leading to severe nephrotoxicity. The mechanisms behind this have been under intense scrutiny and the Megalin-Cubilin receptormediated endocytic pathway has been implicated (for review see: Pahir et al. 2021, Translational Oncology). With the kidney as a major target, it is therefore crucial to fully assess the renal clearance and retention of novel radiopharmaceuticals and their impact on kidney function at an early stage in drug development. Screening for the renal retention and subsequent renal toxicity can be greatly accelerated with cutting-edge in vitro models such as aProximateTM. The aProximateTM PTC model can be instrumental in evaluating this risk by being a predictive model of the proximal tubule environment and crucially expressing the key Megalin-Cubilin receptors. The aProximateTM model provides an ideal platform to screen renal accumulation of radioconjugates and define strategies to reduce renal retention and de-risk candidate radioconjugates for renal retention and renal toxicity.
Assessment of Large Molecule Accumulation
Radioconjugates include a range of structures, including Antibody-Drug Conjugates (ADCs), peptides and bispecific antibodies. A common feature of these constructs is a chelator, a short half-life radioisotope. The short half-life of the isotope has proved a roadblock in studying the renal retention and renal damage of radioconjugates due to the difficult logistics inherent
Modality Detection Method
Peptide 3H-label
ADC Alexa568 Dye
Bispecific AB Lu metal LC/MS IPC
in their use. Newcells Biotech has addressed this by developing a number of different labelling techniques to overcome this hurdle (Table 1). These include measuring a non-radioactive form of the radiolabel, labelling the radioconjugate with 3H or a fluorescent probe.
Cade Studies & Applications
Flux of a Radioconjugated Peptide
The aProximateTM PTC rat model was used to measure the apical and basal uptake of a 3H radiolabelled peptide. This data was fundamental in understanding the kinetics and dynamics within the renal cortex, supporting the development of safer therapeutic agents with minimised renal side effects. The data was complemented with an inhibition study to confirm the active role of Megalin and Cubilin in this transport (Figure 2b).
2: Characterisation of apical uptake of radioconjugate peptide using a 3H radiolabelled peptide in rat aProximateTM. (A) Quantification of Radioconjugate uptake showing apical uptake. (B) Uptake inhibitor study with 3H radiolabelled peptide. Flux inhibition and apical uptake is reduced in the presence of known inhibitors of Megalin/Cubilin by LPS, ROS and RAP.
End Point
Measurement of Apical uptake, transepithelial flux, inhibition of uptake by RAP, single TA
Measurement of Apical uptake, inhibition of uptake by RAP, 3 Tas
Measurement of Apical uptake, inhibition of uptake by RAP, rank order of TAs
Table 1
Figure 1: (C) Immunofluorescence staining of aProximateTM proximal tubule cells for of Megalin and Cubilin.
Figure
Cubilin
Megalin
Cross-species Safety Analysis of Large Molecules
We have developed a number of cross-species models of the proximal tubule; these include human, rat, dog and mouse. These models have been used extensively to understand cross-species differences in the renal handling of small molecules. Figure 4 shows the handling of the protopic anionic molecule-PAH (para-aminohippurate) and the ability to interrogate different preclinical species using the aProximateTM PTC models. This type of study is crucial to ensure the safety and efficacy of therapeutics across different biological systems before clinical trials. This work has also been extended to validate the handling of large molecules – including radioconjugates in rat, mouse and dog.
Conclusion
The aProximateTM PTC model represents a significant asset in the field of renal safety evaluation for large molecules, including radioconjugates. By providing a robust, physiologically relevant
cross-species platform, it enables the detailed analysis of nephrotoxicity, drug interactions, and the specific dynamics of radioisotope and large molecule retention. This white paper underscores how this technology can advance different types of therapy modalities, including peptide receptor radionuclide therapies (PRRTs) and others that utilise radioisotopes. It highlights the model's utility in enhancing drug safety profiles and optimising therapeutic strategies to mitigate renal risks in drug development.
Future Directions
Continued advancements in this area are expected to focus on refining the model to include more complex kidney structures and applying it to a broader range of therapeutic molecules. Additionally, integrating this model with systemic pharmacokinetic models can provide a more comprehensive assessment of drug behaviour across multiple organ systems, further improving the value of in vitro models to define the predictability of human clinical outcomes.
Dr. Colin Brown
Colin was born in Edinburgh, grew up in Fife and was awarded his PhD by the University of St Andrews. He was then a Royal Society European Postdoctoral Fellow at the University of Zurich. Following on from this, Dr Brown was a Wellcome Senior Research Fellow at Manchester University. He was then an Associate Professor in the Institute of Cell & Molecular Biosciences for nearly 30 years. Dr Brown is a leading expert in kidney transport, with research interests in renal, hepatic and GI drug transporters. He has developed and commercialised several primary cell-based assays for measuring kidney transport and toxicity, has authored over 100 publications and several book chapters and has been an invited speaker at numerous conferences around the world. He has been a mentor to 16 PhD students and numerous undergraduate and Masters students. Dr Brown is a past Chair of the AAPS Drug Transporter Focus group and a member of the PPDM executive. He was involved in the organisation of four AAPS DTFG Bench to Bedside workshops, including two as Chair.
Figure 3: Cross-species comparison of PAH, (para-aminohippurate), a derivative of hippuric acid used as a diagnostic agent for measuring renal plasma flow. Upon IV infusion, PAH is extracted from the blood by OAT1 renal transporter in the proximal tubule. PAH excretion was compared in different species, showing a net secretion of PAH in human, dog and rat.
Funct ional & Predict ive Human iPSC Deriv Ret inal Models
Safety & efficacy studies
Disease modelling
Gene therapy vector assesment
Readily available retinal organoids to ship
Predict ive Data from in vitro Studies
Rapid in vitro evaluation of drug efficacy and safety is possible on isogenic human iPSC derived healt hy and diseased ret inal organoid and RPE models, allowing for confident decision making to accelerate lead compound selection or submission of data for your IND
T he models recapitulate t he structure and funct ion of human ret ina tissues and can be generated from healt hy and from pat ients’ iPSCs or gene edited iPSC lines, facilitating predictive advanced in vitro testing of new therapeutics
28-30 October 2025 Messe Frankfurt
The heart of pharma, all in one place
Where the global pharma community gathers to shape the future.
62,000+ attendees
2,400 exhibitors
3 days of innovation, partnerships, and business growth
Experience CPHI Frankfurt
Register now and save 10%
Tackle global challenges with the entire UK lab community
SUSTAINABILITY • AI & AUTOMATION • QUALITY & COMPLIANCE • BIOTECHNOLOGY • SKILLS & DEVELOPMENT
Meet with your peers across the UK lab community, share knowledge with industry leaders in the conference, get-hands on with tech from leading suppliers - and ultimately save time when levelling up your lab practices.
4,500+
200+
NEW lab professionals suppliers networking opportunities
5 conference streams
Page 5
Page 55
IFC
Page 37
Page 49
Page 29
Page 9
BC
Page 76
Page 15
Page 77
Page 75
Page 23
Page 53
Page 43
Page 3
Page 71
IBC
Subscribe today at www.international-biopharma.com or email info@senglobalcoms.com
A&M Stabtest
Aldevron
Asymchem
Biopharma Group
Biosynth
CDD Vault
Cell 2025
CellforCure
CPHI Frankfurt
GenXPro
Lab Innovations
Newcells Biotech
PCI Pharma Services
Polypure
Richter Biologics GmbH & Co. Kg
Senglobal Ltd
Steribar
Taconic
I hope this journal guides you progressively, through the maze of activities and changes taking place in the biopharmaceutical industry
IBI is also now active on social media. Follow us on: