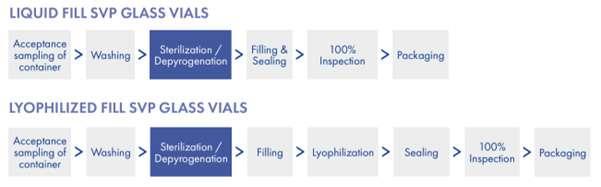
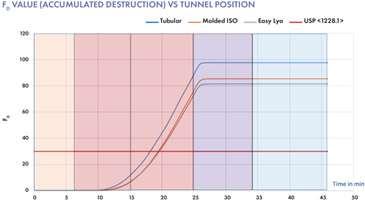
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IPI will be published in Winter 2024. ISSN No.International Pharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
04
REGULATORY & MARKETPLACE
06 Key Steps to Commercialisation Readiness in Europe
The steps to commercialisation readiness are often left too late in the roll out strategy of a pharmaceutical or medical product. In Europe, this can be difficult as the diversity of national legislation can pose challenges to the supply chain logistics. Frederic Pailloux at PLX Healthcare Services, dives into the multifaceted challenges of supplying pharmaceutical products within European markets.
08 GenAI’s Big Safety Proof Point in Life Sciences: Transforming Pharmacovigilance Case Intake
The current approach to pharmacovigilance monitoring treats all cases with the same priority in the early stages, regardless of the case’s risk. Adverse event case intake is one of the most glossed over workflows in the industry. Emmanuel Belabe at ArisGlobal, examines how advanced technologies could potentially pave the way for earlier and efficient conclusions regarding safety events.
10 Step-by-step: Refining Sustainability Performance in Single-use Drug Delivery Devices
Improving sustainability is critical across all industries The healthcare industry – and the pharmaceutical industry which serves it – are certainly no exception to this. Alex Fong of Owen Mumford, discuss why continuous progress is necessary not only for internal sustainability goals but also to reassure pharma companies that manufacturer partners are on the right path.
CLINICAL AND MEDICAL RESEARCH
12 Improved Efficiency for Pharmaceutical Laboratories with Automation of LC Workflows
Recent developments in analytical instrumentation methods have benefited researchers. The pharmaceutical industry has also taken advantage of advancements in automation. Darcy Shave of Waters Corporation examines how the widespread utilisation of automated systems can lead to more efficient pharmaceutical manufacturing.
MEDICAL DEVICES
14 Regulatory Demands in the Medical Device Sector: How are Compliance Strategies Evolving?
The pharmaceutical industry is increasing expectations around the manufacturing and distribution of device due to technology innovations and pressures on health services. Peter Muller and Mike Baird of Schlafender Hase, discuss how the increasing medical devices market is experiencing ramifications as the products must meet certain requirements.
18 Right First Time: What the Medical Device Sector Can Learn from Pharma’s Structured Data Challenges
Teams in regulatory affairs experience overwhelming workloads intensified by continuously changing health authority expectations. Sonia A. Veluchamy of Celegence adds that the medical device sector requires strategic technology investments to streamline regulatory affairs processes.
24 Integrating Medical Devices into Pharmacovigilance Portfolios Part II
When integrating medical devices into pharmacovigilance portfolios, it is crucial that there is an understanding of medical device and drug-device combination product regulation. Humaira Qureshi of Qinesca, explains the current challenges in the pharmaceutical industry and offers a look into best practices and regulatory expectations.
28 Ypsomed's Approach to Platform Products and Strategic Partnerships for Self-Injection Devices
The global pharmaceutical market revenue is expected to grow as new selfinjection devices are being produced for several medical specialties. Ian Thompson of Ypsomed adds how the company has reformed the market for platform-based self-injectables and has a reliable approach in the access of safe and easy to use drug-device combinations.
MANUFACTURING
32 Overcoming AI Anxiety in Medical Affairs: Strategies for Successful Adoption
Artificial intelligence (AI) is adopted in areas of the pharmaceutical industry and promises to transform many facets of medical affairs. Dr. Loubna Bouarfa at Envision Pharma Group, delves into the causes of anxiety surrounding AI in the pharmaceutical industry.
PACKAGING
44 Fake Drugs on the Rise: Addressing Counterfeiting and Diversion in the Pharmaceutical Industry
Counterfeiting and diversion are international issues that affect every industry. In the pharmaceutical industry, counterfeited and diverted products are becoming increasingly apparent. Stephan Von Schilcher at Systec explores the ways in which organisations can collaborate with one another to flag counterfeited and diverted materials and identify illicit distributors.
48 Studies Support Changing Perspectives of Glass Vial Performance Characteristics
In the pharmaceutical industry, glass is the most widely utilised primary packaging material for injectable drugs. The performance of the primary packing is essential in ensuring the stability of the drug. Jingwei Zhang of SGD Pharma describes how manufacturers in the pharmaceutical industry can make better decisions to ensure the safety and efficacy of their products.
HEALTH OUTCOMES
58 The Global Crisis of Counterfeit Pharmaceuticals: A Call to Action Counterfeit pharmaceutical products are a global issue which have a devastating impact on the economy and public health. The effects on the economy lead to a significant loss of jobs in relevant industries. Here, Laura Lopez at MM Packaging et al. discusses the workable solutions to the counterfeit pandemic.
62 The Pivotal Role of Patient Advocacy Organisations in Driving Rare Disease Therapeutic Development
Patient advocacy organisations (PAOs) have an essential role in the rare disease clinical development pathway. Dr. Neena Nizar and Dr. Jana Benesh at ICON’s Center for Rare Diseases, explain how effective engagement and regular interaction with these organisations can lead to better therapeutic outcomes for patients.
64 Listening to the Patient Voice to Improve Eye Care
The growing global ophthalmic eyedroppers market is expected to be driven by the increasing prevalence of eye disease and disorders such as Glaucoma. Zoë Davidson at Nemera, discuss the market growth of agerelated eye conditions.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
68 How Efficient Logistics Can Change the Lives of Rare Disease Patients
With approximately eight thousand rare diseases globally, there is a requirement to find an efficient and cost-effective solution in the transportation of orphan drugs. Dr. Danial Arkwell at Envirotainer, highlights how the barriers to shipping life-saving rare disease treatments can be removed with a patientfocused approach and improved temperature-controlled conditions.
72 Revolutionising Pharmaceutical Logistics: The Path to Sustainable Cooling Solutions
As the pharmaceutical industry rapidly expands on a global level, there is a demand for a more efficient and sustainable approach to cold chain management. Nikolas Nemickas of SpaceWalker Technologies explains how it is a moral obligation for the pharmaceutical industry to find cooling solutions which are environmentally conscious.
74 The Primary Logistics Needs of Life Science Companies
The life science market experiences a sizeable number of logistics-related complexities. John Coleman at YSDS Life Science discusses how four logistical
requirements of the industry have been identified which can positively affect operations, projects and customer relationships.
NASAL & PULMONARY DRUG DEVELOPMENT & DELIVERY
79 In Silico Modelling for Orally Inhaled and Nasal Drug Product Development
Will Ganley of Aptar Pharma company tell us everything you need to know about in silico modelling and how a specific example of physiologically based pharmacokinetic modelling can be used for orally inhaled and nasal pharmaceutical drug development.
84 Keep Breathing – Nebulisers Are Serial Soft Mist Inhalers
The pharmaceutical industry favours the development and use of nebulisers to treat patients with respiratory conditions. Philippe Rogueda at Merxin, explores how there may be advantages to using a soft mist inhaler (SMI) over an industry standard nebuliser.
86 Importance of Pre-coloured ABS in Inhalation Medical Devices
Bright and intense colours on medical devices are necessary for patients as they are associated with helping the patient to feel more positive about undergoing treatment and taking medication. Luca Chiochia at Elix Polymers explains how the use of ABS materials is growing in the healthcare industry.
88 Analytical Considerations when Re-Formulating pMDIs with Next-generation Low GWP Propellant Systems
Carbon emissions can be reduced by switching from current pressurised metered dose inhalers (pMDI) propellants to lower global warming potential (GWP) propellants. Chris Vernall and others at Intertek highlight how reformulating pMDIs can lower the environmental impact.
92 The Preservative Predicament
Regulatory bodies internationally have issued guidelines on how to ensure the safety and efficacy of nasal spray products. The nature of these regulations, particularly regarding preservatives, can often be confusing. Carolyn Berg, David Wilcox, and Mark Ignaczak of Catalent explains the guidance on the use of preservatives in nasal sprays and the manufacturing requirements that are crucial in developing their safety and efficacy.
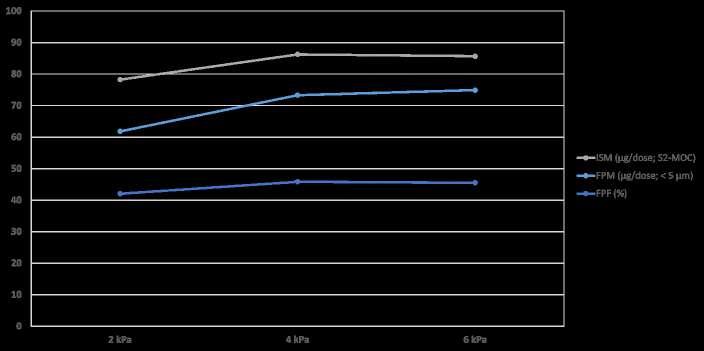
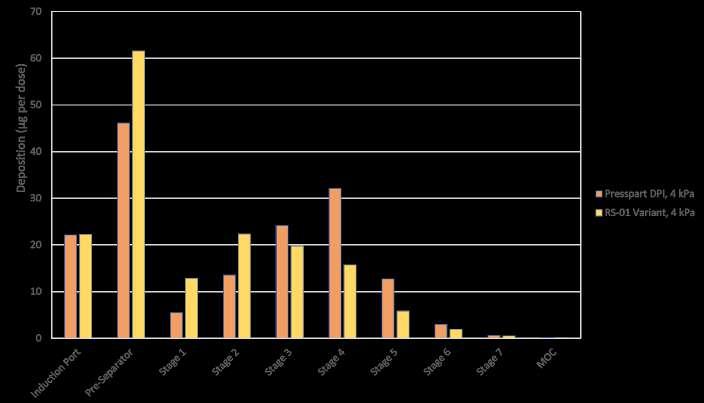
96 Development Approach for a High-performance Capsule-based DPI Device
The pulmonary route is gaining increasing attraction not only for low-dose locally acting therapies, but also for systemic applications often require higher doses or new formulation technologies. Ameet Sule, Sunita Sule and Mirjam Kobler of H&T Presspart explain that to ensure the best performance, the development of the formulation and the device should go hand in hand.
APPLICATION NOTE
36 Sterile and Safe: When Robots Support the Production of Live-attenuated Vaccines
Robot-based cell factory automation is key in the production of live-attenuated vaccinations for conditions like chickenpox. The author, Ralf Högat at Stäubli, highlights how Changchun Keygen Biological Products have been developed to improve the safe and sterile nature of the products.
40 New Product Introduction & Technical Transfer in a High-potency Facility
The core focus of the pharmaceutical market is on medicinal products containing highly potent active pharmaceutical ingredients (HPAPIs). David O’ Connell at PCI Pharma Services, describes how the pharmaceutical industry is advancing in the use of high potent drug product manufacturing.
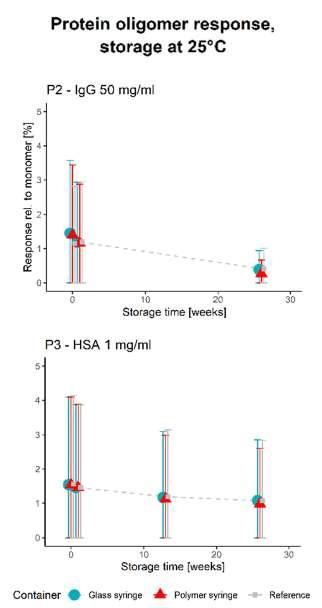
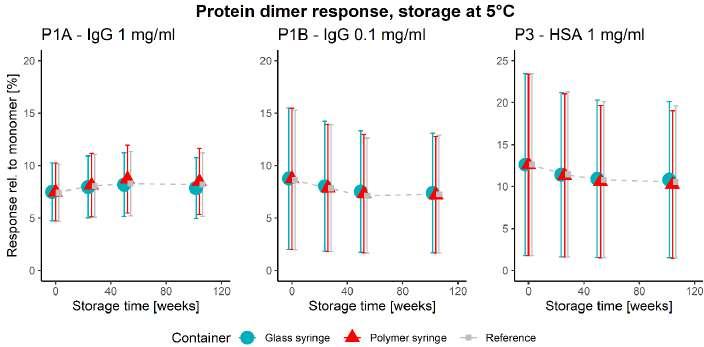
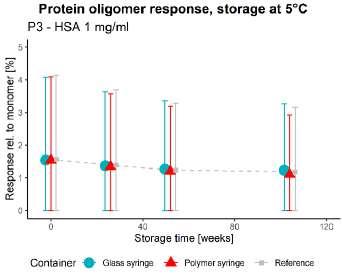
50 Unlocking the Secrets: Long-Term Storage Effects on Pharmaceutical Proteins in Glass and Polymer Prefilled Syringes
The pharmaceutical sector has experienced an increased growth in parenteral products, with new and improved ideas for the use of prefilled syringes. Nina Krautwurst and Jaywant Pawar of SCHOTT Pharma AG & Co. Switzerland, explore how alternative model proteins can be stored in syringes to better understand the container-closure interaction.
Editor's Letter
As autumn falls upon us, Volume 16, Issue 3 of IPI calls. In this issue we delve into the crux of the industry’s latest developments, seeing those in your field pinpointing progressive advancements in technologies, addressing concerns and obstacles and continuing the discussion of the nasal and pulmonary discovery world.
At a time like this Artificial Intelligence is ever evolving and across this issue we hear the benefits and drawbacks, the likes and dislikes, and the optimism and reservations had by those in the industry towards new technological advancements. In the Regulatory and Marketplace chapter Emmanuel Belabe of ArisGlobal positively praises the efficiency and accuracy that both Generative AI and Large Language displayed in improving pharmacovigilance. Dr. Loubna Bouarfa of Envision, however, recognises the worries and anxieties had toward the dependency placed on such unknown technological territory. And so, in her article she addresses these hesitancies, tackling the common misconceptions and ethical issues associated with AI which are not entirely true.
Another recent topic of concern within the industry is that of counterfeit drugs. Both Stephan von Schilcher and the Health and Outcomes roundtable (page 56) explain the alarming rise in ‘fake drugs’ entering the market and the negative impacts this can have on patient safety and company reputations. Thus, these articles look at the best ways to prevent the issue from worsening and how to
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal Content Writer, Clarivate
Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
protect both the consumers and corporations. Building on this idea of consumer care, Jana Benesh and Neena Nizar of ICON look at the importance of Patient Advocacy Organisations (PAOs) in clinical developments. They examine the pivotal role PAOs play in bettering the therapeutic outcomes of the patient, as their influence and effective engagement provides a means of making a patient feel safe.
Returning from our summer issue is our Nasal and Pulmonary Drug Discovery and Development Subsection. In this issue, we cover the latest movements on medical devices from orally inhaled and nasal drug devices, specifically diving into the use of in silico modelling, as well as drug
administration through nebulisers and softmist inhalers. This section highlights the newest discoveries and updates in this field of research, focusing particularly on the safety and efficiency of these tools in ensuring the best human responses.
A progressively informative journal this issue, and as we continue to share our knowledge among each other, we move further forward in bettering the world of all things pharma. I do hope you enjoy reading this spectacular collection of work and I look forward to meeting some of you at the upcoming events.
Chloe Euripides, Editorial Manager
Georg Mathis Founder and Managing Director, Appletree AG
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey Litwin, M.D., F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Stanley Tam, General Manager, Eurofins MEDINET
(Singapore, Shanghai)
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Stefan Astrom, Founder and CEO of Astrom Research International HB
T S Jaishankar, Managing Director, QUEST Life Sciences
Join us at CPHI Milan to explore our advanced solutions for pharma and quality control applications
Discover our GMP, multicompendial CertiPro (low in endotoxin) designed to support and optimize drug development processes.
We are a comprehensive chemical supplier of high-quality Excipients and Fine Chemicals for the pharmaceutical and biopharmaceutical production in Europe, Middle East, and Africa. Our aim is to provide customers with the highest quality GMP raw materials, custom tailored solutions, and sustainable Quality Control endotoxin, non-endotoxin and pyrogen tests.
Include robust animal based as well as sustainable endotoxin and non-endotoxin detection solutions such as PYROSTAR™ ES-F, PYROSTAR™ Neo+ and LumiMAT™ Pyrogen Detection Kit. Quality Control Solutions
Key Steps to Commercialisation Readiness in Europe
The steps to commercialisation readiness are complex, multi-faceted and often left too late in the launch strategy of a pharmaceutical product. This can be particularly problematic in Europe where the diversity of national legislation can make supply chain logistics very challenging. Among the issues faced, particularly for companies outside the EU are defining the product’s logistics, identifying stakeholders who will be involved in the supply chain in Europe, and ensuring that all these individuals are ready to assume their roles soon after the product’s approval (Marketing Authorisation) with the relevant Licences and Quality Technical agreements in place.
Depending on the complexity of the storage and transportation conditions of the product, and the number of stakeholders envisaged for the supply chain, experience shows that preparing all these steps can take up to two years for quality assurance, commercial and supply chain departments. And, while the European Union has harmonised wholesale distribution regulations and introduced good manufacturing and good distribution practices in the pharmaceutical legislation (Directive 2001/83/EC1 and Directive 2003/94/EC2 and Guidelines 2013/C 343/01,3 respectively), which have helped patients to access medicines faster, there are still different post-approval requirements in each EU Member State, and also in the United Kingdom and Switzerland.
Among these differences, the importation, distribution and release of medicines in the EU, UK and Switzerland are still countryspecific requiring national authorisations to permit these activities. Moreover, in our experience, getting the necessary licences to ensure an efficient supply chain in all targeted territories requires significant preparation.
Adopting a Systematic Approach
Before starting the commercialisation journey, it’s important to map out a strategy for the short-, medium-, and long-term that considers the countries where the product should be launched and when. Knowing this
will prevent early missteps that will require costly and complex changes (for example, identifying the EU site of importation and the establishment of the EU supply chain with appropriate low market licences).
From experience, it is advisable to start coordinating commercialisation steps before late-stage development of a product is completed – ideally when there is enough data to give companies greater confidence of a successful marketing authorisation approval by regulators.
One of the first considerations should be to ensure the product can be supplied to the patient once it has been authorised, since a common source of frustration is challenges with the supply chain. In addition, the European Medicines Agency (EMA), through its Medicines Shortages Steering Committee, and some EU agencies, have placed an emphasis on addressing supply chain vulnerabilities and measures to avoid shortages of medicines.4
Companies entering the European market can also struggle with decisions about where to set up their EU headquarters or marketing authorisation holder (MAH) and how best to weigh financial and strategic considerations. Key to those decision is being able to have all the necessary stakeholders and/or partners in place to support the marketed product, including pharmacovigilance, regulatory and medical information professionals, is quality management and compliance. Quality management and compliance teams can support the identification of compliant supply chains and risk assess supply chain challenges.
To determine the right approach, it’s important to consider local regulations together with economic considerations as well as the company’s objectives. The following questions deserve a comprehensive analysis: Which countries is the company targeting for commercialisation? Where is the product likely to be manufactured and how will that impact the supply chain as well as import licences? Has the company identified a local partner or does it plan to apply for an appropriate licence authorising it to manufacture or import, distribute/commercialise the product?
Companies with their sights set on Europe also need to consider whether and how they will manage commercialisation in Switzerland and in the United Kingdom, since these territories represent important marketplaces, though neither are part of the EU or European Economic Area (EEA).
Need for a ‘Commercialisation Licence’
Obtaining a Marketing Authorisation (MA) for a medicinal product is of course a critical prerequisite before placing a product on the market, but the MAH must also ensure that batch testing and release of the product is managed properly by authorised sites. Depending on where the bulk/finished product has been partially or fully manufactured in the EU/EEA or in a third country, the importation and QP batch certification can be complex and the MAH must ensure that all importation/batch certification sites hold a Manufacturing & Importation Authorisation (MIA).5
The MAH must also identify and select distributors who will supply the product to the different targeted EU markets, and ultimately to retail pharmacies and hospitals. These distributors must hold a Wholesale Distribution Authorisation (WDA) defined in the EU Regulation.6
Some EU countries, e.g., France or Germany, also require a national WDA on top of the European WDA granted to Distributors for the commercialisation of medicinal products. These national WDAs are also delivered by national competent authorities that certify that the WDA holder meets GDP requirements.7 WDAs are directly placed under the supervision of a Responsible Person (“RP”) who is named on the license, and who is the Authorities’ single point of contact for most if not all matters related to the commercialisation of the product.
Understanding Country-specifics
One of the most complex countries in Europe from a commercialisation perspective is France, which expects very rigorous oversight of product distribution and lifecycle management. On the other hand, France has put in place attractive early access programmes that offer the hope of faster revenues compared to other EU
markets which experience has shown to have less attractive legislative frameworks in this regard or have lengthy price & reimbursement procedures (which can also be the case in France).8
Companies seeking to market their products in France must have an “Exploitant” Authorisation,9 which is defined under the French Public Health Codex as the organisation responsible for drugs “exploitation”, i.e. commercialisation under the responsibility of a Responsible Person (also known as the “Pharmacien Responsable”). The Exploitant can be a separate entity from the MAH, which can be based anywhere in the EU. “Exploitation” refers to any activity that applies to the commercialisation of medicinal products in France (including quality management and compliance with pharmaceutical legislation, pharmacovigilance activities, market batch release, etc.).
While the definition of the Exploitant is not included in the European regulations, it is highly advisable that a company seeking to market a medicinal product in France hold an Exploitant status locally or partner with a consultancy in France to manage the Exploitant requirements, since many exploitation activities require native French speakers with deep knowledge of the French regulation and specific local requirements.
Being outside of the EU, the United Kingdom also brings additional commercialisation complexities. A national WDA is also mandatory for any company that supplies medicinal products to the UK market. WDAs issued by the national competent authority, the MHRA. In addition, a role of Responsible Person (import) (RPi) unique to the UK, has been introduced. The RPi is responsible for confirming QP certification and oversight of products imported into Great Britain from countries on an Approved Country for Import list (initially, this refers to countries in the EEA).
Just as in France, Germany, and the UK, in Switzerland, there is also a need for a national WDA.11 In addition, companies looking to commercialise their products in Switzerland must have a local entity to apply for marketing authorisation, given that EU approvals are not recognised by Swissmedic, the national competent authority.12 This is because Swissmedic requires a Responsible Person based in Switzerland who can quickly access the site in case there are any issues with a product that need to be resolved quickly.
Regulatory & Marketplace
To obtain a WDA from Swissmedic, the request must be carried out by a company legally established in Switzerland and for which a Responsible Person also based in the country has been nominated to supervise QA activities and maintain the Quality Management System. The RP is also in charge of releasing batches for the Swiss market and is the ‘QA voice’ in contact with Swissmedic and the concerned Cantonal Inspectorate.
Indeed, given that Switzerland is a federation of states, there are also Cantonal considerations in Switzerland, with some regulations being federal, and others Cantonal, which our experience shows adds further complexities for companies seeking to set up a local presence and commercialise their pharmaceutical products in the country. Cantonal inspectors conduct regular inspections, on behalf of Swissmedic, and the MAH must ensure all their procedures and an effective quality management system are in operation under the close supervision of the Responsible Person named on the WDA.13
Preparing for Commercialisation Complexities
Understanding the complexity of supplying medicines within European markets, establishing local/regional entities and the associated licences required does create commercialisation challenges for non-EU companies. However, early planning and a well-executed commercialisation strategy enable companies to better navigate these important markets and expand the reach of their products.
REFERENCES
1. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use.
2. Commission Directive 2003/94/EC of 8 October 2003 laying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use
3. Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use
4. New recommendations to strengthen supply chains of critical medicines, April 2024, EMA.
5. Eudralex, Vol. 4, EU Guidelines for GMP for Medicinal Products. Annex 16: Certification by a Qualified Person and Batch Release
6. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Title VII.
7. German Medicines Act (ArzneimittelgesetzAMG). § 52a (1) of the AMG.
8. How to take advantage of early access programs
for your innovative treatments in France? PharmaLex, 2023. https://pharma-blue.com/ how-to-take-advantage-of-early-accessprograms-for-your-innovative-treatments-infrance/
10. France Public Health Code, Section 1: Scope and definitions (Articles R5124-1 to R5124-15), Legifrance. https://www.legifrance.gouv.fr/ codes/article_lc/LEGIARTI000043761806
11. Ordinance on Licensing in the Medicinal Products Sector, MPLO, RS 812.212.1 https://www. fedlex.admin.ch/eli/cc/2018/786/en
12. Federal Act on Medicinal Products and Medical Devices, TPA, RS 812.21, Art. 10. https://www. fedlex.admin.ch/eli/cc/2001/422/en#art_10
This article is intended to communicate PharmaLex's capabilities which are backed by the author’s expertise. However, PharmaLex GmbH and its parent, Cencora, Inc., strongly encourage readers to review the references provided with this article and all available information related to the topics mentioned herein and to rely on their own experience and expertise in making decisions related thereto as the article may contain certain marketing statements and does not constitute legal advice.
Frederic Pailloux, Pharm.D., M.Sc., is Senior Director, Head Swiss Operations & QA/ Responsible Person, at PLX Healthcare Services (Switzerland) GmbH. Frederic defines and implements strategies for efficient commercialisation readiness of medicines in the EU and Switzerland. As QA and GDP specialist, he also supports companies by helping them build their Quality Management System, and acts as a local contract QA/RP for Switzerland.
Frederic Pailloux
GenAI’s Big Safety Proof Point in Life Sciences: Transforming Pharmacovigilance Case Intake
In pharma R&D, the adverse event case intake process, which takes up so much of Safety/pharmacovigilance professionals’ time and is far from efficient in its support of timely interventions, remains ripe for disruption. And now that promise is finally being honoured by nextgeneration AI technologies. Specifically, Generative AI and Large Language Models are enabling the automation of humanlike decision-making, leading to earlier and more accurate conclusions about Safety events. ArisGlobal’s Emmanuel Belabe explains the tangible difference this has begun to make.
In the modern world it is expected that any authorised medicinal product designed for human use is safe for patients to consume. Pharmacovigilance (PV) processes, which continuously monitor the effects of drugs once on the market, are intended to uphold that position over time once products have been approved for distribution. Approaches to post-market Safety monitoring have changed little in decades, however, despite soaring volumes of available information. Today these are submitted in an increasing array of formats, via a proliferating range of channels.
The approach of “booking” cases, or determining whether the mandatory elements are present, remains prevalent, with a view to quickly assigning an identifier. This approach doesn’t take into account the actual contents of a case, however, which forces PV teams to apply the same treatment to all information. The effect is that all cases are assigned the same priority in the early stages; there is no discretion to allocate teams’ bandwidth according to a potential case’s complexity/risk.
Tracking all the potential signals, assessing their validity, and responding swiftly to relevant cues, is both an absolute mandate and a very costly and labourintensive administrative burden. Adverse event (AE) case intake, in particular, represents one of the most overlooked and broken workflows in pharma in its current form.
Modern AI: Moving Away from Rigid Process Automation Towards More Nuanced Deductions
Technology-enabled process automation has long promised to transform the speed, efficiency, and accuracy of AE case intake and triage, by capturing and assessing relevant Safety signals arising via a wide range of channels (including self- or clinician-reported AEs submitted by email, post, phone call, or web portal, as well as mentions via online forums).
Up to now, machine intelligence has not come close to mimicking human powers of data extraction, filtering, inference, or deduction. Early excitement about this potential, while valid, was premature. Early automation systems had to be highly structured and painstakingly trained to recognise every possible format and variant of how important data might show up – from the basics such as a patient’s date of birth, to richer detail such as the combination of possible contributors to the adverse event (from the individual’s stage of life and overall state of health to other drugs they may be taking). As well as presenting challenges in how systems would recognise and extract the right data, this limited the scope for step changes in Safety process efficiency.
Now, though, Generative AI (GenAI) and Large Language Models (LLMs) are beginning to fundamentally transform AE data collection and their associated workflows, with powerful results. In early pilots, data extraction accuracy and quality have exceeded 90 percent, and overall efficiency gains related to the intake process have topped 65 percent. And that’s from a standing start; results will only improve with human oversight and AI adjustments.
The Rise in Prioritisation of Advanced Automation
New advanced automation solutions, which transform the data collection part of the AE case intake process and associated workflows, are resonating hard in an industry that has been crying out for a modern, more efficient way to execute case intake/safety data collection, as volumes of case data soar and pressure mounts to accelerate analysis times.
A recent industry survey1 confirmed the industry’s growing interest in AI-powered automation, revealing that over 75% of pharma R&D organisations already use some form of advanced automation within daily processes today, and more than 70% plan to expand business process automation over the next 18 months.
This appetite for viable solutions has intensified in line with a maturation of AIpowered process automation technology, from early robotic process automation aligned to regimented processes (guided by strict structure and rules, and specified workflow around exceptions management), to a less inhibited approach where the technology understands much more about what it is looking for (irrespective of format), and what to do with it.
GenAI technology, using LLMs, can quickly identify and infer what’s relevant and important and reliably summarise key findings for the user – and even make predictions. All without the need for painstaking ‘training’ (from scratch) by overstretched teams, as well as lengthy system validation. Rather, specialised applications can now be developed that can apply GenAI-type techniques, contextually, to data they haven’t seen before – learning from and processing the contents on the fly.
This is a significant advance that has seen pharma companies start to put GenAI AE intake solutions to the test in their operations, under the watchful eye of their Safety professionals. The ability to simply instruct a system to “Scan X document for Y contents” paves the way to faster, higherquality extraction of more relevant data, no matter how much greater in volume this is, or how much more diverse or complex the sources – reducing the risk of something significant being missed, and improving downstream efficiency.
Applying Appropriate Controls
A strong aspect of the business case for harnessing GenAI in AE case intake management comes from the scope for handling first-line capture and processing of very high volumes of data – relieving Safety professionals from that labour intensity
and allowing them to delve deeper into the findings and what they might mean.
However letting GenAI take the strain of case intake also removes human limitations such as fatigue, mental overload, distraction, data blindness, and unconscious bias. An AI-powered tool can more efficiently detect patterns and determine trends, with reliable consistency using approaches that are based on precedence. It can draw on the findings of millions of prior cases and assessments, to make credible predictions and unbiased assessments regarding causality (the likelihood of a direct link between a product and a reported adverse event), that are based on probability rather than a gut feel.
Next-generation cognitive computing in the form of GenAI and LLMs – might be considered to be in an adolescent state of maturity currently (largely ready for the world, with some guidance and controls still needed), but the early output is proving very encouraging. Teams are now seeing that the level of oversight, quality review, and sampling that is required to satisfy regulators, develop a track record, and build trust in the technology (e.g. its process of learning and decision-making) – is a relatively low hurdle to clear. It helps that the links back to the sources are readily traceable for checking.
Remaining Open and Agile, Primed for Next Opportunities
From here, as pharma companies look to capitalise on GenAI and LLMs to advance
their process automation goals, they mustn’t focus solely on the potential bottom-line benefits. After all, this is a much-needed chance to re-allocate resources; to elevate Safety professionals’ roles from data management to adding new, strategic insight-driven value to R&D decisionmaking. This then requires provision for change management and transformation ‘readiness’, not just a choice of the right technology for the job.
In the short term, it is AE case intake that has captured companies’ imagination – where unprecedented new insights as well as greater process efficiency promise to revolutionise the function, and its role and value, starting right now. But over time there will be other powerful use cases too, so it’s a good idea to allow scope for additional applications in due course (e.g. by deploying an enabling ‘platform’ rather than a single-use application). Strong next contenders for GenAI/LLM treatment include real-time pharmacovigilance assessments and associated decision-making (e.g. the earlier identification of unexpected benefits/ discovery of new indications); harnessing international Regulatory intelligence to transform marketing authorisation applications and maintenance; and clinical trial modelling, reducing the reliance on traditional clinical studies.
The key to whether GenAI/LLM treatment is appropriate will be the high volumes of data involved in the target processes. Certainly, the more opportunities there are
for the advanced automation system to be exposed to information, the faster it will learn to identify, categorise, assess, and deduce what to do, driving ever greater trust in – and reliance – on the technology to do the heavy lifting.
A recommended first step for pharma companies not yet on the path to intelligent automation would be to break down how current processes are currently managed, the core requirements driving those processes, and where any pain points are. The next priority should be to review and rewrite standard operating procedures so that they can evolve with and be improved by advanced technology, both currently and over time as capabilities continue to evolve.
Belabe is Senior Vice-President for Customer Success, Global Customer Support, and Solution Consulting at ArisGlobal, an innovative life sciences technology company. He is an experienced Safety director with a strong record in applying the latest IT innovations in healthcare and life sciences.
Refining Sustainability Performance in Single-use Drug Delivery Devices
Improving sustainability is critical across all industries as we look to combat rising emissions and ensure global temperatures do not exceed 1.5 C above pre-industrial levels. The healthcare industry – and the pharmaceutical industry which serves it – are certainly no exception to this and have a critical role to play in a drive towards a more sustainable future. In the UK, the National Health Service (NHS) is responsible for around 4% of total emissions so huge strides are needed to reach its ambitious goal of net zero by 2045. As of April 2023, all new NHS contracts above £5 million per annum require suppliers to publish a Carbon Reduction Plan for their UK Scope 1 and 2 emissions and a subset of scope 3 emissions as a minimum.
Remaining a viable partner in this industry is, therefore, likely to necessitate significant changes in the immediate future. Companies are already taking measures to improve everything from the energy efficiency of buildings to weight and plastic content in packaging. However, as readers are aware, there are unique challenges in the medical industry: any efforts to reduce carbon footprint must not be at expense of treatment effectiveness or the safety of patients.
Transitioning to Plastic Substitutes
In parenteral drug delivery, drug stability, anti-contamination and infection control are paramount, so changing materials requires rigorous suitability testing and regulatory scrutiny. To address plastic waste, the logical place to start is, therefore, with products that pose less risk to patients or healthcare professionals, such as packaging, disposable masks, gloves, and coverings and wound care. In fact, commodity plastics from items including tubing, films, packaging, connectors, labware, IV bags, catheters, face masks, housings, luers, membranes, sutures and more make up the majority (70%) of medical plastic waste.4 Meanwhile, syringes form just one part of the remaining 30%. Commodity plastic alternatives are the low hanging fruit
for medical products, where the easiest gains could be made, and with highest impact.
This is not to say we should not seek a substitute material for the petroleumderived plastics used in most single-use drug delivery devices. However, the industry needs a transition plan that considers the current level of need for delivery devices and pre-filled syringes. Given the impact on patient health, there must be a steady supply of such devices in their current form while research into recycled materials or bio-based plastics is ongoing. It must also be noted that the sector has carefully considered biodegradable options. However, biodegradability can sometimes affect stability and drug integrity in prefilled pharmaceutical products.
Aside from material changes, it is more straightforward to focus on reusable drug delivery products that are also easily remanufactured and where their disposable element is easily recycled. This reusable approach is also appropriate for digital devices, where it is clear that the cost and waste from a disposable electronic component would be unacceptable. In a digital or connected auto-injector, a disposable element is still required to meet safety and regulatory requirements, so the most practical solution is to design a
minimum disposable unit within a reusable ‘shell’ holding the electronics.
Breaking Down Product Design
Though we must continue to develop single-use plastic devices, these can still be optimised to improve their carbon footprint in the interim. Device manufacturers can make sustainability improvements in a number of areas without compromising usability. However, companies must examine the entire product lifecycle to make meaningful modifications. Examining elements of a device in isolation is not enough; tweaks intended to improve sustainability in one area can have unintended consequences in another.
Taking a holistic view of the product involves looking at concept development, material selection, design and engineering, manufacturing, packaging, transportation, sales, use, and end-of-life disposal. Optimisation in these areas relies on a collaboration between different segments of a business and ideally, these considerations should be incorporated during the earliest stages of development, creating products that are truly ‘sustainable by design’. While manufacturers have already started this task through evolutions in packaging and transportation, risk reduction, manufacturing efficiency, time to market and safety and regulatory compliance, industry could
go even further. More focus is needed in improving energy efficiency, material usage and recycling, and end of life disposal.
Modifications to individual drug delivery devices will of course vary depending on the product itself and the needs of users. Creating products that are easier and cheaper to recycle, for example, relies on the simplicity of the design, for easier disassembly or remanufacturing.
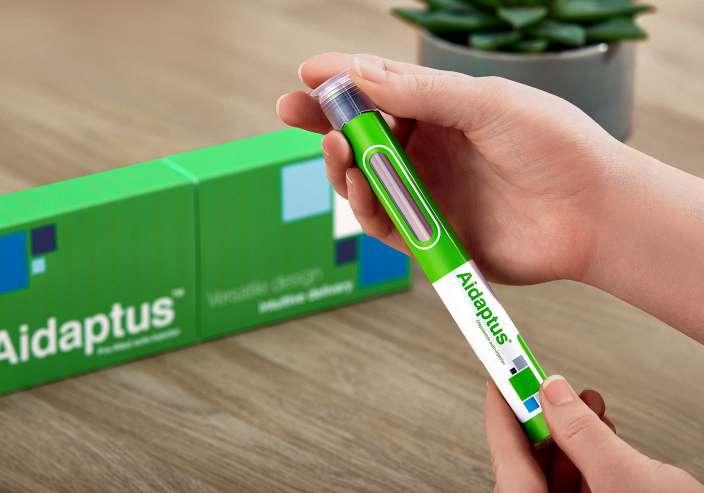
Reducing waste and transportation costs can be achieved by optimising device size and reducing the weight and plastic content of packaging. Owen Mumford Pharmaceutical Services’ Aidaptus® auto-injector, for instance, weighs just 28g (without the syringe). Replacing metal components with other suitable materials can also make an impact on the environmental burden of processing and shipping devices. However, any moves to reduce or replace a material or component must still prioritise patient safety and usability.
With the Aidaptus product, the design sought to derisk device choice for pharmaceutical partners with an approach that streamlines processes and therefore contributes to reducing impact – all while maintaining ease of use for patients. Aidaptus has a wide design envelope, giving pharma companies the flexibility to make changes to formulation, fill volumes or needle sizes without having to change device. This reduces risk during drug development and life cycle management – removing the need for additional verification testing, human factors studies and regulatory documentation. All this also helps to reduce time to market for the final combination product. Aidaptus’ ability to
Regulatory & Marketplace
support multiple drug formulations means it can be used across a number of products in a company’s portfolio, and having a single platform for multiple applications further reduces impact at the manufacturing level.
Sharing Solutions
As we action closer towards milestone targets for emission reduction, it’s likely we will move on from single-use plastics within our drug delivery devices. But until materials are developed that are able to support the exceptional demand for these products, we can focus on the overall
sustainability performance of plastic-based devices. Continuous progress is necessary not only for internal sustainability goals but also to reassure pharma companies that manufacturer partners are on the right path. It is a balancing act; companies need to show a willingness to introduce more sustainable products, while also remaining competitive and maintaining usability – all the while considering patients, clinicians and regulators. Given this complexity, the pace of this progress will be much quicker if the industry as a whole can find ways to work together to find appropriate materials and solutions.
Alex Fong MBA is an experienced senior manager in the Insight, Analytics and Strategy fields. He has applied these skills in a broad range of Industries including the FMCG/CPG, tourism, investment banking, telecoms and management consulting sectors. For the last eight years, Alex has been leading the market research drive at Owen Mumford, with an ever-increasing focus on sustainability.
Alex Fong
Improved Efficiency for Pharma Labs with Automation of LC Workflows
In recent years, researchers have benefited from rapid advancements in analytical instrumentation, particularly in respect to liquid chromatography (LC). As a result, the use of LC in labs across the globe has increased, with 47% of labs globally now using LC-MS systems.1 Pharmaceutical labs in particular have expanded their use of LC and it has quickly become a must-have for every stage of the drug discovery process, from drug development to quality control. By the close of 2023, 58% of labs in the pharma/biopharma industry were using LC-MS systems.1
The pharmaceutical industry has also taken advantage of recent developments in automation. However, there is scope for automation to be more widely adopted, and pharmaceutical labs should investigate how they can make use of new developments in automation to ease and improve their workflows.
Why Automate the Lab?
Resource management is central to pharmaceutical labs, who must ensure resources are handled in the most time- and cost-effective manner. Automation can bring real benefits to lab resource management, especially in terms of time savings and productivity, as it can take ownership of repetitive tasks and give time back to scientists to focus on life-changing research.
For example, specialist scientists are often required to perform or oversee sample preparation and loading, in addition to interpreting and reporting results. Sample prep and loading consumes valuable time that could be better spent analysing and extracting valuable information from results. The manual input required for sample loading also means that many workflows cannot be left to run overnight, as human input will be required to unload the sample and reload the next batch once a sample run has been complete, which further limits the efficiency of the lab.
Another challenge relates to the identification and elimination of errors. Lab errors can arise from a variety of sources,
including environmental, procedural, and instrumental, but humans are most often the lead cause. In fact, it has been widely speculated that anywhere between 23%2 to 80%+3 of total errors in manufacturing are the result of human error, a statistic which can also be applied to the laboratory setting. In a repetitive, high-throughput daily routine, it is inevitable that scientists will make mistakes – for example, mislabelling vials, introducing contamination into samples, or incorrectly preparing or loading samples. Such errors must be identified swiftly as they can prove to be dangerous and costly. In extreme cases, failure to identify errors can result in FDA warning letters/483 observations, which are used to communicate concerns following an FDA inspection.4 Although 483 observations do not incur a fine, they enter the public domain5 and can consequently have a detrimental effect on company reputation.
Alongside the minimisation of error, it is crucial that pharmaceutical labs ensure that their methods and results are reproducible and meet regulatory guidelines. Workflows should follow the same methods and processes across lab systems and sites, and must be consistent. However, making sure that this is always the case can be difficult and time-consuming.
Solutions to Lab Efficiency
Automation presents a potential solution for error minimisation and improved efficiency, through innovative technologies that reduce human involvement. Currently, automation is not widely deployed in pharmaceutical labs. While it is often used to perform tasks such as sample preparation, error identification, and data analysis, it has not been adopted to its full potential. For instance, labs of the future could employ automated systems to quickly and precisely transfer liquid samples and reagents, or could use robotic arms to handle samples to reduce the need for human input. It is therefore important that the pharmaceutical industry investigates the possibility of expanding the use of automation in its labs.
Pharmaceutical companies incorporating automation into their workflows usually enlist the support of third-party system integrators.
System integrators are independent organisations who can support labs in their automation journey by facilitating the deployment of automation into current lab workflows, and the subsequent integration of data management tools. However, while integrators have a comprehensive understanding of lab workflows, they may not have experience with specific systems such as LC-MS or be well-versed in challenges that are specific to pharmaceutical labs. Lab managers should therefore coordinate with both the instrument vendor and the integrator and discuss how the current workflow operates, so that all parties can work together to automate workflows and solve any potential problems that occur.
LC Workflow Automation
Although the capabilities of automation are well known, pharmaceutical labs have been hesitant in its deployment due to the specific challenges of automating LC workflows.
One of the key hurdles that automation must overcome is traceability. It is crucial that scientists can reliably trace every step of the analysis process to ensure consistency and accuracy, as well as to identify where any problems or variables occur in the LC workflow. Automation has the potential to log data and results, saving scientists from taking notes while conducting experiments and ultimately reducing error.
Instrumentation vendors are working hard to respond to challenges such as these, by finding new ways to automate additional steps in pharmaceutical workflows, including the schedule of events, such as plate loading, ejection, and transfer. Automated sample handling is already improving the efficiency of LC analysis in pharmaceutical labs. The COVID-19 pandemic accelerated the automation of remote lab monitoring, as labs faced the challenge of maintaining critical business operations while enforcing social distancing to keep employees safe. Many labs implemented work practices whereby analysts set up and prepared the LC systems remotely, coming on-site only to prepare samples and start the sample analysis. Since the pandemic, these systems have become more sophisticated, and their use has significantly expanded.
At present, automated systems are being used to follow specific protocols and procedures, as well as generate detailed records of all actions taken and results obtained, which is benefitting pharmaceutical labs in a number of ways. It has not only improved lab efficiency and throughput by allowing scientists the time to focus on real science while the samples run automatically, but has also reduced the risk of sample contamination and analyst error through the limitation of human involvement.
Automation in Action
A biopharmaceutical manufacturing facility in America has recently automated its sample preparation and data analysis. The facility, which conducts important research and development, was experiencing considerable bottlenecks due to labour-intensive sample preparation. It had also found that methods used in its own facility were inconsistent with methods used across other sites within the company. To improve efficiency and standardise, the facility decided to automate some of its workflows. A team dedicated to automation has been working on producing methods for sample preparation; connecting functional to analytical equipment; creating software to control third-party instruments; and automating data-related tasks.
With the goal set to minimise time spent on tedious manual steps, the facility’s initial plan was to integrate a liquid handler and robotic arm into its LC workflow that could automate peptide mapping sample prep. This would be achieved by connecting a mass spectrometer that could receive the plates and then trigger the run without the need for human intervention and plate loading. When interest grew in the project, the team then focused its attention on building a more extensive workstation that could bring together various analytical tools into one automated workflow. The team recognised that the peptide mapping took an hour to run and was slowing down the efficiency of the workstation, so they removed this from the workflow and instead focused their attention on the size exclusion chromatography analysis, which completes a run in 10 minutes. Automating the procedure allowed the team to meet its goal of processing 96 samples in one day, doubling turnaround times and ensuring that KPIs were comfortably met. The workstation can also accommodate other short-form HPLC methods, meaning the team can further boost its productivity and automate more elements of its analytical workflows.
Clinical and Medical Research
The facility has experienced a wide range of benefits following the integration of these automated systems in its biopharmaceutical labs. Most importantly, there has been a marked improvement in lab efficiency. Automated systems require less human involvement, meaning that scientists at the facility can spend more time analysing data rather than performing manual tasks. It also means that systems can be left to run overnight, which has substantially increased the facility’s throughput. The facility has also reported that automation has improved result consistency and accuracy by reducing the opportunity for human error or contamination. The subsequent improved confidence in results has helped the facility safeguard the veracity of experimental findings and ensure that it adheres to regulatory requirements. In terms of employee welfare, automation has helped to protect the health of the facility’s scientists by minimising their interaction with dangerous substances, and preventing the physical strain caused by repetitive tasks.
Conclusion
The development of innovative automated systems, such as those employed by the American biopharmaceutical facility, pave the way for more efficient and reliable pharmaceutical development and manufacturing. Although challenges remain in the automation of LC workflows, the extensive range of benefits afforded by automation is already impressive. Pharmaceutical labs are increasingly adopting automated systems, and in most labs, humans work alongside intelligent instrumentation to alleviate the pressures of repetitive routine testing.
With automation techniques and systems continuously advancing, it’s exciting to
think about the impact automation could have on labs of the future. At present, automation is employed primarily for error identification and prevention, but we’re increasingly seeing pharmaceutical labs deploy automation in areas such as sample preparation, processing, and handling to allow scientists to dedicate valuable time on furthering life-saving research. In the upcoming years, automation may expand and become sophisticated enough to handle more complex processes, such as supporting scientists on analysing and identifying valuable insights within large datasets. Ultimately, the end goal is for automation to alleviate pressures and streamline operations in pharmaceutical labs globally – and some technologies are paving the way towards that future.
REFERENCES
1. Top-Down Analytics, ‘2023 mass spectrometry market survey’.
2. Best methods to reduce human error in manufacturing’ Dozuki, https://www.dozuki. com/blog/best-methods-to-reduce-humanerror-in-manufacturing [accessed June 12 2024].
3. How to Reduce Human Error on the Pharmaceutical Manufacturing Floor MasterControl, https:// www.mastercontrol.com/uk/gxp-lifeline/ reducing_human_error_manufacturing_floor/ [accessed June 12 2024].
4. Inspection Observations Food & Drug Administration, https://www.fda.gov/inspectionscompliance-enforcement-and-criminalinvestigations/inspection-references/inspectionobservations [accessed June 12 2024].
5. ‘Inspections/483 Database Search’ Food & Drug Administration, https://www.accessdata.fda. gov/scripts/483inspsearch/index.cfm?action= search.search [accessed June 12 2024].
Darcy Shave
Darcy Shave is a principal product manager in the Small Molecules group at Waters Corporation (Milford, MA, USA), responsible for the purification and hardware automation products. Darcy joined Waters as an applications chemist 20 years ago and has since worked in a variety of business development roles before becoming Product Manager of the hardware automation portfolio. He has a degree in Agricultural Chemistry from the University of Saskatchewan and worked in value added processing of natural products before joining Waters.
Clinical and Medical Research Medical Devices
Soaring Regulatory Demands In The Medical Device Sector: How Are Compliance Strategies Evolving?
As medical devices become more critical to patient outcomes, regulators around the world are steadily increasing controls around the manufacturing, distribution and monitoring of devices. Drawing on new research, Peter Muller and Mike Baird of Schlafender Hase assess how well Class 2 and 3 medical device manufacturers in Europe and the US are adapting.
Technology innovation, combined with pressures on health services to treat patients more effectively, efficiently and conveniently, has led to sharp growth in advanced medical devices and their prominence within care pathways. Globally, the medical devices market is projected to grow from $542.21 billion in 2024 to $886.80 billion by 2032,1 while medical device sector representatives now account for 50–70% of the attendees at meetings of RAPS, the Regulatory Affairs Professionals Society (just three years ago, delegates were mostly from pharma).
As devices become more critical to patient outcomes, and as safety-related scrutiny is intensified, regulators around the world are steadily increasing their expectations and controls around the manufacturing, distribution and monitoring of devices. The aim is to bring developers and suppliers of these products more closely into line with the requirements around pharmaceutical goods.
This has implications right across the medical device industry internationally, spanning a potential need for new systems and processes, attention to the way these are linked and tracked, and the distribution of appropriate skills across the workforce.
A new international benchmark report2 has set out to determine how well manufacturers and their regional or national partners are adapting to the rising regulatory demands. The 2024 study, conducted with 202 regulatory professionals at Class 2 and 3 medical device companies in the EU (Germany) and North America (the US), highlights the number of challenges currently vying for attention and investment and assesses device companies’ current state of regulatory readiness.
Medical Devices: The Rise of Regulatory Requirements
The research first tested medical device companies’ involvement with a number of increasingly prominent regulatory initiatives.
E-labelling/eIFU
E-labelling is high on the medical device regulation agenda on both sides of the Atlantic. Electronic information provision and management promotes standardisation and consistency (e.g. of format and terminology), making it easier to manage and process the contents in any market. It also plays a key role in product traceability, a critical safety lever.
Providing critical safety and identification information digitally (e.g. under expectations associated with electronic instructions for use, or eIFU) makes it easier to issue prompt updates to information, too. It also simplifies international content and translation management and, in the case of user advice or safety information, facilitates spontaneous online or mobile lookup by clinicians or patients. Crucially, e-labelling allows device manufacturers to provide more detail than can fit on a physical label.
Currently, just under two-thirds (62%) of medical device companies are involved in e-labelling initiatives, and up to a third of these (30%) are ‘very’ involved. EU companies are more likely to be actively involved in e-labelling than those in the US (71% vs 53%, respectively). This makes sense as the EU is ahead of the US with the practice; companies here are also less likely to outsource labelling as a service.
FHIR/Standardised Data Exchange
Fast Healthcare Interoperability Resources is a proposed new global standard, designed to streamline data exchange and facilitate real-time information access for healthcare providers. Once fully supported, FHIR will make many regulatory professionals’ lives easier by shifting the emphasis of content creation and management to ‘publishing’ rather than ‘printing’. It is this kind of development that will help drive process digitisation in the production and management of regulated medical device information and content.
In the survey, three in five respondents (60%) claimed to be involved with the standard, rising to 67% for EU (German) respondents; in the US, only just over half were occupied with FHIR (FHIR is not as high profile in the US), though the FDA is encouraging manufacturers to adopt interoperability standards.
UDI/Device Identification
Unique device identification (UDI) employs a unique numeric or alphanumeric code to identify individual devices across the healthcare supply chain. Although approached slightly differently, a UDI system is advocated by both EMA and the FDA as an efficient and effective means of tracking and identifying medical devices globally. Benefits include expedited and more targeted product recalls, a reduction in product counterfeiting, and a better, safer experience for patients.
In the survey, two-thirds (66%) of respondents (rising to 74% of EU survey participants, but accounting for a much lower proportion in the US at 57%) express involvement in UDI activity.
Anti-Counterfeiting
Taking proactive measures to mitigate the threat to product quality and patient safety posed by counterfeit products is a further expectation and robust product identification and traceability are a cornerstone of this practice, along with vigilant supply chain monitoring.
In the survey, over half (55%) of respondents indicate at least some involvement with anti-counterfeiting. Of these, just under a quarter (24%) are very involved and just under a third (32%) are somewhat involved, while just over a third (39%) say this is not within their remit.
Strategies & Challenges when Navigating Regulatory Demands
Medical device companies are dealing with the impact of increasing regulations in a number of ways, including the implementation of key standards (e.g. ISO); process digitisation and automation; greater use of outsourcing or third-party collaboration; and hiring of more regulatory people – all cited by more than a third of companies.
Precision Revolution
Your quest for perfection ends here. Let’s revolutionize your manufacturing process, redefine precision, and gear up for excellence together. When you partner with KAHLE perfection isn’t a destination—it’s the journey we craft, gear by gear.
Kahle® is dedicated to providing custom automation machinery solutions for the Medical Device, Pharmaceutical, and Healthcare Industries around the world.
Clinical and Medical Research Medical Devices
The difficulty of finding and appointing qualified professionals to alleviate soaring regulatory workloads is a particular problem on both sides of the Atlantic. Over a third (34%) of respondents cited this as the greatest challenge facing their company currently, while almost a quarter (23%) said that staff retention was their biggest issue.
Upcoming Priorities
Asked about the main projects their department would be working on over the next 2–3 years, respondents said projects would primarily involve existing devices (cited by 32%, rising to 35% among US respondents); emerging healthcare trends (28%, rising to 34% of German/EU respondents); and new materials & technologies (27%).
Device companies plan to use a range of technology solutions to support these projects, most notably electronic document management (EDM); content management; proofreading/content comparison; labelling management; and product lifecycle management solutions, each cited by around a third of respondents.
Compared to the pharmaceutical market, the use of regulatory information management (RIM) systems is currently less prominent in medical device companies, featuring for just 29% of respondents, followed by structured authoring/creation tools (27%). The penetration of formal systems in the medical device sector is likely to grow as ambitions rise and regulations expand.
Improving Efficiency in Regulatory & Safety Document Preparation
To keep pace with the rising volume and complexity of regulatory submissions, more than a third (36%) of medical device companies already use software for the
proofreading and content review process for regulatory documents, labelling materials, and promotional content, while 29% still resort to manual proofreading in house, rising to 37% in the US. In the EU, more respondents (41%) use software to help them review content quality.
A third (34%) of all respondents currently outsource their content proofreading, which could be as part of a broader arrangement with an external partner.
Packaging & Labelling Challenges
As tracking and supply chain transparency requirements rise, the challenges of producing compliant and correct device packaging and labelling for each respective market intensify. In the research, the subject yielded particularly strong responses.
Just under two thirds (65%) of respondents said they find translations challenging to manage; 61% find barcodes challenging to manage; 60% struggle with graphics including symbols (shorthand guidance on device sterilisation, for instance); and 59% have difficulty with tables. This is on top of any issues getting the text right (cited as a challenge by 54% of respondents).
Technology could offer a powerful solution here, although enhancements to processes will also be important to get the most from any investment.
Lessons Learned & Next Steps
The study (the full report is available here) ended by asking for device companies’ top five takeaways from the last year that will inform their next regulatory actions. The responses cemented the need for greater investment in company culture (cited by 35%, rising to 43% of German/EU respondents);
bolstered resources/recruitment (34%); more emphasis on wellbeing (33%); more investment in technology (33% – rising to 42% of US respondents); and increased focus on education and training (32%).
The prioritisation of company culture and employee wellbeing is further evidence of the growing pressure that regulatory functions are under, and the criticality of making teams – and the way they work – part of the solution.
REFERENCES
1. Medical Devices Market, Fortune Business Insights, June 17, 2024: https://www.fortune businessinsights.com/industry-reports/medicaldevices-market-100085
2. The independent Censuswide survey, commissioned by Schlafender Hase, was conducted in late May/early June 2024, among 202 regulatory professionals at Class 2 and 3 medical device companies (those deemed of intermediate to high risk in the event of a malfunction or quality/safety issue). The samples were split 50/50 between respondents in the EU (Germany) and North America (the US). Link to full report: https://www.schlafenderhase.com/ebooks/ medical-device-report-how-are-compliancestrategies-evolving
Peter Muller
Peter Muller is Director of the Americas at Schlafender Hase. For more than two decades, he has worked on software and process improvement projects with Fortune 500 companies from life sciences and other regulated industries.
Email: peter.muller@sh-p.de
Mike Baird is Director of Product Management at Schlafender Hase in Europe. He is a specialist in business/process transformation, optimisation, and quality, particularly linked to packaging and print, artwork, and labeling, particularly in life sciences.
Minimize your risks by prioritizing high quality, consistency and reliable GMP manufactured raw materials.
Our Benzalkonium Chloride is manufactured following cGMP guide ICH Q7 for APIs, the highest available quality standard in the industry.
Get peace of mind with full traceability, high product purity, full pharmacopoeia compliance (PhEur, USP/NF, JP, ChP), audit access, and 30+ years of cGMP manufacturing experience.
novonordiskpharmatech.com
Clinical and Medical Research Medical Devices
Right First Time: What The Medical Device Sector Can Learn From Pharma’s Structured Data Challenges
As medical device suppliers respond to rising regulatory scrutiny, they can expedite their preparations by looking at the lessons learned in pharma around product data and associated content management. Here, Celegence’s Sonia Veluchamy distils some best practices.
Life sciences Regulatory Affairs teams and their colleagues in Quality and Safety face already overwhelming workloads, which are compounded each year by evolving health authority (HA) expectations around detailed product information capture, monitoring and reporting. While the pharma industry is steeped in experience of this now, the medical device sector is playing catchup.
In pharma, a steady progression of HA requirements has triggered extensive investment over the last two decades – in IT systems, data standards and data governance preparations, and optimised data and content management. Most recently this has been towards adherence with ISO IDMP, the international framework designed to describe medicinal products using agreed vocabularies.
The wider goal for companies as they adapt to regulators’ evolving needs is to achieve this in an affordable and futureproof way with benefits to their own operational efficiency. In pharma, this has prompted a series of strategic technology investments to streamline regulatory processes and ensure that patients can continue to access the drugs they need.
Growing Controls in a Booming Market
The global medical device market, worth an estimated $518.46 billion in 2023, is set to reach a value of $886.80 billion by 2032.1 As ever more sophisticated devices – from surgical robots to implantable defibrillators and smart diabetes monitors – come to play an increasingly prominent and critical role in everyday patient care, regulators are steadily introducing new rules to ensure their safety. (The global drug device combination products market alone, which includes wearable devices, was worth $138.47 billion in 2023 and is set to grow at a CAGR of 9.0% over the next six years.2)
As device suppliers strive to fulfil growing regulatory expectations, including those set out under the EU’s trailblazing Medical Device Regulations (EU MDR),3 there is an opportunity for these companies to learn from the challenges pharma has strived to overcome. These include overcoming information silos, harnessing unstructured data, and streamlining regulatory information management.
Responding to Rising Regulatory Expectations When Resources are Lacking
Since many rising stars in the medical device industry lack sizeable Regulatory functions, it is particularly important that they maximise their resources.
New research among Regulatory Affairs (RA) professionals across pharma and medical devices4 confirms that time and bandwidth are the entire industry’s primary challenge, followed by costs and budgetary pressures – concerns that are particularly acute for device suppliers. While just over half (57%) of pharma RA teams feel underresourced to meet their 2024 priorities, this rises to more than three-quarters (77%) of medical device RA teams.
Among the best practices being established in pharma is a steady distancing from the traditional document-centric, case-by-case approach to dossier creation; extracting, collating and preparing the right
Vials with Exceptional Inner Surface Durability
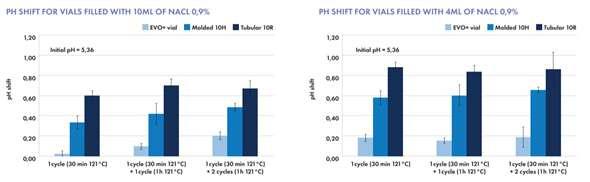
For Biotech
Less interactions of drug molecules and formulations with the inner glass surface
• Optimized lyophilization process (less fogging)
• Reduced risk of glass delamination
For Diluents
Water for injection | Aqueous NaCl solution Lower pH shift
• Reduced risk of glass delamination
Clinical and Medical Research Medical Devices
information each time – a highly repetitive and labour-intensive process that carries a risk of omission or incorrect insertions, and adds little value beyond the immediate purpose.
Modern regulatory information management (RIM) and enterprise information management (EIM) strategies, and supporting systems and processes, help tackle these inefficiencies – first by breaking down silos so that Regulatory, Safety and Quality teams can more readily share data and materials. This means they can avoid creating new content from scratch for each respective set of submission or reporting requirements. Ideally, they will now submit the exact information that is required in each given scenario too – no more, no less.
Minimising Repetitive Tasks
With proportionally fewer Regulatory professionals to defer to compared to pharma, medical device manufacturers can benefit from technology adoption and digitalisation in a number of ways.
A proactive approach to establishing systems and processes for content management and information exchange, for instance, will help companies keep pace as health authorities move forward with plans around electronic information exchange (e.g. under the FDA’s eSTAR; the EU’s EUDAMED; and STED, supported by many countries globally).
More strategically, companies also have an opportunity to leverage content prepared for one country for other markets. They could save a lot of time by reusing existing content components to submit to another countries, supported by appropriate technology. (That potential increases further if they harness niche tools geared to regulatory innovation, such as Generative AI – GenAI – capabilities as an aid in content creation, verification, formatting and change management, as discussed below.)
Identified Priorities For Investment
Medical device companies’ appetite to invest in supporting technology is tangible. In the 2024 survey of Regulatory professionals’ priorities and concerns, the top three targets for planned investment specifically by medical device companies were system capabilities to cope with MDR compliance and MDR maintenance, as well as improvements to regulatory intelligence – to keep track of respective market requirements.
Currently a large proportion of device companies track regulatory developments manually, and respond to changes reactively. This is despite the survey finding that almost half of medical device suppliers indicated ‘knowledge of changing global regulatory landscapes’ as among their top three most critical compliance skillsets for the next 2–3 years.
Digitalisation and cross-enterprise connection rank highest on device suppliers’ wish-lists from RIM systems or platforms, meanwhile, as these companies look to implement something more formal and advanced in this area.
Content Re-use: The Practical Advantages of Structured Data Management
In pharma, where RIM capabilities are more advanced, change management came out as a higher priority in the survey, signalling a capability medical device suppliers are likely to need in future too. All of this places an emphasis on being able to pinpoint where various data and content assets are, and where any cascading interdependencies exist between them.
Many pharma companies are now seeing the benefit of transforming narrative/textbased content from existing documents into data-driven structured/tabular information. Turning flowing text into data or content extracts makes it possible for content to be reliably re-used. This can be achieved via software featuring smart automation –to populate each new template, and fulfil the given set of regulatory submission or publishing requirements – adding just what is needed, and no more. It can also support more efficient change management across the product lifecycle, as part of compliance maintenance.
Taking a more structured approach to content creation also supports ‘lean authoring’, a more direct and to-the-point way of writing that focuses documents on key data – resulting in streamlined documents that are easier to digest; reduced review and quality control time; and increased quality.5 For regulatory dossiers in the life sciences industry, lean authoring involves maximising the scope for reuse of sections of approved content (content modules or building blocks), while keeping the emphasis on what each respective HA ultimately wants. Preparing a new HA submission, report, or compliant labelling/instructions for use, then involves building on content that has already been approved, using technology to help automate
the associated cross-checking and content retrieval.
In the survey more than half of pharma RA professionals highlighted increased consistency across submissions, and reduced time and effort as the two main benefits of automated submission preparation/reuse of content extracts. This was in the context of eCTD 4.0-formatted drug submissions (those adhering to the latest ICH standard, a format that will ultimately also apply to medical device submissions).
Laying The Foundations For AI Use
As pharma’s plans advance for smarter information and content repurposing, the opportunity to harness artificial intelligence (including GenAI) is now coming into focus, again signalling where medical device Regulatory professionals may want to lay foundations now.
Key targets for AI-based task optimisation included automated data extraction from documents and other sources; information summarisation from different sources; submission planning and tracking; document management (including document discovery); and compliance gap analysis. Survey respondents from medical device suppliers identified similar opportunities, but with information summarisation topping the list.
One of the overriding challenges the pharma industry has faced in its RA obligations over the years has been the diversity of approaches, processes, systems and formats in use across global organisations and markets. Bringing visibility, order and consistency to all of this has arguably been the greatest struggle of all.
This is among the strongest reasons for the medical device sector to move forward decisively in its approach both to capturing, managing and sharing its information, and to using this to build, submit and publish regulated content –from HA submissions and safety reports to labelling and instructions for use. If medical device suppliers can capture information from the outset in a form that will be easily retrievable and reuseable in different forms for different purposes, it follows that they will be setting themselves up for maximum process efficiency in future.
Setting Expectations
While exact standards for regulatory content management in the medical device industry
Clinical and Medical Research Medical Devices
are still being set down and harmonised across the major regions of the world, the pharma industry’s progress – with everything from eCTD dossier compliance to AI-supported structured content authoring – maps out a route that the medical device sector is very likely to travel down. So why not prepare for the journey now?
The convergence of the pharma and medical device sectors – driven by drugdevice combinations as well as the growing interest of pharma in its sister sector’s projected market growth – is reason enough for regulatory pathways to dovetail. Since the medical device sector is much more diverse and complex in its product definitions (and, by its own admission, already overwhelmed in fulfilling its existing obligations), it is all the more important that it introduces order, efficiency and sustainability to the way its product and manufacture data and content is managed.
A final recommendation for medical device companies is to cultivate the right mindset internally – towards data and content sharing, re-use, and building resources in a granular and structured way that serves multiple purposes across the
product lifecycle – in contrast to the siloed mentality that has hampered technologydriven process transformation for so long in so many industries.
REFERENCES
1. Medical Devices Market Size, Share & Industry Analysis, By Type (Orthopedic Devices, Cardiovascular Devices, Diagnostic Imaging, In-vitro Diagnostics, Minimally Invasive Surgery, Wound Management, Diabetes Care, Ophthalmic Devices, Dental Devices, Nephrology, General Surgery, and Others), By End-User (Hospitals & ASCs, Clinics, and Others), and Regional Forecast, 2024-2032, Fortune Business Insights, updated July 2024: https://www.fortunebusinessinsights. com/industry-reports/medical-devicesmarket-100085
2. Drug Device Combination Products Market Size, Share & Trends Analysis Report By Product (Transdermal Patches, Infusion Pumps, Inhalers, Drug Eluting Stents), By Region, And Segment Forecasts, 2024 – 2030, Grand View Research: https://www.grandviewresearch.com/industryanalysis/drug-device-combination-market
3. The European Union Medical Device Regulation: https://eumdr.com/
4. Medical Devices Industry Regulatory Readiness & Resources: 2024 Survey Report (Celgence/ Qdossier research conducted with almost 700 members of the Regulatory Affairs Professionals
Society, RAPS, and reflecting differences between Pharma and Medical Device RA teams)
5. Medical writing: regulatory matters – lean authoring, European Medical Writers Association, 2024: https://journal.emwa.org/ social-media/regulatory-matters/
Sonia A. Veluchamy is the CEO and co-founder of Celegence, a company dedicated to improving patient outcomes through intelligent regulatory compliance. Previously Managing Director and a Board Member at ArisGlobal, and with 1.5 decades of industry experience, Sonia founded Celegence in 2017 with COO Punya Abbhi, to support life sciences companies by building technology and solutions to streamline strategic and operational compliance activities and make business more efficient.
The multidose eye dropper for preservative-free formulations
To
Clinical and Medical Research Medical Devices
Integrating Medical Devices into Pharmacovigilance Portfolios Part II
How
to Carry
Out
Post-Marketing Safety Surveillance of Medical Devices and Drug-device Combination Products in the EU And USA.
Understanding medical device and drug-device combination (DDC) product regulation is essential when integrating medical devices into pharmacovigilance (PV) portfolios. This article dives deeper into the reporting essentials and obligations associated with the post-marketing safety surveillance of these products both in the EU and U.S. markets.
The article discusses the current trends and challenges in the industry, offering insights into best practices and regulatory expectations, including how to navigate the complexities of safety surveillance and ensure compliance with international standards.
Global DDC Trends
DDCs are on the increase. The number of combined product submissions to the FDA increased from 317 in 2014 to 518 in 2019.1 The global combination product market continues to grow at a CAGR of 7%,2 with an estimated value of £139 billion by 2025.3 One of the biggest areas of growth is implants, which may have a software component to them, increasing their complexity.
With increasingly complex drug delivery systems, combined products become more common. This complexity impacts all parts of the product life cycle, from technical development to product quality and supply chain integrity. If we look specifically at the regulatory environment, identifying the most efficient regulatory pathway for complex product configuration depends on several factors, including its primary mode of action, regulatory precedents, and market experience with current products.
As awareness of product experience and risk increases, we are also gaining an understanding of the practical challenges we need to address. These include changes and inconsistencies in the classification of devices. This can be anything from
software added to a device to products not described in the guidelines. Ambiguity regarding classification makes it necessary to seek clarification on how elements will be classified in the future.
Other key challenges include:
• Device classification issues and the impact on the Notified Body (NB) opinion requirement.
• Lack of full compliance with relevant general safety and performance requirements (GSPRs) impacting the approvability of the Marketing Authorisation Application (MMA).
• A lack of a clear definition of a "substantial change" and how much additional documentation is required.
To overcome those challenges, we need to increase our understanding of the differences between drug and device safety surveillance and how to integrate medical devices into drug safety portfolios. We must also build effective interfaces between the Quality, Surveillance, and Regulatory departments and establish entity ownership interface agreements.
An Overview of DDCs in the EU and USA
There are four essential types of DDC in the EU based on the authorisation or certification these documents receive. The biggest differentiator is the regulatory pathway and how these products are going to be handled in the future. The four main product categories are:
• integral DDCs
• non-integral DDCs
• devices with an ancillary medicinal substance
• devices intended to administer medicines
An example of an integral DDC is a prefilled syringe or an inhaler that cannot be taken apart. The Marketing Authorisation holder, in this instance, would apply to the European Medicines Agency (EMA) or the National Competent Authority, which might require input from a NB. The output would be Marketing Authorisation with an NB opinion.
In contrast, non-integral DDCs such as dry powder inhalers co-packed with medicine capsules are each approved separately. Here, we would have Marketing Authorisation for the medicinal product and a CE certificate for the delivery device, handled as two distinct products.
For devices with an ancillary medicinal substance or devices intended to administer medicines, you get a CE certificate, sometimes with consultation from a competent authority for medicinal products, or a CE certificate without anything else. The approval documentation determines how these products are handled for safety reporting.
The U.S. concept is similar. Drugs follow drug regulations, which are 21 CFR 314; biologics follow biologics regulations, which are parts 600 and 606; and devices follow 803 and 806. In addition, 21 CFR Part 4 applies to combination products.
Reporting Essentials
Reporting obligations concerning combination products depend on how the product is approved. In the U.S., it is the application and applicant type, while in Europe, it is the regulation pathway. The product's status defines how the safety reporting will be handled.
In Europe, medical device reporting timelines are two days for serious public health threats, 10 days for serious incidents and 15 days for any other reportable incidents. Until the vigilance module of EUDAMED becomes available, national reporting procedures remain in place. Reporting forms for medical devices include a Manufacturer Incident Report, Field Safety Corrective Action, Periodic Summary Report, and Trend Report.
With reporting of combination products in the USA, drug and constituent drug parts have a reporting timeline of 15 days, while device constituent parts have a timeline of 30 days. Safety reporting timelines can get more complex, especially when you have a product available on the market in both the USA and Europe.
In the U.S., the traditional approach allows for separate reporting of regulated
Clinical and Medical Research Medical Devices
components. A streamlined approach will enable us to leverage some common elements in good manufacturing practice and address the provisions that would be different for each device, drug, or biologic. This is an attractive option because it prevents duplication of efforts. However, there can be challenges. For example, if each component is manufactured by a different entity, the device manufacturer may end up with multiple quality management systems. Even though the last entity, the applicant, is ultimately responsible, using a streamlined approach may not be practical if you are dealing with different entities.
Any procedure for reporting DDCs must be able to provide objective evidence that three main areas are covered:
1. There are defined processes to ensure individual device-related adverse events are reported to regulatory authorities as required.
2. Advisory notices are reported to regulatory authorities and authorised representatives when necessary.
3. There are appropriate records of individual device-related adverse events and advisory notices.
Complaint Handling and Vigilance Procedures
Complaint handling is an essential component of any quality management system within the medical device domain. It is described in full detail in ISO 13485 and includes a broad definition of a complaint to cover Quality and Vigilance in all areas. It also includes recording an awareness date to meet vigilance timelines and forms that may differ from device to device.
It is common practice for forms to differ from one device to another and cover the relevant risks. This is different from drugs,
where the forms used to collect adverse events tend to be the same.
There also needs to be a process for determining the reportability of events. With medical devices, this might include malfunctions that did not result in patient injury. A clear description of the escalation process must exist to facilitate communication between all relevant parties. When, for example, a PV department is checking on the portfolio of an acquired company, the interfaces between individual economic operator roles and internal departments can get very complex. It is always important to understand who is responsible for the product itself.
The vigilance procedure covers the applicable vigilance regulations for covered geographies, including definitions. It needs to cover not just the timelines and instructions on how to complete the report but also any investigation and follow-up. This creates a need for interface with the Quality Management Department and, potentially, PRRC.
Post-Marketing Surveillance, Vigilance and Handling
There are some similarities between the post-marketing surveillance, vigilance and handling requirements in the USA and Europe. For example, in both there must be an adequate system for the medicinal product to comply with obligations on the recording or reporting of adverse reactions and with post marketing surveillance requirements.
One of the key differences, however, is that vigilance in the USA for post-market safety reporting is driven by application and applicant type. Application-based reporting is supplemented with specific reporting elements for each of the constituent parts of the combination product. Similar reporting requirements also apply if a reportable event occurs in a similar constituent part of a combination product. There is an expectation that such an event would be reported in the U.S. against the U.S.-marketed product. Hence, the manufacturer has the obligation to monitor similar products.
In the EU, reporting to the competent authority for the medicine product is sufficient. There is, however, no clear recommendation for reporting device complaints with potential impact on drug delivery between the National Competent Authority, where the NB is located, and the Reference Authority of the medicinal
product. There is an assumption that the NB and the National Competent Authority will communicate issues with each other.
To operate in this complex environment, vigilance procedures must cover complaints, escalation and reporting for all constituent parts and products as a whole. We need to update Corrective and Preventative Action (CAPA) systems and reporting procedures and improve processes for complaint handling to ensure vigilance obligations.
Final Thoughts
Integrating medical devices into pharmacovigilance portfolios is an evolving and multifaceted challenge. As DDCs continue to proliferate, pharmacovigilance teams must leverage professional support and stay abreast of diverse regulatory landscapes and industry innovations to ensure the safety and efficacy of their products and keep pace with the dynamic field of medical device integration.
Ensuring compliance requires robust interfacing between quality, surveillance, and regulatory departments, along with welldefined procedures for complaint handling, vigilance, and post-marketing surveillance.
Continuous education and strategic adaptation are crucial for success in this evolving landscape. Expert services and innovative solutions are crucial for navigating these complexities and meeting regulatory requirements effectively.
Qureshi- President of Qinecsa Solutions, with over 20 years of strategic and operational experience in drug safety, Humaira is spearheading a movement to bring together scientific expertise with technology and create world-leading pharmacovigilance solutions.
Humaira Qureshi
Humaira
40 years of building powerful industry partnerships.
Broadest portfolio of self-injection products built on platforms
Serving pharma globally for all their originator and biosimilar needs
Over 70 products launched in 15 different therapeutic areas
Global manufacturing footprint spanning Switzerland, Germany, China and North America
Fully integrated strategic partner network
For more information visit www.ypsomed.com/yds Ypsomed AG // Brunnmattstrasse 6 // 3401 Burgdorf // Switzerland T +41 34 424 41 11 // info@ypsomed.com
Clinical and Medical Research Medical Devices
Ypsomed's Approach to Platform Products and Strategic Partnerships for Self-injection Devices
Ian Thompson, former Vice-President of Account and Business Development at Ypsomed, shares how Ypsomed has reshaped the market for platform-based self-injection devices. Ypsomed has expanded the platform concept beyond the device itself, developing strong partnerships with strategic networks. This approach supports pharmaceutical companies and patients in accessing safe, reliable, and user-friendly drugdevice combination products.
The Outsourcing Dynamics in Pharma
The global pharmaceutical market reached an estimated $1.6 trillion (£1.3 trillion) in 2023, with the top 25 companies making up about half of this revenue. Biologics now represent over $500 billion, or approximately 30% of total sales, and this share is expected to grow as more self-injectable therapies are developed for oncology, neurology, and rare diseases. Additionally, there is an expanding market for autoimmune disease treatments and insulin and GLP-1-based drugs for diabetes.
Pharmaceutical companies typically spend around 15% of their revenue on research and development (R&D) and another 25% on the cost of goods. This equates to roughly $650 billion per year, with up to half of these expenditures outsourced to a variety of organisations, including contract research organisations (CROs) and contract development and manufacturing organisations (CDMOs). Given this significant reliance on outsourcing, pharmaceutical supply chains have become increasingly global and complex, requiring robust networks that can endure unexpected disruptions.
Platform Products for Self-injection Devices: A Strategic Evolution
The platform approach in injectable drug delivery devices first emerged through the use of standardised, ready-to-fill prefillable syringes for heparins and vaccines and prefilled cartridges for insulin and dental lidocaine. These advances coincided with market growth for these molecules in the 1980s and 1990s. During this period, the
self-injection market developed further with devices designed specifically for insulin and human growth hormone (hGH), which needed precise dosing mechanisms for frequent injections.
In the 1990s and 2000s, the development of biologic drugs such as interferons and monoclonal antibodies (mAbs) led to the introduction of spring-driven autoinjectors for less frequent, fixed-dose injections. By 2006, the launch of the first handheld prefilled autoinjectors began to diversify the market, which was primarily dominated by insulin pens from companies like Novo Nordisk, Lilly, and Sanofi, as well as autoinjectors for drugs such as Humira® (adalimumab, AbbVie) and Enbrel® (etanercept, Amgen).
The 2000s were a period of significant change for both injection pens and autoinjectors, marked by the emergence of platform-based business models and a notable increase in drug product launches during the 2010s. A "platform product" refers to a standardised device technology that can be adapted to the needs of various drugs while being manufactured using a shared infrastructure. This strategy, common in other industries, helps reduce risk and accelerate the development and production of different product versions.
Unlike bespoke devices created for individual drugs, platform products are flexible and can accommodate different drug requirements, including packaging configuration, dose volume, and drug viscosity. Recently, there has been a move toward customising platforms to create a range of drug-device combinations, simplifying access to advanced products for a broad spectrum of customers, including small biotech innovators, emerging biosimilar manufacturers, and large global pharmaceutical companies. Some of these companies have even adopted a "platform-in-platform" approach to streamline the sequential launch of injectable drugdevice combinations with established supply chains, partnerships, and manufacturing infrastructures.
Ypsomed's Platform Product Partnership Strategy
To enhance supply chain resilience, pharmaceutical companies have focused on four main strategies:
• Gaining comprehensive visibility into their supply chains to identify potential weaknesses.
• Regularly conducting stress tests and reassessments, often involving routine audits of critical suppliers.
Figure 1: The current Schwerin (Germany) site expansion will increase manufacturing capacity to 500 million injection devices annually.
• Minimising exposure to risks by diversifying supply sources and balancing inventory strategies between just-in-time and just-in-case.
• Ensuring that risk management and supply chain resilience are integrated into strategic planning and daily operations, supported by structured governance.
Ypsomed has incorporated these elements into its delivery systems, aiming to build long-term, trusted partnerships with its clients. Of these strategies, end-to-end transparency and structured governance are especially critical in Ypsomed's strategic partner networks, benefiting both the pharmaceutical companies it serves and their patients.
Collaborative Partnerships and Proactive Auditing
Ypsomed serves a diverse range of customers, from small biotech firms to midsized and large pharmaceutical companies worldwide. To navigate the complexities of the pharmaceutical supply chain, the company emphasises open communication across technical, logistical, and commercial matters. With its extensive experience and cross-industry expertise in drug delivery, Ypsomed guides its customers through the design, development, and regulatory approval processes for self-injection devices.
Ypsomed collaborates with specialised partners such as Interface Analysis Associates, which provides human factors expertise throughout the product development
journey, ensuring the usability and safety of its self-injection devices. This approach ensures robust and reliable delivery systems, supported by routine stress testing, supply chain reassessment, and regular audits of critical suppliers.
All necessary competencies for successful combination product development are centralised at Ypsomed’s headquarters in Burgdorf, Switzerland. This structure facilitates efficient communication between experts, rapid learning across specialist fields, and the establishment and maintenance of robust risk management programmes.
Building A Global Manufacturing Network
Over the past decade, Ypsomed has produced more than 25 million reusable pen injectors and one billion prefilled pen injectors, in addition to supplying over 100 million prefilled autoinjectors since the launch of YpsoMate in 2018. To meet the increasing demand for new product launches and higher production volumes, Ypsomed is expanding its manufacturing capabilities in Switzerland, Germany, China, and North America (Figure 1).
This global expansion reflects Ypsomed's commitment to bringing manufacturing closer to end markets, enhancing efficiency and sustainability.
To free up capacity for core platform products, Ypsomed plans to discontinue contract manufacturing and pen needle production at its Solothurn site in Switzerland. Furthermore, Ypsomed’s in-house tool making capabilities, currently located in Burgdorf, will be duplicated at the Solothurn site over the next 1–2 years. Higher-capacity tooling and
larger sub assembly lines are being added to increase manufacturing efficiency. This expansion is part of an investment plan of CHF 120 million (£107 million) for the 2023/24 business year, with a total of CHF 1.5 billion projected over the next five years.
Producing platform products from multiple sites worldwide reduces risks for Ypsomed’s pharmaceutical partners and supports their largest drug franchises more efficiently and sustainably.
Serving All Customers
Ypsomed’s platform products, such as UnoPen and YpsoMate, are designed to serve all therapy areas and customer segments, with each customised product version supplied on a non-exclusive basis. The shared development and manufacturing infrastructure benefits all clients, enabling more than 70 drug products based on Ypsomed platforms to be available globally (Figure 2). In addition, there are 150 more projects in clinical trials or preparing for commercial launch. YpsoMate autoinjectors have been launched for 20 different branded drug products, and more launches are anticipated in the coming years.
For high-volume commercial quantities, Ypsomed supplies initial volumes through its shared platform product infrastructure and scales its manufacturing capacity according to customer forecasts. The company also considers dedicated manufacturing lines for major drug franchises and, to reduce dependencies, may use third-party contract manufacturing under a royalty scheme.
Figure 2: Ypsomed has over 70 products launched, and counting, serving all therapy areas.
Clinical and Medical Research Medical Devices
Forty Years Of Strategic Partnerships: Beyond The Device
For four decades, Ypsomed has cultivated a network of trusted strategic partners who serve as key suppliers to pharmaceutical customers. Over the last 20 years, these relationships have expanded from cartridgebased peptide therapies for injection pens to include syringe-based autoinjector therapies for antibody treatments. While Ypsomed excels in developing and manufacturing selfinjection devices, it also collaborates with key suppliers to ensure drug-device combination products meet both pharmaceutical and patient needs, delivering them promptly and reliably.
Primary Container Compatibility Specialist
Understanding the primary drug container
system and its interfaces with the injection device is key. Ypsomed collaborates with more than 15 glass and elastomer manufacturers worldwide, testing the container variants at every stage. When a suitable primary container does not yet exist for a new platform injector, Ypsomed works with leading suppliers to define and develop new industry standards, such as those for the YpsoMate 5.5 mL and YpsoDose 10 mL products (Figure 3).
Supporting Final Assembly and CDMOs
Final assembly of the drug with the prefilled pen injector, autoinjector, or patch injector is critical to the success of drug-device combination products. Ypsomed collaborates with leading assembly equipment manufacturers to provide scalable solutions
for clinical and commercial supply, extending to processes like functional release testing, labelling, and final packaging. Ypsomed can make recommendations and facilitate the selection of manufacturers through its extensive global network of fill-finish and final assembly CDMOs (Figure 4).
For YpsoDose, Ypsomed has partnered with SCHOTT Pharma for the 10 mL cartridge and ten23 health to utilise their formulation, process development, filling, assembly, and final product testing expertise.
Engaging Fill-finish and Other Networking Experts
Understanding the dimensions and properties of the primary container is crucial, but so is knowing the intricacies of the filling process and how the filled container functions as a complete system. Ypsomed collaborates with sterile filling equipment manufacturers to stay current with the latest developments. Their equipment is used by both in-house pharma manufacturing and CDMOs involved in over 50% of all pharmaceutical projects. With more than 130 active customers, Ypsomed is a proactive player in the global pharmaceutical and CDMO network.
Whether a client seeks connections within the global drug-device consulting network or needs specialised training devices, Ypsomed offers recommendations from its wide-ranging partner network.
Strategic Networking: An Essential Part Of Ypsomed's DNA
Developing and manufacturing drug-device combinations requires more than just technical expertise – it demands a deep
Figure 3: The main primary drug containers used in combination with Ypsomed’s platform products.
Figure 4: Ypsomed’s strategic partner network includes all leading final assembly equipment suppliers.
Medical Devices
understanding of the entire ecosystem. For over 40 years, Ypsomed has built a network of strategic partnerships with key suppliers, contract manufacturers, and service providers, ensuring that its platform products (Figure 5), like UnoPen and YpsoMate, are seamlessly integrated into the pharmaceutical supply chain (Figure 6). This strategic networking enables Ypsomed to uphold the highest standards of quality,
reliability, and performance across all its offerings.
By fostering close collaborations and maintaining transparent communication with its partners, Ypsomed enhances supply chain resilience and ensures rapid market access for its customers’ products. These efforts have positioned Ypsomed as a reliable partner and a leader in self-injection device innovation.
As Ypsomed looks to the future, it will continue to leverage its expertise, partnerships, and innovative approach to deliver exceptional value to its customers and improve patient outcomes worldwide –making selfcare simpler and easier.
Ian Thompson
Ian Thompson, former Vice-President Account & Business Development, has been with Ypsomed, formerly Disetronic, since 1995 in a number of roles in key account management and business development, working with pharma companies to develop innovative selfinjection systems and bring them to market. He retired at the end of August after a distinguished career. He studied biochemistry and biotechnology in the UK, working initially in commercial roles in fermentation technology. He has worked in medical device companies since moving to Switzerland in 1990. Since 2003, Mr Thompson’s main focus has been business development and new product innovation, leading to the successful development and launch of a range of new pen injector, autoinjector and patch injector customisable platform products for Ypsomed Delivery Systems.
Figure 5: Ypsomed’s comprehensive portfolio of platform products.
Figure 6: Ypsomed employees working together in the production area, focused on ensuring quality in the manufacturing of self-injection devices for drug-device combination products.
Overcoming AI Anxiety In Medical Affairs: Strategies For Successful Adoption
Artificial intelligence (AI) is becoming increasingly integrated across the pharmaceutical industry and promises to revolutionise many aspects of medical affairs. From enhancing drug development and clinical trial design to streamlining real-world evidence generation and optimising patient engagement strategies, AI's potential benefits are undeniable.
Despite this growing enthusiasm, AI anxiety and apprehension over implementing technologies have swept through the medical affairs community. A lack of understanding about AI technologies, data privacy and security concerns and ethical considerations have all contributed to a hesitancy towards embracing AI fully.
In this article, Alice DI Giulio, AI Frontier PR lead at Envision Pharma Group, explores the causes of AI anxiety in the pharmaceutical industry and medical affairs. She addresses the primary sources of concern, debunks common misconceptions and provides strategic solutions for successful AI adoption.
Understanding the Sources of AI Anxiety
To understand AI anxiety in medical affairs it is important to consider the top barriers to AI implementation in the pharmaceutical industry, which have been identified as:1
• Demonstrating value:
Establishing a return on investment (ROI) and demonstrating the tangible benefits of AI helps to justify investments. Medical affairs teams can demonstrate value by showcasing how AI projects translate into measurable outcomes that align with organisational goals.
• Deployment and scaling: Deploying and scaling AI technologies is a complex process. By investing in technical infrastructure and operational experience organisations can overcome challenges and fully integrate AI into existing systems.
• Trust in AI models: Trust in AI can be fostered with trans-
parency and explainability of models. This involves ensuring accuracy and reliability using rigorous validation processes.
• Ethics, fairness and bias:
As biases can arise in AI models, ensuring the ethical use of data and maintaining trust and compliance with regulations helps with implementing AI fairly.
• AI integration:
Integrating AI systems with existing technologies and workflows, while also managing organisational change, leads to successful AI integration.
• Perceived vs. actual benefits: The gap between expected and realised benefits can be bridged by setting realistic expectations and focusing on achievable outcomes. Employee education and training are key to establishing realistic expectations and maximising AI benefits.
As well as the barriers that the pharmaceutical industry faces, medical affairs professionals face additional challenges when dealing with AI-generated insights, which can lead to increased AI anxiety.
AI Anxiety In Medical Affairs
One primary source of anxiety is the difficulty of distinguishing valuable knowledge from the overwhelming noise generated by AI algorithms. Medical professionals are inundated with a constant stream of data from various sources, making it challenging to identify truly important signals. This information overload can lead to missed opportunities and delayed action, hindering the ability to stay ahead in a competitive landscape. Exacerbating this issue is the phenomenon of AI hallucination, where AI systems sometimes produce inaccurate or fabricated insights. This further erodes trust in AI-generated information and makes it harder for medical professionals to confidently act upon it.
Another significant source of AI anxiety in medical affairs is the black hole effect in insight utilisation. Even when valuable insights are successfully identified, they often fail to reach the right audience within
the company and seemingly disappear. This can be attributed, in part, to the opaque nature of AI algorithms. Professionals struggle to understand and trust insights generated by a process they cannot fully comprehend. The lack of explainability surrounding AI's decision-making process further complicates matters, as it becomes difficult to justify actions based on AI recommendations. This lack of transparency and understanding creates a barrier to the effective communication and utilisation of AI-generated insights within medical affairs teams.
Common AI Misconceptions And Strategic Solutions
Understanding the uncertainty surrounding AI in medical affairs and equipping teams with effective solutions is crucial for empowering them and fostering successful adoption.
Large Language Models Vs Large Knowledge Models
A common misconception about AI is that it relies solely on large language models focused on predicting the next word in a sentence. This can lead to the generation of irrelevant or inaccurate information and hallucinations. However, this challenge can be solved by using large knowledge models specifically designed for medical affairs. These models are trained on thousands of medical ontologies, ensuring that the insights generated are more precise, relevant and reliable.
Explainable AI Algorithms And Black Boxes
Another common misconception about AI is that it operates as a black box, with decisionmaking processes that are impossible to understand or explain. As this can increase distrust in AI solutions, designing AI algorithms to provide a glassbox effect improves explainability and transparency in how insights are generated. This clarity allows users to understand the reasoning behind AI-driven decisions, fostering trust and confidence in the technology.
Acting on AI insights
AI-generated insights can often be considered too vague or overwhelming, making it difficult for professionals to take action.
This concern can be directly addressed by deriving specific, actionable insights from the AI model’s knowledge base. Ensuring that the information provided is not only relevant but also clear and concise, enables medical professionals to make informed decisions and take precise actions quickly.
Demonstrating Value and ROI
One of the key misconceptions is that the ROI and tangible benefits of AI are difficult to prove, leading to scepticism about its value. This hesitancy can hinder the adoption of AI solutions, even when they have the potential to significantly improve efficiency in extracting medical affairs insights. The solution to addressing this misconception involves demonstrating clear, measurable outcomes that justify investments in AI. By showcasing the tangible benefits and positive impact on business goals, organisations can build confidence and overcome scepticism surrounding AI adoption.
Ethics, Fairness and Bias
An important misconception to consider is that AI systems inherently possess biases, making them untrustworthy for fair decision-making. This is particularly
relevant to medical affairs as it could impact the outcomes of certain patient populations that are not well-represented in datasets. Addressing biases in AI is a key priority and by actively identifying and mitigating biases during development and implementation, organisations can ensure that AI models operate ethically and comply with regulations. Committing to fairness and transparency helps build trust in AIpowered solutions, allowing them to be used responsibly and effectively in critical applications.
Trust and Transparency in Models
Due to a lack of transparency, AI models are often considered inherently unreliable, lacking the ability to be validated and leading to distrust of outputs. Designing AI models with enhanced transparency allows for greater visibility into their decisionmaking processes. Subjecting models to rigorous validation processes also helps with ensuring their accuracy and reliability. By prioritising transparency and validation, organisations can further build trust in AI models and demonstrate their value as dependable tools for informed decisionmaking.
Integrating with Existing Workflows
Integrating AI technologies into existing workflows can be perceived as difficult. This is often accompanied by concerns about potential disruptions to established processes. However, AI solutions can be designed for seamless interoperability with existing systems. This focus on integration ensures a smooth transition and effective organisational change management, minimising disruptions and allowing teams to quickly realise the benefits of AI technology.
Education and Training
One misconception surrounding AI is that its benefits are often overhyped and fail to meet the high expectations set by organisations. This can lead to disillusionment and scepticism among potential users. Setting realistic expectations and providing comprehensive education and training to bridge the gap between perceived and actual benefits can help with this misconception. Equipping users with the knowledge and skills to effectively leverage AI technology empowers them to realise its full potential and achieve meaningful outcomes.
Successfully Implementing AI in Medical Affairs
Overcoming AI anxiety requires a multifaceted approach that focuses on education, collaboration and strategic implementation. Organisations should focus on large knowledge models, trained on thousands of medical ontologies, to ensure more accurate and relevant insights compared to traditional large language models. Algorithms designed for transparency also provide a glassbox effect that allows medical professionals to understand and validate the insights generated. This transparency builds trust and confidence in the AI-driven decision-making process and actionable insights derived from the knowledge base enable professionals to take swift and effective actions, reducing noise and preventing missed signals.
Implementing an AI platform with bestin-class technology helps with automating the analysis of medical text for insight generation. Platforms should provide custom implementation based on an organisation's data sources and formats, ensuring seamless integration with existing systems. Developing a medical insights playbook, including guidance documents, templates, communications and training for stakeholders also helps to ensure a standardised and effective approach to AIgenerated insights.
Collaborating with strategic partners who specialise in AI solutions and medical affairs can help reduce AI anxiety and accelerate adoption. Medical affairs professionals can leverage the expertise of strategic partners
to implement large language models and transparent AI tools, proactively building trust and confidence in AI technologies.
Strategic partners can also help with demonstrating ROI and the tangible benefits of AI by establishing measurable outcomes that justify investments. Additional support that partners can provide includes prioritising ethical AI use, ensuring rigorous validation processes and designing AI tools that seamlessly integrate with existing workflows and processes. By addressing these key areas, medical affairs teams can implement AI solutions that extract the insights they need with confidence and precision.
A Holistic AI Approach to Drive Medical Affairs Performance
Tailoring AI solutions to the unique needs and challenges of each organisation is crucial for successful implementation and adoption. A one-size-fits-all approach may not address the specific requirements and complexities faced by different organisations, hindering the effectiveness of the AI solution.
The AI insight generation process should be supported by medical affairs expertise to discover, deliver and act on impactful medical insights. Collaboration is key and strategic partnerships can help with establishing the medical insights process and roles and responsibilities across the organisation.
Although the integration of AI in medical affairs holds transformative potential
it is essential to address the anxieties surrounding its adoption. By understanding the root causes of AI anxiety, debunking misconceptions and implementing strategic solutions, organisations can navigate this transformative journey successfully.
REFERENCES
1. Gartner survey: Top Barriers to Implement AI Techniques, Gartner, May 2024
Alice Di Giulio is an experienced communications professional with a specialisation in AI policy within the life sciences sector. In her role as AI Frontier PR Lead at Envision Pharma Group, she focuses on furthering advanced technologies into strategic communication efforts, ensuring innovation aligns with regulatory frameworks and stakeholder expectations. She has been instrumental in establishing the AI Innovation Board at Envision Pharma Group, promoting the responsible use of AI technologies to drive advancements in the industry. Alice has also led policy engagements with the EU Commission, collaborating with key European and international institutions to shape AI policies that foster innovation while safeguarding public interest.
Dr. Loubna Bouarfa
UniSafe® Platform
Delivering the future with safety and simplicity
Developed around UniSafe®, our platform meets the needs of you, our partner, and your varying patients’ needs both now and in the future.
A spring-free, passive safety device for 1mL pre-filled syringes, designed for simple assembly and use.
Proven, on market, in patient use
A spring-free, passive safety device for 2.25mL pre-filled syringes, designed for simple assembly and use.
Fully industrialised and ready to supply
A reusable companion auto-injector for UniSafe® 1mL.
Want to know more?
A reusable connected companion auto-injector for UniSafe® 1mL.
Visit ompharmaservices.com/ipi-sept2024 or email pharmaservices@owenmumford.com
Sterile and Safe: When Robots Support the Production of Live-attenuated Vaccines
Stäubli Robots in Cell Culturing for Chickenpox Vaccines
The leading manufacturer of chickenpox vaccines in China, Changchun Keygen Biological Products relies on robot-based cell factory automation to produce live-attenuated vaccines in a hygienic environment with the highest efficiency. One essential process is the cultivation of the virus in special containers or “cell factories” requiring high-intensity oscillation. Robotic systems integrator Shenyang Great Elites Intelligent Equipment engineered an automated solution with no fewer than 12 Stäubli TX200 Stericlean robots.
Today, chickenpox hardly poses a threat to children or adults – thanks to a vaccine that is used worldwide and consists of live-attenuated varicella viruses. This type of vaccine encourages the body to create antibodies and memory immune cells, thus stimulating a strong, effective, long-lasting immune response. This prevents children from being infected with chickenpox and minimises the risk of developing much more serious complications such as meningitis, pneumonia, and in the course of a lifetime, shingles (herpes zoster).
China’s biggest manufacturer of these vaccines, Changchun Keygen Biological Products (CKBP) in Changchun was established in 2003. With an annual production capacity of eight million doses of chickenpox vaccine, the company is not only supplying the domestic market but also exporting the live-attenuated varicella vaccine to various countries like South Korea and India.
A Major Step: From Manual-assisted to Automatic Operation CKBP was one of the first Chinese companies to utilise robots for vaccine production. The decision for a high level of automation was driven by the need to eliminate all potential xenobiotics, impurities, and contamination caused by personnel to the greatest extent possible and maintain a strict, sterile production environment. A high level of automation is a prerequisite for the safe production of high-quality vaccines.
Given these requirements, robotic systems integrator Shenyang Great Elites Intelligent Equipment – well familiar with the automation of critical processes in biopharmaceutical production – engineered a system that has enabled CKBP to upgrade its live-attenuated varicella vaccine production from traditional manual to automated operations.
Twelve Robots in the Vaccine Factory
Apart from “robotising” CKBP’s varicella vaccine production, the two companies jointly developed a mixing system, quantitative peristaltic pumps, pipeline automation systems, and other equipment. As a result, the entire facility is highly automated.
Core processes of vaccine production include cell factory passaging (including the addition of active ingredients like trypsin and a growth medium) and cell factory inoculation, where the containers filled with the liquid base substance are “fed” the varicella virus and other ingredients. After this, the containers must go through defined movements – like shaking, flipping, oscillating, and recovering – in sequence.
In the first process, cell factory passaging, four robots are involved. In the cell factory inoculation and cultivation steps, eight robots perform multiple tasks, including harvesting and cleaning the cell factories. A robotic tool change system allows for automated filling and emptying of the liquid vaccines and changing of the factories. Each robot is relatively independent and can alternately handle two stacks of cell factories. Changing the cell factories is enabled by quick change tools from the Stäubli portfolio.
Aseptic Robots for Sterile Environments
The automation experts from Shenyang Great Elites opted for 12 identical robots in the cell and virus culturing stages – for good reasons. The six-axis Stäubli TX200 robots are available in the Stericlean version, which means they are designed for the very special environment in which they are working: critical applications in aseptic GMP Class A production areas, where these specialised robots have proven themselves thousands of times over.
The kinematics of the Stericlean six-axis machines are fully encapsulated and sealed
tight, with all cabling on the inside. There are no dead spaces, only smooth surfaces on which there is no opportunity for the formation of impurities. The unique specifications of these outstanding pharmaceutical robots make them resistant to the harshest of regular sterilisation, disinfection and decontamination procedures, for example with hydrogen peroxide in vaporised form (VHP). This method is used in CKBP’s vaccine production.
Precise and Efficient
Apart from the high-grade hygienic requirements, there were other reasons for choosing the Stäubli TX range. Cai Yan, General Manager of CKBP explains: “The robots perform their tasks with high precision, high productivity and availability, and minimal maintenance requirements.” Robust drive systems were another important aspect, according to Cai. “The cell factories oscillate frequently and at high speed. This requires extremely high stability of the robot arm.” Another strong point is the connectivity of the robots.
Their control system is integrated with the pipeline automation system, offering fully automated vaccine production management.
In contrast to the manual operation employed previously, the robots are a great improvement with respect to the health and safety of the skilled staff. “Due to physical exhaustion, we often needed two groups of workers, and they sometimes suffered joint injuries. Now, the robots free our employees from heavy labor, and deliver the highest quality during all shifts,” said Cai.
Introducing the Integrator: An Expert in Highly Automated Biopharmaceutical Production
Shenyang Great Elites Intelligent Equipment is committed to providing customers with professional and intelligent solutions for biopharmaceutical production. It specialises in the biopharmaceutical industry and enables smart digital production lines. Its main products include CFAM series cell factory automation equipment, 10-layer cell factory automation workstations, intelligent cell observation equipment, and aseptic filling equipment.
INTERVIEW WITH GREAT ELITES:
Business Growth in High-end Biopharmaceutical Production with Stäubli Robots
In this short interview, Bian Tianyuan, General Manager of Great Elites Intelligent Equipment, explains how his company and Stäubli collaborate on the development and delivery of automation solutions for biopharmaceutical production.
What are, from the user's view, the main benefits of fully automated vaccine production?
Vaccine production automation involves a high degree of technological integration. It combines numerous advanced technologies such as robotics, sensor technology, and control technology to achieve full automation from raw material processing to finished product packaging. This highly integrated technical characteristic makes vaccine production more efficient, precise, and reliable. Additionally, robots can maintain precision even during long periods of continuous production. Furthermore, vaccine production automation adheres to strict cleanliness standards. When using robots that
meet high hygienic standards, we no longer need to worry about maintaining cleanliness during the production process.
Why did you select Stäubli as a robot supplier?
We have been working with Stäubli in the field of robotics for many years and have established a mutual understanding of cooperation. Stäubli’s life sciences team has accumulated deep technical knowledge and rich industry experience. Whether in earlystage technical consultation, programme design, later-stage installation and debugging or maintenance, they are able to provide timely, professional support. This comprehensive service guarantee allows us to advance our work more smoothly and efficiently during the project implementation process.
What features do you particularly appreciate about the robots?
On the product side, Stäubli’s robots are
renowned for their excellent stability and accuracy, and they maintain excellent performance during long-term continuous operation. At the same time, they also offer high flexibility and scalability. Their robots can be externally integrated with various devices and systems to achieve real-time detection and monitoring of the robot’s status. This integration capability makes our solutions more complete and comprehensive, and better able to meet the needs of our customers. To sum it up, we have established a long-term and lasting partnership with Stäubli. We believe that in future collaborations, Stäubli will continue to provide us with quality products and services, helping us to continuously expand the market and achieve business growth.
Do you use Stäubli robots regularly in medical and pharmaceutical applications?
Our core competence is providing customers
with professional and intelligent integrated solutions for biopharmaceutical production. The main products include CFAM series cell factory automation equipment, 10-story cell factory automation workstations, intelligent cell observation equipment, sterile filling equipment, and others. We have, just to name a few applications, also collaborated with Stäubli on the Vero cell rabies vaccine transfer bottle project of Liaoning Chengda, the cell detection project of the Institute of Human Genetic Resources, and a toothbrush head packaging project for Procter & Gamble.
Thank you very much for sharing these insights!
Dipl.-Ing. Ralf Högel is among the leading authors in the field of robotics and automation. After some years as the editor-in-chief of various automation magazines, he founded his editorial office Industrie Kommunikation Högel in 1994. With a small team of experts, IKH deals with future topics of automation, robotics, AI, AGV, digital transformation, industrial parts cleaning, logistics, etc.
Ralf Högel
ROBOTICS
Next-level pharmaceutical robots for optimal health
Highly aseptic, sterile or Grade C environment, demanding or routine tasks, Stäubli robots meet the highest hygiene and cleanliness standards, while bringing greater efficiency and reliability to your pharma applications.
Stäubli – Experts in Man and Machine
www.staubli.com
Application Note
New Product Introduction & Technical Transfer in a High-Potency Facility
Overview
The pharmaceutical industry is increasingly focused on drug products containing highly potent active pharmaceutical ingredients (HPAPIs). These compounds are highly effective at very low doses, but due to their toxicity they also pose significant risks to operators. The introduction and scaling up of HPAPIs in manufacturing facilities involve complex challenges requiring specialised expertise and stringent safety protocols.
Introduction
Drug products containing HPAPIs account for around 40% of authorised drug products on the market today. In 2023, the OSD market was approximately $36.5bn, and is poised to register a compound annual growth rate (CAGR) of around 6% by the year 2030[RG1] . Within this space, the main drivers for growth include oncology, diabetes and obesity drugs, the latter experiencing a boom due to the role of GLP-1 therapies in weight loss management. As the demand grows, a robust approach to new product introduction (NPI) and technical transfer (TT) becomes increasingly essential. The processes for handling and on-boarding HPAPIs can be difficult due to the inherent risks associated with these substances, with the key challenges including categorisation, risk classification, containment, process flow, and ensuring robust cleaning methods to prevent cross-contamination in a multiproduct facility.
Handling HPAPIs
HPAPIs are designed to elicit a biological response at very low doses, making them highly potent but also highly toxic. Whereas during administration the product’s highly targeted yet toxic profile ensures that fewer side effects are experienced by the patient, they pose a significant risk to CDMO production operators if they’re not handled correctly. Ensuring the safety of production workers and patients therefore requires meticulous control and monitoring, which begins during TT and NPI, but the lack of standardised monitoring tools and methods to ensure
the safety of production environments remains a challenge.
When developing and manufacturing highly potent drug products, there should always be an OEL monitoring programme at each stage of the process. Strict crosscontamination controls are essential, with a risk assessment performed in accordance with four principal modes:
• Airborne;
• Mechanical;
• Personnel transfer;
• Retention of product on contact surfaces.
With traditional non-potent drug products, avoiding contamination from personnel involved in the production process is, of course, critical. However, when manufacturing drug products containing HPAPIs, operating in rooms under negative pressure isn’t enough. High-grade specialised containment equipment is also required to protect employees from the API itself – and to ensure drug product integrity in a multiproduct facility.
Understanding the levels of containment is important when performing TT and NPI as it helps to determine the appropriate handling of any highly potent compounds that are
brought into the CDMO. Primary containment requires suitable contained equipment, running under negative pressure and effectively serving as a ‘clean room’ in its own right, with secondary containment being the facility itself. The necessary solution depends on the product potency and batch size being handled, as there may also be a need for different considerations for laboratoryscale, small-scale, and commercial-scale use. Containment must therefore suit the product potency, with careful consideration of dust extraction (heating, ventilation and airconditioning systems (HVAC) and central dust collection) and breach control procedures.
Technical transfer in the context of HPAPIs revolves around managing risk and layering safety controls. Effective risk management strategies begin with facility design, including designing the ability to segregate materials within the facility to achieve cross-contamination prevention from one product to another.
NPI
When managing potent materials, the introduction of new products (NPI) becomes a critical factor. A leading CDMO should have well-established protocols to evaluate all new molecules for their OEL and PDE before they are approved for site integration. For instance, a COSHH
assessment procedure will delineate the molecule's characteristics, its mechanism of action, and the necessary handling protocols to ensure operator and environmental safety at all times. Conducting toxicological and pharmacological assessments for every molecule is essential not only for safeguarding scientists and operators but also for establishing appropriate cleaning assessments and verification parameters, which are crucial to preventing crosscontamination between products.
Additionally, a GMP Failure Mode and Effect Analysis (FMEA) is performed, utilising data from safety reviews, licensing, equipment, and premises evaluations to ensure the safe processing of the product. For technical transfer projects specifically, Excipient Gap and Process Equipment Gap analyses are conducted to assess the site’s capabilities in comparison with the transfer site, identifying any gaps that need to be addressed before the project begins. A molecule will only be issued its COSHH assessment for handling within the facility after all necessary reviews and evaluations have been completed.
Cleaning Validation
Cleaning Validation (CV) plays a crucial role in the TT process by ensuring that cleaning methods are effectively transferred and adapted to the new manufacturing environment. During TT, the established cleaning procedures from the original site or development stage must be validated in the new facility to confirm their effectiveness in removing product residues, cleaning agents, and potential contaminants. This involves not only replicating the cleaning methods but also tailoring them to the specific equipment, materials, and operational conditions of the receiving site. CV provides immediate assurance that these processes work correctly from the outset, while validation ensures long-term consistency and regulatory compliance. Successful TT and NPI hinge on this robust validation, as it safeguards against cross-contamination and ensures that new highly potent products can be manufactured safely without compromising the quality of other products in the facility.
Each new molecule entering a multiproduct facility adds further complexity, requiring a robust cleaning philosophy.
Starting at NPI, the process should include methods to test and detect detergents and drug substances with swab and rinse samples and any additional cleaning that may be required. Equipment capabilities should be constantly reviewed, with oversight of all cleaning verification and validation, resulting in a science- and risk-based approach to the prevention of cross-contamination.
Employees should be provided with general GMP personal protective equipment (PPE), such as laboratory coats/suits and footwear, hairnets, eyewear, face masks and gloves, whereas powered air purifying respirators (PAPR) should be considered as part of a breach control and system failure programme. But PPE should never be considered as part of the primary protection barrier during potency assessment or normal operations. Operator safety is of critical importance, resulting in a new way of thinking to develop drug products in a controlled facility with programmable logic controller (PLC) equipment.
Emerging Challenges and Opportunities
The future of HPAPI manufacturing presents
several opportunities and challenges, which are fundamental considerations for TT and NPI. One significant area of interest is the potential role of artificial intelligence (AI) in enhancing risk assessments, toxicology evaluations, and process validation. AI could potentially streamline data gathering, protocol writing, and validation report generation, with its predictive capabilities enabling more accurate modelling of toxicological impacts and optimisation of risk management strategies, which could enhance safety profiles and operational efficiency.
Additionally, the rising demand for customised therapeutics presents the need for adaptable manufacturing systems capable of efficiently managing various HPAPIs. This shift towards personalised medicine provides CDMOs the chance to stand out by developing quick prototyping and small-scale production capacities essential for niche, high-potent OSD products. Therefore, despite the demanding nature of the industry, there lies vast potential for innovation and expansion in
specialised CDMO services, propelled by advancements in AI and digital technologies.
Regulatory discrepancies across different regions pose further challenges for CDMOs operating in the highly potent space. Navigating differing regulatory requirements can complicate compliance for CDMOs that operate on a global scale. For instance, the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) might have varying requirements for clinical trials, manufacturing processes, or even the submission of regulatory documentation, which requires CDMOs to adapt their strategies for each market. Such discrepancies may lead to increased cost and time to market, and developing products that meet the specific regulations of multiple jurisdictions can limit the ability to use a onesize-fits-all approach, potentially restricting the ability to standardise processes and leverage economies of scale.
Conclusion
As the pharmaceutical industry continues to advance in high potent drug product
manufacturing, companies must adopt rigorous scientific standards and best practices to ensure safety and efficacy. Embracing innovative technologies and maintaining stringent risk management protocols will be key to successfully navigating the challenges of HPAPI production. The ultimate goal is to deliver ground-breaking therapies while safeguarding the health of workers and patients.
The successful NPI and TT of new products containing HPAPIs are pivotal to meeting the growing demand for advanced therapeutics. The complexity and risks associated with HPAPIs necessitate a meticulous approach to facility design, risk management, and process validation. As the pharmaceutical industry continues to evolve, integrating cutting-edge technologies like artificial intelligence and adapting to regulatory challenges will be crucial for CDMOs. By maintaining stringent safety protocols, embracing innovative solutions, and navigating regulatory landscapes effectively, CDMOs can not only safeguard operator and product integrity but also leverage emerging opportunities for growth and advancement in the high-potency pharmaceutical sector.
David O’Connell is the Director of Scientific Affairs at PCI Pharma Services, an integrated full service provider expertly delivering a seamless transition from development to commercialisation. After graduating with a BSc. in Applied Bioscience, David spent seven years as a Supervisory Scientist working for Aptuit in Edinburgh before moving to Penn Pharma as Head of Formulation Development in 2009. Here he played a vital part in the design of the potent Contained Manufacturing Facility (CMF), which won the ISPE Facility of the Year award for Facility Integration (2014). In 2013 David took on the role of Director, Pharmaceutical Development at the PCI site in Tredegar and in 2017 became PCIs Director of Scientific Affairs.
Combating Counterfeit Drugs: A Multifaceted Approach
Counterfeiting and diversion are global issues that impact virtually every industry, with no organisation immune to the potential damage. Across markets, counterfeit and diverted products pose a threat to manufacturers, retailers and consumers alike. In turn, this damages a brand’s reputation and sales, leading to significant revenue loss.
Although these supply chain threats plague many industries – from medical devices to consumer packaged goods – the issue is becoming increasingly pertinent in the pharmaceutical sector.
Pharmaceutical Industry a Major Target Drug diversion and counterfeiting generate an estimated €400 billion annually. It is a major concern given that counterfeit pharmaceutical goods pose a great risk to patient safety and cause injuries, illness and, in some extreme cases, death. What’s more, when patients consume fake medication that fails to treat their conditions effectively, they are deprived of the life-saving drugs they need. This not only compromises patient health but also leads to quality control issues, brand erosion and reduced customer loyalty for the affected brands.
This surge in counterfeit and diverted pharmaceuticals comes amid a period of ever-increasing demand for such products. For instance, the World Health Organisation (WHO) recently issued a global alert about fake versions of Ozempic – a prescription drug that has gained popularity as a quick weight loss solution. This surge in demand has resulted in shortages, providing criminals with the opportunity to introduce fake Ozempic into the market.
Further contributing to the growth in pharmaceutical counterfeiting and diversion is a surge in predominantly fraudulent online pharmacies – those selling products through illegitimate supply chains. For instance, a consumer in the UK buying online from what appears to be a French pharmacy may in reality be receiving expired, mislabelled or counterfeit goods from a completely different region. Research from the National
Boards of Pharmacies (NABP) shows that 95 per cent of websites selling prescription drugs are doing so illegally and endangering patient health. There are an estimated 40,000 illegal online pharmacies operating at any one time, according to the NABP. This allows for bad actors to increase their profits in the e-commerce space.
Legislation Introduced to Mitigate Challenges
In the US, the INFORM Consumers Act came into effect in July 2023. The legislation is designed to protect consumers from counterfeit products. Online marketplaces that sell either more than $5,000 a year in products or 200 products must disclose who they are, their address and how many products they have available.
In addition, regulations such as the EU Falsified Medicines Directive (EU FMD) and The Drug Supply Chain Security Act (DSCSA) have been enacted to protect patients and increase the security of the pharmaceutical manufacturing and supply chain process.
Nevertheless, even with the implementation of these regulations, counterfeiting and drug diversion continue to plague the pharmaceutical industry.
Addressing Counterfeit Pharmaceuticals with Technology
Catching counterfeiters and fraudulent distributors is an incredibly challenging task. To effectively combat these supply chain threats in the pharmaceutical sector, manufacturers must utilise a multilayered approach. There is no one-size-fits-all solution.
For example, under the EU FMD, all prescription drugs sold within the European Union are required to have an anti-tampering solution and a unique identifier that can be read by both humans and machines. The serialisation process involves assigning a unique code to each item, allowing for tracking and tracing throughout the entire supply chain, from the manufacturer to the end user. This makes it highly unlikely for diverted products to go unnoticed within the supply chain.
The implementation of track-andtrace technology enables pharmaceutical companies to monitor the entire journey of a product within the supply chain. Supply chain visibility requires tracking and documenting the movement of products and materials from the initial production stage to the end consumer. This includes recording relevant information such as the origin, manufacturing processes, transportation, storage conditions and distribution details at each change of ownership and movement. By implementing robust traceability systems, stakeholders can verify the integrity, safety and authenticity of pharmaceutical products, which is crucial for ensuring patient safety.
Moreover, pharmaceutical organisations are seeking innovative product authentication solutions, such as digital fingerprinting, to identify counterfeits and detect diversion. This technology works with existing package codes as well as packaging and is capable of detecting counterfeit products immediately. Brands and their trading partners can use smartphones to instantly verify product authenticity anywhere in the supply chain.
Pharmaceutical manufacturers increasingly seek complete, real-time visibility into supply chain activity to gain a better understanding of ongoing operations.To hyper-accelerate the detection of counterfeiting and diversion across their supply chains, pharmaceutical brands require real-time alerts of suspect activity, in addition to robust item details –where the product is, when it was moved and who it was moved by.
Another crucial step pharmaceutical brands can take to protect themselves is to raise awareness and educate staff about the counterfeiting and diversion issues prevalent in the industry – providing information about the characteristics of counterfeit and diverted pharmaceuticals and how to identify fake products. It is also important to provide regular training sessions to update staff on the latest developments in pharmaceutical counterfeiting and diversion.
Additionally, brands and healthcare professionals have a responsibility to educate patients. Patients should be taught how to differentiate between genuine and
Packaging
counterfeit drugs, encouraged to purchase medications from trusted sources, and advised to exercise caution when using online pharmacies. This education will empower patients to recognise signs of counterfeit pharmaceuticals and verify the authenticity of products before use.
Next-generation Solutions
As the search for efficient and effective solutions to combat supply chain threats, the anti-counterfeiting packaging market is expected to grow significantly in years to come. In fact, the market is projected to reach nearly €300 billion by 2029, at an annual growth rate of 12.7 per cent between 2024 and 2029. Cutting-edge product authentication solutions are already available which empower individuals to identify counterfeit and diverted products for themselves. And with the recent surge in the use of artificial intelligence (AI) and machine learning (ML), pharmaceutical manufacturers are looking for solutions which incorporate these advanced technologies to further assist in the fight against counterfeit and diverted pharmaceutical goods.
Elsewhere, digital product passports (DPPs) are also emerging as a revolutionary solution. They provide a comprehensive, traceable product history from source to end consumer – leveraging advanced technologies such as QR codes and blockchain. A DPP encapsulates all relevant product information, including origin, manufacturing process, ownership history and more. As they are accessible via QR codes, consumers can use their smartphones to instantly access product details.
Counterfeiting and diversion are undoubtedly wreaking havoc in the pharmaceutical industry, with no brand safeguarded from the damage being caused. To effectively overcome this major challenge, technology deployment alone is not enough – a programmatic approach is needed. This requires a combination of people, processes, technology and time to ensure consumer safety and loyalty, as well as to protect brands and revenues. It is also imperative for companies to collaborate with one another to help flag counterfeits and root out illicit distributors.
Stephan von Schilcher is a seasoned global strategic account manager with over two decades of expertise in anticounterfeiting technology consulting and operations. He brings a wealth of knowledge to the table, stemming from his diverse background which spans various industries including consumer packaged goods (CPG), pharmaceuticals, health & beauty, food & beverage, fashion and electronics. At Systech, Stephan plays a pivotal role in setting priorities and guiding strategies to deliver clear solutions that enhance consumer confidence and safety. He specialises in implementing overt and covert authentication technologies as well as ensuring robust protection against counterfeit products and diversion tactics.
Stephan von Schilcher
PYROSTAR™ Neo+
Recombinant Endotoxin Detection Reagent Plus...
PYROSTARTM Neo+ is based on a genetically engineered approach to produce reactive factors required for endotoxin detection at pharmaceutical and medical equipment manufacturing sites. With greater stability of the negative control and better endotoxin recovery in heparin and heparin-based compounds, Neo+ facilitates the same sensitivity of endotoxin detection as our traditional limulus amebocyte lysate (LAL) reagent, through a more sustainable and environmentally friendly method.
Studies Support Changing Perspectives Of Glass Vial Performance Characteristics
The global market for glass pharmaceutical containers is projected to exceed $10.5 billion within the next three years, solidifying glass as arguably the most widely used primary packaging material for injectable drug products.1 In pharma manufacturing, the performance of the primary packaging is critical to ensuring drug product stability, safety and efficacy. Pharmaceuticalgrade glass vials are widely used for parenteral therapies, however, not all glass vials offer the same performance, especially for small volume parenterals, or SVPs, which are defined as <= 100mL. A primary distinction is whether a vial is made by converting previously produced glass tubing, or whether it is moulded directly from the glass furnace.
SVP Vials
Tubular vials are produced in two separate consecutive manufacturing operations. First, a glass tube with the desired outer diameter (OD) and internal diameter (ID) is drawn from a glass furnace, hardened, and cut into lengths of approximately two metres. Then in a separate operation, usually in a different factory, these tubes are converted into vials by heating and forming the vial opening (known as the finish), the vial shoulder, and then the vial heel, before remelting the glass to close the bottom of the vial. For tubular vials, this remelting process can alter the glass composition in specific areas, particularly at the bottom and heel. Tubular vials are wellknown for their high optical quality and uniform wall thicknesses, most commonly in the 0.8–1.2 mm range.
In contrast, moulded vials are formed in a single melting process. Molten glass exits the furnace with the consistency of thick honey, but at temperatures exceeding 1,500⁰C. The molten glass flows through a round orifice where the stream is cut into individual “glass gobs”, each equal to the weight of the vial it will form. These gobs are guided by gravity into a two-stage moulding process where the vial takes shape. The glass transitions from a molten state at over 1,000°C to a fully formed vial at around 600°C in under
ten seconds. Since moulded vials are not remelted, they exhibit exceptionally high interior surface homogeneity. Moulded vial wall thicknesses are almost always greater than 1 mm, and typically ranging from 1.2–1.8 mm, and they are renowned for strength and lower susceptibility to breakage.
Technical Case Studies
Looking more closely at moulded vials, pharmaceutical manufacturers today are increasingly recognising the importance of qualifying multiple vial options for their drug products. Glass vial manufacturers, including SGD Pharma, frequently conduct technical studies to support customers in qualifying their alternative containers, or resolving issues with their current primary packaging and/or container-closure system (CCS).
This article will look at two such studies that examine the comparability of moulded and tubing vials running on the same equipment and depyrogenation cycle, and the impact of fill-volume on pH shift in an aqueous drug product.
The Role of Depyrogenation in Aseptic Filling
The first study examined three 20mL vial designs, two moulded and one tubular, running through the same depyrogenation cycle, on the same equipment. Depyrogenation is a common step in preparing glass SVP vials, whether they will be used for liquid fills, or lyophilised product (see Figure 1).
Standard depyrogenation of glass vials removes dangerous endotoxin and assures sterility by passing the vials through a high temperature, HEPA filtered tunnel oven, exit of which is directly into the sterile filling environment.
A commonly made assumption is that moulded glass vials require higher tunnel temperatures, or longer tunnel residence time (slower belt speed), to achieve the required level of sterility and endotoxin reduction (thermal destruction). This assumption is logical considering the heavier mass of moulded vials vs. traditional tubing vials. Indeed, the study found this not to be the case.
Two designs of moulded vials, processed in the same equipment and depyrogenation cycle as tubing vials, each achieved the minimum total endotoxin destruction factor equivalent to the minimum requirement of six-log reduction. In fact, each moulded vial far exceeded the minimum requirements on the same cycle, (see Figure 2).
The results, which may be surprising to many, were concluded to be consistent with a heat-transfer model wherein the primary mode for thermal degradation on the inside surface of the vials is due to convective heat transfer from hot air entering the open vial from the top, rather than conductive heat transfer through the solid glass base and side walls. This finding thereby neutralises negative impact from the heavier mass of the moulded vials. As long as the tunnel oven is capable of maintaining the temperature profile of the cycle, thorough depyrogenation is achieved without extending the cycle time.
Understanding Glass Container Suitability
The second study examined the influence of underfilling vials (compared to their nominal fill volumes) on pH shift in an aqueous, pH sensitive drug product. The study compared pH shift in three different 10mL, Type I glass container designs. Standard 10R tubing vs Standard 10mL moulded, vs 10mL
Figure 1: A chart showing the steps for preparing SVP glass vials both with liquid fill and lyophilized
Packaging
dealkalised moulded vials were evaluated under conditions of 100% nominal fill and 40% nominal fill. pH shift was evaluated for conditions simulating terminal sterilisation and simulating accelerated heat ageing. Along with pH shift, the study also looked at total extractables coming from the vials.
Although only a short-term study, the pH shift and total extractables results have implications on long-term results being influenced by fill volume (vs nominal volume), post-fill secondary processing, such as terminal sterilisation, long-term storage, and vial design. In all cases, the dealkalised vials had the lowest impact on the packaged solution (see Figure 3). Moulded vials with internal ammonium sulphate treatment (EVO+) have the lowest pH shift and the lowest total extractables, indicating the lowest level of glass-to-product interaction.
The implications of this study for similar risk to long-term stability of other drug products is clear. The study underscores that robust glass vials must not only meet the baseline requirements of EP 3.2.1/USP <660>;2 it is important to optimise the entire CCS (container-closure system), including fill volume.
Drug product packaging professionals are encouraged to read the company’s whitepaper,
as well as industry information such as that in the US Pharmacopeia informational chapter on glass durability and drug product compatibility (USP <1660>), which also addresses concerns over severe interactions potentially leading to, in rare cases, the condition known as delamination.
Conclusion
The two studies point to performance characteristics of moulded glass containers compared to traditional tubular glass containers: one study exploring operational characteristics during depyrogenation, and the other exploring potential impacts on drug product stability during storage. The latter study found that in addition to fill volume, choice of vial format (tubular or moulded) and internal surface treatments can dramatically influence drug product and container interactions, seen in the studies referenced above via pH shift. By understanding and controlling these factors, pharmaceutical manufacturers can make informed decisions to better ensure the safety and efficacy of their products, limiting pH shift and product stability which ultimately protects patient outcomes.
fortunebusinessinsights.com/pharmaceuticalglass-packaging-market-104710 (accessed July 29, 2024).
2. European Pharmacopeia (EP); 3.2.1 Glass Containers for Pharmaceutical Use. 11th ed.; Council of Europe: Strasbourg, 2022 United States Pharmacopeia (USP); USP <660> Plastic Packaging Systems and their Materials of Construction. 43rd ed.; United States Pharmacopeial Convention: Rockville, MD, 2024.
Jingwei Zhang is currently R&D director of SGD Pharma, based in France. Prior to this position, he worked for Saint-Gobain for 24 years, as Strategic Projects Manager and R&D Portfolio Manager. He has a large experience in glass packaging, ranging from products and process development to quality inspection. He is inventor of more than 70 international patents. Jingwei Zhang graduated from Harbin Institute of Technology in China and received his PhD in France.
Jingwei Zhang
Figure 2: A graph showing FD value plotted against travel through the tunnel
Figure 3: Bar charts showing pH shift for vials filled with 10m of NACL 0.9% in different Terminal Sterilisation cycles.
Application Note
Unlocking the Secrets: Long-Term Storage Effects on Pharmaceutical Proteins in Glass
and Polymer Prefilled Syringes
Nowadays, the market is increasingly encouraging the use of high-class prefillable COC (Cyclic Olefin Copolymer) or COP (Cyclo-Olefin-Polymer) syringes, for biological applications such as mRNA, protein peptides, and sensitive molecules. This is most likely due to the nature of the material, which provides benefits in pH stability, and completely mitigates the risk of ion exchange for sodium, potassium, and other various heavy metals, that may occur when using other forms of primary packaging. In this study, we evaluated two different syringe materials and the impact that material selection had on protein aggregation over a period at different temperatures. The model formulations utilised in this study were Immunoglobulin G and Human Serum Albumin in a histidine buffer system. The respective protein formulations were filled in glass and polymer syringes as well as in an external reference (clean glass bottle). All were stored at 25°C (up to 26 weeks) and for a complete storage time at 5°C (up to 2 years). For the duration of the study, storage at room temperature as well as at 5°C, demonstrated stability of the protein monomer concentrations in both glass and polymer containers. By qualitative SEC chromatogram evaluation, minor signals for IgG and HSA dimers were observed in all sample solutions and their respective control solutions. Herewith, we conclude that both glass and polymer syringes offer optimal conditions for storing Immunoglobulin G and Human Serum Albumin.
Introduction
In the last few decades, the pharmaceutical industry has experienced rapid growth in parenteral products, leading to easier availability of life-saving drugs in the market. The advances in sterile product formulations like protein-peptides, liposomes, suspensions, emulsions, and micro- and nanoparticles have modernised the design of drug delivery with better safety and efficacy.1,2 New and advanced ideas have been presented for using prefilled
syringes for improved safety, ease in usage, minimising errors in clinical use, and the ability for self-injections. Prefilled syringes have gained strong acceptance for the drug delivery of injectable drugs, especially in the treatment of chronic conditions that require repeated administration of the medications.2,3
Almost half of the new drugs recently approved by the United States Food and Drug Administration (USFDA) are therapeutic proteins, and the drug-pipeline landscape is strongly shifting towards the same class of drugs.4 USFDA has recently approved Moderna’s bivalent mRNA vaccine to be used as a booster dose against COVID-19.5 In addition to mRNA, several cell & gene therapeutic agents such as ZYNTEGLO (betibeglogene autotemcel) by Bluebird bio Inc, CARVYKTI (ciltacabtagene autoleucel) by Janssen Biotech Inc, ZOLGENSMA (onasemnogene abeparvovec-xioi) by Novartis Gene Therapies, Inc. and others have also received the FDA approval for different products, these all are lifesaving treatments made available for patients. The majority of gene therapy pharmaceuticals are administered directly into the affected area; henceforth, the utilisation of prefilled syringes (PFS) has been highly recognised in recent years.6 Intravenous (IV), subcutaneous (SC), and intramuscular (IM) are major routes for the administration of such biopharmaceuticals. This has contributed to
the increased use of PFS for the administration of such biopharmaceutical formulations.7,8
The treatment of numerous pathologies requires the intravenous (IV) administration of medications, using different medical devices, such as catheters, infusion lines, and syringes. Catheters are tubular devices that allow the administration of drugs into the vascular system through the skin. Therefore, they need to be biocompatible and are usually made up of silicone or polyurethane (PUR).9 They can be connected to various delivery devices, such as infusers and extension lines, which are generally made of polyvinyl chloride (PVC), polyethylene (PE), coextruded PE/PVC, or PUR/PVC.9 During the drug administration sequence by IV route, the medications flow from devices such as bags, bottles or syringes. Apart from the administration set-up, syringes can also be used to access medication from vials (when capped with a needle) or as primary packaging for some medications formulated in ready-to-use syringes. The syringes are made up of a barrel and a rod, which are mostly made of PE, polypropylene (PP) or polycarbonate (PC).10 Three-part syringes, which are used for IV infusions, also possess a plunger (added to the rod), which is used to increase water tightness and reduce risk of leakage. Plungers are made of synthetic rubber which generally contains an elastomer (especially bromobutyl and
Figure 1: A typical polymer and glass syringe from SCHOTT Pharma Schweiz AG
chlorobutyl), a filler, a vulcanising agent, a plasticiser, a stabiliser, a pigment, and a lubricant.10,11 All the previously cited medical devices are known to guarantee container-content interactions (CCI), and parenteral administration is considered one of the most endangered routes according to the USP <1664> and the Food and Drug Administration.14,15
Container-content interaction refers to an interaction between one or more molecules in the liquid phase of the preparation and the solid phase of the container (sorption) or the release of a compound out of the material into the content (leachables).14 Sorption regroups three phenomena: adsorption (surface phenomenon mostly reversible) which can be followed by absorption (absorption of the molecule inside the polymeric matrix) and permeation (absorption then desorption of the molecule on the opposite side of the material matrix). This phenomenon can concern both the active pharmaceutical ingredient (API) and the excipients, interacting with medical devices (generally of polymeric nature) used to administer the medications.15, 16,17 One of its first mentions was in 1974, when Moorhatch et al. demonstrated that medical devices could interact with drugs and lead to a loss of active substance or a release of potentially toxic substances in the medication.18 Unlike the flexible materials used in tubing (plasticised PVC, PUR, silicone) that are known to be subject to CCI,19, 20, 21 there is less data available about potential CCI with the polymers used in syringes. Low-density PP is a low-risk material for sorption phenomena, but some studies have shown decreased drug concentrations (diazepam and nitroglycerin) when stored in PP syringes.19,22 A few studies have put forward the hypothetic implication of the rubber plunger lubricated by silicone oil.19,23,9
In 2015, the Food and Drug Administration (FDA) alerted healthcare professionals on interactions occurring between the rubber stopper and some drugs stored in prefilled syringes,12 forcing the supplier to change the rubber material. Moreover, the free silicone oil, used as a plunger lubricant, can induce proteins aggregation by interacting with the free amino groups in active pharmaceutical ingredients (APIs). Siliconisation refers to the treatment of surfaces with polydimethylsiloxane (as silicone fluid). The silicone oil, a polydimethylsiloxane (PDMS), is applied to both stopper and barrel to achieve smooth gliding of the stopper along the barrel. PDMS is the most used lubricant for medical devices and primary
Application Note
packaging systems because of its stability, hydrophobicity, lubricity, and low toxicity. Proteins adsorb on the surface of silicone droplets, causing a denaturation by unfolding.13,17 In comparison to that, glass has been one of the most established materials to be used in packaging of any parenteral formulation, and this is due to the evidence that it evolved much earlier and has been studied more in comparison to polymer syringes.
For increasing the protein stability, many excipients such as salt, sugar and polysorbates help prevent protein adsorption and aggregation by preventing it interacting with air-liquid or solid-solid interfaces.5
Due to unique physical and chemical properties of proteins i.e. small- and largemolecules, they are prone to a number of changes during the preparation, formulation and storage. Majority of these changes includes oxidation, aggregation, adsorption, glycosylation.4,25 In the formulation and filling process, dissolved oxygen occurs naturally in the drug product and to overcome this challenge, techniques such as nitrogen blanketing and others are often used to reduce the effect of oxygen on pharmaceutical product.11
Nakamura et. al. (2015) reported that polymer PFSs are the most preferred packaging material when used in a deoxygenated packaging system against the glass PFSs.11 The control of dissolved oxygen to prevent oxidation can be performed by several methods, including adding antioxidising agents to the drug formulation and/or controlling the residual oxygen within the primary container during storage. Glass PFSs were found to be less effective when used in a deoxygenated packaging system when compared to polymer PFSs.9,11 Beside the impact of oxygen, the amount of free silicone going into the drug solution plays a crucial role for sorption of the API. To formulate any stable biopharmaceutical formulation to be stored in PFS, it is very essential to ensure the quality of pharmaceutical products. In recent decades, advanced analytical characterisation and quality control strategies have been developed, but these strategies still need to be upgraded. For example, if the protein is being adsorbed onto the container or micron-sized particles are formed in the PFS, the researchers have to improve the quality of whole drug products along with the active pharmaceutical ingredients (API) and the interaction between API and packaging
materials, i.e., prefilled syringes5 needs to be addressed.
In this article, we have reported the impact of packaging material of prefilled syringes viz. (glass and polymer) for storing model proteins such as Immunoglobulin G and Human Serum Albumin dissolved in histidine buffer. We have studied the model proteins' long-term stability by analysing the samples by size exclusion chromatography (SEC) and qualitative particle analyses according to EP 2.9.20 and USP <790>.
Material and Methods
Materials:
The Human Immunoglobulin, Human Serum Albumin, Tinuvin-327, Palmitic acid D31 were procured from Sigma Aldrich. The Histidine was procured from Merck. For the preparation of Histidine buffer we purchased from VWR Chemicals: Mannitol, Merck: Sucrose and Alfa Aesar: Polysorbate 20. The Advance bio SEC 300 Å protein standard was produced from Agilent. The standard contains thyroglobulin (bovine; 670 kDa), Ɣ-globulin (bovine; 150 kDa), ovalbumin (chicken; 45 kDa), myoglobin (equine heart; 17 kDa), and angiotensin II (human; 1 kDa).
Moreover, we also used polymer prefilled syringes manufactured from the raw material manufactured by SCHOTT Pharma AG. The COC (Cyclic olefin copolymer) was manufactured by TOPAS Germany and glass prefilled syringes respectively manufactured at SCHOTT Pharma AG.
Container Types
Component of PFS
Polymeric (C1)
Glass (C2)
Syringe barrel SCHOTT TOPPAC® 1ml long FIOLAX® clear glass,
Tip Cap
Dätwyler FM 257/2 grey, bromobutyl rubber West 7025/65 grey, bromobutyl rubber
Plunger stopper 1 ml long plunger, West NovaPure RU SP 4023/50G
Sterilisation method Gamma-sterilised
Experimental Methods
Preparation of buffer solutions: Preparation of 100 mmol/L phosphate buffer pH 7.4 (Eluent for SEC). 34.5 g sodium dihydrogen phosphate monohydrate (NaH2PO4 x H2O) were dissolved in approximately 2400 mL ultrapure water and pH-value of the solution was adjusted to pH 7.4 with sodium hydroxide solution (50 %). Subsequently the solution was filled up to a total volume of 2500 mL with ultrapure water.
Preparation of 20 mmol/L histidine-HCl buffer pH 5.5 (Protein formulation buffer) 25.0 g sucrose, 0.250 g Polysorbate 20 (Tween 20), 100.0 g mannitol and 7.76 g L-histidine were dissolved in 2300 mL ultrapure water and the pH-value of the solution was adjusted to pH 5.5 with hydrochloric acid (37 %). Subsequently the solution was filled up to a total volume of 2500 mL with ultrapure water.
Preparation of Samples
Required amounts of IgG, respectively HSA were dissolved in the protein formulation
1 ml long plunger, West NovaPure RU SP 4023/50G
ETO-sterilised
buffer described above: P1_A–1.0 mg/mL IgG, P1_B–0.1 mg/mL IgG, P2–50 mg/mL IgG, and P3–1.0 mg/mL HSA. Subsequently, the formulations were filled in the respective container closure systems (C1 and C2; see sample description in Table 1) and were provided for storage at 5°C and 25°C. As reference sample the respective protein solutions were stored in a glass bottle to be able to evaluate effects, which are not related to the syringes. A detailed overview of the sampling schedule can be found in the supplemental material.
Methods for Data Evaluation
Quantification of the IgG respectively HSA monomer concentration was performed using an external calibration covering the
concentrations 0.25–2.0 mg/mL. Signals were evaluated based on the UV absorbance at 220 nm. Representative calibrations are displayed in the supplemental material. The monomer quantification method has been validated with the following relative enhanced measurement uncertainties.
As system suitability control for each storage time point a mass calibration and the external calibration for evaluation of concentrations were performed. Additionally, a quality control sample of known protein concentration was injected regularly during measurement sequences.
Experimental Section Size Exclusion Chromatography with Ultraviolet Detection
A mass calibration was performed to attribute UV signals to the corresponding protein monomer, dimer, and oligomer. Therefore, a mass calibration with ‘Advance bio SEC 300 Å protein standard’ was applied.
For analysis of protein fragmentation and agglomeration a non-validated evaluation was performed using the UV response at different wavelengths depending on total signal intensities (210, 220, or 280 nm) of the LC method. For more information on the instrument method see supplementary information (d). The retention time windows with pronounced signals were evaluated and the area subsequently normalised by the area of the protein monomer signals at retention time (RT) ~ 6.6 min (IgG) respectively RT ~ 7.7 min (HSA). For IgG, the investigated retention times include ~4.7 min (oligomer) and ~ 5.6 min (dimer). For HSA the investigated retention times include ~ 6.2 min (oligomer) and ~ 6.8 min (dimer).
Results
Room Temperature Storage – Size Exclusion
Chromatography
with Ultraviolet Detection
Results of the quantitative determination of IgG and HSA concentrations are summarised in Table 2 to Table 5.
Taking into account the relative measurement uncertainties, no significant decrease in protein concentrations was observed by SEC-UV neither in glass nor polymer syringes during up to 26 weeks storage at room temperature.
Figure 2: Protein formulations for evaluation28
Table 1: Specification of primary packaging materials
Qualitative Evaluation of Protein Dimer/ Oligomer Abundance with SEC-UV at 25°C
Results of qualitative evaluations of UVchromatograms for protein aggregation for each pull point at 25°C are shown in Figure 4. The concentrations of protein monomer were found to be constant for all studied protein formulations over the storage time at 25°C.
The concentrations of protein oligomers (relative to protein monomer) were found to be constant for all studied protein formulations over the storage time at 25°C.
The qualitative indicates here that no calibration was performed and therewith no concentrations of the associated substances were estimated. The evaluations here are
only based on peak areas relative to the monomer peak area. We evaluated peak purity based on the absorption spectra measured by the PDA at the corresponding peaks and found no difference in between samples.
Figure 3 (a-d): Recoveries of protein monomer evaluated by UV signal at 220 nm ((a) P1_A: IgG–1 mg/mL, (b) P1_B: IgG–0.1 mg/mL, (c)P2: IgG–50 mg/mL, (d) P3: HSA–1 mg/mL) for syringes stored at 25°C.
Figure 4 (a, b): Relative abundance of protein oligomer to protein monomer evaluated by UV signal at 220 nm (P2: IgG–50 mg/mL, P3: HSA–1 mg/mL) for syringes stored at 25°C.
Table 2: Overview of quantification results for formulation P1_A (1 mg/mL IgG) in mg/mL for syringes stored at 25°C.
Table 3: Overview of quantification results for formulation P1_B (0.1 mg/mL IgG) in mg/mL for syringes stored at 25°C.
Table 4: Overview of quantification results for formulation P2 (50 mg/mL IgG) in mg/mL for syringes stored at 25°C.
Table 5: Overview of quantification results for formulation P3 (1 mg/mL HSA) in mg/mL for syringes stored at 25°C.
Application Note
Low temperature Storage (5°C) Size Exclusion
Chromatography
with Ultraviolet Detection
Results of the quantitative determination of IgG and HSA concentrations are summarised in Table 66 to Table 88. Details regarding sample preparation, measurements, and data evaluation of SEC-UV analysis are described in the enclosure of this report. Taking into account the relative measurement uncertainties, no significant decrease in protein concentrations was observed by SECUV neither in glass- nor polymer syringes during up to 2 years storage at 5°C.
Qualitative Evaluation of Protein Dimer/ Oligomer Abundance with SEC-UV
Results of qualitative evaluations of UVchromatograms for protein aggregation for each pull point are shown in figure 5 (dimer) and Figure 6 (oligomer). The concentrations of protein dimers and oligomers (relative to protein monomer) did not change significantly for all studied protein formulations over the storage time at 5°C but not significantly different in contrast to the reference sample.
Discussion
Regarding the analysis method, Fekete et. al4 have illustratively reported how size exclusion technology (SEC) can be considered as a reference and powerful technique for
qualitative and quantitative evaluation of protein aggregates. The pharmaceutical proteins have an inherent property to aggregate which can be accelerated by various external forces such as long-term storage, temperature variations, freezingthawing, formulation changes, exposure to light and interfaces, agitation e.g.27 Majority of the protein aggregates often vary in many properties such as size, type of intermolecular bonds, reversibility, morphology and hydrophobicity.
The regulatory authorities such as United States, European and Japanese Pharmacopeias (USP, PhEur and JP, respectively) are harmonised regarding the requirements for visible and sub-visible particles in any parenteral dosage form. For subvisible aggregates, there are two main SEC strategies:1 observe the aggregation profile and quantify the percentage of highmolecular-weight species eluted from the column or2 indirectly estimate the fraction of large aggregates (typically >100 nm) as the loss of total peak area. However, there is no clear definition for aggregate acceptance limits in biopharmaceutical products.4
For our analyses, we had implemented the below strategy:4 based on a protein mass calibration we identified UV signals as protein monomer, dimer, respectively oligomers. Based on method verification results the uncertainty in the estimated masses is < 20 %. Furthermore, qualitative evaluation of chroma-
Table 6: Overview of quantification results for formulation P1_A (1 mg/mL IgG) in mg/mL for syringes stored at 5°C.
Table 7: Overview of quantification results for formulation P1_B (0.1 mg/mL IgG) in mg/mL for syringes stored at 5°C.
Table 8: Overview of quantification results for formulation P3 (1 mg/mL HSA) in mg/mL for syringes stored at 5°C.
Figure 5: Relative abundance of protein dimer to protein monomer evaluated by UV signal at 220 nm ((a) P1_A: IgG–1 mg/mL, (b) P1_B: IgG–0.1 mg/mL, (c) P3: HSA–1 mg/mL) for syringes stored at 5°C.
Figure 6: Relative abundance of protein oligomer to protein monomer evaluated by UV signal at 220 nm (P3: HSA–1 mg/mL) for syringes stored at 5°C.
tograms showed no interfering UV peaks with the signals discussed in this work. Therefore, selectivity of the evaluation for the respective protein aggregates of analysed target substances is considered to be fulfilled.
To overcome the challenges associated with varying interactions with stationary phase such as electrostatic and hydrophobic interactions, we have applied a number of quality assurance criteria, for the acceptance of results: For each measurement series, a fresh mass calibration was performed to ensure system suitability including good column performance. Furthermore, to enable accurate protein quantification, in each measurement series a freshly prepared protein concentration calibration was performed and throughout the series quality, assurance samples with a known protein concentration were injected. All protein concentrations in quality assurance samples match the target concentration taking the indicated measurement uncertainty into account.
For estimation of measurement uncertainties for SEC-UV analyses a method verification based on recommendations described in ICH Q2 (R1) guideline (Validation of analytical procedures) was performed. More concretely, the relative measurement uncertainty for different proteins and application ranges was calculated based on precision and accuracy of protein concentration evaluated by external
Application Note
calibration. Limit of quantification was calculated by the signal to noise ratio of chromatographic peaks.
The primary aim of syringe barrel siliconisation is to obtain the most even anti-friction coating possible along the entire length of the syringe to minimise break loose and gliding forces when the plunger stopper is deployed. Both glass and polymeric syringe are coated from the inside by silicone oil. Moreover, the inside surface of borosilicate glass is hydrophilic, whereas the polymeric syringes have plastic surface which is rather hydrophobic. The silicone of the glass syringe is applied by spraying silicone oil onto the inner surface of the barrel. In case of the polymer syringe, a reactive silicone mixture (the “cocktail”) is applied to the inner wall of the cylinder using a wiping technique. Thermally induced cross-linking of the cocktail on the inner barrel wall ensures immobilisation of the lubricating layer. Both syringes including their inner surface structure have proven to guarantee a reliable drug stability over time for these two protein formulations.
After completion of the analysis, the following key finding was observed using the respective analyses methods:
• Protein concentrations – quantified by SEC UV response – were stable in both polymer and glass syringes during the complete storage time at 25°C (up to 26
weeks) as well as the complete storage time at 5°C (up to 2 years).
Conclusions
The key finding of this study reports the effects of both polymer and glass syringes on protein aggregation in the model protein formulations i.e., the Human Immunoglobulin and Human Serum Albumin.
Protein concentrations – quantified by SEC UV response – were stable in both polymer and glass syringes during the complete storage time at 25°C (up to 26 weeks) as well as the complete storage time at 5°C (up to 2 years). Protein dimers were found in all protein formulations before storage, their concentrations were constant over time in reference samples as well as both syringe types. No significant difference was found between polymer and glass syringes.
It is demonstrated that the COC polymer syringe can store Human Immunoglobulin and Human Serum Albumin over long storage time of 2 years at a typical storage temperature of 5°C. This study provides a deeper understanding for COC polymer syringe as primary packaging product as no increased amount of agglomeration or sorption was found over the shelf lifetime of two years. It is known that COC polymer material does not release any type of metals into the solution and that no change of pH takes place over time of storage which has stabilising effect on the stability of highly
Application Note
sensitive proteins.29 The cross-linked silicone layer used in COC polymer syringe, in addition to a stable pH and no induction of metal ions into the solution, support the long-term stability of the proteins analysed in this study as no increase of dimer or oligomer concentration was found.
It can be concluded that the COC polymer syringe is a stable primary packaging container in addition to the glass PFS and to be used for storage of these two specific model proteins over time for a comparable shelf life.
More alternative model proteins can be explored to be stored in glass and polymer syringes to increase the understanding of container-content interaction and to support the correct drug-device choice.
Acknowledgments
The authors want to acknowledge SCHOTT Pharma AG & Co. Switzerland.
REFERENCES
1. O'Brien, M.N., Jiang, W., Wang, Y. and Loffredo, D.M., 2021. Challenges and opportunities in the development of complex generic long-acting injectable drug products. Journal of Controlled Release, 336, pp.144-158.
2. Panchal, K., Katke, S., Dash, S.K., Gaur, A., Shinde, A., Saha, N., Mehra, N.K. and Chaurasiya, A., 2022. An expanding horizon of complex injectable products: development and regulatory considerations. Drug Delivery and Translational Research, pp.1-40.
3. Siew, A., 2017. Delivering biologics in prefilled syringes. BioPharm Int, 30(11), pp.14-7.
4. Fekete, S., Beck, A., Veuthey, J.L. and Guillarme, D., 2014. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. Journal of pharmaceutical and biomedical analysis, 101, pp.161-173.
5. Coronavirus (COVID-19) Update: FDA Authorizes Moderna, Pfizer-BioNTech Bivalent COVID-19 Vaccines for Use as a Booster Dose | FDA (Accessed on 24th September 2022)
6. Approved Cellular and Gene Therapy Products | FDA (Accessed on 24th September 2022)
7. Yoneda, S., Torisu, T. and Uchiyama, S., 2021. Development of syringes and vials for delivery of biologics: current challenges and innovative solutions. Expert Opinion on Drug Delivery, 18(4), pp.459-470.
8. Chow, E.J., Kitaguchi, B. and Trier, M., 2012. Effects of subzero temperature exposure and supercooling on glass vial breakage: risk management and other applications in cold chain distribution. PDA Journal of Pharmaceutical Science and Technology, 66(1), pp.55-62.
9. Tokhadzé N, Chennell P, Pereira B, MailhotJensen B, Sautou V. Critical Drug Loss Induced by Silicone and Polyurethane Implantable Catheters in a Simulated Infusion Setup with
Three Model Drugs. Pharmaceutics. 2021 Oct 16;13(10):1709.
10. Nakamura, K., Abe, Y., Kiminami, H., Yamashita, A., Iwasaki, K., Suzuki, S., Yoshino, K., Dierick, W. and Constable, K., 2015. A strategy for the prevention of protein oxidation by drug product in polymer-based syringes. PDA Journal of Pharmaceutical Science and Technology, 69(1), pp.88-95.
11. Sacha GA, Saffell-Clemmer W, Abram K, Akers MJ. Practical fundamentals of glass, rubber, and plastic sterile packaging systems. Pharm Dev Technol. 2010 Feb;15(1):6–34.
12. De Zélicourt Y. Caoutchoucs : méthodes d’obtention et propriétés. Plast Compos (Internet). 2015 Jan (cited 2022 Aug 29); Available from: https://www.techniques-ingenieur.fr/ doi/10.51257/a/v1/am7705
13. The United States Pharmacopeial Convention. (1664) Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/ Delivery Systems. 2022.
14. FDA. Guidance for Industry Container Closure Systems for Packaging Human Drugs and Biologics. 1999.
15. Gerhardt, A., Mcgraw, N.R., Schwartz, D.K., Bee, J.S., Carpenter, J.F. and Randolph, T.W., 2014. Protein aggregation and particle formation in prefilled glass syringes. Journal of pharmaceutical sciences, 103(6), pp.1601-1612.
16. Li, J., Cheng, Y., Chen, X. and Zheng, S., 2019. Impact of electroviscous effect on viscosity in developing highly concentrated protein formulations: lessons from non-protein charged colloids. International Journal of Pharmaceutics: X, 1, p.100002.
17. Christine Bizet, Rigoulot A. Extractibles et relargables: Evolution ou retour du Statut Quo? 2013;(36).
18. Masse M, Maton M, Genay S, Blanchemain
N, Barthélémy C, Décaudin B, et al. In vitro assessment of the influence of intravenous extension set materials on insulin aspart drug delivery. PLoS ONE. 2018 Aug 16;13(8):e0201623.
19. Moorhatch P, Chiou WL. Interactions between drugs and plastic intravenous fluid bags. II. Leaching of chemicals from bags containing various solvent media. Am J Hosp Pharm. 1974 Feb;31(2):149–52.
20. Tokhadzé N, Chennell P, Pereira B, MailhotJensen B, Sautou V. Critical Drug Loss Induced by Silicone and Polyurethane Implantable Catheters in a Simulated Infusion Setup with Three Model Drugs. Pharmaceutics. 2021 Oct 16;13(10):1709.
21. Tokhadzé N, Sahnoune M, Devémy J, Dequidt A, Goujon F, Chennell P, et al. Insulin Adsorption onto PE and PVC Tubings. ACS Appl Bio Mater. 2022 May 13
22. Hacker C, Verbeek M, Schneider H, Steimer W. Falsely elevated cyclosporin and tacrolimus concentrations over prolonged periods of time due to reversible adsorption to central venous catheters. Clin Chim Acta Int J Clin Chem. 2014 Jun 10;433:62–8.
23. McCluskey SV, Vu N, Rueter J. Stability of nitroglycerin 110 mcg/mL stored in polypropylene syringes. Int J Pharm Compd. 2013 Dec;17(6):515–9.
24. 24) Mohr A, Erdnüß F, Krämer I. Physicochemical stability of human insulin 1 I.U./mL infusion solution in 50 mL polypropylene syringes. Pharm Technol Hosp Pharm. 2021 Jan 1;6(1).
25. Krayukhina, E., Tsumoto, K., Uchiyama, S. and Fukui, K., 2015. Effects of syringe material and silicone oil lubrication on the stability of pharmaceutical proteins. Journal of pharmaceutical sciences, 104(2), pp.527-535.
26. Mahler, H.C., Friess, W., Grauschopf, U. and Kiese, S., 2009. Protein aggregation: pathways, induction factors and analysis. Journal of pharmaceutical sciences, 98(9), pp.2909-2934.
27. F. Feutry, S. Genay, C. Velghe, C. Barthélémy, B. Décaudin, P. Odou, 2016. Stability of midazolam and noradrenaline stored in cyclic olefin copolymer AT-Closed Vials® and polypropylene syringes during 365 days, p. 10, 13, 14
Worked on article writing, drafting and compilation of data.
Jaywant Pawar
Worked on article writing, drafting and compilation of data.
Dr. Alexander Filippi
Carried out experimental work and compilation of data.
Dr. Matthias Bicker
Carried out experimental work and compilation of data.
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
Health Outcomes
The Global Crisis of Counterfeit Pharmaceuticals: A Call to Action
MM Packaging gathers experts in their field to lay the foundations for the conversation about counterfeit pharmaceuticals; looking into the realities, impact, and potential solutions to this pandemic of problems. This article provides a detailed overview of the approaches taken to understand, tackle, and prevent the threat of counterfeit drugs – by looking deeper at the two way street between consumers and the corporations.
The issue of counterfeit pharmaceuticals is a critical global problem with significant implications for both public health and the economy. According to the World Health Organisation (WHO), counterfeit products represent about 10% of total market sales in some regions. In the European Union, this figure is around 2%, yet it still costs the economy approximately 10 billion euros annually. This economic impact translates into a loss of around 38,000 jobs within the industry each year.
One of the most alarming aspects of this issue is the impact on health and safety. The WHO estimates that counterfeit drugs contribute to around a million deaths globally per year. The rise of online and e-commerce platforms has exacerbated the situation, allowing consumers to bypass traditional healthcare systems and access drugs from potentially untrustworthy sources. This shift has opened new trade routes for counterfeiters worldwide.
We bring together Yann Ischi, Vice President and General Manager, Product Authentication at Crane Currency, Claire Clark, the director of healthcare engagement at GS1, along with Philip Allen, technical account manager, and Laura Lopez, marketing manager from MM Packaging to discuss and dissect these pressing matters.
The Role of the Packaging Industry in Combating Counterfeits
The packaging industry is at the forefront
of the battle against counterfeit pharmaceuticals, witnessing substantial growth in anti-counterfeiting solutions driven by the urgent need to ensure product authenticity. This sector is expanding at an impressive rate of approximately 13.5% per year and is projected to be worth around 124 billion euros by 2023. The rapid growth underscores the increasing recognition of packaging as a critical defence mechanism against counterfeit drugs.
“How can we actually raise this level of trust? This is the work that we need to do together with the brands to help them to raise that level of trust in a very cost-effective manner,”
Yann Ischi
Counterfeit packaging typically refers to products that are sub-standard, falsely labelled, and do not meet the established standards of the brand or pharmaceutical company. The internal product is often falsified or forged, posing a severe threat to public health. Counterfeit drugs can lead to ineffective treatment, adverse health effects, and even death, making it crucial to address this issue through improved packaging integrity.
Advances
in Anti-Counterfeiting Technologies
One of the significant advancements in the packaging industry is the development and integration of sophisticated anti-counterfeiting technologies. These techno-logies include:
Micro-Optics and Optical Variable Devices (OVDs): These are difficult to replicate and provide a visible layer of security that consumers can easily recognise. Microoptics, a branch of optics, are focused on very small optical components. The miniature scale of these components impacts various aspects such as fabrication methods, the choice of optical materials, the physical effects involved, performance constraints, and the practical aspects of handling them.
Radio Frequency Identification (RFID) and NFC Tags: RFID tags can store and transmit information about the product, allowing for real-time tracking and verification
throughout the supply chain. This technology enables manufacturers and distributors to monitor the movement of products, ensuring that they have not been tampered with or diverted.
Standardised DataMatrix codes: which are typically generated by GS1, a non-profit organisation. This is comparable or similar to a government-issued ID, as it has specific characteristics that ensure reliability – GS1 DataMatrix codes ensure the correct and standardised identification of data through a reliable coding system.
“From a GS1 perspective, that's why we try to harmonise around the world: we don`t lobby regulators, we engage with them when they ask, we are aware that for pharmaceutical organisations is costly, but the more they harmonise the more then they have the ability to have multi-market packs; if the data matrix is on the pack and it's on the pack in all 66 countries that the pharma manufacturer supplies to, it gets a lot easier.”
Claire Clark
The Importance of Serialisation
Serialisation is another critical component in the fight against counterfeit pharmaceuticals. By assigning a unique serial number to each package, serialisation allows for individual tracking and verification. This system enables manufacturers, distributors, and consumers to verify the authenticity of the product at various points along the supply chain. Serialisation also supports the implementation of trackand-trace systems, which are essential for identifying and recalling counterfeit products quickly.
Philip Allen goes on to say that in contrast to other industries, Pharmaceuticals take longer to get to market and once they do, packaging changes are rarer. Packaging teams will work towards a launch date and part of the brief is to make sure the appropriate protection features are in place.
"Typically, what we see is that serialisation is part of the mix - for traceability, a second aspect is tamper evidence to secure the outer packaging (what we call the secondary packaging) so the consumer can spot if it's tampered with and a third aspect is the
authentication feature, to allow either brand teams or consumers to judge if it's genuine or not.
Philip Allen
This is a multi-layered approach, so you can layer lots of things within the packaging. And to do so cost-effectively, this is part of the need.
Integration with Track-and-Trace Systems
Serialisation is a foundational element of track-and-trace systems, which are essential for monitoring the movement of pharmaceutical products through the supply chain. Track-and-trace systems use the unique serial numbers to record every transaction and movement, creating an auditable trail that can be reviewed to identify and address any discrepancies.
• Detection of Counterfeit Products: By maintaining detailed records of each product’s journey, track-and-trace systems can quickly identify counterfeit products. If a serial number is found in an unauthorised location or duplicated, the system can flag it for investigation.
• Efficient Recalls: In the event of a product recall, serialisation allows for precise identification of affected products. Instead of recalling entire batches, manufacturers can recall only the specific products with the compromised serial numbers, reducing costs and minimising disruption.
• Regulatory Compliance: Serialisation helps pharmaceutical companies comply with regulatory requirements such as the Drug Supply Chain Security Act (DSCSA) in the United States and the Falsified Medicines Directive (FMD) in the European Union. These regulations mandate the use of unique identifiers and track-andtrace systems to enhance drug safety.
Collaborative Efforts and Regulatory Compliance
The effectiveness of anti-counterfeiting measures in packaging relies heavily on collaboration among stakeholders, including pharmaceutical companies, packaging manufacturers, technology providers, and regulatory authorities. Regulatory frameworks such as the Drug Supply Chain Security Act (DSCSA) in the United States and the Falsified Medicines Directive (FMD) in the European Union mandate stringent measures for serialisation and traceability. Compliance with these regulations ensures that anti-
Health Outcomes
counterfeiting technologies are standardised and widely adopted across the industry.
The packaging industry plays a pivotal role in combating counterfeit pharmaceuticals by developing and implementing advanced anti-counterfeiting technologies. Ensuring packaging integrity is crucial for protecting public health and maintaining consumer trust. Through continued innovation, collaboration, and compliance with regulatory standards, the packaging industry can significantly reduce the prevalence of counterfeit drugs and safeguard the integrity of the pharmaceutical supply chain.
The Broader Implications of Counterfeiting
Counterfeiting is a global pandemic that erodes brand value and consumer trust, presenting significant challenges to industries worldwide. When consumers purchase a product that fails to meet the brand's promises, trust is swiftly shattered, undermining the brand's reputation and market position. Restoring this trust is a complex task that requires a concerted effort from all stakeholders in the supply chain.
Erosion of Brand Value
Brand value is built over time through consistent quality, reliable performance, and positive consumer experiences. Counterfeit products jeopardise this value by introducing substandard and potentially harmful imitations into the market. When consumers encounter counterfeit products, their negative experiences are often attributed to the legitimate brand, leading to a decline in brand equity. This erosion of brand value can have long-term financial implications, affecting sales, market share, and overall profitability.
Loss of Consumer Trust
Consumer trust is the cornerstone of successful brands, particularly in industries like pharmaceuticals, where product efficacy and safety are paramount. Counterfeit drugs not only fail to deliver the promised therapeutic benefits but can also cause severe adverse effects, including death. Such incidents lead to widespread fear and scepticism among consumers, who may become wary of purchasing not just the counterfeit product but also legitimate ones. Rebuilding this trust requires transparency, rigorous quality assurance, and effective communication from the brand.
The public health implications of counterfeit drugs are profound. The World Health
Organisation estimates that counterfeit drugs contribute to around a million deaths globally each year. These drugs often contain incorrect dosages, harmful substances, or no active ingredients at all, resulting in treatment failures and health complications. The proliferation of counterfeit drugs undermines public confidence in healthcare systems and can lead to a public health crisis.
“Patients don't want to risk their healththey would like to have the tools to check if a medicine is real or not. They need to understand how to use the tools. We don't have access to the information as patients, so if the information is deviated from the official channel and you`re getting it online, you cannot be sure that is the real one. No one wants to risk their health. People are aware of the situation, it's all about the tools that we give them.”
Laura Lopez
Economic Consequences
The economic impact of counterfeiting extends beyond the pharmaceutical industry. In the European Union alone, counterfeit goods are estimated to cost the economy around 10 billion euros annually and result in the loss of approximately 38,000 jobs. These losses are not confined to direct sales but also affect associated industries, including packaging, logistics, and retail. The resources required to combat counterfeiting, such as legal actions and enhanced security measures, further strain financial resources.
Regulatory and Technological Responses
In emerging markets, the adoption of anticounterfeiting technologies is happening rapidly. Smartphone scanning, for example, is being utilised to verify the legitimacy of products, as these markets cannot build comprehensive systems quickly enough to tackle counterfeiting.
Pharmaceutical companies have been addressing counterfeiting for decades, but the challenge persists. The industry must prioritise brand protection alongside other critical issues like sustainability. Awareness and demand from stakeholders can drive companies to invest in robust anticounterfeiting measures.
Regulations like the Drug Supply Chain Security Act (DSCSA) in the US and the Falsified Medicines Directive (FMD) in the EU are crucial steps toward mitigating the counterfeit drug problem. However, closing one route for counterfeiters often opens
Health Outcomes
another, highlighting the need for ongoing vigilance and adaptation.
Educating Consumers and Enhancing Supply Chain Security
The rise of e-pharmacies and the convenience of online shopping have transformed how consumers access medications, but they have also created new opportunities for counterfeiters. Counterfeit drugs pose significant health risks, making it crucial to increase consumer and patient awareness while enhancing supply chain security.
The corporations and the businesses in place that develop these drugs must work mindfully with the authorities to ensure they’re complying and keeping people as safe as they can. The awareness must be promoted on both ends, not just in the hands of the clients and consumers.
Increasing Consumer and Patient Awareness
Educating consumers and patients about the dangers of counterfeit drugs is essential. Public health organisations, governments, and pharmaceutic al companies need to collaborate on comprehensive education campaigns. These campaigns should leverage various platforms, such as social media, television, radio, and community events, to disseminate information on identifying counterfeit drugs.
Developing and promoting smartphone applications that allow consumers to scan QR codes or barcodes on medication packaging can significantly aid in verifying authenticity. These apps can provide detailed information about the product, its manufacturer, and its distribution path, ensuring consumers have the necessary tools at their fingertips. In addition to this, clear labelling and instructions on packaging play a vital role in consumer education. Packaging should include stepby-step guides on using verification apps or websites and explanations of the security features embedded in the packaging. This kind of transparency helps consumers understand how to check their medications and recognise authentic products.
Healthcare providers, including doctors, pharmacists, and hospitals, are crucial allies in educating patients. Training healthcare professionals to identify counterfeit drugs and advise patients on safe purchasing practices can greatly expand the reach and impact of awareness efforts.
Enhancing Supply Chain Security
Implementing robust track and trace systems
ensures that every step of a product’s journey, from manufacturer to consumer, is meticulously monitored. Technologies like blockchain can offer immutable records of the entire supply chain, making it easier to detect and prevent the introduction of counterfeit drugs.
As mentioned, serialisation, or assigning a unique serial number to each package of medication, is another critical measure. This allows for the verification of the product's legitimacy at various points throughout the supply chain, from manufacturing to dispensing.
Tamper-evident packaging, incorporating features such as seals or special adhesives, makes it evident if a product has been opened or altered. These features act as a deterrent to counterfeiters and provide consumers with visible assurance of the product’s integrity.
Advanced authentication technologies, including colour-shifting inks and RFID tags, should be integrated into packaging to facilitate verification. Consumers can check these features using simple tools or apps, further ensuring the authenticity of their medications.
Pharmaceutical companies must also collaborate closely with regulatory authorities to ensure compliance with anti-counterfeiting regulations. Sharing information about counterfeit trends and methods can help authorities develop more effective enforcement strategies. Rigorous vetting and regular audits of suppliers and distributors can prevent counterfeit products from entering the supply chain. Establishing stringent standards for partners and ensuring compliance through continuous monitoring are essential steps in maintaining supply chain integrity.
Investing in the latest anti-counterfeiting technologies and continuously updating them is crucial. As counterfeiters become more sophisticated, staying ahead of their tactics requires ongoing innovation and adaptation.
Future Strategies and Technological Innovations
To effectively combat counterfeiting, the industry must adopt a multi-layered approach, including serialisation, tamper-evidence, and authentication technologies. These measures should be integrated into the packaging to ensure that consumers can easily identify genuine products.
Harmonisation and standardisation of product identification are essential. The integration of digital technologies, such as blockchain and artificial intelligence (AI), offers promising solutions. AI can analyse vast amounts of data to verify product authenticity and enhance supply chain transparency.
Conclusion
Combating counterfeit drugs requires a comprehensive approach that includes increasing consumer and patient awareness and enhancing supply chain security. By equipping consumers with the knowledge and tools to verify medication authenticity and implementing advanced security features throughout the suppl y chain, the pharmaceutical industry can protect public health and maintain trust in its products. The role of innovative packaging is crucial in this effort, as it incorporates tamperevident seals, QR codes, and RFID tags to help ensure product integrity and authenticity. Collaboration among all stakeholders, including pharmaceutical companies, healthcare providers, regulatory authorities, and consumers, is essential to effectively address this global challenge.
Laura Lopez Head of Marketing at MM Packaging
Philip Allen Technical Account Manager at MM Packaging
Claire Clarke Director of healthcare engagement at GS1 Yann Ischi
Vice President and General Manager of product authentication at Crane Currency
Unique bottom design
Designed to be pressed on the bottom of the bottle
— 60% less finger force to create a drop
— Excellent choice for users with limited mobility in hands
Ergonomic arm and wrist position
Health Outcomes
The Pivotal Role of Patient Advocacy Organisations in Driving Rare Disease Therapeutic Development
Throughout the rare disease clinical development pathway, patient advocacy organisations (PAOs) can and do play crucial roles that exceed supportive functions like raising awareness around clinical trials. These organisations are instrumental in shaping drug development processes, from initial research phases to postmarketing evaluations, contributing anywhere from study design to regulatory guidance documents and beyond. Understanding their influence, engaging with them effectively, and integrating their insights throughout the drug development lifecycle can significantly enhance therapeutic outcomes.
Understanding the Role of Patient Advocacy Organisations
Patient advocacy organisations are pivotal in rare disease therapeutic development due to their unique position at the intersection of patient needs, scientific research, and regulatory processes.
Their contributions span all stages of drug development.
Patient Engagement in Preclinical Drug Development
In drug development, the preclinical phase is fundamental for setting the stage for successful clinical trials. Integrating patient engagement during this phase significantly influences research priorities and pipeline decisions, ensuring that they align with realworld patient needs. Here are some ways in which patient engagement can enhance the preclinical stage:
• Landscape Analyses: Collaborating with PAOs helps identify research gaps and set priorities based on patient insights. Surveys and interviews with patients and caregivers provide crucial data on the most pressing issues, guiding research focus. Landscape analyses help sponsors identify areas where existing treatments are inadequate or non-existent. By evaluating current therapies and patient outcomes, sponsors can pinpoint
gaps that their drug candidates might address. For instance, communicating with PAOs may help uncover barriers to current therapies or their mode of administration. This ensures that research efforts are directed toward areas with significant unmet needs, increasing the potential impact of new treatments.
Tip: Every PAO is uniquely built with their own specific priorities and goals for engaging in the therapeutic development process. Conducting landscape analyses of PAOs is a helpful best practice in guiding your collaborations.
• Pipeline Selection: Engaging patients through advisory panels and utilising data from patient registries helps evaluate and select drug candidates that address significant needs. This ensures that the chosen candidates are aligned with the real-world challenges faced by patients.
• Setting Research Priorities: Patient focus groups and collaborative research development with PAOs help set research priorities that reflect patient concerns and needs. This alignment ensures that research efforts are directed towards areas with the highest potential impact.
• Registries and Natural History Studies: Developing patient registries and conducting natural history studies with patient involvement provide valuable guidance and data on disease progression and treatment impacts. These data support informed decisionmaking and more relevant research.
• Documenting Patient Journeys: Mapping out patient and caregiver journeys through detailed interviews and surveys offers deep insights into their experiences. This understanding helps design therapies that address realworld challenges and improve patient outcomes.
• Pre-IND Support: By incorporating patient insights into the development process, a sponsor can design a more
relevant and patient-centred clinical trial, addressing key concerns, and enhance the likelihood of successful outcomes.
Tip: Patient registries are often managed by PAOs and can span from regional to global participation. They may include information on patient demographics, clinical outcomes, treatments, and disease burden.
Patient Engagement during Clinical Trials
Phase I–IV
Engaging patients throughout clinical trials is crucial for creating studies that are effective and aligned with patient needs. During Phase I, which focuses on safety and dosing, Patient Advisory Boards (PABs) play a vital role in shaping the schedule of assessments and providing input on the most comfortable route of administration. They can also review Informed Consent Forms (ICFs) and patientfacing materials to ensure clarity and relevance.
Tip: Surveys and focus groups are impactful ways to understand the patient and caregiver experience. For example, this survey of rare disease patients highlighted the vast need for and importance of participation support during clinical trials: https://www.iconplc.com/insights/therapeutics/ rare-and-orphandiseases/crossing-the-finishline
In Phase II, which examines efficacy and dose-response, PABs can contribute by identifying meaningful endpoints and appropriate Patient-Reported Outcomes (PROs). Their input helps ensure that trial measures reflect real-world benefits.
During Phase III, which is critical for confirming efficacy and obtaining regulatory approval, PABs reassess the schedule of assessments and endpoints to ensure they meet patient needs and regulatory standards. Updated ICFs and patient-facing materials are reviewed to maintain transparency and comprehensibility.
In Phase IV, the post-marketing phase, PABs help ensure that patient-facing materials about long-term monitoring and surveillance are clear and supportive. Engagement strategies include leveraging social media,
hosting conferences, and conducting listening sessions to address ongoing patient concerns. Ad hoc strategies may be developed to tackle specific challenges faced by patients, ensuring continuous support throughout their treatment journey.
PABs will often suggest strategies to reduce burden including regular study communications and educational initiatives to keep participants informed, providing travel and accommodation assistance, offering counselling services and promoting diversity, equity, and inclusion (DEI) to ensure the trial is accessible to all eligible participants.
Tip: Patient-focused design improves study success, and the Patient Centric Trial Develop-ment Toolkit is a free resource designed to help all stakeholders identify and overcome barriers in rare disease clinical development: ICONplc.com/rare-diseasetrial-development-toolkit
Patient Engagement During Post-Approval and Market Authorisation
After a drug receives market authorisation, ongoing patient engagement is critical for ensuring its continued effectiveness and addressing any emerging needs. Effective post-study engagement involves several key components:
• Communication: Maintaining clear and consistent communication with patients helps keep them informed about new developments, potential side effects, and updates related to the drug.
• Personalised Support Teams: Establishing dedicated support teams provides patients with personalised assistance, addressing their specific needs and concerns regarding the drug.
• Access to Care/Reimbursement Hurdles: Addressing barriers to accessing the medication, including reimbursement issues, ensures that patients can obtain and afford their prescribed treatments.
• Patient Empowerment Initiatives: Implementing programmes that educate and empower patients helps them manage their condition effectively and participate actively in their care.
• Ongoing Data/Experience Assessments: Continuously collecting and analysing patient data and experiences postapproval allows for the identification of
Health Outcomes
long-term effects and the refinement of treatment protocols.
By focusing on these areas, sponsors can enhance patient satisfaction, improve treatment outcomes, and support the effective long-term use of new therapies.
The Impact of Patient Advocacy on Drug Development
Engaging with PAOs is not just about checking a box or meeting regulatory requirements; it is about embedding the patient's perspective into the fabric of drug development. This engagement has several significant impacts:
1. Improved Study Design: By incorporating the patient voice, drug developers can design more effective and patientcentred trials, potentially leading to more meaningful outcomes and better recruitment and retention rates.
2. Enhanced Data Collection: PAOs can assist in gathering real-world data through registries and natural history studies, which can provide critical insights into the disease and treatment impacts.
3. Long-Term Benefits: Engagement with PAOs extends beyond the drug development phases and into post-marketing efforts. Continued collaboration helps in addressing issues related to access, personalised support, and patient empowerment, thereby enhancing the overall therapeutic impact.
Patient advocacy organisations are essential partners in the rare disease therapeutic development process. Their involvement from pre-clinical stages through to post-marketing evaluations ensures that drug development is aligned with the needs and experiences of patients. Effective engagement with these organisations –through early discussions, advisory boards, and continuous interaction – can drive more meaningful therapeutic advances and ultimately lead to better outcomes
for patients. In a field as challenging as rare disease research, their role goes far beyond supportive to transformative, helping to navigate the complexities of drug development with a patient-centred approach.
Jana Benesh, PhD, is a Director of Therapeutic Expertise in ICON's Centre for Rare Diseases where she focuses on strategic development for rare neurological and neuromuscular diseases. She is a neuroscientist by training, with over 15 years of experience in basic and clinical research, and also serves as a scientific advisor, educator, and community board member to multiple advocacy groups supporting patients and families affected by rare neurological diseases.
Neena Nizar, Ed.D, is Director of Patient Advocacy Strategy at ICON's Centre for Rare Diseases. Neena has 20 years of rare disease patient advocacy strategy experience focused on development of nano-rare therapies and patient-centred outcomes. She has deep expertise in diversity, equity, and inclusion strategy with experience across multiple healthcare settings and academia. Neena has strong expertise in the Middle Eastern/ Asian patient advocacy landscape and is a highly regarded leader in the rare skeletal disease area. She is founder of The Jansen's Foundation and was named a WebMD Healthcare Hero in 2023.
Jana Benesh
Neena
Nizar
Health Outcomes
Listening To the Patient Voice to Improve Eye Care
Growing Markets in Glaucoma and Dry Eye Disease Fuelled by the Aging Population
The growing global ophthalmic eyedroppers market is expected to be driven by the increasing prevalence of eye disease and disorders such as Glaucoma, Dry Eye Disease, and Age-Related Macular Degeneration. Moreover, the increasing ageing population further propels the market growth as there is higher incidence of age-related eye conditions.1
Glaucoma, which is the leading cause of irreversible blindness worldwide, represents a group of optic neuropathies characterised by the progressive degeneration of retinal ganglion cells (RGCs). Open-angle Glaucoma (OAG) is the most common form of Glaucoma, carrying a chronic prognosis and making up 75–95% of primary cases.2 Angle-closure Glaucoma (ACG) can either be acute or chronic, but typically has a faster progression than OAG and, therefore, requires more drastic interventions. These two main types often require the use of eyedrops to reduce intraocular pressure and prevent further damage. With the ageing population, the demand for Glaucoma medications administered via eyedroppers is also expected to increase, thereby driving market growth. For example, elderly people are six times more likely to develop Glaucoma after the age of 60.3
The Dry Eye segment dominated the market in terms of revenue in 2023, owing to its rising prevalence.1 The definition of a Dry Eye according to the Tear Film and Ocular Surface Society Dry Eye Workshop II is: "Dry Eye is a multifactorial disease of the ocular surface characterised by a loss of homeostasis of the tear film, and accompanied by ocular symptoms, in which tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities play etiologic roles."4 One increasingly common extrinsic risk factor for Dry Eye is digital screen use (eg, computer, laptop, tablet, and smartphone use), which is thought to contribute to its development by affecting blinking dynamics.5
Other factors contributing to Dry Eye include ageing, air pollution, and hormonal changes. Eye drops or artificial tears are a primary treatment options to alleviate symptoms by lubricating the eyes and providing moisture.1
Given the growing ophthalmic eyedroppers market, owing largely to the increase in ageing population, drug delviery device designers and manufacturers must strive not only to understand but to provide solutions for patients encountering administration challenges and to improve at home eye care management. One way of gaining insight in to the patient journey is via testimonials and reviews of real patients using existing products on the market.
Acknowledging Patient Pain Points to Improve Eye Care Management
As patients become increasingly informed
and discerning, they heavily rely on the experiences and opinions of others to make well-informed choices. A key advantage of Novelia®’s strong position as a leading multi-dose eye dropper for preservative-free formulations, is having access to a cornucopia of verified patient reviews on e-commerce websites and social media platforms. This is especially true for over-the counter products treating Dry Eye.
Marketed products using the Novelia® device rate consistently high amongst patients. For example, Systane™ COMPLETE PF Multi-Dose Preservative Free Dry Eye Drops have maintained an average star rating of 4.5/5, across +10K verified customer reviews since its market launch in 2022, making it the #1 product in the Moisturizing Eyedrops Category on Amazon US.a (Figure 2)
Figure 1: The Novelia® system uses a non-return valve that removes the need to filter the liquid.
Health Outcomes
Online reviews by patients are an invaluable asset for Nemera, providing insights into these real patient experiences. Even less than positive aspects offer valuable feedback for improvement that Nemera embrace as an opportunity to identify weaknesses and make meaningful improvements to our products and services.
Preservative-free
The majority of eye drops today contain preservatives to maintain the formulation’s sterility. The most used preservative is Benzalkonium Chloride (BAK), which has been known to damage the cornea over long term use. Preservatives can also cause allergies or ocular irritation, and some can even cause a toxic response.6 Any such reactions are issues for patients who rely on the long-term use of eye drops for chronic conditions.
“Very good quality eye drops, preservative free which I needed as I use eye drops frequently and I wanted to limit the irritation that can be caused by more additives to the eye solution.”b
Control
Patients and HCPs alike desire a bottle that allows patients to control the number of drops delivered, consistently delivering a single drop with each actuation. A study of Glaucoma patients found that 90% utilised an erroneous technique, with many patients missing the eye entirely.7
Novelia®’s patented PureFlow™ Technology not only serves as a venting system but also controls the medication flow (Figure 2). We have adapted the flow-control within
Novelia® that avoids multiple drop delivery into the eye and ensures that only one calibrated drop is dispensed at a time. Nemera offers three different PureFlow® versions, each tailored to formulations of differing viscosities, from highly liquid to highly viscous. In addition, five different valve sizes are available, each one delivering a different calibrated drop size. This allows Nemera’s team to customise the drop size depending on specific product requirements. This improved control leads to increased patient confidence (of accurate dosing), reduced frustration and medication waste.
“I like the fact that these eye drops are preservative free. The dropper is a little different but very easy to use.”c
Ease of Use
Patients, even those with dexterity issues and tremors/ shaking, must be able to effectively handle/ manipulate the delivery system and administer a drop.
Contributing factors to Novelia® being the multidose eyedropper for preservativefree formulations preferred by 76% patients included the intuitiveness of the screw-on cap and the associated reassurance and the squeeze force required towards the end-oflife the product. Novelia® required only 6% more pressure to squeeze the bottle from the beginning to the end of the treatment, compared to 35% for the other device.8
“So delighted with this packaging. Have used a competitor's product and I had to
use 2 hands to squeeze the bottle. (I am 83 with very little grip strength.) This bottle is much softer, and I am able to use 1 hand which easily leaves my other hand free to "open" my lower lid and instil the drops in the appropriate way. I don't care for the individual PF drop containers as they are more expensive when using throughout the day and I found those containers were also difficult to squeeze.”d
Novelia®’s patented blue tip is also a favourite feature of the device. It helps patients target the eye before drop administration and anticipate the angle of the drop on to the ocular surface.
“The blue dot on the dispenser helps line up the bottle with the eye. Because I can see how far away the bottle is from my eye, I am less threatened by it than a pointed dispenser. Thus, I can relax more when applying drops to my eyes (whether I do it or someone else does). This makes the application process a lot easier.”e
Transparency
Patients need more visibility into their medication supply, so they can replenish as needed and don’t find themselves without.
A full range of bottles is available in terms of size, material, and sterilisation type (5 mL, 7.5 mL, 11 mL, and 15 mL. All sizes are available in low density polyethylene (LDPE) either in white or natural (transparent), allowing patients to know when their medication is running low. Nemera has also developed a polypropylene (PP) 11 mL bottle for specific formulation compatibility. (Figure 3) Novelia® bottles been validated using both gamma and ethylene oxide (EtO) sterilisation.
“The bottle is transparent, so you know how much you have remaining.”f
Portability
A patient must be able to easily transport their medication. Note that for DES patients this is likely to be daily, while for Glaucoma patients this is likely only for overnight/ travel.
The Novelia® device features a Screw-On cap which fits tightly on to the device nozzle which is optimal in terms of portability. Other marketed devices comprising a Snap-On cap have been found by patients to be less robust with several instances of leaking during transport, be it in a handbag or pocket. As such, Novelia® outperformed
Figure 3: A full range of bottles is available in low density polyethylene (LDPE), 5 mL, 7.5 mL, 11 mL, and 15 mL as well as 11 mL polypropylene (PP).
Health Outcomes
another marketed device in terms of cap opening, hermetic sealing, and nomadic use, with a mean score of 4.5 out of 5 for the three features.9
“Love the portability. It’s nice not needing to carry around small individual containers for each use in your pocket and around the house. I do like that this one (as) you can twist the cap on and off (so it) feels more secured.”g
Sustainability
Patients also report concerns regarding unit doses. These include cost (as more packaging and eye drop solution per dose is required), waste, and convenience, as it is easier to store a multi-use bottle in a preferred location than to ensure the patient has the correct number of unit dose pipettes with them every day.10 Difficult handling has also been pointed out regarding (unit doses) and their use by elderly patients and with inappropriate finger manipulation could be associated with an increased risk of contamination.11 Due in part to the rapidly aging population, the number of people having Glaucoma worldwide is expected to increase to over 111 million by 2040.12
A comparison analysis was conducted by Nemera comparing Novelia® with unit dose packaging for a Glaucoma type regimen (one drop per eye twice per day) over a onemonth treatment. The results conveyed that with Novelia® there is eight times less plastic used, 25 times less drug waste and nine times less energy needed for transportation compared to a unit dose.13
“Not having to carry around all the little vials of drops is great. I went on a 10-day vacation and this little bottle were the only drops I took with me.”h
In 2021, Nemera decided to subscribe to the Science-Based Target initiative (SBTi) to define and develop best practices for carbon reduction. One of our first objectives is to reduce our scope 1 and 2 emissions by 90% by 2030 from a 2019 base year. In addition, since 1st February 2024, the manufacturing facility in La Verpillière, France, where Novelia® is manufactured, has been certified ISCC PLUS. This certification scheme for biobased, renewable and circular raw materials reinforces Nemera’s implementation of sustainability goals (Figure 4).
Supporting Customers in Supporting Patient Needs Worldwide Nemera offers a range of laboratory
services for Novelia®, including the testing of customers’ bulk formulation. This testing comprises usage simulation over a two-week period, drop size analysis (variable depending on valve diameter), flow control and squeeze force testing (beginning and near end of life). The culmination of these tests results allows Nemera to determine the best Novelia® configuration for a particular customer formulation. Nemera can recommend the most suitable PureFlow™ control, bottle type and valve size to achieve the desired drop calibration.
Nemera’s regulatory team is on-hand to support customers with their submission filing, providing guidance on supportive documents for registration. Nemera can also assist customers in finding the right readyto-go dossier available for private labelling certain molecules with the Novelia® delivery system. Nemera has a substantial list of partners, formulation licensors and fillers, all working in collaboration to bring to customers a finished drug device combination product with Novelia®.
While it is important that containers be user friendly, it has been found that educational resources instructing patients to apply their eye drops correctly mitigates many issues with unintentional noncompliance.14 Nemera can support
customers with product market launch, for example, in educating sales teams and HCPs on the delivery device, dedicated trainings, and materials to assist in promotional material creation. Customisable patient guidance videos are also available in several languages to increase patient compliance around the world.
Today, Novelia® has approximately 300 references on the market for prescription and over-the-counter products in over 55 countries across Europe, Latin America, North America, Oceania, Middle East, and Asia Pacific.
To serve customers in supporting patient needs, Nemera has extended its manufacturing capabilities, in the US and in doing so has doubled its capacity to produce Novelia® multidose eyedropper for preservative-free formulations (Figure 5).
REFERENCES
1. 2023, Grand View Research, Inc., USA “Ophthalmic Eye Droppers: Market Analysis Segment Forecast From 2018 To 2023.”
2. 2023, GlobalData_GlaucomaCompetitive Landscape_061123, date accessed 26/01/2024
3. Allison K, Patel D, Alabi O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus. 2020 Nov 24;12(11):e11686. doi: 10.7759/cureus.11686. PMID: 33391921;
5. Al-Mohtaseb Z, Schachter S, Shen Lee B, Garlich J, Trattler W. The Relationship Between Dry Eye Disease and Digital Screen Use. Clin Ophthalmol. 2021 Sep 10;15:3811-3820. doi: 10.2147/OPTH.S321591. PMID: 34531649; PMCID: PMC8439964.
6. Report of the International Dry Eye Workshop. Ocul Surf (2007); 5[2]; 65-204.
7. Tatham AJ, Sarodia U, Gatrad F, Awan A. Eye drop instillation technique in patients with glaucoma. Eye (Lond). 2013 Nov;27(11):1293-8. doi: 10.1038/eye.2013.187. Epub 2013 Aug 23. PMID: 23970024; PMCID: PMC3831141.
8. “User study performed for Nemera by GfK to understand the Novelia® market opportunities versus competitors”. GfK report, Paris, France, (2015).
9. “User study performed for Nemera by QUALIQUANTICI, Novelia® and a marketed pump product testing”. Marketing Espace report, Lyon, France, (2018).
10. Erb, C., n.d. “Glaucoma and Dry Eye”, (2020), p37.
11. Campolo A, Crary M, Shannon P A Review of the Containers Available for Multi-Dose Preservative-Free Eye Drops. Biomed J Sci & Tech Res 45 (1) -(2022). BJSTR.MS.ID.007130.
12. Tham, Y., Li, X., Wong, T., Quigley, H., Aung, T. and Cheng, C., (2021). Global Prevalence of Glaucoma and Projections of Glaucoma Burden Through 2040.
13. “Comparison analysis conducted by Nemera comparing Novelia® multidose eyedropper for preservative-free formulations with unit dose packaging,” Nemera, La Verpillière, France, (2020).
14. Davis SA, Carpenter DM, Blalock SJ, Budenz DL, Lee C, Muir KW, Robin AL, Sleath B. A randomized controlled trial of an online educational video intervention to improve glaucoma eye drop technique. Patient Educ Couns. 2019 May;102(5):937-943. doi: 10.1016/j. pec.2018.12.019. Epub (2018) Dec 18. PMID: 30583913.
a) Amazon US customer reviews January (2024) Systane COMPLETE PF Multi-Dose Preservative Free Dry Eye Drops, https://www.amazon.com/ Systane-COMPLETE-Multi-Dose-PreservativeDrops/dp/B09NRYCNDC (accessed 26/01/2024)
b) Verified Amazon US customer review, 7th January 2022 (Systane™ Hydration PF Lubricant Eye Drops 10ml)
c) Verified Amazon US customer review, 7th January 2022 (Systane™ Hydration PF Lubricant Eye Drops 10ml)
d) Verified Amazon US customer review, 3rd July 2021 (Systane™ Hydration PF Lubricant Eye Drops 10ml)
e) Verified Feefo UK (Butterflies Eyecare) customer review, 1st July 2023 (Evolve SOOTHE & RENEW eye drops 10ml)
f) Verified Amazon US customer review, 10th September 2023 (Systane™ COMPLETE PF MultiDose Preservative Free Dry Eye Drops 20ml (Pack of 2 – 10mL bottles)
g) Verified Amazon US customer review, 15th September 2022 (Systane Ultra PF PreservativeFree Eye Drops 10ml)
h) Verified Amazon US customer review, 3rd August 2021 (Systane™ Hydration PF Lubricant Eye Drops 10ml)
Zoë Davidson has held the position of Global Category Marketing Manager for Nemera’s ophthalmic franchise since 2019. In this role, Zoë is responsible for the strategy management of Nemera’s flagship Own-IP multidose eyedropper for preservative-free formulations, Novelia®. With over seven years’ experience in the pharmaceutical and medical device industry, Zoë is motivated about gaining insights into patients’ needs and wants within the ophthalmic space. Hailing from Glasgow, Scotland, Zoë now calls Lyon, France home, where she lives with her husband and their two young sons.
Email: zoe.davidson@nemera.net
Zoë Davidson
Figure 5:
capacity to produce Novelia® by creating two additional assembly lines in their US site, Buffalo Grove, IL.
How Efficient Logistics Can Change the Lives of Rare Disease Patients
Dr. Danial Arkwell, Head of Global Key Accounts, Pharma at Envirotainer highlights the vital role of efficient logistics in mitigating risk and finding the right solutions to get orphan drugs to patients in desperate need.
There are approximately eight thousand rare diseases in the world and many of these are chronic, life-threatening conditions. To fight these elusive illnesses, the medical community focuses on developing specialised medications known as orphan drugs. Originating from the United States Orphan Drug Act (ODA) in 1983, the term “Orphan Drug” reflects the lack of attention and investment necessary for their development.
The diseases targeted by orphan drugs often have low prevalence rates, making them economically unattractive to develop treatments for, due to the limited potential market. The result is that many of these diseases are being historically neglected as a result of the perceived low profitability.
The Orphan Drug Act and similar legislation aim to progress the development of drugs for rare diseases by offering various incentives
to drug manufacturers. This includes tax credits for clinical research costs, seven years of market exclusivity upon approval, and assistance with clinical trials, design and funding.
Despite government support, orphan drugs have been priced significantly higher than non-orphan drugs, owing to the inherent complexities and high costs of production. The significant upfront investment required to bring these much-needed treatments to market not only slows down their development but also contributes to incredibly high prices for patients, potentially putting them out of reach for many.
Yet, there are other barriers to patients receiving life-saving treatments. One that’s frequently overlooked is the complexity of transporting these highly sensitive drugs from the lab to the end patient. If manufacturers cannot ensure safe, costeffective delivery, then access to these rare treatments will remain out of reach for many patients. Efficient logistics must take centre stage.
Logistical Barriers Stand Tall
Companies developing orphan drugs and other rare treatments often do so at a
net financial loss. This cost is not solely placed on the research and production, the safe transport and distribution of these specialised treatments is also a consideration. Unfortunately, there is a lot that can impact delivery.
One of the most significant challenges in shipping orphan drugs is their temperature sensitivity. Many of these products are biologics, gene therapies, or other advanced therapies that require strict temperature control to protect their efficacy. Some require storage at extremely low temperatures, often below freezing, while others may need to be stored at controlled room temperatures or other conditions.
Even a small temperature deviation during shipment can compromise the product and render the drug ineffective or even harmful to patients. Furthermore, when developed as personalised medicines, the transportation complexities and logistics costs increase further, often resulting in higher total landed cost (TLC) for shipping.
Despite the small patient pool, orphan drugs often need to be delivered to different, sometimes hard-to-reach, locations around the world, adding another layer of complexity
TEMPERATURE TEMPERATURE
&
Temperature protection of pharmaceutical and healthcare products in airfreight (+15°C + 25°C°) and (+2°C + 30°C) (+15°C + 25°C°) and (+2°C + 30°C)
Multilayer thermal blanket for PMC-ULD - Euro and Block pallets
Stress-tested in summer (+46°C) and winter (-15°C) profiles
Airfield Tarmac tested on solar power and greenhouse effects
Tel. (Belgium): +32-11.26.24.20
E-mail: info@krautz.org
Website: www.krautz.org
Logistics & Supply Chain Management
to the process. Import and export regulations and differing international standards can create bottlenecks and delays, increasing the risk of temperature deviations if a shipment is stuck in customs.
Last-mile delivery is another critical challenge when ensuring patients receive their treatments safely and efficiently. Unlike many medications, orphan drugs often require direct delivery to patients' homes or specialised medical facilities, introducing additional requirements to consider. Challenges such as geographical remoteness, limitations in infrastructure (e.g. storage space, electricity, and cooling capabilities), and the need for personalised handling, add further to the complexities of last-mile delivery.
All these costs and logistical challenges only push the cost of treatment up higher, creating significant barriers to affordable treatments and potentially pricing outpatients who need the treatment most. For manufacturers, however, the safe shipping of these drugs is non-negotiable, no matter how much it costs.
Finding the Right Balance
For the pharmaceutical industry, efficient logistics isn’t just about cost savings; it’s about ensuring life-saving medications reach the right patients, at the right time, in the right condition.
Finding the optimal shipping solution involves balancing performance, environmental impact, service level and cost.
For strict temperature-controlled drugs, it’s crucial to choose packaging that’s designed to maintain a consistent temperature range throughout the shipping process. It’s vital that the optimal packaging for the shipment mitigates the risk of temperature deviations and prevents potential product loss and any consequential increase in transport costs and drug usability at the destination.
In addition, as many as 20% of these products are spoiled and unable to be delivered due to failures in the cold chain, resulting in severe implications for patients. However, there are strategies to mitigate risk, such as ‘smart’ secondary packing solutions with real-time temperature and location monitoring, which send alerts in real-time if an unexpected event occurs during transit, enabling swift corrective action - preventing costly product spoilage and re-shipping and saving money in the long run.
Both active and passive containers have their role to play, but the best choice depends on varying factors including product temperature requirements, tradeline complexity, TLC and ultimately the level of risk one is willing to take with one's medicine.
Because orphan drugs are manufactured in smaller quantities, these drugs have often been seen sharing cargo space with other pharmaceutical products, potentially increasing the risk of exposure to unsuitable conditions. To mitigate this risk, manufacturers can prioritise collaboration
and coordination with logistics partners to optimise trans-portation. By leveraging shared logistics networks and implementing Just-In-Time inventory strategies, distribution processes can be streamlined while also minimising costs and maximising efficiency.
The Bottom Line of Efficient Logistics
The journey of an orphan drug, from research lab to patient delivery, is a complex process and often an expensive one. However, the cost of rare disease drugs isn’t just in development, it’s in delivery. Inefficient logistics can eat up resources and can even lead to patients not getting the treatment they so desperately need.
Yet, with the right packaging choice, ongoing collaboration between pharma manufacturers and specialists in temperaturecontrolled logistics, and a patient-centric approach, the barriers to shipping rare disease treatments can be removed. This means that more patients get access to the drugs they need, regardless of how rare their condition is. After all, it’s not just about saving money, it’s about saving lives.
Dr. Danial Arkwell has a Ph.D in Molecular Biology, and with over 20 years commercial experience in healthcare, clinical diagnostics and pharmaceuticals, he is an industry expert in temperature controlled logistics. Working with Envirotainer, he believes that close collaboration, open communication and a passion for patient safety are key to navigating the challenges facing this industry.
Dr. Danial Arkwell
Revolutionising Pharmaceutical Logistics: The Path to Sustainable Cooling Solutions
The healthcare industry is at a pivotal moment in its journey toward sustainability, as the demand for efficient cold chain management in transporting and storing medical and pharmaceutical products continues to grow.
As the pharmaceutical industry continues its rapid global expansion, the demand for efficient and sustainable cold chain management can no longer be pushed aside.
From vaccines to insulin, medications require precise and accurate temperature control to ensure their efficacy and safety. Traditional cold chain models rely heavily on fossil fuels, generating a high carbon footprint.
This current approach poses significant challenges, particularly in light of the global push for reducing greenhouse gas emissions and combating climate change.
The energy-intensive nature of traditional cooling systems puts pressure on the already strained energy infrastructure in many regions, particularly in developing countries which often need medications the most. Sustainable cold chain practices can enhance the access to medications and vaccines in these regions, ensuring that vital treatments remain safe and effective during transport. This can help address healthcare disparities and improve patient outcomes and health on a global scale.
As the healthcare industry strives to meet the growing demand for storing and transporting pharmaceutical products, it must confront the environmental impact of its practices. Sustainable cooling solutions are essential for preserving the integrity of medications while also reducing the industry's environmental footprint.
This article will explore how re-evaluating our approach to cooling in the pharma sector is not just a necessity; it is a moral imperative.
What is the Significance of Cold Chain Management?
Cold chain management is crucial in the
pharmaceutical industry, directly affecting our ability to maintain the quality and integrity of temperature-sensitive medications.
Certain products must be stored and transported within specific temperature ranges to prevent degradation, spoilage, or loss of safety. Maintaining the integrity of these products throughout the supply chain requires a highly controlled environment, often involving refrigeration or freezing. This is known as the cold chain – a temperaturecontrolled system that encompasses storage and transportation.
Failure to control the conditions and temperatures that medications are stored and transported in can result in ineffective treatments and even pose serious health risks to patients. Therefore, maintaining a reliable cold chain is paramount to preserving the life-saving or life-changing capabilities of these medications.
What are the Challenges of Traditional Cold Chain Models?
Traditional cold chain models rely heavily on fossil fuels for transportation and storage, contributing to significant greenhouse gas emissions and environmental degradation.
This reliance on non-renewable resources worsens the industry's carbon footprint, hindering the efforts to combat climate change. Additionally, the use of singleuse packaging materials and refrigeration technologies with harmful refrigerants burdens the environment further.
Apart from the environmental impact, these conventional methods have additional limitations. The available cold chain model can be costly and inefficient, particularly in regions with inadequate infrastructure or limited access to resources, as alluded to previously.
The expanding and adapting landscape of the pharmaceutical industry demands a more sustainable and reliable approach to cold chain management.
Is it Time to Rethink the Cold Chain Model?
The cold chain model needs a fundamental and dramatic transformation to achieve
sustainability. This involves re-evaluating existing cooling practices and exploring innovative alternatives that prioritise energy efficiency and environmental responsibility.
One vital aspect is the adoption of renewable energy sources to power cooling units. Solar, wind, and other renewable energy technologies can significantly reduce the carbon emissions associated with cold chain logistics.
Through the utilisation of clean energy sources, the healthcare industry can evolve toward sustainability without compromising the quality of medical products.
Another important approach is the development of advanced cooling technologies that maximise efficiency while minimising energy consumption. For instance, phase change materials (PCMs) can be used in cooling units to maintain stable temperatures over long periods without the need for constant power. These materials absorb or release heat during phase changes, providing a reliable and energy-efficient cooling solution.
Additionally, the development of smart sensors and IoT-enabled monitoring systems allows for real-time tracking and control of temperature conditions, optimising logistics and reducing waste.
What
is the
Role of Sustainable Cooling Units in Improving Cold Chain Efficiency and Sustainability?
Sustainable cooling units play a pivotal role in preserving the integrity of life-saving medications and products.
These units are designed to maintain precise temperature control, ensuring that pharmaceutical products remain within the required temperature range throughout the supply chain. Proper temperature management is critical for maintaining the efficacy and safety of medications, particularly vaccines and biologics.
It is also vital that cooling units incorporate advanced monitoring and tracking technologies within their system. Real-time data on temperature, humidity, and other environmental factors can be
Logistics & Supply Chain Management
used to ensure that products are stored and transported under optimal conditions.
This data-driven approach allows for early detection of potential issues, enabling proactive measures to mitigate risks. This again, provides a solution to the current challenge of spoilage and waste when it comes to the storage and transportation of medical products.
Addressing Challenges and Seizing Opportunities
The benefits of sustainable cold chain management are clear however, adopting these practices can be challenging. Initial investment costs in new technologies and infrastructure can be substantial, alongside the need for a technically skilled and motivated workforce to maintain and operate these advanced systems. Transitioning away from traditional methods may require changes in operational practices and an industry mindset.
Although the transition to sustainable cooling solutions presents challenges, it also offers significant opportunities for the healthcare industry. By embracing sustainability, companies can enhance their reputation, appeal to environmentally conscious consumers and investors, and contribute to the global effort to combat climate change.
Moreover, sustainable practices can lead to cost savings in the long run. Energy-
efficient cooling units can lower operational expenses, while the use of renewable energy sources can shield companies from volatile energy prices. Additionally, reducing the industry's carbon footprint aligns with emerging regulations and policies aimed at curbing emissions.
Regulatory bodies worldwide are now placing increasing emphasis on the importance of sustainability in the pharmaceutical cold chain. This is made evident through guidelines such as the Good Distribution Practices (GDP), which ensures that medicinal products are stored and transported safely, while also considering environmental impacts. Adhering to these regulations not only guarantees the integrity of medications but also aligns with broader efforts to minimise the industry's carbon footprint.
This transition towards sustainable cold chain management will almost certainly require collaboration across the entire supply chain. Pharmaceutical companies can work with logistics providers to optimise transportation routes and reduce emissions. By sharing best practices and developing joint sustainability strategies, these partnerships can lead to more efficient and environmentally friendly cold chain solutions.
Change is Required: A Moral Imperative
The journey toward sustainable cooling solutions in the pharmaceutical industry is
not only a strategic business decision but also a moral imperative. As the healthcare industry continues to expand its reach, it must take responsibility for its impact on the planet and future generations.
By adopting renewable energy sources, advanced materials, and eco-friendly practices, we can preserve the efficacy of life-saving medications while mitigating the environmental impact.
Policies and incentives could play a crucial role in driving the adoption of sustainable practices in the healthcare industry. Governments can offer tax credits or financial support for using renewable energy sources in cold chain logistics, encouraging companies to invest in green technologies, something that is now becoming more common in central Europe. Policy frameworks that prioritise sustainability can also motivate industry-wide change and set a standard for best practices.
Ultimately, the healthcare industry is at a crossroads, facing the urgent need to transition to sustainable cooling solutions for medical and pharmaceutical products. By prioritising energy efficiency, renewable energy, and advanced cooling technologies, the industry can preserve the integrity of life-saving medications while reducing its environmental impact.
This transformation is essential for meeting the challenges of the future and ensuring a healthier world for all.
Nikolas has 20 years plus of experience in the agribusiness industry focusing on farming, processing, supply chain management and integration of new technology into business operations. He has most recently completed the development of a patent pending token technology that accommodates the exchange and liquidity of precious metals utilising block chain. In 2020 Nikolas shifted to a leadership ship role as CEO and co-founder SpaceWalker LLC, a super-cold chain solution that utilizes NASA patented technology to enhance the efficiency of the global cold chain.
Nikolas
Nemickas
The Primary Logistics Needs of Life Science Companies
The low volume life science market has significant complexities when it comes to logistics. Shippers must have an educated understanding of Good Distribution Practice (GDP) methods and processes, and detailed knowledge of how to ship the pharma related product in question. These products can range from novel prototypes up to final stage development pre-trial products, including component ingredients and failed items being returned for analysis.
As a consequence of the complexity, the need for add-on features when shipping are high on a per shipment basis. Unique packaging, individual insurance and special requests in-transit are often what is required for a temperature-sensitive, high-value shipment to be delivered in its desired state and on time. Additionally, customs teams in each country do not regularly see items like these, so it’s easy to see why most life science companies have major concerns when shipping. It’s therefore essential to work with a specialist logistics partner to effectively outsource the process beyond standard logistics provisions.
YSDS Life Science sat down with four prominent UK life science companies, all identifying four different but equally important aspects of the logistical needs of the life sciences industry.
A Close Collaboration Between The Shipper and Logistics Provider
Products shipped in the Life Science industry are often referred to as “high value”. This is generally speaking a misnomer however, as the products themselves have little monetary value. The value lies in the role they play in the bigger picture; these are business-critical shipments for companies
often in the start-up, spin-out or SME phase. The success of a project and conformity to agreed timelines can have significant impacts on proof-of-concept, funding conversations, and meeting critical strategic markers with mentors. So, what’s the most important factor for shipping success in this low volume-high value market?
LocateBio specialises in developing next generation orthobiologic products to relieve symptoms for people with musculoskeletal conditions. Its products are novel, with complex shipping requirements. “As a startup company, we require a fast turnaround from collection to delivery to ensure that project timelines are adhered to and lastminute requests are processed quickly and efficiently,” says Lyndsey Johnson – Scientific Programme Manager at LocateBio.
What Johnson highlights as the most important factor when working with a logistics provider is regular communication with a dedicated contact who can handle their account throughout the entire shipping process: “This saves us having to bring new people up to speed each time a shipment is raised, and makes the process so much easier.”
Backup Solutions, Proactivity and Flexibility
Whilst clear communication is a must, the logistics provider’s ability to solve problems is equally as important. This according to Dr. Peter De’Ath, Head of Client Account Management at HistologiX: “The importance of high value insurance cover and timely solutions for when things do go wrong cannot be highlighted enough.”
HistologiX is a CRO specialised in tissuebased analyses. The company receives precious tissue samples from its clients, which are then processed and redistributed. Their predominant shipping needs are for ambient shipments of immunohistochemically stained tissue mounted on glass microscope slides and formalin-fixed paraffin-embedded (FFPE) tissue blocks, as well as refrigerated and frozen tissue shipments. “Due to our fastpaced research environment, we are not able to hold fixed shipment dates and we often need to arrange reduced temperature shipments on short notice,” says De’Ath.
Shipping high-value, temperaturecontrolled or time-dependent items has always been a strategic challenge, not only because of the flexibility required, but also because of external conditions the shipper cannot control, i.e. weather, staffing issues, strikes, transport problems, national holidays, conflicts, political events, etc.
Summarising HistologiX’ logistics needs, De’Ath says: “It has to be adaptable to whatever is currently happening in our laboratories and have clear backup solutions in order to deal with all unexpected delays.” Johnson agrees: “A logistics provider needs to show great willingness to think outside the box and actively contribute with solutions to get things back on track as quickly as possible.”
The Highest Quality
The biotech company Emergex highlights the need for logistical quality. Emergex is pioneering the development of a range of 100% synthetic CD8+ T cell-priming immune set-point candidates. The candidates are designed to harness the body’s natural cellular immune response to destroy pathogen-infected cells, and to provide broad and robust immune protection for some of the world’s most urgent infectious diseases.
Emergex’ shipping requirements vary over different temperature-controlled conditions. So how do you ensure quality in the shipping process? By working with GDP certified partners.
GDP – Good Distribution Practice –describes the minimum standards a wholesale distributor must meet to ensure that the quality and integrity of goods is maintained throughout the supply chain. “It’s of critical operational importance that we have access to GDP-certified solutions for time and temperature-sensitive products,” says Chad Zaloumis – Purchasing and Warehouse Manager at Emergex.
De’Ath continues this argument, “A GDP certification gives us complete confidence that our precious and highly valuable materials will be handled with care and the utmost quality of service.”
Logistics & Supply Chain Management
Full Visibility and Transparency
The rapidly growing cell and gene therapy subsegment within the life sciences industry is putting increased demands on the entire logistics chain, of which visibility and transparency is a huge part, for an optimal vein-to-vein process.
Pharmaron is a cell and gene therapy CDO contract manufacturer. Its predominant shipping needs are small-scale shipments, mostly on dry ice and -80°C, and raw material sampling. As its products are mostly classified as controlled substances, Pharmaron needs to acquire pre-authorisation from the receiving sites when shipping abroad. Full transparency in this process is something the company values highly: “Since starting to work with our specialist logistics partner, we’ve come to understand that gaining pre-authorisation isn’t a six week process (as it was for us with our previous logistics provider). It’s all a matter of communicating clearly with the consignee and being fully transparent with us, the shipper, so that we can work collaboratively to get all the necessary pieces in place for a successful and smooth shipment process,” says Simon Davies – Shipping and Logistics Specialist at Pharmaron.
Knowing where your product is and how it’s doing at all times is key for making the right decisions. If the condition of a business-critical shipment changes anywhere on its journey, sufficient data and information is needed to make the necessary changes. “Knowing that we will get regular updates for expected movements, and receive a notification if something goes wrong, along with suggested solutions, is very important. Being transparent and taking ownership when things do go wrong allows us to know the full picture so that we can make the right decision to correct it. Having access to all of the information available allows us to share it with interested parties, whoever that is – whether they are scientists or our customers,” says Davies.
LocateBio, HistologiX and Emergex all agree on the importance of traceability. As Johnson summarises, “Traceability of product throughout the shipping process is of paramount importance to us.”
Common Factors
Although LocateBio, HistologiX, Emergex and Pharmaron are all different types of companies within the life science industry, all have similar logistics needs that are complex
and multifaceted, requiring specialised knowledge, proactive problem solving, and stringent quality controls. Close collaboration, clear communication and full visibility are essential for ensuring the successful delivery of high-value, temperature-sensitive shipments. Partnering with a specialist logistics provider that understands these unique requirements can significantly impact projects, operations, customer relationships, and ultimately business success.
John Coleman, Business Unit Director for YSDS Life Science, has worked with temperature-sensitive logistics for the past 20 years. His expertise spans cold chain logistics, quality assurance, risk assessment and clinical trial logistics, making him a leading authority in ensuring the successful shipment of temperature-sensitive products.
STEAMING SOLUTIONS FOR ALL INDUSTRIES
John Coleman
Subsection: Nasal & Pulmonary Drug Development & Delivery
Nasal & Pulmonary
In Silico Modelling for Orally Inhaled and Nasal Drug Product Development
Q&A
with Will Ganley, Senior
Specialist, Nanopharm
Can you Explain ‘in Silico Modeling’ and What a PBPK Model is?
In silico modeling involves using computer simulations to predict how drugs behave in the human body. An example is physiologically based pharmacokinetic (PBPK) modelling, which uses drug property, physiology, and biochemical data to predict absorption, distribution and excretion (ADME) processes using pharmacokinetic models.
The outcome is a system of equations employing a 'bottom-up' strategy that begins with modeling each specific process that moves the drug throughout the body. Starting from these foundational elements and progressing to a comprehensive model that integrates data from various sources, such as in vitro tests and clinical trials, we can replicate observed clinical outcomes. By developing a model that assigns significance to input data based on physiological relevance, we create solutions that accurately predict clinical outcomes, resulting in a trustworthy PBPK model.
How is the PBPK Model Applied Specifically to Inhaled Drug Products?
A typical whole-body PBPK model can be regarded as a large flow diagram, which maps mass transport processes across different types of tissue and blood vessels. If all the drug mass starts in the lung after a subject has inhaled, the model shows that the drug mass moves through the rest of the body and is exposed to the different tissues and blood compartments before it is ultimately metabolised and eliminated. For orally inhaled products, we are particularly interested instead in the details of what happens in the lungs.
The lung can be divided into two broad regions: the central region, which encompasses the upper airways where the blood flow goes from the arterial blood through to the venous blood, and the lower peripheral airways where the blood flow
is reversed. We can further segment both regions into more granular compartments.
Within each compartment, the mucus layer is where the drug will typically start its journey as either a completely dissolved system, for example from a nebuliser, or as undissolved particles. For the latter, we directly simulate the particles’ dissolution in the mucus layer before they permeate down into the lung tissue.
In the mucus layer, dissolved drug will passively diffuse into the epithelium, then subepithelium, before entering the blood vessels and being carried away to the rest of the body. Knowing this, it is important that we consider the inputs to the model when developing and designing studies that allow us to generate the model’s input parameters.
There are two key types of input parameters for orally inhaled PBPK models. Deposition data that is obtained through deposition models or imaging data. Commonly used
deposition models include computational fluid dynamics (CFD) or semi-empirical methods (such as the National Council on Radiation Protection and Measurements model). The advantage of CFD models is that computed tomography (CT) scans of healthy volunteers and patient airways can be used in the simulations to generate realistic and subject specific deposition predictions in the lungs.
The other form of input data is dissolution data, typically through in vitro studies. At Nanopharm, we have developed a dose collection system called Dissohale™ that allows us to deposit the lung dose uniformly and measure its dissolution. We then use a modelling approach that allows us to determine the dissolution of deposited particles that are irregular in shape and agglomerated. We take the fitted equivalent spherical representation of these particles and scale down the dissolution medium volume, matching the mucus volume in each region of the lung, which can be input into our PBPK model.
How Do You Use This Information to Generate Data?
Once we have the input parameters for the PBPK model, the next step is to address the questions posed by clinical product development.
To start, we define the problem – whether it be as simple as observing the interplay between two competing processes, through
Figure 1: High Level Lung Structure
Figure 2: Detailed Lung Region Structure
Nasal & Pulmonary
to a complex scenario of simulating a clinical trial. This information allows us to determine the number of lung compartments required, the dissolution model, and whether we will obtain the input parameters from pre-clinical data or literature.
Following on from that, we verify the prediction feasibility to check whether the predictions follow the right time course, or the right curve shape and ensure they are not producing wildly unexpected results. To do this, we recommend gathering clinical data either generated as part of the drug development programme or from literature. For any uncertainty with initial data, we recommend that parameters be refined until the model is producing the data that is expected within the model.
Can You Introduce Simhalation™, and Discuss How it is Being Used in Drug Development?
Simhalation™ is an in-house PBPK model, developed at Nanopharm. Our software will generate a set of equations representing the system and build executable computer code.
The model will ask us for two things: physiological specifications (for example, the lung surface areas of the subjects we are interested in) and the molecular specification (such as log pKa and the rate of hepatic clearance). This allows us to input parameters for a range of patients and a range of formulations or molecules, run our simulations and finally generate the output we are looking for.
The Simhalation PBPK model can be used across the entire product development life cycle for orally inhaled drug products, but first
it is important to understand how different ‘levers’ in the model will affect the exposure of a drug at the site of action.
Factors such as how the angle of insertion from different device variants impacts the deposition within the nose, dissolution rates and mucociliary clearance (MCC) which can be propagated through to the local or systemic exposure in the PBPK model to understand the effect on the exposure of your target drug at the site of exposure. Using this information, we can run multiple simulations for repeated dosing and assess things like inter- and intrasubject variability as well.
How Can You Ensure The PBPK Model is Credible Enough to Provide the Answers You Need?
There are two key concerns I see with PBPK models:
The first concern is that PBPK models are so highly parameterised that users think it is possible to achieve any answer by tuning the parameters accordingly, and secondly, are we
able to justify using the model if we are going to have to perform a clinical study anyway?
Before starting any work, it makes sense to address both concerns by revisiting how the model is going to be used. Are we using it to answer some simple drug product development questions, or are we trying to replace a clinical study or use the predicted data to aid regulatory decision making?
Because these two things require different levels of credibility to be established, it can be useful to understand the model constraints, and the data available.
Even if we do not know the exact value of parameters for a given model – such as the lung epithelial permeability – for the types of molecules we are looking at, we might know their values to within a few orders of magnitude. Applying all the known or estimated model parameter ranges constrains the prediction space, meaning that the scope of possible predictions is quite limited.
Secondly, look at the data available or consider how it could be generated. Can we access data from available literature, or do we need to generate this for ourselves? Once a model is verified for a particular type of molecule, say a lowly soluble and highly soluble molecule, the model validation data should be applicable to other molecules and products of the same type. The regulatory landscape surrounding this is evolving rapidly now and should allow the use of validated modelling “platforms” in submissions soon using frameworks such as the Model Master File, which is current being developed by the United States Food and Drug Agency.
What is useful about this framework is that we can do it as a paper-based exercise before modelling commences to understand what
Figure 3: PBPK Model Building Process
Figure 4: Simhalation Modeling Process
we can and can’t use the model for, whether we need to perform any lab work or if we can already answer our questions of interest.
Can You Share Some Examples Where You Have Used PBPK Modeling For OINDPs at Nanopharm?
There are a few examples of how we’ve used PBPK modeling for orally inhaled drug product development here at Nanopharm.
The Influence of Dry Powder Inhaler Dissolution on Systemic Pharmacokinetics
We wanted to look at the dissolution of dry powder inhalers (DPIs) to understand what the influence of the dissolution of fluticasone propionate is on the systemic exposure of these drug products.
We were able to parameterise the model for fluticasone propionate using data from a literature study to reproduce three different doses by tweaking the dose in the model but keeping all other parameters the same.
Within the study data, three different dry powder formulations of fluticasone propionate had been developed with different aerodynamic and dissolution rates. Using this, we refined our model parameters against one of them and then using the in vitro data that was generated in that study, we extracted the dissolution rates and input that in the model for the other two formulations.
We found that with the PBPK model, we were able to reproduce the shapes of the systemic pharmacokinetic curves by tuning the dissolution parameters and show that dissolution was the driving factor and make credible predictions by using the data. Also, we predicted the systemic exposure of different parameters as if we were developing a product.
The model ended up concluding that you can deliver different doses fluticasone propionate to the lung with different dissolution rates, and if the total dose is fixed, then the total exposure that the subject received will be the same across different formulations but the maximum concentration will be different. This is an important consideration for drug product safety and would be useful information at the start of a new development program.
Nasal & Pulmonary
How Disease State Attribute Influences Local Drug Exposure for Cystic Fibrosis Patients
In another example, we were looking specifically at the delivery of spray dried amikacin sulphate powder to cystic fibrosis patients using a spray dried powder characterised using the anatomical mouth-throat models and realistic breathing profiles. We were very interested in the difference in exposure for both cystic fibrosis patients and healthy volunteers.
We parameterised the model using literature data and then worked to generate CFD deposition data using two lung geometries from our database. When we ran these simulations comparing a cystic fibrosis patient and a healthy volunteer, we found that when the same dose was delivered to these two subjects, the healthy volunteer would receive a lot more of it to their peripheral lung whereas the cystic fibrosis patient would receive a lot more to their central lung. Inputting this data into our PBPK model and running a sensitivity analysis similar, we were interested in disease state differences rather than differences in performance parameters.
When we input some other aspects of cystic fibrosis into the model – such as mucus thickness – we're able to see what effect that might have on systemic exposure. In this case, it effectively dilutes the mucus layer and means less of the drug can get through to the tissue as quickly. But due to the thick and sticky mucus of cystic fibrosis patients, the cilia beating is inhibited resulting in mucus clearance that isn't particularly effective. So, in the model, we can represent this by effectively turning mucus clearance off.
What we found is that this gave us a negligible change compared to having mucus clearance on, and our hypothesis is to this might be the case is that the spray dried amikacin sulphate actually dissolves very quickly and permeates through the lung, before the mucociliary clearance would have been able to have an effect.
How Do Regulators View These Models?
It depends on the regulators that we work with, but in our experience at Nanopharm, we've had a lot of interaction with the US
FDA at various points over the past couple of years.
They have been interested in how simulation approaches might be used to form part of alternative bioequivalence approaches for orally inhaled and nasal drug products. The feedback we have gotten during our interactions has been very positive because they are really interested to see how this technology could be used to reduce the burden on clinical testing and complexity required to get drugs to market.
The FDA are currently working on the concept of a model master file which would allow technology providers like ourselves to keep validation data on file at the FDA for drug developers to reference. This has the potential to reduce duplication of effort and broaden access to modeling and simulation for drug submissions.
I think we're getting there, and over the next few years I expect to see more drug products approved using simulations like CFD and PBPK as a key part of the data package.
Nasal & Pulmonary
Is The Model Able to Make Predictions About Repeated Dosing?
Repeated dosing can refer to multiple doses during a day, or multiple doses over a predetermined time. But with the PBPK model, we can inject doses at any time point during the simulation (Figure 5). If a drug is to be delivered once daily, we can simulate how it might build up in the body over time.
This is particularly useful when considering repurposing a drug that is taken via a different route – intramuscular injection or intravenous infusion, for example – and how that builds up in the body over time. Using the PBPK model, we can try to replicate this with our orally inhaled product before proceeding with clinical development.
How Do You Validate Your Model? Do You Have to Validate Every Molecule?
We need to make sure that the data we produce using these models is robust and valid. The US FDA are quite public about their expectations, and this is making the process of validation easier.
At Nanopharm, we perform validation typically through a risk-based framework called the ASME V&V 40, that has been provided to us by the US FDA. We identify how a model is going to be used and the credibility level is required, so that we can design a validation package based on that.
The validation framework is used to verify that your model solves the numerical problem to an appropriate degree of accuracy, and that your model outputs can be compared to clinical data. Currently we perform this to validate each individual molecule that we investigate.
Recently, the term ‘platform validation’ has been used a lot – this is where you take a simulation methodology, such as PBPK or CFD, and demonstrate that it is valid for typical use cases. This could be where the model works for varying solubilities and permeabilities,
and a combination of the two. Provided that we can show that the model predicts well for the extremes of these cases, we can claim validity for the cases in between too.
Could Simulation Replace Certain Clinical Studies for Orally Inhaled Products?
We can say that PBPK model structures for orally inhaled and nasal drug products are very well established. The main challenge in making them work is generating the required parameters and input data from your preclinical and formulation development work.
There are many different applications for PBPK models in the product development for OINDPs – spanning from early development where you're interested in identifying your critical parameters, all the way through to clinical validation where you're trying to either derisk or replace clinical studies.
Understanding what it is that you are using your model for means that you can establish what validation data are required and what the possibilities for using that model are. It is worth noting that most of the applications that we have seen fall somewhere in between those requiring that robust clinical validation, such as the granting of biowaivers, and those requiring only inputs literature studies, such as addressing low risk product development decisions.
Will Ganley
Will Ganley, Senior Specialist at Nanopharm, an Aptar Pharma company, is a Physical Chemist with a PhD from the University of Bristol, UK. He started his career as a postdoc in Pharmaceutical Surface Science Lab at the University of Bath, UK. His focus was on advancing physical characterisation and simulation techniques for dry powder inhaler formulations, aiming to better understand the connection between physical attributes and delivery to patients. In 2019 Will joined Nanopharm as Head of Computational Pharmaceutics where he led the development of a number of statistical and mechanistic modelling methodologies, notably Nanopharm’s Simhalation PBPK platform. Will is now a senior member of the Science & Technology department at Nanopharm where he supports Nanopharm’s customers in product development and regulatory strategy and manages a portfolio of internal research and development projects aimed at advancing Nanopharm and Aptar Pharma’s scientific excellence in the use of advanced physical characterisation and digital technologies in inhaled and nasal drug product development. Will has authored a number of peer reviewed publications on pharmaceutics, statistics and physical chemistry, has presented his work at a range of international conferences and is a Scientific Advisor for the Drug Delivery to the Lungs conference.
Figure 5: Simulated Repeated Intranasal and Intravenous Dosing
Nasal & Pulmonary
Keep Breathing! SMIs Deliver More SMIs Deliver Higher Single Breath Doses Than Nebulisers
Takeaway Message
MRX004 SMI delivers single breath doses 3 to 5 times higher than nebulisers.
We contend that nebulisers are serial SMIs and only achieve mg delivered doses because of repeat dosing.
In short, Keep Breathing! SMIs deliver more – Q.E.D.
For many years, nebulisers have been the preferred choice for respiratory drug developers needing an inhaler to deliver high doses to the lungs. This preference is largely due to nebulisers' ability to deliver a continuous stream of aerosol that can be inhaled via natural, tidal breathing over several minutes, thereby seemingly facilitating the delivery of high doses. However, a shift in perspective reveals a superior alternative: the soft mist inhaler (SMI), particularly the MRX004. SMIs deliver medication as a fine mist, which can be deeply inhaled in a single breath, offering greater efficiency and convenience (www.mrx004.com).
Delivering High Doses To The Lungs –Nebulisers Are A Legacy Technology
To understand why an alternative to nebulisers is desirable, it is important to recognise their limitations. Nebulisers are bulky devices that require an external electric power source to aerosolise a drug solution. The patient sits and inhales for several minutes, making the use of a nebuliser a time-consuming process. This is compounded by the high level of manual handling required through the assembly of the device, dispensing of the dose, and cleaning and maintenance of the nebuliser. Nebulisers are not portable inhalers, unlike pMDIs, DPIs and SMIs.
Nebulisers are comparatively wasteful. The need for electricity creates a demand for power that is unnecessary for purely mechanical devices like SMIs. In addition to spare parts requiring changing (metering chambers, tubing, masks), the aerosol is continuous, meaning that the patient is only breathing in half of the emitted dose. The other half is exhaled, wasting valuable
drug product and contaminating the air around the patients. Nebulisers rely on tidal breathing to deliver the drug to the lungs; this is characterised by shallow breathing with a breathing cycle of about 15 breaths (inhale/exhale cycle) per minute, each characterised by an inhalation/exhalation cycle, or 6 effective inhalations per minute. This translates into a 4 seconds breathing cycle and an inhalation duration of 2 seconds. A single dose from a nebuliser therefore takes 2 seconds to be delivered; bear that number in mind.
The efficiency of a nebuliser is characterised by the fine particle fraction delivered to the deep lung, i.e. the proportion of droplets below 5 mm. Typically this is about 30%, in some exceptional cases 50%, once the losses from exhalations are discounted.
Are Nebulisers Serial SMIs? – The Challenge Of Comparing SMIs And Nebulisers SMIs, such as Merxin Ltd's MRX004, deliver drugs to the lung by producing a slow-moving mist that patients inhale via a bolus or deep inhalation. Unlike nebulisers, SMIs deliver a discrete metered dose upon activation rather than a continual stream. In the case of MRX004, each dose is delivered in 1.5 seconds. No drug is wasted during exhalation, as an entire unit dose is delivered in a single, deep inhalation, rather than requiring the patient to sit and breathe with the device for an extended period as with a nebuliser.
The precise dosing capability of SMIs allows developers to accurately determine the amount of drug delivered with each activation. This precision has sometimes led to the perception of SMIs as a low-dose format. However, this is only the case if we limit the SMI to single doses. By administering multiple activations in sequence – similar to how nebulisers deliver medication over multiple continuous breaths – SMIs can effectively deliver high drug doses to the lungs. In this context, it is possible to see a nebuliser as a serial SMI, delivering drug over multiple continuous breaths where an SMI delivers doses one breath at a time.
What matters therefore is to compare the single dose delivered by an SMI with
the single dose delivered in one inhalation by a nebuliser.
Thinking in this way, a fair comparison of SMIs and nebulisers requires comparing them breath-for-breath, single dose for single dose, metered dose for metered dose. However, doing so from existing literature is difficult as there is a wide variety of performance characterisation across nebulisers, droplet size, formulation concentration, physical properties, and drug wastage during exhalation, as well as a paucity of single breath/dose measurements. To make the task of accurate comparison even more difficult, the in vitro performance testing requirements for nebulisers and SMIs in the EU and US Pharmacopoeias are not aligned; nebulisers are averaged over the therapy’s duration (a timescale of minutes), while SMIs are assessed on a single spray (a matter of seconds). Traditionally the output of nebulisers is given in mL/min, not corrected for fine particle fraction and breathing cycles, while the output of portable inhalers (pMDIs, DPIs and SMIs) is quoted in weight of the fine particle dose (droplet/particles below 5 μm) of API per metered dose ex-device delivered at a fixed flow rate.
Taking these limitations into account, the best way to make a direct comparison of a nebuliser with an SMI is to calculate the fine particle dose (FPD) of a single breath for each. For an SMI, this value can be calculated from a single activation and is already standardised. For nebulisers, a new calculation is required, and this is what we are going to use in this article.
Let us assume that the entire content of a nebule is delivered in a single sitting, so we can use the following formula to calculate the FPD per breath:
Here, TIME is the time taken for the entire dose to be delivered during nebulisation (usually of the order of minutes). DOSE is the dose contained in the nebuliser (typically 1 mg in a 1 mL nebule). The DOSE/TIME ratio is in fact the output of the nebuliser and is a physical limitation of the nebuliser. FPF is the fine particle fraction of the delivered dose (typically 30% for nebuliser due to
inefficiencies and tidal breathing that do not promote deep lung deposition). DURATION is the length of one breath, which as mentioned above is defined as 2 seconds by the US Pharmacopeia and ISO standards.2
Using this formula in conjunction with available literature, one can calculate a theoretical FPD per breath emitted from a nebuliser. Assuming a nebule concentration of 1 mg/mL and a nebule volume of 1 mL to normalise the calculations, the DURATION of the inhalation is fixed at 2 seconds as per the pharmacopoeia. One can calculate the FPD by varying the TIME and FPF. The theoretical range of nebuliser FPDs are listed in Table 1.
Assuming a 50% FPF and a TIME of 8 minutes, the FPD per breath of an average nebuliser is 1.67 μg.
Nasal & Pulmonary
Where CONCENTRATION is the API bulk concentration in the cartridge and MC is the volume of the metering chamber (15 µL).
MRX004 has a FPF of 70%. If we assume an API bulk concentration of 1 mg/mL, the unit dose per breath is 10.5 µg, or about 5 times the single breath FPD of a nebuliser (cf 1.67 μg).
Theoretical FPDs of MRX004 for a range of FPFs are listed in Table 3.
Recent publications at DDL 2023 on the FPD of MRX004 can be compared with theoretical values. The experimental FPD per inhalation of MRX004 SMI for Dornase Dornase Alfa (2.5 mg/ 2.5 mL) was 6.28 μg4 and Salbutamol 5 mg/mL was 35.1 μg, or 7 μg
inhaler MRX004 SMI is and that SMIs can achieve much higher doses than nebulisers.
We therefore contend that nebulisers are serial SMIs and only achieve mg delivered doses because of the repeat dosing. In short, Keep Breathing! SMIs deliver more.
More work is required to establish a direct experimental comparison between SMIs and nebulisers, but this initial review demonstrates that by viewing nebulisers as serial SMIs and comparing them on a breath-for-breath basis, most available nebulisers struggle to compete with SMIs. If patients just keep breathing and take multiple doses from an SMI, they can benefit from all the advantages of SMIs as a patientfriendly delivery device,1 along with excellent efficiency for delivering high doses to the lungs.
2. US Pharmacopeia. (1601) Products for Nebulization – Characterization Tests.
Table 1: Theoretical FPD per inhalation range of nebulisers as a function of the required nebulisation TIME (fixed by the nebuliser hardware) and a range of FPFs. The nebule concentration is assumed to be 1 mg/mL in a 1 mL nebula.
Nebuliser
MicroBase µSMI 44.3 ± 1.7 0.26 ± 0.01
Aerogen Solo 47. ± 5 0.30 ± 0.04
Philips Innospire Go
PARI eRapid
± 1 0.21 ± 0.02
± 2 0.10 ± 0.01
Most nebulisers are not so efficient; assuming an FPF of 30% and 12 minutes required nebulisation time, the FPD becomes 0.83 μg per breath. This can be compared with the data from Table 2 with effective experimental FPDs for a small selection of commercial nebulisers. The experimental FPDs of these commercial nebulisers is much lower than the theoretical ones calculated in Table 1.
Keep Breathing – SMIs Are The Portable Inhaler Of Choice For High Doses
Let us now turn to the MRX004 SMI. The FPD per breath for an SMI is calculated thus:
when normalised to 1 mg/mL (assuming no effect on the aerosolisation process from the dilution). This is 3 to 4 times higher than nebuliser single breath FPDs.
This demonstrates that MRX004 SMI outperforms nebulisers on a single inhalation basis by delivering a unit dose up to 5 times higher. Add to this that the plume duration of MRX004 is 1.5 seconds but a nebuliser single breath is 2 seconds, and that SMIs deliver their payload to the deep lung when nebuliser aerosols deposit mostly in the upper respiratory track, it is then obvious what a superior
3. Hsiao S et al, “A Novel Contact-triggered Vibrating Mesh Nebulizer: Aerodynamic Performance and Drug Distribution of Suspension Drug Delivered with MicroBase μSMI”. RDD 2018, 2018, Vol 2, pp 459–462.
4. Antoniak D et al, “Inhaled biologics: from nebules to SMIs – Dornase alfa in MRX004”. Poster Presentation at DDL 2023.
Philippe Rogueda is the Chief Business officer and a co-founder of Merxin Ltd. Merxin Ltd makes inhalers. He is an inhaled formulation expert with a global reputation and an enviable track record of developing pMDIs, DPIs, Nebules, Nasal and soft mist inhalers for both generic and originator companies. Philipe trained in Physical Chemistry at the Universities of Bordeaux (France) and Bristol (U.K.), where he obtained his PhD. He subsequently worked for AstraZeneca, Novartis, Actavis before founding Inhalation Asia (Hong Kong) and Merxin Ltd (U.K.).
www.linkedin.com/in/philipperogueda
Table 2: Experimental FPDs per inhalation for commercial nebulisers.3
Table 3: Theoretical FPD range for MRX004 SMI as a function of FPF.
Philippe Rogueda
Nasal & Pulmonary
Importance Of Pre-coloured ABS in Inhalation Medical Devices
Medical device bright and intense colours are relevant for patients because they stimulate a positive attitude to take the drug medication, they help the distinction of different device part and facilitate the way how to use the device itself. In addition, they help to distinguish different types of drugs that are targeting to different types of diseases. Despite colour in medical device applications is such a key property, there are strict regulatory limitations in the type of pigments admitted in the colour formulations, and in their maximum allowed concentrations. When the biocompatibility requirement according to ISO 10993 must be met, not only the base ABS material must be biocompatible but also all the additives compounded with the material, including the colour formulation with all its different pigments. No biocompatible pigments must be directly excluded, and maximum allowed pigment concentrations must not be exceeded. Furthermore, special attention must be given to possible mutual interactions among different pigments, ABS material and other additives. ELIX Polymers eliminates those risks to OEMs, processors and developers providing a medical precoloured ABS material formulation that includes the complete colour recipe and does not need any further material modification. The complete formulation has been fully reviewed and pre-tested to meet biocompatibility according to ISO 10993 and other regulatory requirements. This is a much safer approach for medical OEMs and moulders instead of choosing a medical ABS in natural colour and compound it themselves with a masterbatch colour, during the injection moulding process.
The ABS market offers mostly natural ABS, forcing processors, OEMs and moulders to buy natural ABS and assume most responsibilities and risks, additional quality control costs and regulatory compliance verifications at different development and production stages.
As mentioned, in the case of pre-tested precoloured medical ABS, the material
formulation must not be modified by the customer, reducing responsibilities, supporting the medical device approval process, and avoiding the risk of compounding mistakes during the injection moulding production process. All required medical compliance certifications are already provided by ELIX and are referring to the complete material formulation, including all included colour pigments, additives, and related concentrations.
Regulatory compliance is a pre-requisite for inhalation medical device, but there are also other important quality properties that are strictly related with colour: its homogeneity along the complete device part, its consistency from lot to lot productions, or required colour target contrast in case of surface laser marking (typically for traceability reasons to comply with UDI EU MDR and US CFR regulations). In all these cases colour deviations are not admitted, and a pre-coloured ABS can bring again relevant advantages when compared with natural material post coloured (with masterbatch during the injection moulding process).
Pre-coloured ABS is obtained during a compounding extrusion process, mixing the ABS intermediate materials directly with colour pigments in powder form. Three important elements to consider come into
play at this point to optimise dispersion homogeneity in the material compound: the type of technology used (extrusion compounding), the fact that the colour pigments are in powder form, and the mixing step that happens when the base ABS material is not already a compound but still a “set of ingredients” made of different ABS intermediates raw materials, like for example the ABS rubber phase, which also comes in powder form, and the ABS matrix phase (SAN). This combination of factors is not possible in the case of an injection moulding process, as injection moulding machines are not specifically designed to mix optimally different ingredients together but to melt, feed and inject specific types of materials into a mould in a reasonable cycle time. On the other hand, compounding extrusion machines can handle powder recipes and have a specific double screw design that optimises dispersion homogeneity. Twin screw extruders have the right length, L/D relation and helical elements shape to provide adequate compound mixing and better interaction of all ABS intermediate raw materials, but also better interaction with pigments and additives employed. In this way pre-coloured ABS offers better colour pigment dispersion and homogeneous distribution, which is consistent from lot to lot, and also laser marking enhancers benefit of this optimal compounding ability.
Nasal & Pulmonary
In the case of natural ABS post coloured during the injection process, there is also an additional product needed which does not exist in the situation of pre-coloured ABS: a colour masterbatch. This includes a carrier (an additional material to the ones mentioned until now) and a concentration of colour pigments. The carrier is needed to incapsulate the colour pigments and help the pigment distribution with the natural ABS during the injection moulding process. Due to the mentioned compounding limitations during the injection moulding process, the targets of colour dispersion and lot to lot consistence are more difficult to achieve compared to pre-coloured ABS. Masterbatch carrier compatibility with base material and other additives must be assured, and production personnel needs additional training and competences for colouring with masterbatch and managing possible unexpected situations (e.g. colour differences in different injection cycles, production stop, colour trouble shooting, specific interactions between ABS with
MB carrier or colour formulation). Even when colour targets may be achieved with a masterbatch, there is still the doubt of biocompatibility compliance of the final compound ABS + Masterbatch for the easy reason that no biocompatibility test according ISO 10993 will be conducted on the resulting compound. Such test must be conducted on the final devices for medical approval but not before. Instead, in the case of ELIX pre-coloured medical ABS, the biocompatibility tests are already conducted and passed on the complete compound ABS + colour formulation + additives.
When it comes to sustainable design for medical devices, colour quality turns into an even more critical and sensitive property that needs to be preserved. The demand for new sustainable ABS materials for drug delivery devices applications is growing in the healthcare sector. Due to the risk of cross contamination, medical regulatory compliance cannot be fully fulfilled with mechanically recycled ABS materials. On the other hand, the new scenarios of chemically recycled and bio-based ABS materials are already available and offer the same exact chemical composition and properties of virgin medical ABS, fulfilling the same drug delivery applications along with medical regulations requirements. All the colours that are available in the virgin medical ABS version can be also used in the bio-circular version, guaranteeing not only regulatory compliance, but also the availability of bright and intense colours in chemically recycled ABS formulations. These types of
colours cannot be achieved in any case with mechanical recycled content.
ELIX vision is to be a driving force of the new plastics economy in the next years, participating in the redefinition of plastic waste as raw material. The mission is offering top-of-the-line sustainable solutions in our markets, promoting the transformation of the value chain towards a circular economy model.
The company was the first ABS manufacturer to get the International Sustainability and Carbon Certification (ISCC+ certification) for sustainable materials. The certified raw materials content of ELIX M203FC and M205FC medical grades can be adapted according to the customer OEMs’ sustainability targets.
ELIX medical ABS formulations with chemically recycled and/or bio-based content have been approved by the FDA for the inclusion in the same Drug Master Files (DMF) of standard virgin ELIX medical ABS formulations M203FC and M205FC. This will support an easier transition towards the use of more sustainable ABS medical materials in drug delivery devices in the coming years.
Graduated in management engineering at the "Politecnico di Milano" University (Milan, Italy), Luca has 20 years’ experience in the fields of plastics, composites and OEMs devices. Luca joined ELIX Polymers in 2017 in the position of Business Development Manager for the healthcare strategic sector. Since 2020 he is actively involved in the development of ELIX E-LOOP sustainable solutions and circular innovations, that include a new growing sustainable ABS and blends material portfolio, with chemically recycled, bioattributed, bio-based and mechanically recycled content. Luca wrote several technical articles on behalf of ELIX about specialties and sustainable ABS for medical applications that were published on several renowned medical and pharmaceutical magazines. He lived in different European countries and speaks fluently 6 languages (Italian, English, German, Spanish, French and Catalan).
Luca Chiochia
Nasal & Pulmonary
Analytical Considerations when Re-Formulating pMDIs with Next-Generation Low GWP Propellant Systems
Switching from current pressurised metered dose inhaler (pMDI) propellants to new propellants with lower global warming potential (GWP) could help to reduce the carbon footprint of pMDIdelivered medicines significantly. When looking at the reformulation of an existing product, there is also a need to review the analytical methodology and overall chemistry, manufacturing and controls approach.
With changes being made to the formulation, a suitable validation gap analysis of methods will be required as these methods will underpin the generation of critical data supporting product characterisation, stability and in vitro bioequivalence work. This presents opportunities to address key analytical issues associated with contaminants such as nitrosamines or leachables which represent significant risks to patient safety.
Nitrosamine Formation in pMDIs
Since the EMA and FDA introduced guidelines in 2020 on controlling nitrosamine impurities in medicinal products, industry stakeholders have undertaken extensive risk assessments and testing. These efforts are time-consuming and costly, requiring the development of sensitive detection methods and comprehensive root cause analysis, potentially leading to significant reformulation to comply with new intake limits.
The transition to low GWP propellants in pMDIs provides a chance to proactively address nitrosamine formation early in formulation development. Utilising advanced analytical techniques, such as Liquid Chromatography Mass Spectrometry (LC-MS) and Gas Chromatography Mass Spectrometry (GC-MS) can help streamline efforts to meet regulatory requirements for nitrosamine levels. Tandem LC-MS/MS provides high sensitivity and specificity for detecting APIspecific nitrosamines, even in complex pMDI matrices. GC-MS is effective for analysing volatile, non-API-specific nitrosamines and potential degradation products from
propellants. Utilising both LC-MS and GCMS in early trials enables comprehensive screening, identifies nitrosamine sources and ensures regulatory compliance.
Nitrosamines primarily form through reactions between secondary or tertiary amines and nitrosating agents, like nitrous oxides or nitrites, under acidic conditions. In pMDIs, nitrosamine formation can result from interactions among the propellant system, active pharmaceutical ingredients (APIs), excipients, and device materials. The introduction of low GWP propellants to pMDIs necessitates thorough analytical evaluation to minimise the risk of nitrosamine formation.
Next generation low GWP propellants, such as HFO-1234ze and HFA-152a, differ chemically from traditional propellants. As unsaturated compounds, HFOs can potentially react with other formulation components or degrade over time, forming reactive species that may contribute to nitrosamine formation. The interactions between these new propellants and other formulation components, especially in the presence of moisture, heat, and light, are not yet fully understood.
Each component of the pMDI formulation, including APIs, excipients, and device materials, must be evaluated for its potential to contribute to nitrosamine formation. Therefore, reformulation efforts should include rigorous testing with robust and validated methods at all stages of development, to assess any increased risk when using these new propellants.
Nitrosamines: Environmental and Storage Considerations
The conditions under which pMDIs are stored and used (temperature, humidity, and exposure to light) can significantly influence nitrosamine formation and should be carefully controlled. Accelerated stability studies can help predict how formulations might behave over time and under different environmental conditions, providing essential data for formulation decisions.
The new low- GWP propellant systems could mean changes in recommended long
term storage conditions or packaging to provide the re-formulated product with the necessary stability to remain a viable commercial product.
Nitrosamines: Analytical Strategies and Regulatory Compliance
Reformulating pMDIs with low GWP propellants allows addressing nitrosamine concerns early in the drug development process (something that wasn’t considered when many of the current marketed pMDI products were being developed in the 1990s and 2000s). However, analytical strategies must align with regulatory guidelines and include robust risk assessments that consider all potential sources of nitrosamine contamination, including the supply chain, raw materials, and manufacturing processes.
Extractables and Leachables
The different chemical and physical properties of the new low GWP propellants will mean that adjustments to existing formulations, different drug and excipient combinations and inhaler designs will be necessary to account for the different characteristics of the low GWPs.
Due to the new production processes of the low GWP propellants, a unique impurity profile is to be expected. These impurities must be tested for their concentration and toxicity before the new propellants are used in clinical or commercial products.
The physical and structural properties such as density, vapor pressure, polarity and viscosity (Figures 1–3 and Table 1)1 undoubtedly affect the extraction behaviour and interaction with contact materials or storage containers, resulting in different profiles of extractables and leachables after long-term storage in pMDIs.
In a recent publication from Faucard et al., three fluorinated propellants (HFO-1234ze, HFA-152a and HFA-134a) have been compared regarding their leaching behaviour.
In the study by Faucard et al, a typical pMDI valve, consisting of the following materials: PBT (polybutylene terephthalate), EPDM (ethylene propylene diene monomer) and COC (cyclic olefin copolymer), was exposed to these propellants and tested after intervals of T=0, T=1, T=3 and T=6 months’ storage time with different analytical screening techniques.2 The leaching of target compounds was investigated with multiple analytical techniques, such as GC-FID and LC-UV.
Nasal & Pulmonary
The PBT dimer and trimer was investigated as a representative compound for a nonvolatile leachable. HFA-152a showed the largest increase in leaching of the dimer after 6 months, and the leaching of the trimer was also higher compared to the other blowing agents, but generally at a much lower level.
As representatives for the semi-volatile leachables, the “antioxidants” and “other semivolatile leachables” have been investigated and reported as individual compound-groups. The extracted concentration of each group was investigated per time point. THF was chosen as representative example of a highly volatile leachable.
The total concentration of leached species was below the toxicological limits, but there were differences reported in the leaching behaviour between the individual propellants.
Both the extractable concentration of the individual substance groups and the speed of extraction appear to depend strongly on the structures and physical properties of the gas molecules. If we look at the properties in Table 1, the different structure is also associated with differences in the physical characteristics.
HFA 152a shows a higher extractability of the non-volatiles and THF as highly volatile. While for the antioxidants and other volatiles, the HFA 152a and HFO 1234ze showed a slightly stronger and partially faster extraction behaviour which indicates that more parameters than just the volatility of the leachable compounds play a role for the leaching behaviour. Unfortunately, only sum parameters or specific target compounds were investigated in this study, which prevents a correct correlation of the leaching behaviour with different molecular structures of the leachable compounds. There could be an increased selectivity of the new propellants, which leads to a pronounced leaching of compounds with special structures.
In a pre-study performed by Intertek in Switzerland, a rubber material component used as an inhaler valve gasket was analysed by Thermodesorption Gas Chromatography – Mass Spectrometry (TDS-GC/-MS). TDSGC/-MS is a powerful technique often used for extractables profiling.3 As the thermal desorption approach does not involve a solvent and trapping of compounds, it covers multiple compound classes which could be thermally desorbed including volatiles and many semi-volatile species.
Due to the trapping of desorbed compounds, even species with very low concentration can be detected. Table 2 shows a list of the compounds detected and identified in the rubber seal. Many different compounds from various substance classes were detected, such as hexane, a residual solvent, butylated hydroxytoluene, a stabiliser, partially halogenated rubber oligomers, a by-product of rubber production, and diethyl phthalate, a plasticiser. It is obvious that the rubber gasket contains many small molecules of varying structure and polarity, all of which could be selectively leached out by formulations including the new low GWPs.
As already shown in the previous systematic studies, the extraction behaviour indeed depends on the physical properties and structures of the new low GWPs.2 Since pMDI systems, and especially the valves, are typically composed of many different materials, all with unique additive sets and other low molecular weight impurities, there is some risk of leaching of new compounds or higher levels from the traditional materials used in pMDIs if low GWPs are introduced.
The unique extraction selectivity of the new fluorinated propellants must be investigated in more detail to understand the differences and to rule out a toxicity issue for dedicated compounds. In the so far published literature, only a few targets or sum parameters have been investigated.2 The next step will be a dedicated generic
Adapted from Buttini, F., Glieca, S., Sonvico, F., & Lewis, D. A. (2023). Expert Opinion on Drug Delivery, 20(8), 1131–1143. https://doi.org/10.1080/17425247 .2023.22641841
Figure 1: Structure of HFA 134a
Figure 2: Structure of HFA-152a
Figure 3: Structure of HFO-1234ze
Table 1: Physico-chemical characteristics of propellants used for the formulation of pMDIs.
Table 2: Compounds in a bromobutyl rubber material detected and identified by TDS-GC/MS, sorted according to increasing retention time
extractables study (performed by Intertek in Switzerland) which compares the extraction behaviour of traditional propellants (including also HFA 227) against the new low GWP candidates in further detail. In particular, the effects on a wide range of extractable compounds with different
structures and polarities should and will be investigated in order to assess the risk of pronounced leaching for certain compounds and the potential toxicity problem associated with this.
According to USP <1663>, extractables screening of individual construction materials is helpful to assess complex container closure systems, such as pMDI products.4 Techniques such as thermal desorption are cited as the method of choice to detect a wide range of compounds with little effort, as direct analysis is possible without prior extraction.
This material-specific extractables screening study is important for tracing the extractables detected after incubation of the combined pMDI device. Quantitative screening techniques, such as GC-MS and LCMS, need to be optimised to characterise the extract and capture a wide range of different compounds.
As the extractables content in most plastics is typically very low, even a small variation in the additive, as would be expected in different batches of material, could distort the results and make it difficult to adequately compare the different propellants.
For the planned exemplary comparative study on leachables, it is important to compare the same type of inhaler, ideally from the same batches of material incubated with the different types of propellants.
The pure propellant gas should be used as the incubation medium to also exclude variations in the drug formulation. Alternatively, a suitable control sample or blank sample of the formulation must be stored in an inert container under the same conditions and analysed using the same techniques to exclude effects not caused by the inhalation system.
Nasal & Pulmonary
After the quantitative evaluation of the results, the differences in extraction behaviour and extraction selectivity of the new low GWPs compared to the traditionally used propellants will be investigated peak by peak to prove whether there are major differences for specific compounds, compound classes or whether the extraction behaviour is similar for a wider range of compounds and has a similar selectivity to the old solvents.
After the results of this more substancespecific extractables study are known, a general indication could be given if big differences in extraction behaviour should be expected for the new low GWPs. As a “best case”, there is only a small difference over all classes of extractables and no selective effect in specific plastic ingredients. In such a case, the traditional construction materials could be further used without concern.
Conclusion
The transition to next-generation low GWP propellants in pMDIs is a crucial step toward reducing the environmental impact of respiratory treatments. However, this introduces new analytical challenges in ensuring product safety, particularly concerning nitrosamine formation and leachables. A comprehensive analytical approach is necessary to understand and mitigate these risks. By optimising formulation components and employing advanced analytical techniques, the pharmaceutical industry can successfully reformulate pMDIs with low GWP propellants while ensuring
patient safety and compliance with regulatory standards.
REFERENCES
1. Buttini F, Glieca S, Sonvico F, Lewis DA. Metered dose inhalers in the transition to low GWP propellants: what we know and what is missing to make it happen. Expert Opin Drug Deliv. 2023 Jul-Dec;20(8):1131-1143. doi: 10.1080/17425247.2023.2264184. Epub 2023 Oct 16. PMID: 37767756.
2. Faucard P, Fontaine I, Rives S, Le Corre B, Cannette C, Ferrao J. Leachables Assessment from a New Generation of pMDI Using Low Global Warming Potential Propellants.
Dr. Tino Otte, Managing Director at Intertek Switzerland, is an expert for extractables and leachables studies. He holds a degree in polymer chemistry from the University of Halle/Saale and a Ph.D. from the Darmstadt Technical University. Prior to joining Intertek, he worked with different research, development and manufacturing companies where he served in several functions in product management and development of analytical services. He has over 20 years of experience in GMP regulated environment within multiple areas of product analysis, including method development, validation, and QC.
Respiratory Drug Delivery 2023: 163-168.
3. Scherer, Nicole Marion Doris (2019): Leachable and extractable studies on single-use system technologies in commercial scale drug filling lines. Dissertation, LMU München: Faculty of Chemistry and Pharmacy. 10.5282/edoc.24707
4. United States Pharmacopeia (2024). General Chapter, (1663) Assessment of Extractables Associated with Pharmaceutical Packaging/ Delivery Systems. USP-NF. Rockville, MD: United States Pharmacopeia. DOI: https://doi. org/10.31003/USPNF_M7126_03_01
John McLaughlin
John McLaughlin is a Principal Scientist at Intertek Melbourn, where he specialises in LC-MS analysis for the Biologics team. He manages LC-MS work, covering a wide range of samples with expertise in small molecule impurities in pharmaceuticals and large molecule proteins and oligonucleotides in inhaled formulation products. His focus is on method development and validation in GxP regulated environments. John earned his master's degree in Chemistry with Medicinal Chemistry from the University of Warwick in 2018. Prior to joining Intertek, he has worked in various research organisations, focusing on bioanalysis of small and large molecules.
Chris Vernall
Chris Vernall is the Commercial Director at Intertek Pharmaceutical Services, leading the business development team at the inhaled and nasal drug product center near Cambridge, UK. With a master's degree in Medicinal and Pharmaceutical Chemistry from Loughborough University, he started his career as an Inhalation Scientist, specialising in formulation development and analytical testing, before moving into commercial roles. Passionate about advancing respiratory drug product development, Chris fosters partnerships and collaborations across the industry. He advises companies on technical and commercial strategies and chairs the Inhaled and Nasal Biologics and DNA Forum, an annual conference in Cambridge, now in its third year.
Dr. Tino
Otte
Nasal & Pulmonary
The Preservative Predicament
Nasal sprays have become an important route of delivery for a variety of indications and as their use becomes more widespread, regulatory bodies across the globe have issued various guidelines and recommendations to ensure their safety and efficacy. However, the landscape of these regulations, particularly concerning preservatives, remains complex, leading to confusion and inconsistency of application. This article delves into the current guidance on the use of preservatives in nasal sprays, explores the market trends, and discusses the manufacturing requirements essential for producing safe and effective nasal spray products.
Regulatory Perspectives on Preservatives in Nasal Sprays
In Europe, the regulatory landscape for nasal sprays is influenced significantly by the recommendations of the European Medicines Agency (EMA) and the German Federal Institute for Drugs and Medical Devices (BfArM). Both agencies have recommended the removal of one preservative – benzalkonium chloride (BAK or BAC) – in nasal sprays, citing potential long-term adverse effects on patients. However, it is crucial to note that these are recommendations rather than mandates. The EMA has also updated its requirements for patient labeling on formulations containing BAK, reflecting a growing awareness of preservative-related concerns.1,2
Despite these recommendations, there has been no definitive guidance mandating the development of preservative-free nasal formulations. This regulatory ambiguity has led to a perception that the trend towards preservative-free formulations is more of a regulatory compulsion, rather than an industry-driven tendency.
Similarly, the BfArM has echoed the EMA's stance on BAK, recommending its removal but stopping short of enforcing a mandate. This cautious approach highlights the ongoing debate within regulatory circles about the necessity and feasibility of eliminating preservatives from nasal formulations.
Across the Atlantic, the U.S. Food and Drug Administration (FDA) has provided more detailed guidance on various aspects of nasal spray development. According to the latest FDA guidelines, numerous factors need to be considered during the development program of nasal sprays. These include formulation, container closure systems, manufacturing processes, stability, and controls of critical steps and intermediates. The FDA emphasises that any changes in these aspects can significantly impact the product's ability to deliver reproducible doses to patients throughout its shelf life but stops short of recommending or issuing guidance on removing preservatives in formulations.3,4
The FDA acknowledges that if a formulation is preservative-free, it requires the use of a preservative-free device designed to prevent microbial ingress. This can be achieved using .22 sterile filters and other closure mechanisms in the nasal devices.5
The global regulatory landscape for nasal sprays reflects a cautious and evolving approach towards preservative-free formulations. Although there is a tendency towards such formulations, there is still no clear mandate from regulatory authorities. This ongoing debate underscores the need for continuous review and adaptation of manufacturing practices to meet both regulatory expectations and market demands.
Current Market Landscape for Nasal Sprays
Until recently, nasal sprays have been primarily used for treating allergies, rhinitis and sinusitis, with products ranging from over the counter (OTC) products to prescription (Rx) decongestants, antihistamine and antiinflammatory products. The largest class of commercially available medicines are the nasal corticosteroids, or nasal steroid sprays, with drugs such as fluticasone, mometasone, and triamcinolone.
Since the late 1990s, intranasal drug delivery for systemic indications, especially for rescue therapies, have overshadowed all commercial approvals. This is due to a rapid onset of action of the drug, ease of administration and convenience as opposed to injections, oral delivery or other routes of administration. Indications range from
treatment of opioid overdose, migraine, seizure, anaphylaxis and a variety of other indications. Many of today’s leading Rx nasal sprays, some of which are now OTC, utilise preservatives like benzalkonium chloride and sodium phosphate, to ensure their stability and efficacy over time (Table 1).
In the United States, there are only five products that are preservative-free: Migranal®, Noctiva®, Nayzilam®, Rivive® and Zavpret®. Of these five only one appears to be manufactured in sterile conditions –Noctiva. Specifically, the approved labeling states “nasal sprays are not typically required to be sterile. Noctiva is manufactured under aseptic conditions but becomes non-sterile once in use. The device adequately prevents ingress of bacteria, which is important because the product does not contain a preservative and bacterial contamination could degrade desmopressin”.7 The other products specifically state they are “non-sterile” and/or submitted to the appropriate microbial growth controls during the manufacturing process.8,9,10,11
While it may appear that there is a shift towards preservative-free nasal sprays, it is evident that this trend is more driven by industry innovation and consumer preference rather than stringent regulatory requirements. The perception of a strong movement towards preservative-free formulations needs to be balanced with the reality of regulatory guidance and market reality.
Manufacturing Requirements for Nasal Sprays
Manufacturing nasal sprays involves adhering to several key requirements to ensure product safety, efficacy, and quality. The FDA's guidance on nasal spray manufacturing emphasises the critical need to ensure that the product can deliver consistent and reliable doses with the highest quality throughout its shelf life. This involves rigorous attention to formulation, container closure systems, manufacturing processes, stability, and process controls at every stage of production. Each of these aspects must be tailored and maintained to the specific product requirements to sustain its efficacy and safety.
Developing preservative-free nasal sprays introduces additional complexities, which
Nasal & Pulmonary
Reference Drug Molecule Indication Preservative Year Approved
Flonase® and generics fluticasone propionate perennial allergic or nonallergic rhinitis benzalkonium chloride 1994 OTC in 2003
Nasarel® flunisolide seasonal or perennial rhinitis benzalkonium chloride 1995 (discontinued)
Afrin® and generics oxymetazoline hydrochloride relief of nasal congestion
chloride 1966 OTC in 1975
Nasacort® and generics triamcinolone seasonal and perennial allergic rhinitis benzalkonium chloride 1996 OTC in 2013
Nasonex® and generics acetonide seasonal allergic or perennial rhinitis benzalkonium chloride 1997 OTC in 2022
Migranal® and generics mometasone furoate acute treatment of migraine none 1997
Imitrex® and generics sumatriptan acute treatment of migraine Sodium phosphate 1997
Rhinocort® and generics budesonide seasonal or perennial allergic rhinitis
Zomig® and generics zolmitriptan acute treatment of migraine
sorbate 1999 OTC in 2015
phosphate 2003
Dymista® and generics azelastine hydrochloride and fluticasone propionate seasonal allergic rhinitis benzalkonium
Narcan® and generics naloxone opioid overdose
Xhance® fluticasone propionate chronic rhinosinusitis without nasal polyps.
Noctiva® desmopressin nocturia due to nocturnal polyuria
Tosmyra® sumatriptan acute treatment of migraine
Spravato® esketamine hydrochloride treatment resistant depression and depressive symptoms in adults with MDD with acute suicidal ideation or behavior
midazolam
diazepam
2015
nalmefene
Neffy® epinephrine emergency treatment of allergies/ Anaphylaxis
Table
require adherence to stringent guidelines to ensure patient safety. These formulations have a unique set of requirements that pose significant challenges. These challenges include the following: advanced formulations techniques, sterile environmental handling, processing, controls, and the use of delivery devices capable of maintaining sterility. Additionally, these formulations require thorough risk assessments and validation studies to demonstrate that the product can maintain sterility and efficacy throughout its shelf life.
Nasal sprays can be confidently manufactured utilising a preserved formulation or a preservative free formulation with a sensible approach to risk. The following techniques minimise bioburden to ensure one can produce a product of the highest quality.
• Aseptic Techniques: Mimicking conditions before and during the transfer of formulations into nasal delivery devices is critical. This includes the use of single-use disposable vessels and tubing during compounding and filling processes.
• Low Bioburden Grade C Environment: Manufacturing in a controlled Grade C environment helps minimise microbial and particulate contamination. This baseline allows for limited to no environmental impact to the formulation.
• Dedicated Single-Use Product Contact Pieces: Using dedicated single-use contact pieces wherever possible further reduces contamination risks.
• Closed System and Sterilisation Filtration:
The manufactured solution poststerilisation filtration remains within a sanitised system (hold vessel, single use tubing, filling needles) until moment of fill. Ultimately, limiting exposure to the open Grade C environment to seconds prior to stoppering/capping. between filling and stoppering/capping of finished product.
• Personnel Practices: Personnel must adhere to strict aseptic techniques, including the use of personal protective equipment (PPE) and rigorous cleaning practices.
• Terminal Sterilisation: Techniques such as radiation can
1. Rx and OTC Nasal Spray with Type of Preservative in the Formulation6
Nasal & Pulmonary
be used to inactivate any potential microbial contamination post-filling.
Cost Implications and Benefits of Low Bioburden Manufacturing
Drug development companies need to evaluate how the chosen production pathway of their drug candidate and the related costs of such will impact its commercialisation success. For example, the cost implications of manufacturing nasal sprays, particularly preservative-free formulations in sterile conditions, are substantially higher than those of preserved formulations and will lead to a higher cost of goods (COGS) and can directly impact the profitability of the product for commercialisation. Sterile manufacturing of nasal products is also not as widely available, thus limiting third-party manufacturing options. Low bioburden manufacturing, as before, is generally less expensive than sterile manufacturing, offering a cost-effective alternative while still ensuring high levels of safety, quality, and efficacy.
Adopting low bioburden manufacturing techniques can provide several benefits for drug companies. It allows them to meet regulatory and market demands for preservative-free products without incurring the higher costs associated with sterile manufacturing. Also, it supports producing high-quality nasal sprays safe for acute and chronic use by patients.
Conclusion
In the evolving landscape of nasal spray manufacturing, regulatory recommendations and market trends are shaping the development of preservative-free formulations. While the debate on preserved or preservative-free continues, the industry must assess what is best for the drug candidate considering current recommendations and consumer preferences. As a contract development and manufacturing organisation (CDMO), our role is to support innovators in navigating these complexities, offering expertise in both traditional and preservative-free nasal spray manufacturing. By leveraging advanced aseptic techniques and ensuring low bioburden levels, CDMOs help clients achieve their formulation goals while maintaining the highest standards of safety and efficacy.
The future of nasal sprays will see continued innovation and adaptation as regulatory bodies refine their guidelines and the market evolves. In the meantime, a comprehensive understanding of current requirements and best practices remains
essential for successful product development and patient care.
REFERENCES
1. Riechelmann H et al, “Nasal toxicity of benzalkonium chloride”. Am J Rhinol, 2004, Vol 18(5), pp 291–299.
2. European Medicines Agency. Questions and answers on benzalkonium chloride used as an excipient in medicinal products for human use. Accessed online March 14, 2023 at: ema. europa.eu/en/documents/scientific-guideline/ questions-answers-benzalkonium-chlorideused-excipient-medicinal-products-humanuse_en.pdf
3. Guidance for Industry Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products – Chemistry, Manufacturing, and Controls Documentation. Center for Drug Evaluation and Research (CDER) July 2002.
5. Ehrick JD, Shah SA, Shaw C, Kulkarni VS, Coowanitwong I, De S, Suman JD. Considerations for the Development of Nasal Dosage Forms. Sterile Product Development. 2013 Jun 22;6:99–144. doi: 10.1007/978-1-4614-7978-9_5. PMCID: PMC7120012. article
6. The above information was compiled using the information from each respective product on the Drugs@FDA database: Drugs@FDA: U.S. Food & Drug Administration: Drugs@FDA [Internet]. Silver Spring, Maryland. 1938-present [accessed August 7, 2024]. Available at: www.fda.gov/drugsatfda
7. Acerus Pharmaceuticals. Noctiva (desmopressin acetate) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata. fda.gov/drugsatfda_docs/labe/2027/201656lbl. pdf. Revised March 20, 1027. Accessed August 07, 2024.
8. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 211321 [Memorandum, May 14, 2019]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2019/211321Orig1s000ChemR.pdf.
9. Center for Evaluation of Drugs and Research. Chemistry Review, Application Number: 20148 [Memorandum, October 6, 1992]. https:// www.accessdata.fda.gov/drugsatfda_docs/ NDA/97/20148_Migranal_chemr_EA.pdf
10. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 217722 [Memorandum, June 13, 2023]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2023/217722Orig1s000ChemR.pdf
11. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 216386 [Memorandum, May 12, 2022]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2023/217722Orig1s000ChemR.pdf
Carolyn Berg
Carolyn Berg has over 25 years of experience in pharmaceutical sales, marketing and business development. She is currently Vice President, Business Development for Catalent's inhaled drug delivery solutions where she is responsible for growing the Inhalation business in North America and Europe. Carolyn holds a Bachelor of Journalism in Public Relations and a Bachelor of Arts in French from the University of Texas at Austin, and an MBA from the University of South Carolina.
David Wilcox
David Wilcox, Director of Inhalation Product Development, has over 25 years of pharmaceutical industry experience, with substantial expertise and knowledge in formulation, development, and manufacturing of orally inhaled and nasal drug products. In his role, he is responsible for providing operational, scientific, and technical leadership for inhalation product development activities. David holds a Bachelor of Science degree in Chemistry from Wofford College in Spartanburg, South Carolina.
Mark Ignaczak
Mark Ignaczak, Director of Innovation and Partnerships for Nasal Delivery at Catalent, has over 18 years of pharmaceutical experience and over 15 years of direct experience with nasal spray products. He has worked on more than 75 nasal programs at varying stages of the project lifecycle and held various roles within bioprocessing engineering, product development, supply chain and nasal delivery program management and strategy. He holds a Bachelor's degree in biochemical engineering from Rutgers University, New Brunswick, US.
spray drying
encapsulation & blistering
Catalent is your preferred CDMO for pulmonary and nasal delivery, with clinical to commercial-scale development and manufacturing capabilities for dry powder inhalers, unit-dose, bi-dose and multi-dose nasal sprays.
With over 30 years of inhalation product experience and state-of-theart facilities in Boston, Massachusetts and Morrisville, North Carolina, Catalent can handle the most complex projects and help bring your inhaled therapy to market, faster.
characterization
development & manufacture of spray dried & carrierbased powders
spray drying expertise for small & large molecules
scale up from lab to psd-1, psd-4 & psd-7
nasal liquid spray & powder
unit-dose, bi-dose & multidose filling & assembly
Nasal & Pulmonary
Development Approach for a High-performance Capsule-based DPI Device
The pulmonary route is gaining increasing attraction not only for low-dose locally acting therapies, e.g. Asthma and COPD, but also for systemic applications often require higher doses or new formulation technologies.
High-performance devices need to be developed to accommodate the requirements to deliver these new drugs/ formulations efficiently. To ensure the best performance, the development of the formulation and the device should go hand in hand.
Dry Powder inhaler devices are used to deliver the medication in powder form to the lungs via oral inhalation. Medication in powder form can be filled in either a capsule, blister, reservoir or cartridge based on the drug product configuration. The device can be either an active or a passive device. In many cases, capsule-based inhalers are preferred solutions for new applications because they offer several interesting features, e.g. possibility to use a wide range of different APIs and doses generally ranging from 5 mg to 50 mg. Further advantages include ease of use and a good feedback mechanism for the patient.
Due to the complexity and cost of manufacturing multidose DPI devices, pharma companies want to minimise cost and risks in the new applications by using a simpler and more affordable device.
For some new applications, e.g. pain relievers or antibiotics, a reusable dose is beneficial; for others, e.g. vaccines, a disposable single-use is required. In both cases, capsule-based inhalers can be a good solution.
Covid 19 has made it imperative that an easy-to-use vaccine is available worldwide and remains stable at ambient conditions. Research continues to happen to deliver the vaccine by the inhalation route.1
However, there are some drawbacks associated with current capsule-based DPI devices, e.g. Low performance is an inherent
feature of many capsule-based DPI devices on the market. Even newly developed integrated solutions for antibiotics, e. g, Tobramycin, do not reach a higher FPF than 35%. In order to achieve the therapeutic dose of the antibiotic, the patient has to inhale 4 capsules a day.2
Many of the strategies for overcoming the inherent challenges of most existing devices are done by focusing on improved dispersion properties of the formulations. Not a lot of attention was paid to improving the devices either by technological advancements of the device performance (higher deagglomeration and less retention of powder in the device/ capsule) or on increasing patient adherence for better handling and thus, higher deposition in the deep lungs.3
The novel Presspart DPI device addresses the above problems and challenges of capsule-based inhalers. The most important features of an ideal capsule-based DPI device are, the ease of use with a good feedback mechanism to the patient coupled with a cost-effective design. In addition, dose delivery with high reliability and consistency, and high-performance efficiencies for a wide range of applications are desired.
Device Engine and Development Approach
While developing the novel capsule-based inhaler, the main focus was to create a high-efficiency engine to de-aggregate and aerosolise the powder formulation: a medium resistance and relatively flowrateindependent device. Several prototypes have been tested and studied throughout the development journey.
The airflow through the capsule chamber was designed so that the capsule oscillates. This causes impaction of the capsule within the capsule chamber leading to the breakdown of big powder aggregates inside the capsule and, consequently, an efficient release of the powder from the capsule.
After the initial de-aggregation of the powder in the capsule, the powder evacuates from the capsule and reaches the swirl chamber. Further de-aggregation takes place due to the shear forces caused by the turbulent airflow created in the swirl chamber and the impaction of the powder on the walls
of the swirl chamber. The combination of the capsule movement and powder flow in the swirl chamber leads to a highly dispersed powder. This powder exits into the tubular mouthpiece through the mesh.
The powder de-aggregation and dispersion potential in a device are crucial parameters to achieve high efficiencies of the device. Various techniques can be used to study airflow and powder de-aggregation behaviour within a device. Here, a combination of a simulation with CFD (computational fluid dynamics) modelling in a steady state and an experimental approach was used to develop the new device.
In one experimental design CFD was used to study the airflow structures within the device. Two prototypes were tested prototype 1 (Figure 1a) without a flow straightener and prototype 2 (Figure 1b) with an integrated flow straightener in the mouthpiece. The test of fine particle assessment by NGI was conducted on the prototypes. As seen in Figure 1c, the CFD data of Prototype 1 exhibits a swirling flow that proceeds out of the mouthpiece, reflecting a higher deposition and a swirl pattern observed in the induction port of the NGI. However, with the introduction of the flow straightener in Prototype 2, the CFD data shows a high reduction of the swirl exiting the mouthpiece (Figure 1d). This is also in correlation when comparing CFD data with our NGI data (Figure 2). There was a higher deposition in the induction port at both the flow rates for prototype 1, and it was significantly higher @ 30 LPM(p<0.05). The statistical analysis performed using an independent Student t-test gave probability values of less than 0.05 which was considered as a significant difference. The flow straightener reduced the swirl of the airflow exiting the device, thereby reducing the deposition in the induction port. There was no significant difference in Fine Particle Fraction (FPF) between the two devices indicating that major deaggregation occurs within the device. CFD proved to be a valuable tool for studying the air flow dynamics of the dry powder inhaler. NGI testing provided the supporting data and visual observation of the drug deposition in the induction port, which indicates/simulated probable oropharyngeal deposition.
Nasal & Pulmonary
A modular engine set-up during development allowed for adapting small changes in the prototypes. Significant improvements could be achieved by changing different engine parameters. Figure 3 demonstrates the improvement in performance of the test data from Prototype 1 to 4. The NGI testing demonstrated the reduction in deposition in the Induction Port and pre-separator stage and increase in the lower stages of the impactor thereby increasing Fine Particle Fraction as we moved along our design development.
Resistance
The intrinsic resistance of a device is often discussed controversially in the literature. For Asthma/COPD applications, often lowresistance devices seem to be more beneficial for the patient struggling to achieve high flow rates. However, low resistances of a device often come along with a lower deposition of the fine particles of the API in the deep lung. Medium resistances have the big advantage of creating a deeper lung deposition.4 The inspiratory resistance of the new Presspart device was determined to be designed as a medium resistance device.
Another development target was to achieve a relatively flow rate-independent device. The airflow path was designed and optimised such that there was no significant difference in the fine particle fraction for flow rates ranging from 30ltr/min to 90ltr/ min (Figure 4).
Evaluation of Different Formulation Types
Two types of formulations were studied a binary mixture and spray-dried engineered particles.
Budesonide a glucocorticoid is known for its property of its sticky nature, and its difficulty to de-aggregate was chosen as the candidate formulation. A binary mixture of a marketed formulation of lactose and budesonide 200 mcg per dose with a capsule fill weight of 25mg was selected to test the prototype. In addition, the performance was compared to a marketed Plastiape RS01 equivalent device. The novel Presspart device exhibited a high fine particle fraction compared to the marketed Plastiape RS01 device confirming the engine's efficiency (Figure 5).
In a study in a corporation with the University of Bonn, the performance of the novel DPI Device was assessed by testing a spray-dried Rifampicin formulation. More detailed information on that can be
Figure 1a–d: Schematics of DPI device prototypes: (a) Prototype 1 without flow straightener and (b) Prototype 2 with flow straightener configuration. CFD simulations for (c) prototype 1 and (d) prototype 2 configuration.
Figure 2: Comparison of NGI deposition of Prototype 1 and Prototype 2 at 30 lpm.
Nasal & Pulmonary
found in the recently published article.5 The formulation was used to benchmark the Presspart device performance against devices already well introduced in the market. As shown in Table 1, the FPFs generated are highest for the PP device when compared to the commercially available standard devices Handihaler and RS01 equivalent.
Conclusion
Several key factors influence the performance of the dry powder inhaler device. The most important ones are the patient's inhalation technique, device handling and the device engine i.e. the efficiency of de-aggregation mechanism of the device. Targeting a medium resistance and a relatively flow rate independency were critical factors within the device development approach.
Various complementary techniques were used to study airflow and powder
Figure 3: NGI Deposition profile of a Salbutamol Sulfate carrier-based formulation with different prototypes of the Presspart capsule-based device.
Figure 4: Fine Particle Delivery of Budesonide 200 µg Powder for Inhalation at 2, 4 and 6 kPa.
Figure 5: Comparison of NGI deposition profile of Presspart DPI and RS-01 Variant with Budesonide 200 µg Powder for Inhalation.
Table 1: Comparison of FPF at different Flowrates for Presspart (PP) Device, RS01 equivalent and Handihaler for the Rifampicin particle-engineered formulation.
Nasal & Pulmonary
de-aggregation behaviour within a device. Combining different development approaches of fast-paced prototyping, CFD technique and laboratory data for verification can effectively develop cost-effective high-performance DPIs for the inhalation market. These techniques enabled H&T Presspart to develop a higperformance prototype independent of the formulation type tested (carrier-based as well as an engineered formulation).
REFERENCES
1. MIT technology review September 8, 2022
2. Geller DE, Weers J, Heuerding S, “Development of an inhaled dry-powder formulation of Tobramycin using PulmoSphere™ technology”. J Aerosol Med Pulm Drug Deliv, 2011,24, pp 175–82.
3. Hoppentocht M, Hagedoorn P, Frijlink HW, de Boer AH, “Technological and practical challenges of dry powder inhalers and formulations”. Advanced Drug Delivery Reviews, 2014, 75, pp 18–31.
4. Dal Negro RW, “Dry powder inhaler and the right things to remember: a concept view”. Multidisciplinary Respiratory Medicine,2015, pp 10-13
5. Groß R, Berkenfeld K, Schulte C, Ebert A, Sule S, Sule A, Lamprecht A, „State of the Art in CapsuleBased Dry Powder Inhalers: Deagglomeration Techniques and the Consequences for Formulation Aerosolization”. Pharmaceutics, 2022, 14, pp 1185
Ameet Sule, Director, Inhalation Product Technology Centre, H&T Presspart, is a pharmaceutical professional having worked in the industry for more than 20 years, specialising in the development of inhalation products and devices. Mr Sule works closely with H&T Presspart’s customers around the globe, understanding and mitigating the frontend development challenges of new and generic products for inhalation drug delivery.
Sunita Sule
Sunita Sule, Inhalation Product Consultant, is a pharmaceutical professional who has over 25 years of experience in the pharmaceutical industry, with core expertise in formulation and device development of inhalation products.
Mirjam Kobler, PhD, is currently Global Business Development Manager for H&T Presspart. Dr. Kobler oversees the management and business development activities for H&T Presspart’s DPI device portfolio. Before joining H&T Presspart Dr. Kobler worked at Meggle, where she headed the R&D Department of Meggle’s Excipients and Technology business group. Dr. Kobler’s background includes seven years of experience in various areas of lactose excipients, especially for DPIs.
Ameet
Sule
Mirjam Kobler
Advertisers Index
Page 83
Page 21
Page 3
Page 45
Page 25
Page 95
Aptar
Aurena Laboratories
Austria Wirtschaftsservice Gesellschaft mbH
Biopharma Process Systems Ltd.
Biosynth Ltd
Catalent Pharma Solutions
IFC EyeC GmbH
Page 5
Page 47
Page 61
Page 76
FUJIFILM Wako Chemicals Europe GmbH
FUJIFILM Wako Chemicals U.S.A. Corporation
Gerresheimer AG
H&T Presspart
Page 15 Kahle Automation
Page 69 Krautz Temax
IBC Merxin Ltd
Page 23 Nemera
Page 19 Nipro
Page 17 Novo Nordisk A/S
Page 35 Owen Mumford
Page 43
Page 57
Page 39
PCI Pharma Services
Senglobal Ltd.
Stäubli International AG
BC Stoelzle Glass Group
Page 78
Page 33 & 75
UPC Cambridge Ltd
Valsteam ADCA
Page 71 The Wool Packaging Company Limited (Woolcool)
Page 27
Subscribe today at www.international-pharma.com or email info@senglobalcoms.com
Ypsomed AG
I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry
IPI is also now active on social media. Follow us on: