IBI - International Biopharmaceutical Industry Journal
Navigating the Future: Insights from Leading Indian CDMOs on the Impact of the BIOSECURE Act
5 Key Considerations for Designing Relevant Cell-Based Assays to Screen Antisense Oligonucleotides
Advancing Gene Therapy Research with Trapped Ion Mobility Spectrometry
Accelerating Drug Discovery: High Throughput Screening with C. elegans
Sponsor Company:
MISSION
#OneStopShop for stability and release testing
Since 1995 A&M STABTEST has been a household name for highest quality analytical services “Made in Germany” Our 340 strong team of dedicated specialists and stateof- the - art equipment get your projects done on time and in budget. We are your #OneStopShop solution for:
• Stability and release testing of small molecule and biological drug substances and drug products
• Analytical development and method validation
• mRNA -LNP ready methods acc. to draft USP guideline
• Dedicated cell-based potency assay group
• ICH- stability storage and logistics service
• Laboratory dedicated to inhaled drug testing (DPI, MDI, Nebulizer)
• GMP compliant LC -MS service for extractable and leachable testing, protein characterization, identification of unknowns and quantification of impurities and excipients
• Functional testing of container closure systems, prefilled syringes and injection devices (CCIT, force measurements)
• Analysis of ATMPs
DIRECTOR: Mark A. Barker
INTERNATIONAL MEDIA DIRECTOR: Anthony Stewart anthony@senglobalcoms.com
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IBI will be published in Autumn 2024.
ISSN No.International Biopharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
06 Navigating the Future: Insights from Leading Indian CDMOs on the Impact of the BIOSECURE Act and the Shift from Chinese CDMOs
The pharmaceutical and biopharmaceutical sectors are undergoing transformative shifts driven by geopolitical tensions and regulatory changes. Among these changes, the BIOSECURE Act and the 2032 decoupling deadline from Chinese CDMOs stand out. Chloe Eurpides of IBI speaks with Ramesh Subramanian of Aragen Life Sciences, Himanshu Gadgil of Enzene Biosciences and Alex Del Priore at Syngene, on how these companies are adapting to the new landscape and positioning themselves as competitive alternatives to Chinese CDMOs.
REGULATORY
& COMPLIANCE
08 On the Cutting-edge of Drug Metabolism
Determining metabolic fate is crucial in drug discovery. Horror stories are often shared around expensive, late-stage failures due to unexpected metabolism. Many challenges crop up, including poor metabolic stability, unforeseen drug-to-drug interactions and the formation of reactive or toxic metabolites. Dr. Tamsin Mansley of Optibrium Inc. breaks down all you need to know about the challenges of drug metabolism, and the in silico approaches to overcome these problems.
RESEARCH / INNOVATION / DEVELOPMENT
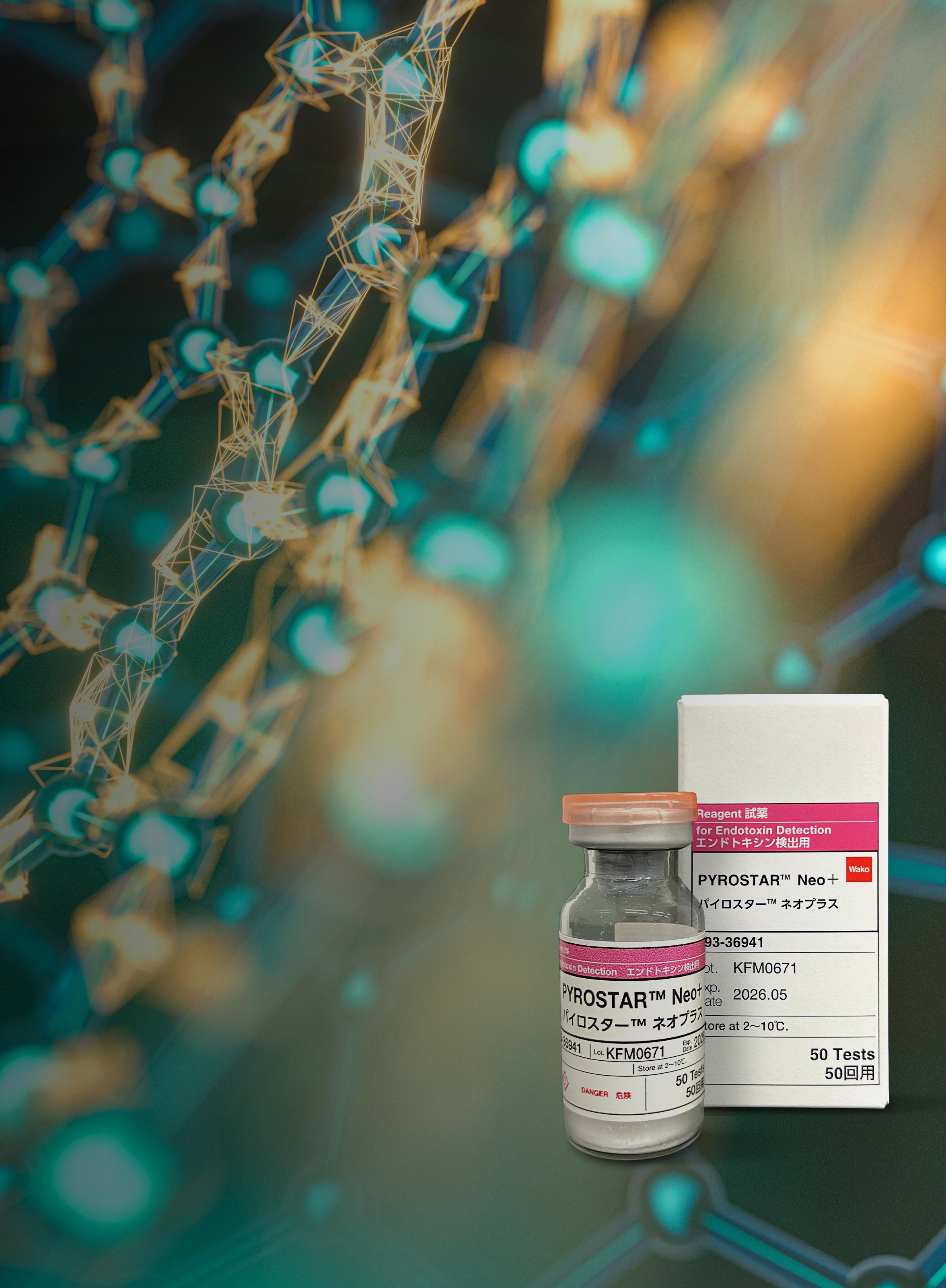
14 The Expanding Need for Endotoxin Testing
Since the acceptance of the gel clot technique as a compendial test in 1980, endotoxin testing has become a crucial anchor for the expansion and development of safe pharmaceuticals and medical devices. With recent developments, sustainable testing reagents have only ensured the permanency of the Bacterial Endotoxin Test for the next generation of pharmaceutical and medical advancements. Timothy Francis at FUJIFILM Wako Chemicals U.S.A. Corporation provides an overview of the areas that are experiencing growth in their need of endotoxin testing.
16 5 Key Considerations for Designing Relevant Cell-Based Assays to Screen Antisense Oligonucleotides
With newly developed antisense oligonucleotide (ASO) molecules, early efficacy and toxicity assessment is crucial to prevent costly failures in later stages. Noelia Muñoz-Martín and Shushant Jain of Ncardia explore the 5 key steps you need to consider how to successfully develop an ASO in vitro screening assay and the
extra-advantage of designing a physiologically relevant assay that can shorten your timelines.
TECHNOLOGY
24 Advancing Gene Therapy Research with Trapped Ion Mobility Spectrometry
Gene therapy presents a promising future for disease treatment across a broad spectrum of diseases. Its potential to revolutionise biopharmaceutical research offers significant hope for patients battling genetic disorders. Susumu Uchiyama and Dr. Satoshi Oshiro of Bruker Japan K.K. explores how trapped ion mobility spectrometry (TIMS) is advancing research into gene therapy products and progressing the development of new treatments for diseases that were previously considered untreatable.
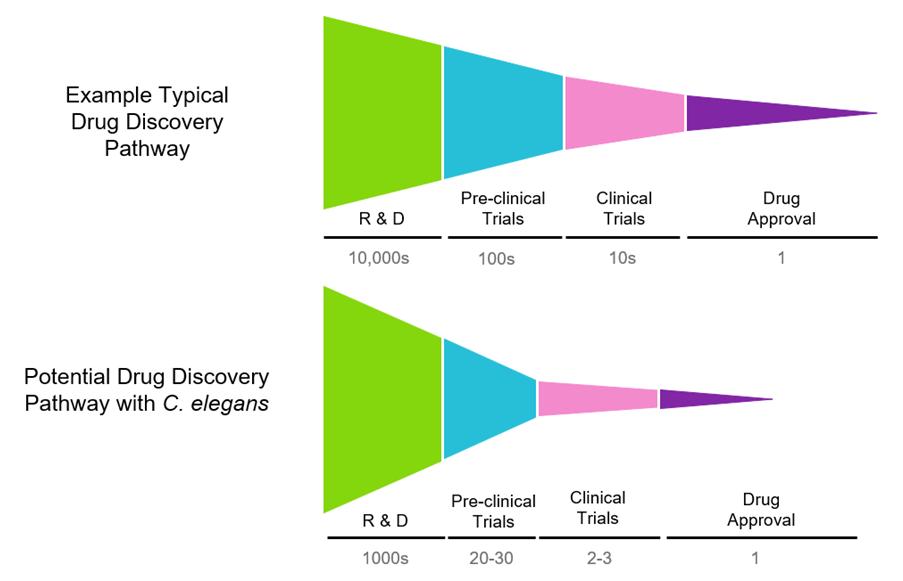
28 Accelerating Drug Discovery for Diseases of Ageing: In Vivo High Throughput Screening with C. elegans
High throughput screening (HTS) emerged in the 1990s as a significant development, allowing for the swift and effective identification of active compounds and deeper insights into biological pathways. This process involves testing large libraries of compounds against biological targets to identify potential drug candidates. David Weinkove of Magnitude Biosciences, explores recent advancements in HTS, highlighting emerging trends, technological innovations, and the potential of using C. elegans as a scalable in vivo model for more effective drug discovery.
LOGISTICS & SUPPLY CHAIN
34 Packaging: The First Line of Defence in Cell and Gene Therapy Logistics
Packaging has a multi-faceted role in supporting cold chain logistics to ensure transit times are met and the shipment is protected during handling. Unique packaging solutions have been developed to transport anything from sensitive biological materials during a clinical trial to life-saving medicines and cell and gene therapy treatments.
Christopher Good of Biocair explains how temperature-controlled packaging plays a vital role in cell and gene therapy logistics.
APPLICATION NOTE
12 Safeguarding your IP: Thermo Fisher Scientific Pharma Services' Global Commitment to Confidentiality
Intellectual property (IP) rights and confidentiality protections play a critical role in biopharmaceutical development and manufacturing. Appropriate protections enable developers to drive innovation, safeguard investments, and accelerate the development of new drugs and therapies, while creating a favourable environment for research, development, and commercialisation. Experts at Thermofisher Scientific explains that partnering with a CDMO with the global expertise to navigate complex landscape requirements, and with global frameworks in place to standardise protections, can alleviate potential challenges for drug developers and IP owners.
20 Understanding Roller Compaction in Pharmaceutical Development
Roller compaction is a critical process in pharmaceutical manufacturing, offering numerous benefits, especially for moisture-and heat-sensitive compounds. David O'Connell of PCI Pharma Services explores the advancements in dry granulation and the fundamentals of roller compaction, presenting it as a viable alternative to traditional wet granulation for producing oral solid dosage forms like tablets and capsules.
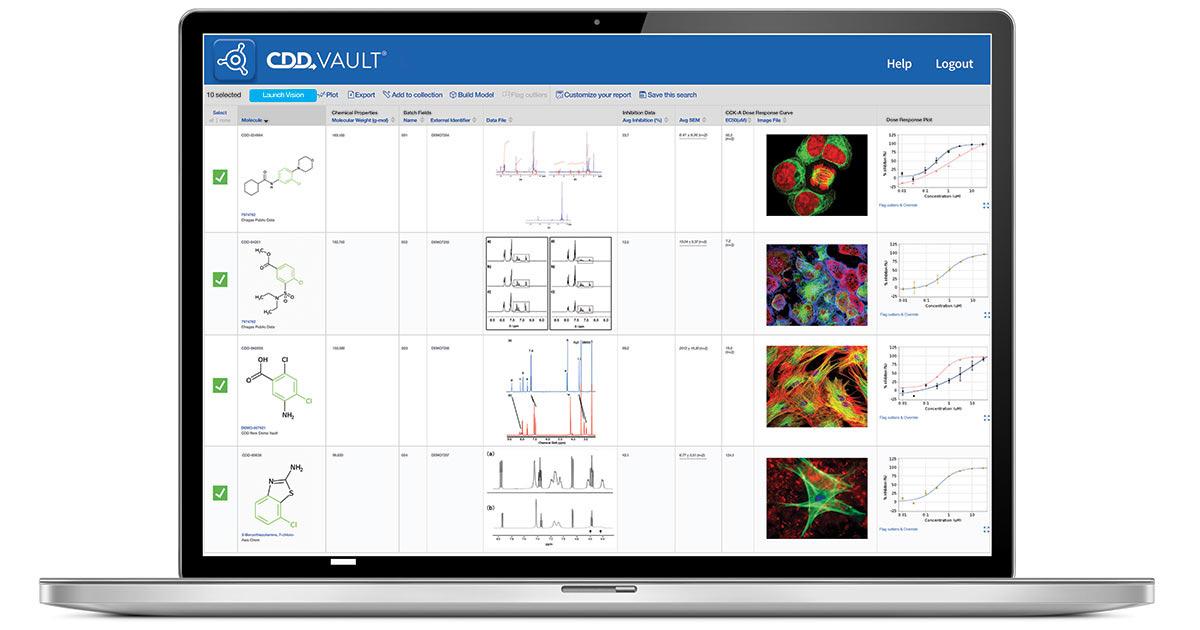
32 Isosterix Uses CDD Vault to Expedite Drug Discovery & Securely Collaborate
Isosterix was created to develop small-molecule inhibitors of KAT6A, an epigenetic oncogene that is implicated in multiple cancers. Roopa Rai of Isosterix shows how effective the deployment of Collaborative Drug Discovery (CDD) Vault is, at hosting drug discovery informatics and securely managing both internal and external biological and chemical data.
GREAT FLUIDIC SOLUTIONS
BIOTECH LIQUID µFLOWMETER
Continuous Flow Measurement 10 nl – 80 µl/min
Software compatibility with leading CDS
Efficient degassing at µl/min flow rates
Widest chemical compatibility
Welcome to the 2nd issue of IBI. The pharmaceutical and biopharmaceutical sectors are undergoing transformative shifts driven by geopolitical tensions and regulatory changes. Among these changes, the BIOSECURE Act and the 2032 decoupling deadline from Chinese CDMOs stand out, prompting companies to reassess and revamp their supply chain strategies. Companies must have a strategy in place to balance this wider range of demands, which are often competing, or face being cornered on all fronts. A report by the Medicines Patent Pool (MPP), in partnership with Boston Consulting Group (BCG) released in May 2024, highlights how voluntary licensing advances global health while being commercially viable for biopharmaceutical companies. The study, co-funded by the World Intellectual Property Organisation (WIPO) along with the Government of Canada presents evidence that voluntary licensing is an effective mechanism for addressing global health disparities, especially in low and middle-income countries. Using both qualitative and quantitative analyses, it illustrates how voluntary licensing advances access to medical products across multiple geographies while offering economic benefits for biopharmaceutical companies.
Advancement in precision oncology is driven by a virtuous cycle of innovation – innovations in research and clinical tools, technologies, data, and services contribute to the discovery and development of novel therapies, creating value that gets re-invested in novel tools, technologies, data, and services to fuel the next wave of innovation.
To understand the current state of investment in this innovation ecosystem, we conducted interviews with 25 leaders and decisionmakers in pharma R&D to evaluate critical areas of investment, and perceived ROI, today and in the near term (over the next 3 years). Our discussions reveal that companion diagnostics, genomics research and diagnostic tools, clinical trial enablement solutions, and informatics / AI are the areas of greatest pharma investment and perceived ROI today and are expected to remain so soon, with liquid biopsies and real-world data also playing increasingly important roles in drug development.
IBI – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Senior Medical & Regulatory Writer, Clarivate Analytics
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Hermann Schulz, MD, Founder, PresseKontext
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Over the next few years, multiple market forces and trends, including increasing regulation of diagnostic tests, the shift of precision medicine into earlier lines of cancer care, innovation in early cancer detection, and the emergence of novel therapeutic modalities are expected to influence pharma investments throughout the precision oncology development value chain.
In the increasingly competitive pharma research market, and in an economic environment where capital efficiency is vital, understanding pharma’s priority investment areas and levers is critical to strategic planning for research and diagnostic tools, technology, data, and service providers looking to play a leading role in precision oncology innovation ecosystem.
We have some exciting articles in this issue of IBI. In the article titled, “5 Key Considerations for Designing Relevant Cell-Based Assays to Screen Antisense Oligonucleotides” Noelia Muñoz-Martín and Shushant Jain of Ncardia explore the 5 key steps you need to consider how to successfully develop an ASO in vitro screening assay and the extra-advantage of designing a physiologically relevant assay that can shorten your timelines.
David O'Connell of PCI Pharma Services explores the advancements in dry granulation and the fundamentals of roller compaction, presenting it as a viable alternative to traditional wet granulation for producing oral solid dosage forms like tablets and capsules.
“Packaging: The First Line of Defence in Cell and Gene Therapy Logistic”, Christopher Good of Biocair explains how temperaturecontrolled packaging plays a vital role in cell and gene therapy logistics and David Weinkove of Magnitude Biosciences, explores recent advancements in HTS, highlighting emerging trends, technological innovations, and the potential of using C. elegans as a scalable in vivo model for more effective drug discovery.
I hope you all enjoy the summer holidays and look forward to meeting you all soon.
Dr. Steven A. Watt, CBDO (Chief Business Development Officer) at A&M STABTEST GmbH
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
UniSafe® Platform
Delivering the future with safety and simplicity
Developed around UniSafe®, our platform meets the needs of you, our partner, and your varying patients’ needs both now and in the future.
A spring-free, passive safety device for 1mL pre-filled syringes, designed for simple assembly and use.
Proven, on market, in patient use
A spring-free, passive safety device for 2.25mL pre-filled syringes, designed for simple assembly and use.
Fully industrialised and ready to supply
A reusable companion auto-injector for UniSafe® 1mL.
A reusable connected companion auto-injector for UniSafe® 1mL.
Visit ompharmaservices.com/ibi-june2024 or email pharmaservices@owenmumford.com
Navigating the Future: Insights
from Leading Indian CDMOs on the Impact of the BIOSECURE Act and the Shift from Chinese CDMOs
The pharmaceutical and biopharmaceutical sectors are undergoing transformative shifts driven by geopolitical tensions and regulatory changes. Among these changes, the BIOSECURE Act and the 2032 decoupling deadline from Chinese CDMOs stand out, prompting companies to reassess and revamp their supply chain strategies. Chloe Euripides of the International Biopharmaceutical Industry Journal recently interviewed three key representatives from leading Indian CDMOs: Alex Del Priore of Syngene International Ltd., Ramesh Subramanian of Aragen Life Sciences, and Himanshu Gadgil of Enzene Biosciences. Their responses provide a comprehensive view of how these companies are adapting to the new landscape and positioning themselves as competitive alternatives to Chinese CDMOs.
The BIOSECURE Act and Strategic Shifts
The BIOSECURE Act and the 2032 decoupling deadline are significantly shaping Enzene’s strategic planning. Himanshu Gadgil, CEO, highlights that the shift has driven Western companies to explore alternatives to Chinese CDMOs, resulting in increased interest and customer visits to Enzene’s facilities. To leverage these opportunities Enzene is expanding its operations, including launching a new drug discovery division and investing in innovative technologies like EnzeneX™. This expansion aims to provide end-to-end solutions and meet the evolving demands of their clients.
Ramesh Subramanian, Chief Commercial Officer, notes that the BIOSECURE Act is prompting Western pharma companies to reconsider their strategies. Aragen is preparing for these strategic shifts by investing USD 250 million to expand its R&D and manufacturing facilities in Hyderabad. This includes adding biologics manufacturing capabilities and expanding their footprint in small molecules, peptides, oligonucleotides, and ADCs. Unlike the rapid shifts during the COVID-19 pandemic, Subramanian observes a more deliberate approach as companies plan for long-term changes.
Alex Del Priore, Senior Vice President of Manufacturing Services, explains that Syngene is strategically positioned to capitalise on the supply chain shifts accelerated by the BIOSECURE Act. The company has experienced a surge in interest from biopharma companies seeking to reduce reliance on China. Syngene’s strategy involves leveraging its dual growth engines – CRO services and CDMO services – while offering flexible supply chain options to cater to clients’ needs for both China-based and China-independent sourcing.
Unique Strengths of Indian CDMOs
Enzene's unique strengths in the global CDMO market include their proprietary EnzeneX™ technology, which offers significantly greater productivity and cost-efficiency compared
to traditional methods. With operations in India and a planned US site, Enzene focuses on innovation, quality and customercentricity, making it an attractive alternative to Chinese CDMOs.
Aragen’s strengths lie in its skilled English-speaking workforce, strong track record of quality compliance, and experience with Western regulatory guidelines. These factors, combined with rich experience in the West, position Aragen as a reliable partner for Western clients seeking high-quality CDMO services.
Syngene’s integrated services from discovery to commercial scale manufacturing, coupled with their investment in next-generation technologies and a large pool of skilled scientists, make them a strong competitor in the global CDMO market. Their flexible R&D services and long-term strategic collaborations further enhance their attractiveness as an alternative to Chinese CDMOs.
Responding to Increased Interest and Demand
Enzene has seen a significant increase in site visits from big pharma, driven by geopolitical tensions and supply chain disruptions. Gadgil notes that the feedback has been overwhelmingly positive, with clients appreciating their innovative manufacturing solutions. Enzene is launching a new facility in New Jersey equipped with state-of-the-art continuous manufacturing processes to meet the high standards of their clients.
The increasing interest in Aragen’s services from big pharma reflects the importance of geo-diversity in business continuity planning. Subramanian emphasises that Aragen’s facilities have been audited by multiple clients and certified by major global regulatory authorities. The positive feedback from both existing and new clients highlights Aragen’s focus on quality and their ability to operate as an extension of their clients’ teams. To meet the growing demand, Aragen is expanding its capabilities in small molecules and biologics, with significant investments in clinical and commercial manufacturing capacities.
Syngene has also experienced heightened interest from big pharma, driven by the need to de-risk supply chains. Del Priore notes that the company’s ability to deliver end-to-end solutions, its investment in cutting-edge technologies, and its robust quality assurance measures have positioned Syngene well in order to capture new opportunities. The recent acquisition of a biologics manufacturing facility and plans to expand drug substance and drug product capacities, demonstrate Syngene’s commitment to scaling up operations and meeting client expectations.
Scaling Up Operations
To handle increased demand, Enzene is making strategic investments in expanding its capabilities. This includes bringing online new manufacturing lines in Hopewell, NJ, and developing
Talking Point
EnzeneX™ 2.0 to enhance various aspects of their processes. Additionally, Enzene is working on new cell lines designed to achieve high productivity at a lower cost, ensuring they can scale up operations effectively.
Aragen has been proactively investing in expanding its capabilities across small molecules and biologics. The company has added clinical manufacturing capacity, opened a biologics pilot plant in Bangalore, and is set to add GMP manufacturing capacities by the end of the year. With significant investments in R&D and manufacturing facilities in Hyderabad, Aragen is well-positioned to meet the surge in demand for its services.
Syngene’s growth strategy focuses on scaling up and widening its operations through strategic investments. This includes expanding biomanufacturing capabilities, enhancing quality control laboratories, and integrating advanced technologies like AI and ML into their processes. Syngene’s recent acquisition of additional land in Genome Valley, Hyderabad, prepares the company for future growth and expansion, ensuring they can handle the increasing demand resulting from the shift away from Chinese CDMOs.
Future Competitive Landscape for Indian CDMOs
Gadgil foresees a significant evolution in the competitive landscape for Indian CDMOs, driven by the increased focus on biologics and advanced manufacturing technologies. Enzene’s pioneering use of continuous manufacturing technology, commitment to innovation, and focus on cost-effectiveness position them to lead this transformation. As industry standards evolve and regulations support continuous manufacturing, more CDMOs are likely to adopt this approach to meet regulatory and environmental demands.
Subramanian anticipates a major shift in the competitive landscape for Indian CDMOs, particularly with the growing demand for biologics and advanced manufacturing technologies. Indian CDMOs are investing in digitization and digitalisation technologies, including AI and ML tools, to enhance productivity and improve processes. Aragen’s use of AI-based tools to optimise critical process parameters has already yielded significant improvements, positioning the company to lead in this evolving landscape.
Del Priore expects the competitive landscape for Indian CDMOs to evolve significantly over the next five years, driven by advancements in biologics and manufacturing technologies. Syngene’s comprehensive capabilities in areas like CAR-T cells, PROTACs, mRNA, and ADCs, combined with continued government support and strategic investments, will enable the company to capture a substantial market share. Syngene’s strong focus on strategic partnering, alternative supply options, and deep trust from US and European biotechs will further strengthen its position in the global market.
Conclusion
The BIOSECURE Act and the shift away from Chinese CDMOs are reshaping the global pharmaceutical and biopharmaceutical industries. Indian CDMOs like Enzene Biosciences, Aragen Life Sciences, and Syngene International Ltd. are strategically positioning themselves to capitalise on these changes. Through significant investments in innovative technologies, expanded capabilities, and a strong focus on quality and customercentricity, these companies are emerging as competitive alternatives to Chinese CDMOs. As the industry continues to evolve, the unique strengths and strategic initiatives of these Indian CDMOs will play a crucial role in shaping the future of global drug development and manufacturing.
Alex Del Priore
Alex Del Priore has three decades of experience in developing, commercialising and life-cycle management of products in various life science industries. Holding positions in both the US and Europe, his experience includes senior roles with global P&L responsibility. As a member of the Executive Committee Alex plays a techno-commercial role, providing technical expertise to the API plant at Mangalore, while building a sustainable client base for the business in collaboration with the commercial and business development teams.
Dr. Himanshu Gadgil
Dr. Himanshu Gadgil, Chief Executive Officer at Enzene Biosciences Ltd., brings with him 24 years of experience in the pharmaceutical industry. Under his leadership, Enzene has grown from a start-up biotech to a multi-vertical, multi-site product development and manufacturing service-based biopharmaceutical company. Previously, as Sr. Vice President at Intas Pharmaceutical Ltd., he revitalised the commercial product pipeline, launching several biosimilar products globally. During his stint in the US, he led different facets of process and product development at Amgen, spearheading IND, BLA and Market authorisations of various blockbuster biotech products. At the inception of his career, he joined Waters Corporation where he pioneered development of QBD enabling multi-attribute methodologies for biopharmaceutical characterisation.
Ramesh Subramanian
Ramesh Subramanian, Chief Commercial Officer, is responsible for global business growth, leading sales, marketing, strategy and the corporate development function. With over 20 years of experience in leadership roles, Ramesh has built global businesses in Asia and Europe, raised venture capital, established transformational strategies, driven M&A, negotiated cross-border deals and managed alliances. Prior to Aragen, he served as Senior Vice President and was part of the management team at Piramal Pharma Solutions. Ramesh was previously a part of management teams at Chemizon, a firm he led from start-up to successful entry into the equity market and Jubilant Life Sciences.
On the Cutting-edge of Drug Metabolism
Optibrium Inc.’s President, Dr. Tamsin Mansley, breaks down all you need to know about the challenges of drug metabolism, and the in silico approaches to overcome these problems.
Determining metabolic fate is crucial in drug discovery. Horror stories are often shared around expensive, late-stage failures due to unexpected metabolism. Many challenges crop up, including poor metabolic stability, low bioavailability, unforeseen drug-to-drug interactions, issues from genetic polymorphisms, and the formation of reactive or toxic metabolites. Early in silico modelling can help to prevent any problems down the line.
Identifying the Enzyme Culprits
The first step to optimise metabolism is understanding which enzymes and isoforms are primarily responsible for your compound’s metabolism. Then, you can identify the sites on your molecule that these enzymes are metabolising, and how to design your compound to block these.
There are a range of different enzyme families which may be involved in metabolism. For example, cytochrome P450s, aldehyde oxidases and flavin-containing monooxygenases can cause oxidation of your compound's functional groups. Sulfotransferases and uridine diphosphate glucuronosyltransferases cause conjugation of compounds to polar groups. Additionally, within each enzyme family, there are numerous different isoforms which are functionally similar enzymes that differ slightly in amino acid sequence.
Using classical categorisation models, it is possible to quickly determine which enzyme families and isoforms are most likely to metabolise a specific atomic site.1–2 This can indicate compounds which can be metabolised by multiple enzymes, with multiple routes of clearance.
There are two main reasons you might want compounds with multiple routes of clearance. Firstly, genetic polymorphisms between individual patients may mean different isoforms of enzymes are present or absent and in varying concentrations. Therefore, in situations where only one isoform is responsible for drug metabolism, issues related to toxic drug build-up may arise in certain populations.
Similarly, single clearance routes increase risks from drug-todrug interactions. Co-administered drugs may inhibit or induce action by certain drug metabolising enzymes, causing variability in a patient’s exposure to the relevant drug. By ensuring multiple routes of drug clearance, these effects can then be mitigated.
Mapping Metabolic Liabilities
Knowing which enzymes cause your compounds’ metabolism
is only half the battle. To optimise metabolic stability, you also need to identify where your compounds may be metabolised. To model this regioselectivity we can take a dual approach, considering both the reactivity and the accessibility of each atomic site to metabolism.3–6
The reactivity of a specific site on a compound to a particular metabolic reaction can be modelled with quantum mechanical simulations. These physics-based methods take a holistic view of a molecule and the electronic distributions within it and hence the electron flow within a reaction pathway. The reactivity of each site on a molecule will be specific to the enzyme family, but will not vary between isoforms of the same enzyme.
The accessibility component of a regioselectivity model is influenced by the substrate’s molecular shape and functional groups, along with the particular enzyme’s active site structure. This means accessibility will be specific to each isoform and enzyme family. The particular steric and/or polar features within both the enzyme binding pocket and the substrate will determine the substrate’s orientation and whether a particular area can access the active site; thus, some sites will be less vulnerable to metabolism than others. Accessibility effects can be modelled using descriptors rooted on each site of metabolism on the ligand.
Reactivity and accessibility effects for each enzyme and isoform can be combined using robust machine learning models, trained on high-quality data and tested on an independent test set. By applying these regioselectivity models, a good comprehension can be gained into which labile sites need to be blocked for increased metabolic stability.
Too Many Metabolites
In addition to knowing if your structure will be metabolised, you need to be able to understand which compounds will form. Will any toxic or reactive metabolites be generated, causing any serious adverse side effects? Unfortunately, metabolite prediction is not as easy as it may seem.
Any predictive models must satisfy two criteria. Firstly, are the structures generated accurate representations of potential metabolites? Are all the experimentally-observed compounds being predicted? This can involve an added layer of complexity when metabolites are unstable and quickly undergo further reactions such as hydrolysis.
Secondly, and much more challenging, is considering the number of potential metabolites being predicted. Of course, you can generate every possible structure which a compound may be metabolised into, but how easy is it to pick out the subset of metabolites that are actually observed experimentally? Does your method suffer from vast over-prediction, obscuring the important, relevant experimentally observed metabolites?
LumiMAT™ Monocyte Activation Test
• Easy-to-use
• Stable performance (Less lot to lot variation)
• Animal free
• Shorter time to result (4-8 Hours compared to 1-2 days)
• High sensitivity, high accuracy and low CV
• Detects a variety of pyrogens: (Endotoxin and NEPs)
The Monocyte Activation Test (MAT) is an in vitro pyrogen detection assay that can detect both endotoxin and non-endotoxin pyrogens (NEPs). Conventional MAT assays utilize PBMCs (Peripheral Blood Mononuclear Cells) and an ELISA readout, which may lead to lot-to-lot variances, can take up to two days to obtain a result, and require multiple pipetting and washing steps; however, the novel LumiMAT™ assay utilizes the NF-κB reporter gene introduced into a cell line to achieve a rapid, robust, and highly sensitive assay.
The advantages of the LumiMAT™ assay are that it is ELISA free, easy to handle and provides a significantly shorter reaction time, as this system does not need to wait for IL-6 release.
In the near future, the current pyrogen testing method, RPT (Rabbit pyrogen test), will be replaced by MAT (Monocyte activation test) as a more reliable, sustainable and environmentally friendly method of pyrogen detection; however, MAT testing using PBMC (Peripheral Blood Mononuclear Cells) and ELISA (Enzyme-Linked Immunosorbent Assay) raises concerns about data variability, PBMC availability, and required assay time that are effectively mitigated with FUJIFILM Wako’s LumiMAT™ system.
Regulatory & Compliance
Using a heuristics approach to combine regioselectivity models and classification models, it is possible to predict the most likely metabolites with a much higher sensitivity than traditional rule-based methods. This streamlines the discovery process, minimising time wasted sifting through irrelevant metabolite predictions and making interpretation of metabolite ID experiments much easier.
Picking the Best Animals for the Job
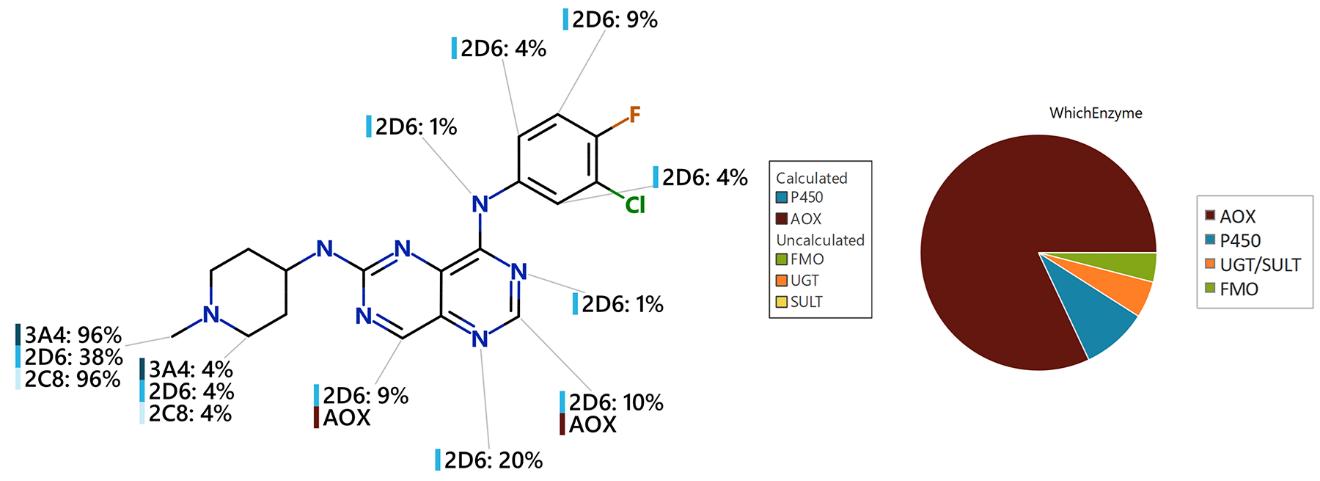
One final common query around drug metabolism is relating to preclinical studies. Selecting inappropriate animal species for these studies can have devastating consequences. Take, for example, the case of Falnidamol. This was a clinical drug candidate under investigation as a cancer treatment, which passed routine pharmacokinetics studies in rats and dogs, but failed in clinical trials, due to extremely low oral exposure in humans. This was due to rapid metabolism by AOX, which was not picked up during the preclinical PK studies, as rats have low aldehyde oxidase activity and dogs are devoid of aldehyde oxidase. More appropriate preclinical species for accurate preclinical trials could have been guinea pig or rhesus monkey, which have high aldehyde oxidase activity.
Had appropriate early-stage modelling been used, the right species could have been selected and labile sites identified and blocked to improve metabolic stability. This would have saved enormous amounts of time, money, and resources for the researchers.
Modelling animal species is more difficult than modelling human enzyme regioselectivity, due to the limited quality data available with which to build models. However, there are models currently available for rat, mouse and dog cytochrome P450s, with the potential for future research pathways into different animal species or enzyme models.
A Future Perspective
There are a few limitations around what we can currently achieve with this type of modelling. The first is data. High quality enzyme specific data is needed to train relevant models, so not every enzyme family or isoform, and not every common preclinical species can be modelled right now. As research continues and data improves, coverage of models can become more comprehensive.
Modelling metabolism using quantum mechanical simulations is also very computationally expensive and time intensive. New
methodology is constantly being developed to improve this, for example machine learning interatomic potentials.7 This will speed up future calculations to make metabolism prediction easier and more commonplace in discovery.
The extent of our current abilities in metabolism prediction has grown exponentially in recent years, with medicinal chemistry teams and DMPK scientists well-supported to create safe, efficacious drugs. As this field continues to progress, the future is looking even brighter.
REFERENCES
1. M. Öeren et al., “Predicting routes of phase I and II metabolism based on quantum mechanics and machine learning”, Xenobiotica, 2023, 1–49
2. P. A. Hunt et al., “WhichP450: a multi-class categorical model to predict the major metabolising CYP450 isoform for a compound”, J Comput.-Aided Mol. Des., 2018, 32, 537–546
3. M. Öeren et al., “Predicting regioselectivity of AO, CYP, FMO, and UGT metabolism using quantum mechanical simulations and machine learning”, J. Med. Chem. 2022, 65, 20, 1406–1408.
4. M. Öeren et al., J. Comput.-Aided Mol. Des., 2021, 35, 4, 541-555
5. M. Öeren et al. , “Predicting regioselectivity of cytosolic sulfotransferase metabolism for drugs”, J. Chem. Inf. Model. 2023, 63, 11, 3340–3349
6. J. D. Tyzack et al., “Predicting reactivity to drug metabolism beyond P450s - modelling FMOs and UGTs”, J. Chem. Inf. Model. 2016, 56, 11, 2180–2193
7. E. Gelžinytė et al., “Transferable Machine Learning Interatomic Potential for Bond Dissociation Energy Prediction of Drug-like Molecules”, J. Chem. Theory Comput. 2024, 20, 1, 164–177
Dr. Tamsin Mansley
Dr. Tamsin Mansley, President, Optibrium Inc. and Head of Application Science, Optibrium. Tamsin holds a PhD in Organic Chemistry from the University of East Anglia in the UK and pursued postdoctoral studies in the labs of Prof. Philip Magnus at the University of Texas, Austin. She is an experienced drug discovery scientist with experience as a medicinal chemist in pharma and biotech and has worked in computational chemistry, supporting discovery organisations for over 20 years. Her interests lie in coupling machine learning and artificial intelligence techniques with generative chemistry approaches to guide decisions and accelerate discovery projects.
Figure 1: The output of the StarDrop™ Metabolism module for Falnidamol. Here the WhichEnzyme™ pie chart clearly shows AOX as the major metabolising enzyme.
Safeguarding your IP: Thermo Fisher Scientific pharma services' global commitment to confidentiality
Intellectual property (IP) rights and confidentiality protections play a critical role in biopharmaceutical development and manufacturing. Appropriate protections enable developers to drive innovation, safeguard investments, and accelerate the development of new drugs and therapies, while creating a favorable environment for research, development, and commercialisation. Partnering with a CDMO with the global expertise to navigate complex landscape requirements, and with global frameworks in place to standardise protections, can alleviate potential challenges for drug developers and IP owners.
At Thermo Fisher Scientific, we are committed to keeping your intellectual property and confidential information secure. We understand the sensitivity of your data and apply the same rigorous controls as we do to our own information. We standardise these strict controls across all sites in our global network to help ensure your information is safe, regardless of where you work with us.
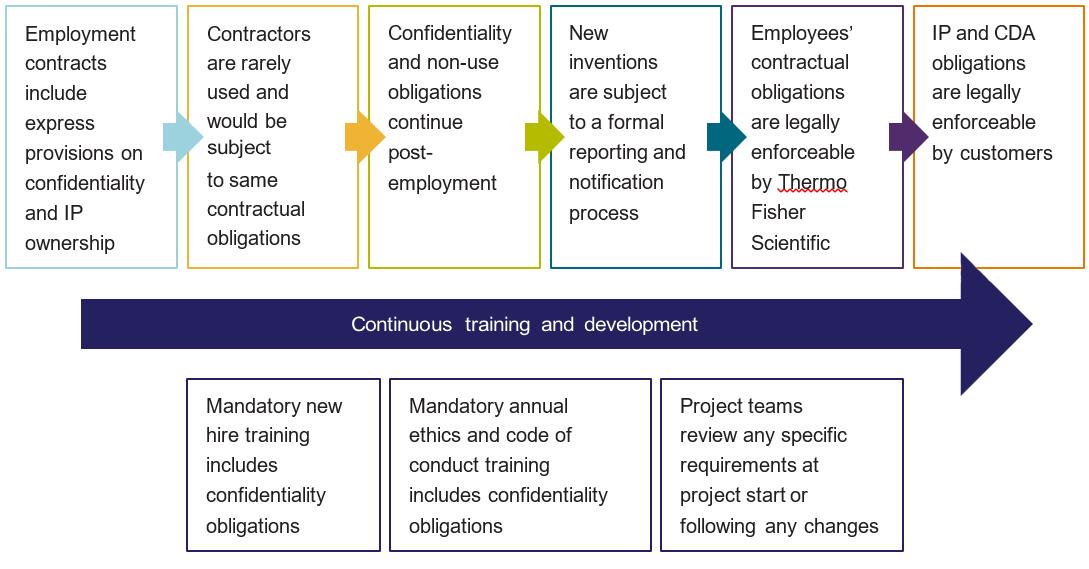
Enforcing Contractual Protections
Our standard contractual terms are consistent with industry expectations and allow customers to legally enforce our confidentiality and IP obligations.
Confidentiality Protections
• Strict processes ensure a valid CDA is in place prior to and throughout the disclosure of confidential information
• Confidentiality obligations are incorporated in the MSA and apply throughout the project and afterwards
• All pharma services sites have rigorous standards in handling confidential information conforming to Thermo Fisher Scientific global policies
IP ownership
• Customer information disclosed to us remains the property of the customer at all times
• New customer IP under the MSA is assigned to customer across all our sites and in compliance with local laws
• Employee agreements, including affiliates subcontractors, are consistent with our IP obligations towards the customer
• We offer technical support as requested by the customer in securing new customer IP (e.g. assistance with patent applications)
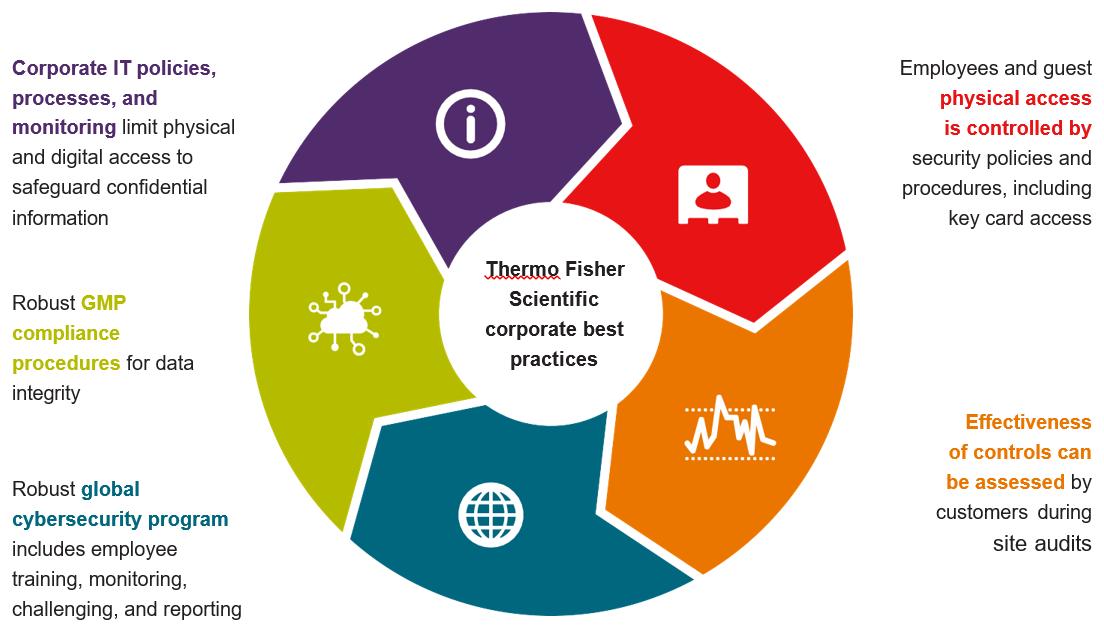
Protecting Your Data
We implement Thermo Fisher Scientific corporate best practices across all pharma services sites to keep your data safe and secure.
Global Framework Overview for Standardised Protections
Our Global Commitment
Thermo Fisher Scientific is dedicated to safeguarding our customers’ confidential information and intellectual property. We prioritise the protection of customer information by treating it with the same level of security as our own. Our commitment to compliance and integrity extends to our employees and contractors, who abide by contractual agreements and policies to ensure adherence to our strict standards. Consistent with this commitment, we implement strong physical and digital access controls across all our sites. Additionally, we provide our customers with auditable and legally enforceable contractual
commitments, further strengthening the security measures in place to protect their valuable information.
Our Team’s Commitment to Your Success
Our project teams are dedicated to helping you reach your goals and abide by the confidentiality and IP obligations consistent with our contractual commitments, including continuous training and development.
The Expanding Need for Endotoxin Testing
Pyrogen testing was borne out of the need to screen medical devices and pharmaceuticals for molecules (pyrogens) that could initiate an intense reaction mimicking septic shock. These pyrogens can survive steam sterilisation. They are endotoxins from bacteria that survive beyond the sterilisation from the microorganisms themselves. The most potent of these pyrogens is the lipopolysacaride (LPS) molecules of gram negative bacterial. In vivio pyrogen testing was replaced by an endotoxin-specific test using a protein cascade found in the hemolymph of the horseshoe crab Limulus polyphemus. The Bacterial Endotoxin Test has become synonymous with QC testing for pharmaceutical products. Since the acceptance of the gel clot technique as a compendial test in 1980, endotoxin testing has become a crucial anchor for the expansion and development of safe pharmaceuticals and medical devices. Although much has changed in the 40+ years, the invaluable benefit of endotoxin testing has only grown. With the recent development, and compendial acceptance anticipated?, sustainable testing reagents have only ensured the permanency of the Bacterial Endotoxin Test for the next generation of pharmaceutical and medical advancements. In this editorial, I will provide an overview of the areas that are experiencing growth in their need of endotoxin testing.
Pharmaceutical Injectables
The Bacterial Endotoxin Test is primarily focused on testing pharmaceutical injectables for product release. The guidelines provide guidance on calculating endotoxin limits allowing for concrete endotoxin limits to be determined. Since the 1980s, the BET has been well established as a requirement for injectables (USP <85>), and so most manufacturers of pharmaceuticals will be well established in endotoxin testing. The primary focus for these clients will be to transition to improved reagents due to its sensitivity or endotoxin specificity. FUJIFILM Wako provides reagents that are formulated to be endotoxin-free using large amounts of b-1, 3-glucan to saturate the protein (Factor G) found in the reagent that reacts to these molecules. This renders the LAL reagent to be endotoxin specific.
However, in recent years, with the withdrawal of the FDA’s definite guidelines on sampling requirements, as well as the increase in customised drug products that are manufactured in small batches, new and expanded requirements have been created for the monitoring and control of the endotoxin manufacturing process and raw materials. Emphasis is placed on more continuous monitoring; allowing for sampling numbers to be lowered for end-product testing. Among contract drug manufacturers, often one form of endotoxin testing will be the standard practice, but they may be looking to expand their testing to different methods to better suit the needs of their customers.
As a result, rapid, affordable, quantitative data at the point of test has gained in popularity among pharmaceutical manufacturers. The primary focus is on the compliant, end-process testing that the reagents allow. However, a new market opening will be for the monitoring that rapid, economical testing provides.
For raw materials, excipient manufacturers generally have set endotoxin limits set by pharmacopeial monographs. However, API manufacturers, especially if they are new technologies such as cell and gene therapies, will often not have endotoxin limits. These areas are becoming more familiar with the endotoxin level needs of their clients and are a good market for adopting endotoxin testing.
The products in the pharmaceutical industry that are well established or expanding in their need of endotoxin testing are active pharmaceutical ingredients (API) manufacturing, excipient production, production and monitoring of sterile water for injection, expanded need for in-process product monitoring, contracted drug manufacturing, raw materials, injectable veterinary pharmaceuticals, PET tracers, and custom medical devices.
Dialysis Devices and Solutions
For Dialysis products, the dialysis equipment itself being endotoxin from the manufacturer’s site is imperative. For manufacturers of dialysis equipment, tubing, grafts, catheters, membranes, and replacement fluids, as medical devices, it is well known and accepted that these will comply with endotoxin testing. Any manufacturer of these devices will be performing endotoxin testing.
A special need for developers of dialysis membrane tubing, whether cellulose or synthetic, will be for endotoxin testing of the membrane permeability to endotoxin as this is a critical factor in the amount of endotoxin reaching the bloodstream.
However, the primary focus at the forefront is endotoxin levels of dialysis water. Traditionally, dialysis water was held to a more lenient standard than WFI and is typically produced onsite. However, increased emphasis on the purity of the dialysate in addition to the permeability of the dialysis membrane has led to the definition of ultrapure dialysate with more stringent endotoxin limits. Practically, this has led to the implementation of WFI-quality filtration systems onsite. However, this has led to the need for an ability to effectively test microbial and endotoxin levels in the filtration systems to comply with the endotoxin levels as well as mitigate the growth of biofilms in the water production.
The release of ANSI/AAMI/ISO 23500-3:2019 was a regulatory push to bring all dialysis water to a uniform standard rather than being an advisory recommendation.
These guidelines push for monitoring of the water produced for dialysis for endotoxin testing, using a product such as the
Research / Innovation / Development
aBET system or the gel clot single test for low volume, low-cost testing.
Manufacturers of dialysis equipment are targets for BET adaptation as well as any dialysis center that filters their water onsite.
The dialysis devices and solutions well established or expanding in their need of endotoxin testing include dialysis water/ultrapure dialysis water/buffers and solutions, cellulose acetate membranes/tubing/synthetic tubing, heparin and enoxaparin solutions/blood thinners, drain bags and lines, catheters and grafts, and water treatment systems.
For a client performing testing for environmental samples, the AAMI guidelines put an emphasis on the need for a rapid test method. Traditionally, the LAL test takes 60 minutes for incubation to occur. However, a reader such as the αBET endotoxin detection system can provide results in approximately 15 minutes for monitoring purposes.
Testing Synopsis: Cell and Gene Therapeutics
Currently, there are no binding endotoxin limits for cell and gene therapy. However, the FDA has released a 2019 draft guidance on “Setting Endotoxin Limits during Development of Investigational Oncology Drugs and Biological Products.” Although not binding, the advice has the potential to become binding in the future. Specifically for Cell and Gene Therapy, it gives the following recommendations.
For early clinical products, all agents that are going to become a part of an investigational drug, including cell and gene therapy products, should have endotoxin levels within accepted BET requirements. Although not required, screening of these ingredients will ensure the safety and and purity of the final product.
For late-stage clinical development, endotoxin limits should be systematically validated and implemented to ensure that by the time of the marketing application, the entire validated procedure is in place. This limit should reference USP General Chapter <85>’s requirements for the risk and exposure that this investigational drug and concurrent additional administrations of ancillary drugs., to ensure that the patient dose remains in compliance of the pharmaceutical threshold pyrogenic dose.
Initially, performing a risk assessment and assigning a preliminary endotoxin will allow for the adoption of monitoring to take place during the early clinical development stage to fulfil these FDA recommendations. This should lead to the full validation of the endotoxin testing procedure being completed during the late-stage clinical development. As the reagent
manufacturer, FUJIFILM Wako provides our clients with validation guides and technical guidance through this process.
Since monitoring is all that is recommended during this procedure, clients may find the aBET system appealing. The client could then continue to use the aBET system for their compliant testing or use the same reagent in transitioning over to more large-volume testing.
Some of the major products in the growing development of cellular therapeutics that are well established or expanding in their need of endotoxin testing are cell products, excipients, viral particles, DNA plasmid material, and cell culture media.
The main concern for these products is the complex molecules found in the API’s and excipients of these products. The guidance does allow risk assessments to provide alternatives to traditional sampling and testing when it is shown that all the ingredients coming together are endotoxin free and the process is under control. Apart from that, various solutions such as heat treatment, dilution, and a treatment such as Predictive Oncology’s EndoPrep can mitigate interferences from proteins and potential endotoxin masking in these complex products.
Conclusion
As the need for the safety screening of new pharmaceutical products and medical solutions becomes apparent as the clinical trials approach, the need for endotoxin testing of the products becomes imperative?. A revolutionarily beneficial product is not beneficial if it is not also safe from environmental contamination. Thankfully, the Bacterial Endotoxin Test is readily implemented in nearly any laboratory situation. The availability of traditional and simple qualitative testing, effective and resilient quantitative test reagents, as well as sustainable recombinant reagents provide an endotoxin solution for every need. Please reach out to FUJIFILM Wako’s LAL division for support and guidance through every step of the way of your testing needs.
REFERENCES
1. USP. Bacterial Endotoxin Test, Chapter <85>
2. ANSI/AAMI/ISO. Preparation and Quality Management of Fluids for Haemodialysis and Related Therapies, 23500-3:2019.
3. FDA. Setting Endotoxin Limits during Development of Investigational Oncology Drugs and Biological Products, 2019.
Timothy Francis
Timothy Francis is the Senior Technical Specialist for the LAL Division of FUJIFILM Wako Chemicals U.S.A. Corporation. He holds a B.S. in Biochemistry and a M.S in Science Education. He comes into the Technical Specialist role with 5 years of experience teaching the natural sciences at a college level. He is proficient at taking the complex, technical aspects of a topic and breaking them down into clear, understandable pieces that all connect back to the big picture. He draws upon this experience to provide professional technical support and training for the PYROSTAR™ line and to help you with your technical needs.
Research / Innovation / Development
5 Key Considerations for Designing Relevant Cell-Based Assays to Screen Antisense Oligonucleotides
With newly developed antisense oligonucleotide (ASO) molecules, early efficacy and toxicity assessment is crucial to prevent costly failures in later stages. But, what are the assays that will give you the best predictions to advance your drug candidate with confidence?
Here you can find 5 key steps you need to consider to successfully develop an ASO in vitro screening assay. Read till the end to discover the extra-advantage of designing a physiologically relevant assay that can shorten your timelines.
Antisense oligonucleotides (ASOs) are a short, synthetic nucleic acid molecule that can bind to specific RNA sequences and can thereby modulate gene expression. This modulation can happen either through inducing degradation of the complimentary sequence, blocking translation or modulating splicing.
This trait can be incredibly valuable in the treatment of genetic disorders and hereditable diseases by either reducing target RNA transcript levels or restoring protein function.
Actually, some ASO therapeutics already made it to the market: Vitravene (Cytomegalovirus (CMV) retinitis),1 Kynamro (Familial Hypercholesterolemia),2 Tegsedi (TTR Polyneuropathy)3 and Waylivra (Familial Chylomicronemia Syndrome).4
Nonetheless, safety concerns due to off target effects and limited efficacy are obstacles that need to be overcome, to deliver on the potential of ASOs as an effective therapeutic.
To enhance the likelihood of achieving late-stage success, early adoption of relevant cell-based in vitro screening assays can significantly influence subsequent outcomes.
Delve into 5 essential considerations to design relevant cell-based in vitro assays that empower you to obtain more accurate predictions regarding ASO efficacy and safety.
How to Design a Successful Cell-based In Vitro Assay for ASO Screening
1. Choosing a Cell Model Relevant to Your Target Disease
For the development of an in vitro assay, the basis lies on the selection of the right cell model. In principle, any cell line can be used to develop an assay, but you should look for the one that best fits your research questions and goals.
Each cell model has different (dis)advantages which you must know in advance to be able to get a balance between its physiological relevance, genetic stability, availability, reproducibility, scalability and cost.
One of the most common models used are immortalised cells. The advantage of an immortalised cell is, as the name suggests, the unlimited proliferative capacity.
However, they are not genetically stable and lack the capacity to replicate important cell functions or disease phenotypes. This considerably limits the options of getting accurate predictions for efficacy and toxicity.
Another option is to use primary human cells, which offer a high physiological relevance and enable researchers to mimic a diseased condition in vitro
The disadvantage of this model is the limited proliferation capacity which hinders their application in primary screenings where large cell batches are required. Primary cells also present inter-donor variability, reducing the robustness of the assay, especially in the context of high-throughput screening.
Overall, primary human cells can be a valuable addition for validation studies where the need for scalability is much lower.
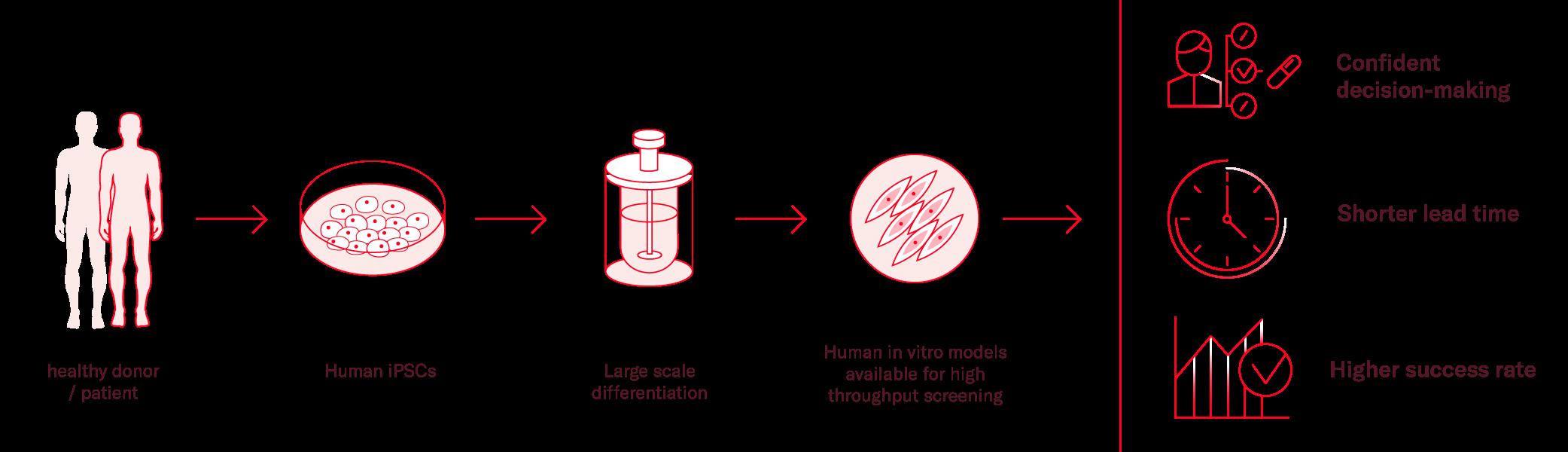
As an alternative, induced pluripotent stem cells (iPSCs) can be a useful tool in many cases, especially when investigating diseases where primary material is difficult to obtain, such as heart and brain related diseases. Availability of primary cells for those tissues is particularly limited, but iPSCs can be differentiated into neural and cardiac cells, as well as any body cell type.
Additionally, iPSCs have unlimited proliferation capacity and genetically stable. They can be patient-specific and recapitulate main disease phenotypes in vitro. To further enhance the relevance of the model and its translatability, different cell types can be cocultured to build more representative environments.
With the right expertise, iPSCs can be differentiated at a large scale producing one single source of cells for the full screening cascade, considerably reducing the variability of your results.
As there is no perfect model, the downside of iPSCs is the required technical expertise to develop robust differentiation procedures, produce large batches and model disease phenotypes in the context of miniaturised assays. Therefore, many researchers partner with expert companies to accelerate pipelines and save resources.
2. Selecting a Relevant Readout Technology
There are three options you can consider to quantify the changes in the expression of your target.
Firstly, based on the abundance of transcript, you can choose either qPCR or ddPCR, selecting the latter when the
Enabling our customers to harness the full potential of iPSCs
We help you increase the probability of downstream success by easily integrating our relevant iPSC platforms early in your research. As pioneers in iPSC technology, you can work with us to obtain dependable cell models, clinically relevant data of your candidate’s activity or develop robust large-scale processes for cell manufacturing. With a focus on clinical success, reproducibility, and efficient timelines, we enable you to confidently progress in your research.
Let’s start a conversation about how we can work together. Contact our experts at:
support@ncardia.com or visit www.ncardia.com
Supporting our clients with:
Discovery Services
Cell supply
Regenerative Medicine
Custom iPSC Services
Research / Innovation / Development
abundance is low. Quantification of the RNA allows for the direct measurement of target engagement and is often the method of choice for many ASO screens.
The assay used to quantify the target mRNA needs to be optimised by adjusting the various parameters, such as thermocycling conditions, primer and probes designs and RNA quality and quantity, among others.
At this point, it is important to identify and evaluate at least two to three housekeeping genes (controls to normalise gene expression) prior to deciding on a specific one for your assay.
By systematically optimising these parameters, you can enhance the sensitivity, specificity, and reliability of qPCR assays for quantifying target nucleic acid sequences.
Another option for quantification is high-content imaging (HCI) where either the RNA species can be visualised by fluorescence probes (FISH) or the target protein levels –end-product of your target gene – can be quantified.
However, this technique is only applicable when there are specific antibodies available for your target. If that’s the case, you might consider HCI instead of PCR for the following advantages:
• Quantifying protein expression with HCI allows the evaluation of the effectiveness of ASO treatment in achieving the desired alterations in protein expression.
• HCI can be multiplexed with additional markers for cell health or disease cellular pathways, providing a more holistic understanding of mechanism of action and toxicity.
3. Understanding the Expression Profile of Your Target Gene
After the selection of the target and cell model system, the cellular models need to be cultured under physiological conditions and the kinetics of the target gene established.
This will ensure that the cells express the target mRNA at detectable and robust levels and the readouts are performed at a time point when the target levels (whether that be mRNA or protein) have stabilised.
Next step is to ensure that the cellular model is amenable to oligonucleotide transfection or delivery.
4. Selecting a method for ASO delivery
The most prevalent method is gymnotic uptake, where the ASOs enter the cell without the use of transfection reagents just following the principle of passive diffusion through the membrane.
This technique is highly dependent on properties of the ASO molecules, as well as the target cell and its membrane permeability. Therefore, you may need to optimise the delivery method to achieve efficient oligonucleotide uptake and intracellular delivery.
If the gymnotic method does not deliver enough ASO, transfection reagents or nucleofection can be considered. This technique is very suitable for delivering nucleic acids across the cell membrane, but it is vital to optimise it to prevent cytotoxicity while achieving modulation of the target gene.
For optimisation purposes, it is recommended to perform qPCR of the target gene with positive control ASOs to ensure that most efficient delivery method of the ASOs is selected.
5. Optimise Assay Conditions
The screening protocol is further optimised to maximise sensitivity, specificity, and reproducibility by determining the appropriate:
• Oligonucleotide concentrations – normally determined by performing an 8 or 10 point concentration response curve (CRC).
• Duration of the ASO exposure – different study designs can be applied, but frequently the main decision is between an acute treatment (3–5 day exposure) or a chronic treatment (with multiple ASO additions through media refreshments).
• Overall assay conditions – performing pilot experiments that would mimic the workflow of the primary screen.
It is advised to identify both positive and negative control ASOs as it helps with the assay optimisation and benchmarking the performance of the assay.
To effectively and robustly screen large libraries, it is recommended to automate the different steps of the screening process: e.g. cell seeding, media refreshes, ASO addition, assay readout, data analysis, etc. This helps you make a more confident selection of the hits before moving into potency determination or functional studies.
Figure 1. Benefits of iPSC technology in drug discovery
Research / Innovation / Development
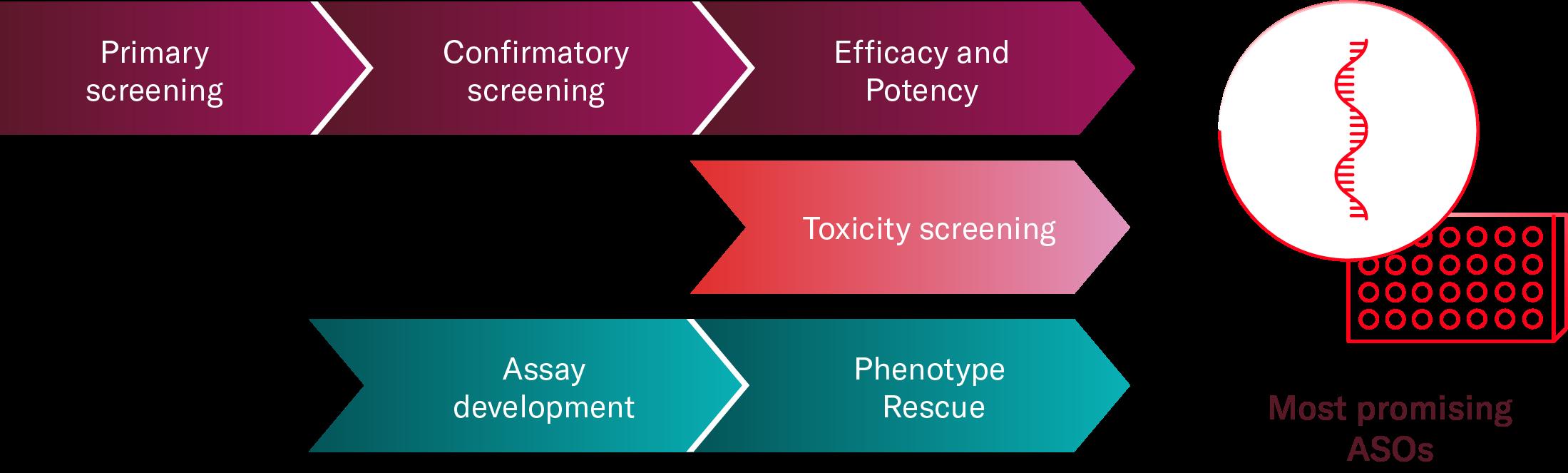
The Advantage of a Physiologically Relevant Design – Save Time By Running Complementary Assays Simultaneously
Once your assay is designed your next question is how to make accurate predictions on ASO in vivo efficacy and toxicity.
In both scenarios, the use of clinically relevant readouts is paramount for generating accurate predictions and advancing your therapeutic to next stages. It is important to recognise that the range of available readouts is heavily influenced by the choice of cell model made in step 1.
When selecting a relevant human cell model, there is the additional advantage of using the same system for complementary assays, saving time and resources.
For example, you can consider adding early toxicity readouts where ASOs are added to neuronal or cardiac cells to predict potential in vivo cardiotoxicity or neuronal liabilities. The toxicity of the ASO can be estimated through changes in calcium signalling or cellular metabolism, among other readouts.
Another range of assays that can help you select the most promising ASOs earlier, focuses on evaluating efficacy to rescue the disease phenotype. This can determine if the knockout or knock-down of the target gene is actually reversing or reducing important disease hallmarks.
Once more, this is only relevant if the cellular model can recapitulate disease phenotypes.
Through these assays, you can evaluate the direct effect of the therapeutic candidate on the disease phenotype.
Conclusions
When developing an ASO for therapeutic applications, early identification and validation of safety and efficacy are pivotal for success in both preclinical and clinical studies.
You must be aware of the profoundly influence that the cell model, readout and assay setup of choice have on the resulting data and the trajectory of your research. Therefore, it is crucial to carefully select those that will give you the most adequate answers to your questions.
Understandably, answering the questions is a challenge on its own! In complex areas like cardiovascular or neurological
diseases, navigating these challenges becomes even more intricate.
REFERENCES
1. Fomivirsen approved for CMV retinitis. (1998, October 1). PubMed. https://pubmed.ncbi.nlm.nih.gov/11365956/
2. Geary, R. S., Baker, B. F., & Crooke, S. T. (2015). Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (KynamRo®): A Second-Generation Antisense oligonucleotide inhibitor of apolipoprotein B. Clinical Pharmacokinetics, 54(2), 133–146. https://doi.org/10.1007/s40262-014-0224-4
3. Gales, L. (2019). Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharmaceuticals, 12(2), 78. https://doi. org/10.3390/ph12020078
4. Paik, J., & Duggan, S. T. (2019). Volanesorsen: First global approval. Drugs, 79(12), 1349–1354. https://doi.org/10.1007/s40265-019-01168-z
Noelia Muñoz-Martín
Noelia Muñoz-Martín obtained her PhD in biomedical research in 2019 and worked as postdoc before joining Ncardia. She has extensive experience in cardiac and neurological diseases as well as in vitro and in vivo models. Noelia has contributed to science communications of several organisations and companies from different angles, writing and editing peer-review articles and blogs, and creating website and social media content.
Email: noelia.munoz-martin@ncardia.com
Shushant Jain
Dr. Shushant holds a PhD in Molecular Genetics of Neuroscience and has significant expertise in developing high-content/ high-throughput assays, automation and data analysis with a strong focus on neurodegenerative diseases. He has led large, multiyear research programs for drug discovery initiatives in several therapeutic areas. Shushant’s research has been published in Neuron, Genome Biology, the Journal of Biomolecular Screening and the Journal of Biological Chemistry, among others.
Email: shushant.jain@ncardia.com
Figure 2 Using disease relevant cell models in the early stages of ASO discovery can shorten your timelines as it offers the possibility of running complementary assays in parallel to your confirmatory and potency screening. Additionally, it helps selecting the most promising candidates before progressing into in vivo models.
Application Note
Understanding Roller Compaction in Pharmaceutical Development
David O'Connell of PCI Pharma Services explores the advancements in dry granulation and the fundamentals of roller compaction, presenting it as a viable alternative to traditional wet granulation for producing oral solid dosage forms like tablets and capsules.
Q: What Exactly is Roller Compaction?
A: Roller compaction is a crucial process in the pharmaceutical industry, particularly in the formulation of solid dosage forms. This dry granulation technique involves the aggregation and densification of dry powders components into a uniform solid mass, known as a ribbon, which is subsequently broken down into specific granule sizes via a milling system. In this process, powder particles adhere to one another and form larger compacts, without the addition of a liquid binder, distinguishing it from wet granulation methods such as high shear or fluid bed granulation. Essentially, dry granulation compacts a powder blend by applying force, increasing the density, preventing powder segregation, improving compaction and flow properties of the resulting granules.
Q: So How Does this Form of Dry Granulation Work?
A: In roller compaction, a powder blend is fed, either by gravity or through a feeding system (auger feeder and tamp auger), into a set of directly opposed rotating rollers. The powder is then introduced into a narrowing gap between the rollers, where it is subjected to high pressure. This pressure increases the bulk density and particle size uniformity of the granules. The primary goal of dry granulation is to enhance the bulk density of powders and improve particle size uniformity to ensure better flow properties, which is crucial for high-speed tablet and capsule manufacturing.
Q: Can You Provide a Little More Detail?
A: The roller compaction process begins with feeding the powder blend into the rollers. The gap between these rollers is carefully controlled, and the powder is subjected to high pressure and a specific roller speed. This compaction forces the powder to form a dense ribbon. The pressure applied, roller speed and the gap dimensions are the most critical parameters, as they directly influence the bulk density and uniformity of the resulting granules. Precise control of these parameters is essential to ensure consistent granule properties, which are vital for downstream processing into dosage forms like tablets and capsules.
Q: Why is the Pressure or Force Applied so Important?
A: The pressure applied during roller compaction is crucial because it determines the degree of densification and compaction of the
powder blend. The applied pressure compacts the powder into a ribbon with predetermined thickness. The required pressure depends on the material, process parameters and product specification. This defined pressure must be carefully controlled to ensure uniformity in the ribbon and the resulting granules. Variations in the applied force can lead to inconsistencies in granule density and particle size distribution, affecting the overall quality and performance of the final dosage form.
Q: Does Roller Compaction Offer Any Benefits Over Wet Granulation?
A: Roller compaction offers several advantages over wet granulation, particularly for moisture-sensitive compounds. Unlike wet granulation, roller compaction does not require the addition of an aqueous solvent to aid in the binding of primary formulation components, making it suitable for compounds that may degrade or form toxic impurities when exposed to moisture. Additionally, roller compaction eliminates the need for a drying stage, making it ideal for heat-sensitive compounds. This results in a more efficient process with shorter production times. Moreover, roller compaction can support continuous batch processing, enhancing overall production efficiency and reducing operational costs.
Q: Do Other Forms of Dry Granulation Exist?
A: Yes, another method of dry granulation is slugging. In slugging, a tablet press compacts the powder into large tablet compacts, or "slugs," using large flat tooling. The resulting slugs are then milled into granules using an oscillating or conical mill. However, slugging presents several challenges. The pre-slugged blend does not consistently fill into the tablet die (poorly flowing materials with low bulk density materials), leading to inconsistencies in tablet weight and compaction force. These variations can cause differences in the mechanical strength of the slugs, resulting in differential granulate production. Due to these challenges, slugging is less commonly used, with roller compaction being the preferred method for dry granulation.
Q: Do Different Types of Roller Compaction Exist?
A: Yes, there are two primary types of roller compaction systems: fixed rolls and floating rolls. In fixed roll systems, the distance between the rollers remains constant, which can lead to inconsistencies in the compaction force if the powder feed varies. In floating roll systems, the gap adjusts dynamically based on the amount of powder being fed, maintaining consistent compaction force and resulting in more uniform granules.
Q: Does One Offer Any Advantage Over the Other?
A: Floating roll systems are generally considered superior due to their ability to adjust the roller gap based on powder feed, maintaining consistent compaction force. This minimises fluctuations in ribbon thickness and granule properties, leading to a more homogeneous granulate. Technologies like GERTEIS®
employ this floating gap system, providing better control and uniformity in the granulation process, ensuring high-quality and consistent granules suitable for further processing.
Q: After Compaction, What Happens Next?
A: Following compaction, the resulting ribbons are milled into granules using a screen with a specific mesh size to control particle size. Depending on the type of roller compactor the milling system can be online or performed off line. The milling should be gentle to avoid generating fines, which can negatively impact granulate quality and therefore oscillating millings are commonly used, rather than conical mills. The granules produced through roller compaction are then processed to extra-granular blending (additional excipients i.e. diluent disintegrants and lubricants) and then into tablets or capsules or bottle/sachet filling, ensuring dose uniformity and improving product consistency. This continuous process ensures efficiency and high throughput, essential for large-scale pharmaceutical manufacturing.
Q: How Does Roller Compaction Fit into the Development of Specialised Medicines, such as Those with Increasing Potency?
A: The development of highly potent compounds requires specialised handling to ensure safety and efficacy. Traditional reliance on personal protective equipment (PPE) is being supplemented with contained engineering solutions to enhance operator protection. CDMOs must incorporate fully contained roller compaction systems capable of processing potent molecules with occupational exposure limits (OEL) as low as 0.01 μg/m³. At PCI Pharma Services, for example, we have invested in
state-of-the-art contained roller compaction facilities to address the growing demand for processing highly potent compounds. This ensures we can offer both wet and dry granulation solutions while maintaining the highest safety standards.
Q: How has PCI Pharma Services Responded to the Increasing Demand for Dry Granulation of Potent Molecules?
A: PCI Pharma Services recognised the need for specialised facilities to handle potent compounds and invested in a fully contained roller compaction solution within our purpose-built manufacturing facility. This investment allows us to process potent molecules with low OELs without relying solely on PPE. Our facility's design ensures that we can handle both dry and wet granulation with best-in-class containment, offering our clients comprehensive solutions for the development and manufacture of highly potent drug products.
By leveraging cutting-edge technologies and maintaining stringent safety and quality standards, PCI Pharma Services continues to lead in the development and manufacturing of oral liquid dosage forms, ensuring that we meet the evolving needs of the pharmaceutical industry.
The Future of Roller Compaction in Pharmaceutical Manufacturing
Q: What Future Trends Do You See in the Roller Compaction Process?
A: The future of roller compaction in pharmaceutical manufacturing looks promising with advancements in technology and a growing focus on specialised medicines. We anticipate continued
Application Note
improvements in roller compaction equipment, including more sophisticated control systems that enhance precision and uniformity. Additionally, the integration of real-time monitoring and process analytical technology (PAT) will further optimise the roller compaction process, ensuring consistent quality and reducing the risk of deviations.
Q: How is the Industry Addressing the Need for Continuous Processing?
A: The pharmaceutical industry is increasingly adopting continuous processing to improve efficiency and scalability. Roller compaction is well-suited for continuous processing due to its ability to provide consistent granules in a streamlined manner. By integrating roller compaction into continuous manufacturing lines, companies can achieve higher throughput, reduced downtime, and lower production costs. This shift towards continuous processing aligns with the industry's goal of enhancing manufacturing efficiency and flexibility.
Q: What Role Does Sustainability Play in Roller Compaction?
A: Sustainability is becoming a key consideration in pharmaceutical manufacturing. Roller compaction supports sustainability by eliminating the need for solvents and reducing energy consumption associated with drying processes. The ability to process heat- and moisture-sensitive compounds without additional energy-intensive steps contributes to a greener manufacturing process. Furthermore, advancements in roller compaction technology aim to minimise waste and optimise resource utilisation, aligning with the industry's sustainability goals.
Q: How is PCI Pharma Services Positioning Itself for the Future?
A: PCI Pharma Services is committed to staying at the forefront of pharmaceutical manufacturing by continually investing
in advanced technologies and infrastructure. We focus on developing innovative solutions that meet the evolving needs of our clients. Our state-of-the-art facilities and commitment to quality and regulatory compliance ensure that we remain a trusted partner for pharmaceutical companies worldwide. As the industry advances, we will continue to leverage our expertise and capabilities to deliver high-quality, efficient, and sustainable manufacturing solutions.
Conclusion
Roller compaction is a critical process in pharmaceutical manufacturing, offering numerous benefits, especially for moisture- and heat-sensitive compounds. With advancements in technology and a growing focus on specialised medicines, roller compaction is poised to play an even more significant role in the future. PCI Pharma Services, with its state-of-the-art facilities and commitment to quality, is well-positioned to lead in this evolving landscape, providing comprehensive solutions for the development and manufacturing of high-quality pharmaceuticals and delivering life changing therapies to patients.
David O’Connell
David O’Connell is the Director of Scientific Affairs at PCI Pharma Services. After graduating from Glasgow Caledonian University with a BSc. in Applied Bioscience, David spent seven years as a Supervisory Scientist working for Aptuit in Edinburgh before moving to Penn Pharma as Head of Formulation Development in 2009. Here he played a vital part in the design of the potent Contained Manufacturing Facility (CMF), which won the ISPE Facility of the Year award for Facility Integration (2014). In 2013 David took on the role of Director, Pharmaceutical Development at the PCI site in Tredegar and in 2017 became PCIs Director of Scientific Affairs.
talkfuture@pci.com pci.com
•
•
•
•
•
Building
Advancing Gene Therapy Research with Trapped Ion Mobility Spectrometry
Gene therapy is a rapidly developing field of therapeutic treatment that holds the potential to treat a wide range of diseases, including cystic fibrosis, sickle cell, cancer, heart disease, diabetes, and human immunodeficiency virus (HIV)/ acquired immunodeficiency syndrome (AIDS).
In 1988, the first clinical trial of human gene therapy was conducted for the treatment of Gaucher disease.1 Since then, significant strides have been made in the field of gene therapy, propelled by enhancements in analytical technology, genomics, and molecular biology. For example, the United States Food and Drug Administration (FDA) has approved 37 cell and gene therapy products which have been licensed to treat several conditions2 and currently, there are over 1,100 open gene therapy clinical trials and ribonucleic acid (RNA)-based therapy clinical trials globally.3
This article explores how trapped ion mobility spectrometry (TIMS) is advancing research into gene therapy products and progressing the development of new treatments for diseases that were previously considered untreatable.
History of Gene Therapy
Speculation about gene therapy began in the 1960s when scientists hypothesised that introducing deoxyribonucleic acid (DNA) sequences into patients’ cells could hold the key to curing genetic disorders.4 Though without protection from a carrier, the nucleic acid material was rapidly eliminated from the body.
It wasn’t until the 1980s when the first notable advancements in gene therapy were made with the discovery of vectors as a delivery method and a paper was published demonstrating the use of a virus to insert genes into blood-forming stem cells in mice.5 Then, in 1990, came the first human success story in a patient who was born with a severe combined immunodeficiency (SCID) due to lack of the enzyme adenosine deaminase (ADA).6 Doctors delivered a healthy ADA gene into the patient’s blood cells, using a disabled virus unable to spread in the body. Despite initial setbacks, including the death of a patient in 1999, subsequent trials demonstrated promising results, heralding the potential of gene therapy for treating genetic diseases.
Having acknowledged the crucial need for a delivery system to protect the nucleic acid, usually small interfering ribonucleic acid (siRNA) and on occasion DNA, as it travels through the body to reach the target cells, researchers are now investigating a variety of delivery options, including viral vectors and non-viral vectors (such as nanoparticles and liposomes) to improve gene delivery efficiency and specificity.
The Rise of Recombinant Adeno-associated Virus (rAAV) Vector AAV is a non-enveloped virus belonging to the genus Dependo-
parvovirus in the family Parvoviridae that is non-pathogenic, replication-defective and packages a single-stranded viral DNA.7 As a small virus (approximately 20–25 nanometer in diameter), rAAV can carry a small payload of single-stranded DNA (ssDNA) which can be used for therapeutic purposes. The ssDNA is encapsulated in viral proteins and transported to the site of action in the body.
rAAV has emerged as a leading gene delivery vehicle (vector) for in vivo gene therapy due to several advantages, namely high infectivity, efficient delivery of therapeutic genes into target cells, and low pathogenicity,7 minimising the risk of causing disease in the host. AAV also possesses widespread tissue tropism, allowing it to target a broad range of tissues throughout the body.[8] Furthermore, rAAV-mediated gene expression demonstrates long-term persistence, even in non-dividing cells.
Recent advancements in developing clinically desirable rAAV capsids have contributed substantially to the growth of the gene therapy field. Researchers continue to refine rAAV vectors to enhance their targeting specificity and reduce unwanted immune response to further improve the efficacy of gene therapy treatments. Developments in vector design and engineering to provide reliable and robust delivery look set to advance targeted gene delivery, while minimising adverse effects. However, final preparation, formulation, and characterisation of rAAV drug substances are fundamental to support preclinical and clinical applications.
Solutions for rAAV Characterisation
To ensure the safety and efficacy of rAAV drugs for human gene therapy, the quality of rAAV vectors should be carefully monitored and controlled. Characterisation of rAAV preparations includes the determination of identity, potency, purity, stability, and safety of a batch.
The characterisation of rAAV-based gene therapy products represents significant challenges owing to their extremely large molecular sizes, structural complexity, heterogeneity, and limited sample amounts.1 Mass spectrometry (MS) is one of the key analytical tools that can overcome these challenges and serves as an important technique for the analysis of multiple attributes.
For quality control, MS is used to identify the amino acid composition and sequence of viral proteins, and to monitor the post translational modification (PTM) status of viral proteins, leading to determine the exact stoichiometry of viral proteins in rAAV.9 MS is also used to identify the host cell protein (HCP). Typically, rAAV is composed of three different viral proteins: VP1, VP2 and VP3, each with distinct functions. Accurate determination of the stoichiometry is crucial for researchers as a higher number of VP1 and VP2 proteins usually correlates with greater efficacy.10 Therefore, precise quantification of these proteins is essential for quality control. PTM at certain amino acids in rAAV have impact on
CONTRACT DEVELOPMENT AND MANUFACTURING OF BIOPHARMACEUTICALS
Richter-Helm is a Germany-based GMP manufacturer specialized in products derived from bacteria and yeasts, with a proven 30-year track record.
Count on us to flexibly provide a comprehensive range of services and customized solutions. Clients worldwide have already benefited from our commitment to good manufacturing practice and total transparency. Our work focuses on recombinant proteins, plasmid DNA, antibody fragments, and vaccines.
Richter-Helm consistently works to the highest standards of pharmaceutical quality. Contact us
+49 40 55290-801 www.richter-helm.eu
LEARN MORE ABOUT OUR
the transgene expression level thus quantification of PTM is also essential for rAAV quality control.11 The viral vector output is then used by a clinical group who adds the genetic material and takes it into a clinical trial.
The TIMS Advantage MS has become an indispensable tool for proteomic research. However, alone, its ability to comprehensively characterise protein complexes, particularly regarding complete sequence coverage and detailed analysis of molecular interactions and downstream effects, remains limited.
TIMS has emerged as a novel technique that addresses these limitations. TIMS offers significant improvements in sensitivity, selectivity, and separation speed for peptides and proteins. This enhanced capability is particularly valuable for analysing low-abundance species that are often crucial for understanding disease mechanisms. By offering a new dimension of separation based on size and shape, TIMS provides valuable insights into protein conformation and dynamics, furthering understanding of protein function in complex biological systems.
TIMS uses an electrical gradient to separate ions based on their size and shape in the gas phase. This allows for the selective manipulation of ions within the TIMS tunnel, enabling their controlled release, fragmentation, identification, and quantification through a process known as parallel accumulationserial fragmentation (PASEF). With TIMS and PASEF, the ion mobility measurements can be used to determine ion specific collisional cross section (CCS) values. The incorporation of CCS values introduces a critical fourth dimension to proteomic analyses, transitioning the field from reliance on the traditional 3D-proteomics approach (retention time, mass-to-charge ratio (m/z), and tandem MS (MS/MS) fragment ion spectra) to more comprehensive 4D-proteomics. This additional dimension of CCS data significantly enhances the system's selectivity, leading to more reliable quantitation of proteins within complex biological samples.
Conclusion
Gene therapy presents a promising future for disease treatment across a broad spectrum of diseases. Its potential to revolutionise biopharmaceutical research offers significant hope for patients battling genetic disorders. Continued advancements, fostered by collaborative efforts and guided by ethical considerations, position gene therapy as a cornerstone of personalised and precision medicine. Using advanced characterisation methods, such as TIMS, will deliver safe and effective treatments to individuals, holding the promise of improved health outcomes and a significant enhancement in quality of life.
REFERENCES
1. Serrano, M. A. C.; Furman, R.; Chen, G.; Tao, L. Mass Spectrometry in Gene Therapy: Challenges and Opportunities for AAV Analysis. Drug Discovery Today 2023, 28 (1), 103442. https://doi.org/10.1016/j. drudis.2022.103442.
2. Approved Cellular and Gene Therapy Products. U.S. Food and Drug Administration. Available online: https://www.fda.gov/ vaccines-blood-biologics/cellular-gene-therapy-products/approvedcellular-and-gene-therapy-products (accessed on 10 May 2024).
3. Kevin W. Doxzen et al. The Translational Gap for Gene Therapies in Lowand Middle-Income Countries. Sci Transl Med 2024, 16 (eadn1902).
https://10.1126/scitranslmed.adn1902.
4. What is biotechnology? “Gene Therapy.” (2023). https://www. whatisbiotechnology.org/index.php/science/summary/gene-therapy/
5. Williams, D.; Lemischka, I.; Nathan, D., et al. Introduction of New Genetic Material into Pluripotent Haematopoietic Stem Cells of the Mouse. Nature 1984, 310, 476–480. https://doi.org/10.1038/310476a0.
6. Naam, R. ‘More Than Human’. The New York Times, July 1, 2005, Available online: https://www.nytimes.com/2005/07/03/books/ chapters/more-than-human.html (accessed 13 May 2024).
7. Bijlani, S.; Pang, K. M.; Sivanandam, V.; Singh, A.; Chatterjee, S. The Role of Recombinant AAV in Precise Genome Editing. Frontiers in Genome Editing 2022, 3, 799722. https://doi.org/10.3389/fgeed.2021.799722.
8. Colón-Thillet, R.; Jerome, K. R.; Stone, D. Optimization of AAV Vectors to Target Persistent Viral Reservoirs. Virol J 2021, 18, 85. https://doi. org/10.1186/s12985-021-01555-7.
9. Oyama H, Ishii K, Maruno T, Torisu T, Uchiyama S. Characterization of Adeno-Associated Virus Capsid Proteins with Two Types of VP3-Related Components by Capillary Gel Electrophoresis and Mass Spectrometry. Hum Gene Ther. 2021 Nov;32(21-22):1403-1416. doi: 10.1089/hum.2021.009.Epub 2021 Jul 16. PMID: 34082578; PMCID: PMC10112878
10. Ohnishi et al., Enhancement of recombinant adeno-associated virus activity by improved stoichiometry and homogeneity of capsid protein assembly. Mol. Ther. Meth.Clin Dev. 31:101142 (2023).
11. Yamaguchi et al., Glycosylation of recombinant adeno-associated virus serotype 6.Mol. Ther. Meth. Clin Dev. in press (2024).
Susumu Uchiyama
Professor Uchiyama studied at Nagoya University and completed his PhD at Osaka University in the Pharmaceutical Department. Following this he worked as a researcher at the RRF institute Inc. in Japan, and became an assistant professor in the Department of Biotechnology in Osaka University, where he studied proteomics analysis and identifying whole sets of proteins in cells and biological substances using MS. In 2006, he launchedU-Medico Inc, providing analytical services on therapeutic proteins and viral vectors. Uchiyama has published over 250 peer-reviewed papers/reviews, as well as edited the Analytical Ultracentrifugation published by Springer. He has also been on the advisory board for Coriolis Pharma since 2012, a globally operating contract research and development organisation (CRDO), leader in formulation research and development of biopharmaceutical drugs, including cell and gene therapy products and vaccines.
Dr. Satoshi Oshiro
Dr. Satoshi Oshiro has a background in biophysical analysis of proteins. He studied protein engineering and biophysical analysis of proteins including surface plasmon resonances (SPR), spectroscopy, and thermodynamics at the University of Tokyo, Graduate school of Frontier Sciences. After completing his PhD, he worked as postdoctoral fellow at Gunma University and Kyoto University from 2014–2017, then joined Nitto Analytical Techno-Center Co., Ltd. as an analytical engineer and dedicated to surface analysis of polymers and materials with TOF-SIMS for 4 years. In 2021, he joined Bruker Japan K.K. as an application engineer and supports Biopharma applications both in Mass spectrometry and SPR.
Expand your horizons
CPHI Milan connects you to the global pharma community to level up your business at the heart of our industry.
8-10 October 2024
Fira Milano, Italy
SCAN ME
Accelerating Drug Discovery for Diseases of Ageing: In Vivo High Throughput Screening with C. elegans
In an era where rapid drug discovery is crucial, high throughput screening (HTS) emerged in the 1990s as a significant development, allowing for the swift and effective identification of active compounds and deeper insights into biological pathways. This process involves testing large libraries of compounds against biological targets to identify potential drug candidates. It relies on robotic automation and the use of standard format 96, 384 or 1536 well plates to screen tens of thousands of compounds per day.
Despite its transformative impact, current HTS methods face significant challenges in studying complex in vivo responses and age-related diseases. This article explores the potential of using C. elegans as a scalable and effective in vivo model for high throughput drug discovery in these therapeutic areas.
The Uses and Challenges of Traditional Models in HTS
In Vitro Screening
In vitro screening methods are widely used due to their cost-effectiveness and suitability for initial compound screening. These methods involve testing compounds in biochemical assays or with cultured cells to identify those that exhibit desired biological activity. However, in vitro models have limitations. They often fail to replicate the complex interactions that occur in living organisms, making it difficult to predict the efficacy and safety of compounds in humans. This limitation is particularly problematic for studying age-related diseases and other conditions where the cellular environment plays a critical role.1
Traditional 2D cell cultures, for instance, do not adequately mimic the three-dimensional architecture and microenvironment of tissues, which can significantly influence cell behaviour and drug responses. Moreover, cells in culture do not experience a comparable ageing process to whole organisms, which further limits their utility in age-related disease research.2
The only cells that show ageing are human primary fibroblasts and they are difficult to obtain in high numbers and vary between individual donors. Using these cells fails to replicate the systemic and tissue-specific ageing processes observed in whole organisms. This discrepancy can lead to misleading results when evaluating the efficacy of compounds to interfere with chronic diseases of ageing.
3D cell culture techniques have been developed to address some of these limitations by better replicating in vivo conditions. These advanced models incorporate more than one cell type and extracellular matrices to create a more physiologically relevant environment. However, despite their
advantages, 3D cultures are less amenable to HTS, mostly because consistent establishment of cultures and microscopy is challenging in multi-well plates to the level required for large throughput screening.1,2
A related development is organoids, which are mini clusters of cells that mirror many properties of organs. Scaling this approach to HTS is possible but constrained by issues of consistently sorting large numbers into multiwell plates, and then performing microscopy on them.
Current Routes to In Vivo Data
For most drug discovery, the use of mammalian models, such as mice, provide the first time that a compound is tested in vivo. Mammalian models provide a closer approximation to human biology, making them invaluable for assessing drug efficacy and safety in complex living systems. These models can mimic human disease conditions more accurately than in vitro models. However, they are for obvious reasons, not amenable to HTS. Mammalian models are costly and time-consuming, requiring extensive resources and lengthy study periods just for experiments with a handful of conditions. For example, rodent testing in cancer therapeutics alone can add an estimated 4 to 5 years and cost $2 to $4 million per study.3
Additionally, the use of animals in research raises ethical concerns and involves complex regulatory requirements. These factors limit the scalability of mammalian models for high throughput applications. Ethical considerations and regulatory compliance add layers of complexity, making it challenging to expand the use of mammalian models in a high-throughput setting.
The average cost of developing a new drug, including the use of rodent models, is around $900 million, and the time to market is typically 10–15 years.4,5 Moreover, the attrition rate in drug development is notably high. Out of every 5000–10000 compounds, only 250 make it to preclinical trials, five enter human trials, and just one reaches the market.6 This high rate of failure further underscores the inefficiency and high cost associated with mammalian models.
Despite the significant advancements and potential of HTS in drug discovery, traditional methods face several critical challenges. in vitro models, while cost-effective, fail to replicate the complex interactions and ageing processes found in living organisms, limiting their predictive power for human efficacy and safety. Mammalian models, though more representative of human biology, are costly, time-consuming, and involve ethical and regulatory complexities that hinder their scalability.6 These challenges underscore the need for innovative approaches that can bridge the gap between in vitro and mammalian models, offering more efficient and scalable solutions to bring in vivo research earlier in drug discovery.
C. elegans as a Developing Model for High-Throughput Screening
The nematode worm Caenorhabitis elegans is being increasingly explored as a HTS model that bridges the gap between in vitro and mammalian models. With adults only a millimetre in length, several of these worms can easily fit into one well of 96 or 384 well plates and advanced microscopy or spectroscopy used to monitor them.
Historically used in basic biological research, C. elegans offers several distinct advantages, making it an attractive model organism in drug discovery. One significant advantage is its human translatability. C. elegans have significant homology with human cell types and organs, and it uses most of the neurotransmitters found in humans, such as dopamine, serotonin, acetylcholine, GABA, and glutamate. This biological similarity makes C. elegans a relevant model for studying human diseases. High conservation at the amino acid level of many targets makes it suitable for investigating drug responses.
Understanding age-related processes is another area where C. elegans excels because it ages fast, and genetics can be used to determine which genes influence ageing. It was the first model organism in which single gene mutations were found to slow ageing, with key pathways such as insulin/PI3K/FOXO and mTOR, modulating ageing processes. In drug discovery, researchers can quickly observe the effects of drug treatments on the ageing process, accelerating the pace of discovery.
In addition to studying natural decline of function with age, several models have been developed to study the consequences of overexpressing human genes implicated in neurodegenerative diseases. This makes C. elegans particularly useful for studying the genetics of ageing and identifying compounds that influence longevity.
This makes C. elegans particularly useful for studying the genetics of ageing and identifying compounds that influence longevity.
Scalability is a crucial factor in HTS, and C. elegans excels in this regard. These worms can be easily maintained in large numbers, can be sorted using automatic sorting machines, fit into tiny wells and are transparent for microscopy. Ageing is intrinsically heterogeneous, so large test populations are required for robust data when studying the diseases of ageing. The C. elegans system and HTS can address this challenge and has been used to screen drugs that extend lifespan.7,8
Cost-effectiveness is another key advantage of C. elegans Maintaining these worms is inexpensive compared to mammalian models, reducing the overall cost of large-scale screening projects. This economic benefit, combined with the other advantages, positions C. elegans as a powerful tool for HTS and drug discovery. The ability to bring in vivo testing earlier in the drug discovery pipeline can significantly de-risk the transition from in vitro studies to mammalian studies, providing a more efficient path to identifying promising drug candidates.
This year’s Bioprocessing Summit features main conference sessions, in-depth training seminars, a 1-day investor conference for C-level executives, a workshop dedicated to acquisition and retention strategies, an engaging exhibit hall, and a plethora of networking opportunities.
Cambridge Healthtech Institute’s
By leveraging these advantages, C. elegans studies can provide parallel in vivo data that significantly enrich and complement data from in vitro experiments, enhancing the overall robustness and predictive power of drug discovery efforts.
From Hits to Mechanisms
High throughput screens using C. elegans can identify compounds with potential, which can then be further studied to understand their mechanisms of action. This is particularly useful in areas like neurodegeneration, gut health, muscle health, weight management, toxicity, and reproductive health. C. elegans offers various disease models to test compounds, including models for ALS, Alzheimer's, and Parkinson's disease. By studying these models, researchers can gain insights into how compounds interact with specific biological pathways, facilitating the development of targeted therapies.
Successful Applications
C. elegans has been used in numerous studies to identify neuroprotective compounds, study aging processes, and assess toxicity. Research using C. elegans has led to the discovery of compounds that extend lifespan and improve health-span by modulating various genetic pathways.
One notable example of where C. elegans was used to find a new therapy is the PPM2-CDG case study. PPM2-CDG is a rare genetic disorder that affects multiple systems in the body. In this study, a model of PPM2-CDG was developed in C. elegans using mutations homologous to those found in humans. This mutant could not grow properly in the presence of a stress-inducing drug. Using HTS method, researchers conducted a drug repurposing screen of 4,000 drugs in 384 well plates using this model, and identified aldose reductase inhibitors as a class of drugs that modified the PPM2-CDG phenotype. One of these inhibitors, Epalrestat, was repurposed to treat a patient named Maggie, who had PPM2-CDG. Treatment with Epalrestat led to significant improvements in Maggie's speech, walking, fine motor skills, balance, and growth. The study has progressed to a Phase III clinical trial to evaluate the safety and efficacy of Epalrestat for PMM2-CDG patients, demonstrating the potential of C. elegans in drug discovery and the repurposing of existing drugs to treat rare diseases.9
Regulatory and Ethical Considerations
The regulatory landscape for HTS involves ensuring data integrity and reproducibility, particularly when using advanced technologies like AI. C. elegans models, due to their simplicity and ethical acceptability, face fewer regulatory hurdles compared to mammalian models. This makes C. elegans an attractive option for high throughput applications, as they align well with regulatory requirements while providing valuable in vivo data.
Ethical considerations in HTS include the use of animals and data privacy concerns related to AI. C. elegans provides an ethical advantage as a non-mammalian model, reducing the ethical concerns associated with higher animals. The use of C. elegans aligns with the principles of the 3Rs (Replacement, Reduction, and Refinement) in animal research, promoting more humane and responsible scientific practices.
Additionally, AI applications in HTS must address issues of data privacy and algorithmic transparency to ensure ethical integrity.
Emerging Trends in HTS
Technological Innovations: Recent advancements in HTS have significantly improved the efficiency and accuracy of the screening process. Automation and miniaturisation are key innovations that reduce costs and increase throughput. Automated systems can handle large libraries of compounds with minimal human intervention, ensuring consistent and reproducible results. Miniaturisation techniques, such as microfluidics, allow for the testing of smaller sample volumes, conserving valuable reagents and reducing overall costs.
AI and Machine Learning: AI-driven HTS leverages advanced machine learning algorithms to accurately predict active compounds, optimise lead compounds, and seamlessly integrate diverse datasets.10 These technologies address traditional HTS challenges by reducing false positives and improving data analysis. Machine learning models can analyse complex biological data, identifying patterns and relationships that may not be apparent through traditional methods. This
enhances the efficiency and accuracy of HTS, making it a powerful tool in modern drug discovery.
Data Integration and Analysis: Advanced data processing techniques are crucial for extracting meaningful insights from the vast amounts of data generated by HTS. Automation and AI significantly enhance the efficiency and accuracy of data integration and analysis. By combining data from various sources, researchers can obtain a more comprehensive view of biological interactions and identify potential drug candidates more effectively. This integration enables better-informed decisions and accelerates the drug discovery process.
Integrated Research Frameworks: Integrating C. elegans models with other HTS frameworks could provide a more holistic approach to drug discovery, leveraging the strengths of various models and technologies. This integration would enable more comprehensive and accurate drug discovery, benefiting from the unique advantages of each model. For example, combining C. elegans with mammalian cell cultures and AI-driven data analysis can provide a multi-faceted understanding of drug effects, enhancing the overall robustness of the screening process. By leveraging AI, researchers can process large datasets more efficiently, identify patterns, and make data-driven decisions that accelerate the drug discovery process.
Alternative Model Organisms: In addition to C. elegans, other model organisms like zebrafish and fruit flies (Drosophila melanogaster) are gaining traction in HTS due to their genetic similarities to humans and ease of maintenance. Zebrafish, for example, are particularly useful for studying developmental processes and genetic mutations due to their transparency and small size during larval stages, allowing for non-invasive imaging and high-throughput screening.11 Fruit flies offer advantages in studying neurological diseases and genetic interactions due to their well-mapped genome and short life cycle.
Human Cell Cultures and Organoids: Human cell cultures and organ-on-a-chip technologies are being increasingly adopted to replicate human tissues and organs, providing more relevant human data while reducing the reliance on animal models. These technologies allow for the study of complex cellular interactions in a controlled environment, improving the predictive power of HTS. For instance, 3D organoids can mimic the architecture and function of human organs, providing a more accurate model for drug testing and disease research.12
CRISPR and Gene Editing: Advances in gene editing technologies, such as CRISPR-Cas9, are revolutionising HTS by enabling precise genetic modifications in various model organisms and cell cultures. This allows researchers to create specific disease models and investigate the effects of genetic variations on drug responses, leading to more targeted and effective therapies.
Ethical and Regulatory Evolution: As the field evolves, so too will the ethical and regulatory landscape. There is a growing emphasis on reducing animal use in research and improving the ethical standards of HTS. Innovations like C. elegans, zebrafish, and human cell-based models align with these goals by providing alternative methods that meet regulatory requirements while minimising ethical concerns.
Conclusion
High throughput screening has revolutionised drug discovery by enabling rapid and efficient screening of compounds. Emerging trends and technological advancements, particularly in AI and machine learning, continue to enhance the effectiveness of HTS. C. elegans, and presents a scalable, in vivo alternative that, when integrated with advanced technologies, holds great promise for the future of drug discovery. The ongoing evolution of HTS methodologies will undoubtedly lead to more successful therapeutic discoveries and improved healthcare outcomes.
REFERENCES
1. Badr-Eldin, S. M., Aldawsari, H. M., Kotta, S., Deb, P. K., & Venugopala, K. N. Three-Dimensional In Vitro Cell Culture Models for Efficient Drug Discovery: Progress So Far and Future Prospects. Pharmaceuticals 15, 926 (2022)
2. Langhans, S. A. "Three-Dimensional In Vitro Cell Culture Models in Drug Discovery and Drug Repositioning." Frontiers in Pharmacology 9 (2018): 6. doi:10.3389/fphar.2018.00006.
3. DiMasi, J. A., Grabowski, H. G., & Hansen, R. W. "The price of innovation: new estimates of drug development costs." Journal of Health Economics 22, 151-185 (2003).
4. Tufts Center for the Study of Drug Development. "Tufts CSDD R&D cost study." (2016).
5. Baldrick, P. "Pharmaceutical Toxicology: Special Topics." (2008).
6. Paul, S. M., Mytelka, D. S., Dunwiddie, C. T., Persinger, C. C., Munos, B. H., Lindborg, S. R., & Schacht, A. L. "How to improve R&D productivity: the pharmaceutical industry's grand challenge." Nature Reviews Drug Discovery 9, 203-214 (2010)
7. Weinkove, D., & Zavagno, G. "Applying C. elegans to the Industrial Drug Discovery Process to Slow Aging." Frontiers in Aging 2, (2021). doi:10.3389/fragi.2021.740582.
8. Petrascheck, M., Ye, X., & Buck, L. B. "A High-Throughput Screen for Chemicals that Increase the Lifespan of Caenorhabditis elegans." Annals of the New York Academy of Sciences 1171, 1, 95-106 (2009). doi:10.1111/j.1749-6632.2009.04377.x
9. Maggie's Pearl. "Maggie's Pearl Phase 3 PMM2-CDG Trial." Maggie's Pearl (2024). Available at: https://www.maggiespearl.com/maggiespearl-phase-3-pmm2-cdg-trial
10. Ofenbauer, Andreas. (2019). Characterisation of the KASH domain gene unc-83 and the pseudogene F55A3.7. 10.18452/20457.
11. NC3Rs. "Five Reasons Why Zebrafish Make Excellent Research Models." NC3Rs (2024). Available at: https://www.nc3rs.org.uk/news/ five-reasons-why-zebrafish-make-excellent-research-models
12. Cruelty Free International. "Alternatives to Animal Testing." Cruelty Free International (2024). Available at: https://crueltyfreeinternational.org/ about-animal-testing/alternatives-animal-testing
David Weinkove is well respected in the academic fields of C. elegans and ageing biology. As a postdoc, he worked with David Gems, Ronald Plasterk and Erik Jorgensen. He is now Associate Professor at the Department of Biosciences, Durham University and Chair of the British Society for Research on Ageing. David is passionate about applying the strength of C. elegans research to industrial application and he co-founded Magnitude Biosciences to bring reliable automated technology together with experienced C. elegans scientists to bring increases in productivity to the whole field.
Email: david@magnitudebiosciences.com
David Weinkove
Isosterix Uses CDD Vault to Expedite Drug Discovery & Securely Collaborate
Situation
Roopa Rai is the Founder and CEO of Isosterix. With over 25 years of experience, Roopa has held leadership positions in discovery research in various therapeutic areas including oncology. She has a successful track record of driving programs from discovery to IND-ready compounds including six clinical candidates. Past positions include SVP, Discovery Research at Myoforte Therapeutics and Senior Director of Chemistry at Medivation and Assembly BioSciences. She is a co-author/ inventor on 70+ patents and publications. She has a Ph.D. in Organic Chemistry from the University of Illinois at UrbanaChampaign and has done postdoctoral research at Cornell University and Stanford University.
Rai founded Isosterix to develop small-molecule inhibitors of KAT6A, an epigenetic oncogene that is implicated in multiple cancers including breast, prostate, lung, ovarian and hematological cancers. KAT6A/B are histone acetyl transferases (HATs), a new class of epigenetic regulators. KAT6A is a promising new oncology target particularly in endocrine-resistant breast cancer. Isosterix has inhibitors that are selective for KAT6A over other HAT family members, including its paralogous protein Kat6B.
Solution
As Rai created her new company, she deployed Collaborative Drug Discovery’s (CDD) Vault, the hosted drug discovery informatics platform that securely manages both internal and external biological and chemical data.
This was an easy decision, as she had been using CDD Vault for more than a decade while working at other biotech
companies. Rai has also consulted with companies that don’t use CDD Vault, which reminds her of how much she values it.
''I have used CDD Vault for years and couldn’t do the work that I do without it,” Rai says. “CDD Vault helps speed time to discovery.” Isosterix uses CDD Vault to store some 350 compounds. Rai uses CDD Vault to register compounds, view assays, perform data searches, SAR analysis, and to securely collaborate with the company’s consultants and offshore CRO.
Benefits
Isosterix has found a number of benefits since adopting CDD Vault, including:
• Providing a “single source of truth”
• Speeding time to drug discovery
• Serving as a powerful tool for startups
• Supporting collaboration with consultants and CROs
• Protecting intellectual property
Providing a “Single Source of Truth”
Having worked widely within the world of pharmaceutical research, Rai appreciates the significance of CDD Vault providing what many refer to as a “single source of truth.” She notes that the alternative is too often a collection of spreadsheets or other forms of decentralised data stores which can make it difficult and time consuming to determine where the most recent data resides.
“With CDD Vault, the freshest data is always just a click away,” Rai says. “The moment new data is uploaded into CDD Vault, it's available to all scientists registered on your CDD Vault.”
This immediacy of data availability, combined with CDD Vault searchability and SAR analysis helps drive discovery. “New data elicits new ideas, such as how the next molecules might be made,”
Rai says. “As a central data repository, everyone on the team can securely access the newest data to guide their work.” With her long background working with CDD Vault, Rai finds it frustrating when working with organisations that don’t have a centralised repository.
“I’m consulting with a company now that doesn’t have CDD Vault and a lot of time can be wasted looking for data that would be just a click away with CDD Vault,” Rai says. “I recently spent 30 minutes pulling together data that I could have had within moments using CDD Vault. So, it can be frustrating to work without it.”
Speeding Time to Drug Discovery
The powerful SAR analysis and robust searchability of CDD Vault helps speed time to discovery. “The SAR analysis in CDD Vault helps you to think of new molecules and generate new hypotheses for whatever problem you are trying to solve,” Rai says. “In drug discovery you go through iterative cycles of structure, activity relationships. You look for trends. You might say: I need a hydroxy group, but at another position on the molecule. That's my next hypothesis. We improve whatever it is we're trying to improve in that particular cycle of SAR.”
“CDD Vault expedites drug discovery by giving you the ability to quickly visualise your data, and then propose the next molecule to make,” Rai says. “CDD Vault helps in the thinking process. It gives you the ability to sort data and ask questions, and to quickly propose the next, most promising, molecule to make. This saves you from having to make 50 molecules to get to the next iteration of improvement in potency or improvement in oral bioavailability. CDD Vault helps you get to the next step more quickly by querying the data and getting the answers.”
CDD Vault is a Powerful Tool for Startups
After working for a number of biotechnology companies, Rai decided to create her own, initially with just herself as the sole employee, augmented by consultants and a CRO. From this experience, she suggests that every biotech startup make CDD Vault part of their work foundation, or at least to do so by the time they begin to do their research. “CDD Vault is a great organiser,” Rai says. “It should be an essential part of any new drug discovery venture because otherwise the data becomes overwhelming in a hurry. You are tracking multiple kinds of data for each compound, and that complexity grows with each new compound. The moment you start making compounds and gathering data, which is what drives your progress, that's when you need CDD Vault. I couldn’t do without it.”
CDD Vault Is Easy to Use: “I Don’t Have Any IT Specialists. With CDD Vault, I Don’t Need Any.”
CDD Vault has long been praised for its ease of use, which is especially important for startups and other small biotech businesses that might not have IT specialists on staff. “CDD Vault is extremely easy to use,” Rai says. “I don’t have any IT specialists. With CDD Vault, I don’t need any. When I have a question, I just contact CDD and they respond almost immediately, providing all the help and guidance I might need.”
Rai also appreciates built-in protections to ensure data integrity, including automated duplication protection which prevents registering the same compound twice. Registration of
Application Note
new compounds is simplified through use of custom templates that CDD creates for clients.
“I just send CDD a description of my current program and they create a custom template for me,” Rai says. “With the template, it’s just a few clicks and your compounds are registered into CDD Vault.”
Supporting Collaboration with Consultants and CROs
Rai has long valued the ease with which CROs can be given secure, granular access to CDD Vault, enabling them to directly upload their work, making it immediately available to others from anywhere around the world. “Working with biotechs prior to starting my own business I might have one CRO doing biology and another CRO doing chemistry, and I would set them up with licenses so they could directly interact with permissioned areas of CDD Vault,” Rai says.
“This is a very efficient way to collaborate across the world”. Now with her own company, Rai has found that even without providing licenses to her CRO, she is able to simply upload their data through the CRO’s use of the CDD Vault template she sends them.
“My CRO returns the template and with just a few clicks I can register new molecules and related data into CDD Vault,” Rai says. “I also use CDD Vault to provide access to my consultants. For example, if a biologist needs a molecular weight or something, they can get the data immediately.”
Protecting Intellectual Property
The tight, granular, security of CDD Vault helps protect intellectual property, which is absolutely essential for successful drug discovery.
As Isosterix prepares to license a compound to a pharmaceutical company, it is able to demonstrate IP protection through its use of CDD Vault. Along the way, the company can securely grant specific data access on an as-needed basis to collaboration partners. Rai also likes that using CDD Vault makes it easy to securely share data rather than sending material as attachments.
“I communicate with one of my consultants every day through emails,” Rai says. “If I want her to see a certain SAR, I just send her a link from CDD Vault. She can click on the link and securely see the data from within CDD Vault, because she's a registered member. This means we can share data in a confidential way without sending structures attached in an email.”
Collaborative Drug Discovery
Collaborative Drug Discovery provides a modern approach to drug discovery informatics that is trusted globally by thousands of leading researchers. The flagship product, CDD Vault, is a hosted informatics platform that securely manages both private and external biological and chemical data. It provides core functionality including entity registration, structure activity relationship, inventory, visualisation, ELN, AI and lab automation. For more information, visit us at www.collaborativedrug.com.
Logistics
Packaging: The First Line of Defence in Cell and Gene Therapy Logistics
Temperature-controlled packaging plays a vital role in cell and gene therapy logistics.
Packaging has a multi-faceted role in supporting cold chain logistics to ensure transit times are met and the shipment is protected during handling. Unique packaging solutions have been developed to transport anything from sensitive biological materials during a clinical trial to life-saving medicines and cell and gene therapy treatments.
The appropriate packaging solution will ensure internal temperatures are maintained throughout global supply chains and that consignments remain viable to their final destination.
Biocair’s Director of Cell and Gene Therapy Logistics, Christopher Good, shares his insight: “As advanced treatments such as cell and gene therapy have become more complex and prevalent, there is an increased need for comprehensive packaging solutions that can maintain the integrity of a shipment in a wide range of conditions and, most importantly, alert supply chain specialists to any breach in the cold chain. It is the first line of defense against temperature breaches and damage sustained during handling or tampering.”
Industry Challenges
The global rollout of the COVID-19 vaccine demonstrated the need for robust cold chain logistics solutions in the face of a pandemic, with successful delivery and treatment reliant on packaging and transportation for a vast range of terrains and temperatures.
Research from the World Health Organisation (WHO) underlines the global challenges the industry needs to overcome. WHO reports approximately 50% of vaccines globally are wasted, including the COVID-19 vaccine, and a proportion of those are damaged during transit and storage due to cold chain malfunction.
Good identifies “A greater risk of future pandemics, the climate crisis and an increasing reliance on personalised treatments, such as cell and gene therapies, are accelerating the need for innovative, sophisticated and validated packaging solutions that can help to mitigate these issues.”
Packaging may be the first line of defense against temperature excursions, but how can specialists ensure thermal performance is maintained throughout the consignment’s journey?
“As global temperatures rise, and supply chains become more complex, cold chain packaging will inevitably have to work harder,” explains Good.
The 10 warmest years in the 174-year record have all occurred during the last decade (2014–2023), according to data from the National Centers for Environmental Information.
“Identifying where cold chain malfunction occurs is the key to reducing the industry's biggest issues, including the occurrence of temperature excursions,” says Good.
Cold Chain Packaging
Packaging for cold chain life science shipments is evolving to meet the growing demand for specialist treatments. Vacuuminsulated packaging and phase change materials are just some of the complex packaging components that are being used to maximise the thermal performance of cold chain packaging during transit.
What does the packaging selection process look like?
Good describes, “Packaging is carefully selected based on each consignment’s unique requirements, with temperature profile, transit time and shipping lane data all analysed and evaluated to identify the right packaging requirement. Every packaging option must also be GDP compliant and ISTA7 tested – the temperature test for transport packaging managed by the International Safe Transit Association (ISTA).”
He continues, “Solutions have been developed to meet and sustain a comprehensive range of temperatures, from cryogenically frozen (-150ºC and below) to refrigerated (+2ºC to +8ºC) and every temperature in between, while also providing protection from external factors like shock.”
Packaging Designed to Protect
Packaging manufacturers must prove that packaging for sensitive CGT materials will withstand temperature and other environmental changes throughout a shipment’s journey.
But, while packaging manufacturers are required to perform some tests on their products, additional validation testing must be carried out before a packaging solution is used – and through its lifecycle.
Using packaging that has been validated for a specific supply chain route is vital if a product is to arrive safely and on time. This means having an expert understanding of the environmental and human factors to which the packaging may be exposed.
Packaging validation starts with the manufacturer, but third-party logistics providers (3PLs) are typically tasked with creating and implementing a validation plan. 3PLs often have higher internal testing standards, including between-use testing.
Validating packaging to protect against these risks, across the supply chain, is a detailed and scientific process.
Validating Packaging
Validation involves a comprehensive set of testing protocols that prove a packaging solution reliably meets a unique set of standards.
It is essential that any validation protocol is fully agreed before the validation process begins and data is collected. Protocols may need to be updated throughout a package’s lifecycle.
The protocol must be compliant with government regulations and take into consideration any company-specific parameters. For example, liquid nitrogen dewars used in the transport of cell and gene therapy treatments are typically validated between uses. GPS positioning and remote condition monitoring are required to monitor location and temperatures in real-time during transit.
All validation protocols should detail the following:
1. A test plan
2. A sample size
3. Frequency of tests
4. Method justification
5. Aging conditions
6. Acceptance criteria (i.e. the degree of packaging failure that is acceptable)
Validation plans often involve mock shipment tests that provide dynamic routing information. The data from mock shipments allows senders to understand the worst-case scenario in transit.
Test shipments should ideally be performed in triplicates and meet acceptance criteria laid out in the validation plan before being used for actual shipments.
Acquiring data from real-world mock shipments is just one step in validating packaging.
Validation, Qualification and Verification Tests
To successfully validate packaging, systems, materials and
people must all be qualified, as set out in the validation protocol.
‘Qualification’ can refer to a number of different tests for different purposes, but they are all intended to show that a package will perform again and again across the supply chain, in changing circumstances and over time.
Tests for qualification are often annual or semi-regular. First, verification tests determine the capability of materials, equipment, processes and products to meet the requirements. A combination of verifications provides a qualification. Four primary qualifications are:
• Design qualification (DQ)
• Installation qualification (IQ)
• Operational qualification (OQ)
• Performance qualification (PQ)
Whenever a material or system is modified, requalification is necessary. Requalification can also be triggered when a packaging option needs to be tested for a specific purpose, supply route, or other parameter.
Navigating Unique Supply Chains
Packaging and validation processes must always comply with global standards, regulations and recommendations. However, validation must go beyond manufacturer testing to meet unique shipping requirements. Specialist logistics providers can take the burden away from the life science research or manufacturing organisations, leading to packaging options that outperform market standards.
Whether it’s small consignments for targeted cell and gene therapies, or the need to move bulk consignments during the COVID-19 pandemic, having a cold chain logistics partner that can supply fully validated packaging solutions is vital in a fast-changing world.
New risks and challenges are already on the horizon – for example, extreme weather and supplier consolidation. As that occurs, it will be important to develop standard procedures that account for any potential variation in distribution. These include delivery delays, differences in handling practices, distribution models and transportation, climatic variations and more.
Logistics
Utilising Accurate Temperature Profiles to Maximise Supply Chain Efficiency and Reduce Risk
Along with mock shipment testing, developing seasonal temperature profiles custom to the shipping lane adds an additional level of confidence that packaging solutions will withstand route-specific temperature extremes.
These profiles are important to include in packaging validation plans, not only to comply with strict regulations, but also to minimise the risk of temperature fluctuations in a parcel.
ISTA provides standards for testing a parcel in different thermal conditions, as well as a host of temperature data for common route profiles. Winter and summer profiles help operationally qualify packaging for effective cold supply chain management.
As thermal packaging technology evolves, and takes various forms using different materials, it is necessary to rework current sustainment methods and find more quality-control mechanisms that can be followed throughout the entirety of a product’s lifecycle. For accurate results, third-party logistics experts run mock shipments to create these profiles.
ISTA 7E Temperature Profiles Inform Packaging Selection
Along with enabling accurate assessment of risk throughout the shipment journey, temperature profiles inform which insulated shipping container should be selected for use. The International Safe Transit Association (ISTA) cites, “From design to design and manufacturer to manufacturer, it is very difficult to know if the performance of one insulated shipping container is better or worse than another for a particular cold chain challenge.” For this reason, the 7E profiles have been created by the ISTA and adopted as the global standard. The best practice when selecting an insulated shipping container is to ensure that it has been tested by the manufacturer according to the 7E profile.
Tracking and Data
Increased connectivity through monitoring and tracking devices, as well as clear communication between supply chain partners and logistics coordinators, are one of the best tools the life science supply chain has in understanding where temperature excursions occur and how to overcome them.
Good explains, “Location tracking and the 5G wireless network allow for comprehensive live monitoring of a shipment,
identifying delays on route that may increase transit time, and therefore, contribute to consignment damage.”
Where do temperature excursions occur? What external factors have impacted the performance of the packaging? These are important questions that only data can answer.
Having full sight of a shipment during transit enables specialists to intervene at the time of an excursion or take preventative measures, while improved customer visibility means increased customer confidence in the security and On-Time In-Full (OTIF) arrival of their materials.
This level of detail enables the logistics team to develop solutions that reduce or prevent the likelihood of an excursion happening by identifying preferred transit lanes and making informed decisions when selecting airlines and couriers.
“Complex location tracking and condition monitoring in conjunction with predictive modeling technologies are the future of life science logistics,” notes Good.
He continues, “These tools will become an essential component of life science packaging in the near future, as they can provide clarity where human communication falls short.”
Ultimately, increasing industry-wide availability and use of condition monitoring technologies will help to prevent cold chain malfunction and increase OTIF rates to improve patient safety.
Building Supply Chain Resilience
Data is central to the future of cell and gene therapy packaging, and its ability to increase resilience.
Climate change and its impact on global temperatures will place more strain on the cold supply chain, meaning that packaging will need to undergo more extreme testing if it is to deliver the same thermal performance in the future.
Predictive technologies that support at the pre-planning stage will become vital in forecasting what will happen during a shipment and where the biggest risks lie.
Digital models that can analyse temperature in real-time and predict the impact of extreme conditions will enable specialists to calculate a more accurate picture of how different packaging solutions will perform.
Adopting increased data gathering will also lead to digital decision-making, with programs designed to evaluate red flags and re-route where it predicts issues are likely to occur.
Having the technology to predict delays, high traffic, or even grounded flights due to adverse weather, would provide greater insight and improved decision-making to identify the shortest route with the least risk.
Overall, greater investments and developments in tracking technology will create more robust supply chains and improve patient safety as the industry grows.
Christopher Good
Christopher, director of Cell and Gene Therapy Logistics, has played a pivotal role in the company's evolution since joining in 2002, from specialising in pre-clinical and small molecule products to focusing on complex clinical biological material logistics. Christopher shaped the company's growth in many ways, including establishing global processes, managing the world's largest implanted medical device recall program and overseeing one of the first commercial launches for a gene therapy. Currently, his focus is on further enabling the complex supply chain needs necessary for commercial distribution of FDA approved therapies at scale.
November 12–13, 2024 Leipzig, Germany
Register now and join us at #LIONconf24 for a stimulating scientific program and networking opportunities.
www.LION- conference.com
Events Preview & Review
Drug Delivery to the Lungs 2024
DDL2024 will be live at the Edinburgh International Conference Centre (EICC) and virtually for those unable to travel to Scotland, on Wednesday 11th, Thursday 12th and Friday 13th December 2024.
Celebrating its 35th year, the Drug Delivery to the Lungs Conference will again showcase the latest research in the area of inhaled drug delivery. Alongside the talks, industry exhibition and posters, there will be the usual convivial hospitality and networking opportunities for which the conference is renowned, with a complimentary Gala Dinner for all delegates in the spectacular National Museum of Scotland, Edinburgh.
DDL2024 provides a balance of coverage across key areas of aerosol science and the development of inhaled medicines e.g. CMC, clinical and device development – to appeal to both academia and industry – using a single conference stream meaning no difficult decisions as to which presentation to attend.
The conference programme will include talks from invited speakers as well as those who have submitted papers to be considered for podium presentations.
The Session Titles for DDL2024 are:
• Transition to new propellants – current solution and future threat
• Advances in nasal drug delivery
• Inhalation across the ages
• Gene therapy and biologics for the lungs
• Developing Inhalation technology – Lessons learnt and current innovations
The call for papers is now open and the deadline to submit your paper is Friday 12th July. For full details and to submit your paper, please visit https://ddl-conference.com/ddl2024/ submit-conference-paper/
The Pat Burnell Young Investigator Award is also now open for submissions. This award encourages junior researchers from either academia or industry to present their work at the DDL Conference. Shortlisted Authors will be asked to deliver a 10-minute oral presentation live in Edinburgh at DDL2024. The award will be given to the researcher who, in the opinion of the DDL Organising Committee, has not only produced the best research but has shown that they understand and can explain their work. The winner of the Pat Burnell Young Investigator Award receives an award and £500.00 prize funds.
As a “not for profit” organisation, we would like to extend our thanks to our valued conference sponsors, without whom
we would not be able to bring you the great value, high quality DDL conference experience.
Delegate registration costs will continue to be held at a level that enables attendance across the breadth of industry, with attendance from academia actively encouraged, to facilitate cross fertilisation of knowledge across the sector. Students will continue to enjoy complimentary registration, providing the next generation of aerosol and drug delivery researchers with an extra-ordinary opportunity to engage with world-renowned scientists from across the aerosol and respiratory delivery field, to promote their own work, and to gain experience participating in a premier conference in their research field.
The New Researcher Network (NRN). Following their successful launch in 2018, the conference will once again host onsite networking events organised by the Network. The aim of the NRN is to develop a broad community of early career scientists and facilitate discussion surrounding research ideas/challenges, exchanges, joint projects and personal and professional development. New and early career researchers from both academia and industry, as well as PhD students, are encouraged to participate in the NRN, which has expanded to include a networking LinkedIn Group. – https://www.linkedin. com/groups/8942643/.
Events Preview & Review
Drug Discovery 2024: Connecting Minds, Transforming Science, Empowering the Community
2–3 October 2024, ExCeL London
ELRIG’s flagship conference is once again back in London with a packed two-day schedule with four auditoria, three auxiliary theatres, scientific poster sessions and an exhibition hall with suppliers showing their latest technologies.
Whatever your role in drug discovery and development, there is something for you and it’s free to attend!
The
Science
Each day starts with an inspiring and insightful talk from a keynote speaker, this year ELRIG are welcoming Prof. Ijeoma Uchegbu and Prof. Marcus Schindler.
Prof. Ijeoma Uchegbu from UCL researches the mechanisms of drug transport, which led to the discovery of transformational peptide nanoparticles that can be delivered across the blood-brain barrier and she will be explaining how nanoparticles can be used to enhance efficacy and minimise off-targets effects.
Prof. Marcus Schindler is Executive Vice President for Research and Early Development and CSO at Novo Nordisk and is Adjunct Professor of Pharmacology at the University of Gothenburg. His keynote presentation will focus on therapeutic approaches for metabolic disorders, such as obesity and diabetes.
ELRIG is delighted to once again work with scientific partner organisations including: The Cell & Gene Therapy Catapult; the Royal Society of Chemistry; SBi2 and SLAS to provide eight tracks of world-class science.
This year’s scientific tracks include:
• Advances in Assay Development and Screening – supported by SLAS
• Translating ideas into Therapies
• Mechanism and Therapeutic Strategies of Ageing
• Small Molecules beyond Rule of 5
• High-Content Imaging in Drug Discovery – supported by SBi2
• Target Discovery and Disease Modelling
• Cell & Gene Therapy – supported by the Cell & Gene Therapy Catapult
• Chemistry in Drug Discovery: Innovation, Collaboration and Inspiration – supported by RSC
Posters sessions run throughout the event showcasing the wide array of disciplines and technologies used in our industry.
Sustainability
Both in the lab and at the conference, sustainability is a feature of ELRIG events. The Tech Theatre will highlight the ways your labs can embrace sustainability and make changes to your work
patterns. While at the conference, ELRIG continually strives to reduce the carbon footprint and food waste from the event.
Careers and Learning
The Learning Hub sits at the centre of the conference, providing a space for early careers professionals to meet, gain advice to progress their career and participate in the ELIG ‘Network like a boss’ session, a fantastic opportunity to have conversations with senior members of the community to learn more about the opportunities in the industry.
ELRIG also supports the scientists of the future through the ‘Adventures in tiny lab coats’ programme inviting local schools to visit the conference and learn more about the possibilities in life sciences.
Innovation
ELRIG’s Breakthrough Zone has market-place feel with space to chat, with a dedicated theatre for young, innovative and dynamic companies wanting to make a splash with presentations and educational content on how to launch your own scientific innovation. A must for all budding ideas people!
Exhibition
At the centre there is the exhibition hall, which is a great opportunity to meet over two hundred drug discovery suppliers and to learn about all the latest products and services you can use in your research. Whether you’re involved in early drug discovery research or translating your promising therapy to the clinic, you’ll be sure to discover something that can help you at this year’s event.
Accessibility and Inclusion
ELRIG aims to remove as many barriers to conference attendance as possible, so all events are free to attend and with a prayer room, family room and quiet room available, ensuring our promise of inclusivity is at the heart of all our events.
REGISTRATION IS FREE
To learn more, to register or to enquire about exhibition and sponsorship opportunities, visit www.elrig.org
Events Preview & Review
Dive into the Heart of Pharma at CPHI Milan 2024
8–10th October 2024
Join us at CPHI Milan 2024, the premier global event for pharmaceutical professionals, from October 8–10 at Fiera Milano. Celebrating 35 years of excellence, this event promises unparalleled opportunities for networking, innovation, and professional growth. With over 62,000 attendees and 2,500 exhibitors, CPHI Milan offers a unique platform to connect with industry leaders, explore cutting-edge exhibitions, and discover new awards such as ‘Woman of the Year’ and ‘Future Leader’. It’s where suppliers, innovators, and distributors converge, promising unmatched opportunities for networking and partnership.
Event Overview
CPHI Milan 2024 returns with expanded opportunities for networking, innovation, and collaboration in the pharmaceutical industry. This three-day exhibition immerses attendees in every facet of pharmaceuticals, emphasising supply chain resilience and sustainable practices. It’s a pivotal opportunity to connect with industry professionals and forge lasting partnerships.
“This year is particularly significant for CPHI as we celebrate our 35th anniversary. It will be our largest event yet, featuring new awards and enhanced bioproduction zones and content. As we return to Lombardy, a hub for pharmaceutical manufacturing and API production, I eagerly await welcoming the global pharmaceutical supply chain, its partners, and drug innovators to Italy. The partnerships forged here will drive future drug development and ultimately improve patient access and treatments," remarked Orhan Caglayan, Group Director, CPHI Milan.
With over 2,500 exhibitors and 62,000+ industry professionals, CPHI Milan provides unparalleled opportunities for networking and partnership-building essential for enhancing supply chain resilience in an increasingly dynamic market. Attendees can expect a dynamic agenda designed to inspire collaboration and facilitate discussions on the latest trends, innovations, and sustainable practices shaping the industry today.
What’s New at
CPHI Milan 2024
Celebrating its 35th anniversary, CPHI Milan introduces several enhancements to elevate the attendee and exhibitor experience:
Supply Chain Resilience
Stay ahead with insights into new technologies and strategies essential for diversifying and stabilising the supply chain. CPHI Milan 2024 showcases the latest innovations and practical solutions to navigate the complexities of the pharmaceutical landscape. The event will focus on strategies for mitigating
risks, ensuring continuity, and optimising operations within the supply chain, crucial in today's volatile market environment.
Awards Ceremony at CPHI 2024
On October 8th at Alcatraz Milano, the CPHI Awards Night will commence with a glamorous drinks reception, celebrating excellence and innovation in the pharmaceutical industry across 14 categories. Among the highlights is the new "Woman of the Year" award, honoring women leaders who inspire colleagues, champion diversity and inclusion, and advocate for industry progression. Nominees are evaluated on their leadership, collaboration, and ability to create opportunities for women while challenging the status quo.
Equally anticipated is the novel "Future Leader" award, recognising rising stars under 35 with at least five years of experience in pharma, biopharma, or academic institutions. This award celebrates those who bring fresh perspectives, manage teams effectively, and introduce innovative ideas, marking them as the future leaders set to shape the pharmaceutical industry with their dedication and vision.
Key Highlights of CPHI Milan 2024
Unmatched Networking Opportunities
At CPHI Milan 2024, anticipate connecting with over 62,000 pharmaceutical professionals and 2,500 exhibitors, including industry giants like Lonza, Samsung Biologics, Thermo Fisher, and Pfizer CentreOne. It's expected to be the largest gathering yet, offering unprecedented networking possibilities. The event serves as a unique platform to establish new partnerships, strengthen existing relationships, and explore potential business ventures in a dynamic setting.
Conference Program Highlights
The CPHI Milan 2024 conference program is meticulously curated to inspire and educate attendees. Industry thought leaders lead insightful sessions on critical topics such as regulatory trends, technological innovations, and sustainable practices. From visionary keynote addresses on pharmaceutical R&D to interactive panel discussions on emerging therapies and market dynamics, the agenda promises a wealth of learning opportunities. Engage with over 150 expert speakers across 5 content tracks over 3 days at CPHI Milan, ensuring a comprehensive exploration of the latest advancements and trends in the pharmaceutical industry.
Events Preview & Review
Snapshot Overview of Some Key Sessions:
• Keynote: Advancing Immunisation with Next-Gen mRNA Vaccines Tuesday, October 8, 2024, from 11:00 AM to 11:30 AM
• Navigating Regulatory Landscapes: Ensuring Quality and Compliance in Pharma Ingredients Tuesday, October 8, 2024, from 1:15 PM to 1:40 PM
• Panel Discussion: Why the Middle East Can Be the New Frontier for Next-Generation Medicine Tuesday, October 8, 2024, from 4:15 PM to 5:00 PM
• Panel: Excipient Excellence: The Power of Excipient Grade Selection Wednesday, October 9, 2024, from 11:35 AM to 12:45 PM
Final Thoughts
CPHI Milan 2024 is more than an exhibition – it's a catalyst for change and advancement within the pharmaceutical industry. With a renewed focus on supply chain resilience, attendees gain actionable insights into navigating challenges,
optimising operations, and ensuring supply continuity. By embracing these trends and focusing on strategic imperatives, the pharmaceutical industry can achieve greater efficiency, resilience, and success in the years ahead. The future of pharmaceutical outsourcing is increasingly shaped by strategic, integrated, and collaborative partnerships. As the industry evolves, CDMOs and pharmaceutical companies must collaborate to navigate regulatory challenges, optimise supply chains, and deliver innovative therapies worldwide. CPHI Milan 2024 offers the perfect platform to explore these opportunities and stay at the forefront of industry trends. Don't miss your chance to be part of the conversation and drive the future of pharmaceuticals forward.
Join Us
Join us at Fiera Milano, Italy, and immerse yourself in a dynamic environment where ideas flourish and partnerships thrive. Let's shape the future of pharma together. Visit CPHI Milan 2024 to learn more and register today!
Page 29
Page 3
Bioprocessing Summit
Biotech Fluidics
Page 11 Collaborative Drug Discovery Inc.
Page 27 CPHI Milan
Page 39 DDL 2024
Page 41 Elrig
Page 37 Fraunhofer IZI
FC & 9 FUJIFILM Wako Chemicals USA
Page 17 Ncardia
IBC Newcells Biotech
BC Novo Nordisk Pharmatech A/S
Page 5
Owen Munford Ltd.
Page 23 PCI Pharma Services
Page 25 Richter-Helm Biologics GmbH & Co. Kg
Subscribe today at www.international-biopharma.com or email info@senglobalcoms.com
I hope this journal guides you progressively, through the maze of activities and changes taking place in the biopharmaceutical industry
IBI is also now active on social media. Follow us on:
For more information scan the QR code or visit: newcellsbiotech.co.uk/retina
Wondering which retinal cell types are transduced by your viral vector or how safe it is?
Human iPSC Derived Retinal Organoids and RPE Models That Give You Confidence to Progress In Vivo
Gene therapy vector assessment
Disease model development
Drug testing
The only functionally validated human cell-based models of the retina to study efficacy and safety of new gene therapy vectors1 and licensed for commercial development of therapies.
A unique preclinical platform for ocular drug discovery, offering tailored solutions for retinal gene therapy viral vector evaluation and drug development for retinal disease.
We can design and deliver a customised study such as determining if new viral vectors mainly transduce photoreceptors or if they target a broader range of cells in the retinal organoids. The organoids are available for purchase live or frozen to conduct your studies internally.