
10 minute read
Publication Highlights


Intercellular Transmission of Hepatic ER Stress in Obesity Disrupts Systemic Metabolism
Amir Tirosh, Gurol Tuncman, Ediz S. Calay, Moran Rathaus, Idit Ron, Abdullah Yalcin, Grace Yankun Lee, Rinat Livne, Sophie Ron, Neri Minsky, Ana Paula Arruda, Gökhan S. Hotamışlıgil Cell Metabolism, December 2020 (online) and January 2021 (in print) https://doi.org/10.1016/j.cmet.2020.11.009
Advertisement
This study demonstrates that regulation of intercellular communication is a critical component of tissue adaptation to metabolic stress. In the liver tissue of obese mice, Cx43, a ‘building block’ of network of channels between adjacent cells, is increased in both amount and activity under endoplasmic reticulum (ER) stress, a common insult in obesity and diabetes. Such an increase in Cx43 results in increased communication between cells, allowing cell-cell transmission and propagation of stress signals. Upon such cell-cell transmission between neighboring cells, the ER stress-naïve cells also become ‘stressed’ and dysfunctional, and further aggravate the liver impairment at the level of the tissue. Increase in Cx43 under ER stress conditions in hepatocytes, leads to abnormal Cx43-mediated intercellular communication. The increased cell-cell coupling allows transmission of harmful signals from ‘stressed’ to neighboring stress-naïve ‘bystander’ cells, resulting in impaired ER function and insulin resistance. Blocking Cx43 in hepatocytes prevents this transmission and protects the cells from overnutrition-induced stress, ER dysfunction and metabolic problems.
Endoplasmic reticulum (ER) stress and dysfunction is an important mechanism in the pathophysiology of obesity-associated metabolic pathologies such as insulin resistance and diabetes. While some of the cellular events linking ER stress and insulin resistance are well characterized, how an organ coordinates its adaptive response to stress, and the role of communication between its cellular constituents remains unclear. Increased gap junction (GJ)— intercellular communication, primarily involving connexin Cx43, plays a key role in the maladaptive tissue response to various stresses and has been implicated in the pathogenesis of atherosclerosis and neurodegenerative diseases, which, like obesity, are characterized by chronic low-grade inflammation and ER stress. In this study we demonstrate that in hepatocytes, ER stress results in increased expression of Cx43 mRNA and protein, and in increased GJ-mediated cell-cell coupling. Co-culture of ER stressed donor cells with ER stress-naïve recipient cells resulted in intercellular transmission of stress signals leading to impairment in the ER function (‘bystander response’). The propagation of ER stress required cell-cell contact, and genetic suppression of Cx43 prevented the transmission of ER stress from donor to recipient cells. In accordance with the in vitro cellular model, we observed that diet-induced obesity resulted in hepatic ER stress and upregulation of Cx43 in the liver. Remarkably, mice lacking Cx43 specifically in the liver were protected from HFD-induced liver ER stress, insulin resistance, glucose intolerance and NAFLD. Importantly, hepatocytes from HFD-fed mice were able to transmit ER stress to intact hepatocytes from lean mice in a Cx43- dependent manner. Taken together, our results indicate that in obesity, the increased Cx43-mediated cell-cell coupling may cause tissue propagation of ER stress. This novel maladaptive response to over-nutrition exacerbates ER stress in the liver, promoting fatty liver disease and impairing whole-body glucose metabolism.
Given the strong evidence that increased ER stress in obesity plays a key role in the pathogenesis of metabolic dysfunction, interfering with Cx43-mediated cell-cell coupling in hepatocytes may block tissue propagation of ER dysfunction and thus represent a novel approach to alleviate fatty liver disease, systemic insulin resistance and abnormal glucose homeostasis.
Cardiolipin Deficiency in Barth Syndrome Is Not Associated with Increased Superoxide/H2O2 Production in Heart and Skeletal Muscle Mitochondria
Renata L.S. Goncalves, Michael Schlame, Alexander Bartelt, Martin D. Brand, Gökhan S. Hotamışlıgil FEBS Letters, October 2020 https://doi.org/10.1002/1873-3468.13973
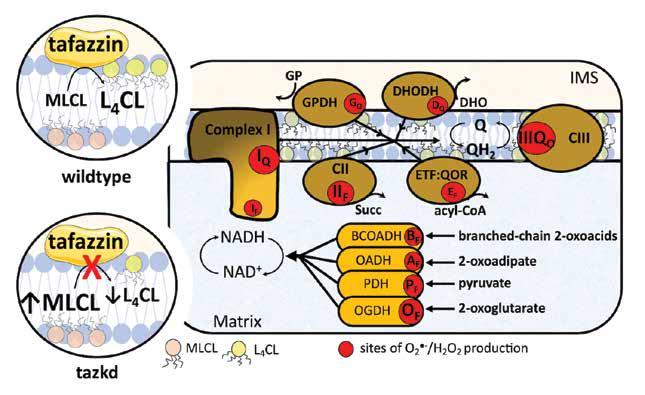
Barth Syndrome (BTHS) is a rare X-linked genetic disorder caused by mutations in the gene tafazzin (taz). Taz is an enzyme important for the remodeling of cardiolipin, a phospholipid uniquely localized in the mitochondria. The loss of cardiolipin in the mitochondrial membranes causes severe heart and skeletal muscle disease. The production of mitochondrial oxidants has been implicated in the cardiomyopathy in BTHS and in many other conditions of metabolic pathology. In general, oxidants are examined as one entity, although in the mitochondria, eleven different sites can produce these oxidant molecules at significant rates. Which of these sites generate oxidants at excessive or abnormal rates in BTHS and in most metabolic diseases is unknown. In this study, we measured the mitochondrial oxidants production from each of these sites in the heart and skeletal muscle of a mouse model of BTHS, the tafazzin knockdown mice (tazkd). Despite manifesting the disease’s symptoms, we did not find differences in mitochondrial oxidant production between the tazkd mice and their normal wild type littermates. Therefore, our study raises questions about the involvement of mitochondrial oxidants in BTHS pathology and illustrates the importance of detailed, individual examination of oxidative species.
Aberrant Ca2+ Homeostasis in Adipocytes Links Inflammation to Metabolic Dysregulation in Obesity
Ekin Güney, Ana Paula Arruda, Güneş Parlakgül, Erika Cagampan, Nina Min, Yankun Lee, Lily Greene, Eva Tsaousidou, Karen E. Inouye, Myoung Sook Han, Roger J. Davis, Gökhan S. Hotamışlıgil BioRxiv, October 2020 https://doi.org/10.1101/2020.10.28.360008
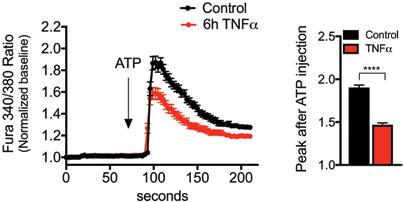
Chronic metabolic diseases including obesity, insulin resistance, diabetes, and atherosclerosis are the greatest threats to human health. The importance of proper metabolic balance has also become vividly apparent during the COVID-19 pandemic and they present the most important risk factor for disease severity and death. A key mechanistic link between these disease states comes from the immunometabolic dysregulation that is a central characteristic of metabolic pathologies. Earlier studies in our group demonstrated that chronic metabolic inflammation is a key feature of obesity, insulin resistance and diabetes. While the importance of chronic metabolic inflammation, or metaflammation, for metabolic disease is well established, there are important gaps in the mechanisms by which metabolic stress or obesity initiates and propagates this abnormal immune response, particularly in the adipose tissue. Also unclear is the integration of various known stress and inflammatory signals to impact metabolic properties of adipocytes.
In this work, we show that in adipocytes, altered regulation of the intracellular calcium handling is a key, adipocyte-intrinsic event involved in the emergence and propagation of inflammatory signaling and the resulting insulin resistance. This discovery was inspired by the observation that in the context of metabolic stress and obesity, there is an abnormal increase in the expression and activity of a calcium channel called IP3R in adipocytes. We also showed that inflammation, either induced by cytokines in cells or by obesity in mice, is responsible for this abnormality. The increased expression of IP3Rs results in increased cytosolic calcium signaling and impaired insulin action. Interestingly, the link between these mechanisms involved activation of a stress signaling enzyme called JNK, which we have shown in earlier studies to be critical to systemic insulin resistance and glucose intolerance. When JNK activity is blocked, the obesity-induced abnormal increase in IP3Rs can be prevented in both adipocytes in culture or in adipose tissue. Finally, we tested whether directly targeting this mechanism can be used to prevent obesity and associated pathologies. In mice, adipocyte-specific loss of IP3R1/2 protected against adipose tissue inflammation and insulin resistance despite significant diet-induced weight gain. Thus, this work reveals that IP3R over-activation and the resulting increase in cytosolic calcium is a key link between obesity, inflammation and insulin resistance, and suggests that approaches to target adipocyte calcium homeostasis may offer new therapeutic opportunities against metabolic diseases, including human obesity.
High Resolution 3D Imaging of Liver Reveals a Central Role for Subcellular Architectural Organization in Metabolism
Güneş Parlakgül, Ana Paula Arruda, Erika Cagampan, Song Pang, Ekin Güney, Yankun Lee, Harald F. Hess, C. Shan Xu, Gökhan S. Hotamışlıgil BioRxiv, December 2020 https://doi.org/10.1101/2020.11.18.387803

The functional diversity of the cells allows responses to changing environment and proper adaptation and survival. In particular, certain metabolic cells, such as hepatocytes, exhibit a wide array of metabolic functions, and in addition to very high capacity for protein production and secretion, they can alter glucose and lipid metabolism to adapt to environmental and nutritional challenges. These, of course, include physiological responses, such as feeding and fasting, and pathological alterations such as the case in obesity and associated metabolic problems. In hepatocytes, this rich functional repertoire is accompanied by a very complex and pleitrophic constellation of architectural features of the subcellular structures. Whether and how these structural features are regulated during metabolic challenges and whether they themselves are a determinant of metabolic output is not understood and represents a critical area of investigation in the field.
In this study, we have conducted a deep exploration of the subcellular architecture of organelles in liver tissue, in the native environment of hepatocytes and related architectural regulation to function. In order to obtain a detailed 3D information of the endoplasmic reticulum’s (ER’s) structural organization and morphology in a hepatocyte under physiological and pathological states, we utilized enhanced Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) imaging, a technique that has the highest spatial resolution and the advantage of large tissue imaging. Using this approach, we were able to image, segment and quantify intracellular organization of intact liver volume, containing around 20 full or partial hepatocyte volumes. These platforms allowed us to visualize inside and through the organellar constellations of the liver cells. Our analysis included 22,035 tissue sections, annotations and segmentation of 16TB of information using machine learning and neural networks in a volume of 935,495 cubic microns containing 1.82 trillion voxels. This work was possible through a multidisciplinary collaboration and the talented graphic art team of Refik Anadol Studios for the construction of video tours inside the cells at an unprecedented resolution.
Through this analysis, we observed that obesity, which is a well-established condition of organelle dysfunction in experimental models and in humans, leads to a striking remodeling of ER structure with a marked alteration in ER sheets and tubules. We also substantiated these findings with an array of biochemical and fluorescence imaging approaches. We then tested whether re-structuring of the ER closer to its native configuration could be achieved through the use of membrane shaping proteins and examined the functional consequences in vitro and in vivo. These experiments provided striking results and showed that by simply regulating structure, the function of the ER can be restored in obesity at a cellular level with systemic repercussions. Experimental restoration of ER sheets in obesity resulted in marked improvement in systemic metabolic homeostasis. Hence, subcellular architecture is a critical rheostat of metabolic function and its regulation is a key determinant of health and disease.
BRIDGING KNOWLEDGE THROUGH COLLABORATION
The Unfolded Protein Response Regulates Hepatic Autophagy by sXBP1-Mediated Activation of TFEB
Zeyuan Zhang, Qingwen Qian, Mark Li, Fan Shao, Wen-Xing Ding, Vitor A. Lira, Sophia X. Chen, Sara C. Sebag, Gökhan S. Hotamışlıgil, Huojun Cao, Ling Yang
Autophagy, June 2020
Adipocytes Promote Interleukin-18 Binding to Its Receptors During Abdominal Aortic Aneurysm Formation in Mice
Cong-Lin Liu, Jingyuan Ren, Yunzhe Wang, Xian Zhang, Galina K. Sukhova, Mengyang Liao, Marcela Santos, Songyuan Luo, Dafeng Yang, Mingcan Xia, Karen E. Inouye, Gökhan S. Hotamışlıgil, Guanyi Lu, Gilbert R. Upchurch, Peter Libby, Junli Guo, Jinying Zhang, Guo-Ping Shi European Heart Journal, July 2020

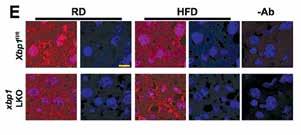
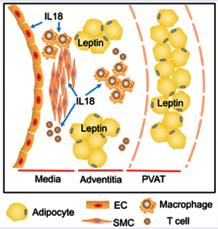
Defective autophagy and endoplasmic reticulum stress contribute to obesity-associated metabolic dysfunction, although the mechanisms linking these pathways remain completely understood. In this study, systematic examination of the targets of these pathways pointed to the regulation of a transcription factor, TFEB, a master regulator of autophagy and lysosome biogenesis. Collectively, our data provide novel insight into how two organelle stress responses are integrated to protect against obesityassociated metabolic dysfunction. It is well recognized that obesity is a risk factor of abdominal aortic aneurysm (AAA) but the mechanisms remain enigmatic. In this study, a mechanism is proposed through the increased binding of IL18 to its receptors demonstration of regions enriched in adipocytes or adjacent to perivascular adipose tissue. Two additional adipokines, leptin and FABP4 were also essential in regulating these interactions demonstrating a critical role for adipose tissue inflammation and adipokines in driving vascular abnormalities such as AAA in obesity.
CREATING A GLOBAL COMMUNITY THROUGH SCIENCE

...BY PREPARING FOR THE FUTURE

In our ongoing fight against the greatest threats to human health worldwide, metabolic diseases, we continue to support global scientific development through supporting young scientists and by bridging knowledge gaps through our research and collaborations.






