7 minute read
Hutchinson Gilford Progeria (HGPS) - Mishaal Faisal, Keya


Hutchinson Gilford Progeria (HGPS)
Mishaal Faisal, Keya Nanavati
Advertisement
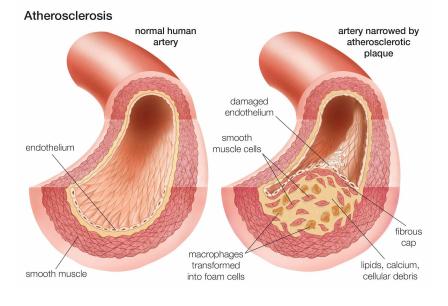
Affecting 1 in 20 million people, Hutchinson Gilford Progeria (also known as Benjamin Button disease) is a rare progressive genetic disorder that occurs in children. These children begin to age rapidly, yet seem perfectly normal at birth, making this disorder very hard to diagnose. During the first year of a child’s life with this disorder, symptoms such as slow growth and hair loss begin. The average life expectancy is approximately 13 years, and there’s no cure. Children die at such a young age because of cardiovascular complications, such as atherosclerotic heart disease- which refers to the build-up of fats and cholesterol in your arteries. Such conditions normally happen at an older age, suggesting how children at a young age with progeria are prone to suffer.
Surprisingly, only one gene is responsible for this complex disease: LMNA, located on chromosome 1. LMNA codes for the lamin A protein that fabricates structural proteins that holds the nucleus together and keeps the genome intact. Lamin A is a key component of the membrane of the cell’s nucleus. A mutated or defected version of this protein, known as progerin, causes the nucleus to become unstable and causes DNA damage. This misspelling on the lamin A gene causes the premature ageing that we associate with progeria.
Normally, the lamin A protein has a farnesyl group (lipids which act as protein anchors) added to the c-terminus (end of an amino acid chain) by farnesyltransferase (an enzyme). In progeria, the farnesyl group at the end of the lamin A protein cannot be removed. The protein is fixed to the nuclear rim instead of being part of the nuclear envelope. This means that the nuclear envelope doesn’t have support to remain in its original state and may die prematurely.
Progerin can cause many cell nuclei anomalies in addition to disturbances in the organisation of heterochromatin, mitosis and gene transcription. Progerin also damages DNA by hindering replication. The stress is sensed by proteins such as cGAS and STING which activate an immune response (STAT1) that leads to cellular decline. The immune response, though seems to be an interferon-like response, is not actually conducted through the release of inflammatory molecules, such as interferon. Interferons are signalling proteins released in the presence of viruses and trigger the immune system. This indicates that the ageing effects of progeria are cell-intrinsic and not external factors to the cell.
While the exact reason as to why premature ageing occurs is not fully understood, research suggests that this is due to the cumulative cellular damage from ongoing chemical processes (metabolic) within cells. The theory implies that compounds called free radicals are produced during chemical reactions in the body and these free radicals eventually accumulate in body tissues, causing damage to cells and impairing their functions.
This causes rapid ageing. Some enzymes, called antioxidant enzymes, are tasked with eliminating damaging free radicals. As enzymes speed up chemical reactions, researchers believe that reduced activity of these enzymes may cause the rapid ageing of HGPS. One study compared the fibroblasts (skin cells) of a person with HGPS with healthy skin cells and found that the activity levels of some primary oxidant enzymes were much lower than those present in healthy fibroblasts. This could highly support the theory from before, assigning the ageing effects of progeria to a decline in the activity of enzymes.
Though it is a genetic disease, progeria does not pass down in families. Other types of progeroid syndromes do tend to be passed down much easier though HGPS is considered to be a sporadic autosomal dominant mutation due to two factors: dominant due to the fact only one copy of the gene needs to be changed in order to be passed down, and sporadic as it is a new change in the family. It is considered to be caused by a random accident during cell division, not a hereditary disease.
Evidence is shown to suggest that parents having a child who already suffers from progeria may have a higher chance (around 23%) of having another child with the same condition. This is because of a condition called mosaicism, where a parent has the genetic mutations required for progeria in some of their cells but fails to have the actual disease.
Children with this condition can have complications related to atherosclerosis, which is when arteries harden. The walls of the arteries that carry oxygen and nutrients from the heart to the rest of the body stiffen and become thick, often restricting proper blood flow. This is why children characterized by this condition result in having heart attacks and congestive heart failure due to inadequate blood supply. Other health issues such as strokes also occur, because of problems with the blood vessels that supply the brain. These are also known as cerebrovascular problems.
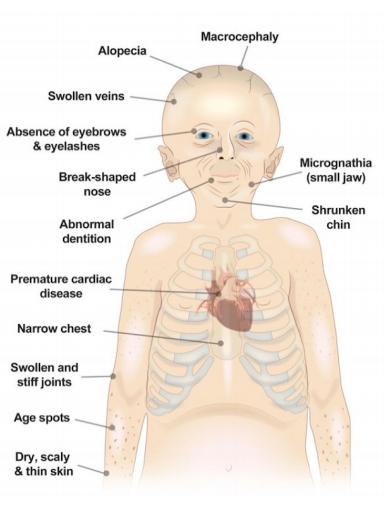
In the first few years, distinct appearances and health issues become evident, but motor development and intelligence remain normal. Some appearance-focused symptoms include a narrower face, head disproportion, prominent eyes and incomplete closure of eyelids, wrinkled skin, visible veins, and a high-pitched voice. In parallel, health issues such as cardiovascular disease, delayed and abnormal tooth formation, hearing loss, skeletal abnormalities, insulin resistance, and hip dislocation coincide. Skeletal defects are also evident, with a thin ‘dome’ portion of the skull as an example.
The prominence of the effects of HGPS develops throughout the lifespan of a patient. While patients are still newborn, they might display instances of shiny, hardened skin over numerous areas in the body. Bluish discolouration of the skin is also common during these times in addition to various other symptoms, though, growth delay is displayed only around 24 months. By 2 years old, patients display underdeveloped cases of bone, with the jaw and facial bones as examples. Here is where most indicators come into play such as loss of scalp hair and the aforementioned symptoms.
A doctor suspects progeria if a child is experiencing such characteristics of the syndrome. From there, a genetic test for LMNA mutations can confirm the diagnosis of progeria. The test requires a blood sample from which doctors can deduce if the child has progeria. Other physical examinations help identify this syndrome since a physical appearance plays a large role in recognising this disease.
Lonafarnib, sold under Zokinvy is a medication used to reduce the risk of death from HGPS. This is the first-ever treatment the FDA approved in order to cure this disease. Lonafarnib inhibits farnesyltransferase, an enzyme that hosts the production of progerin. This farnesyltransferase inhibitor (FTI) has demonstrated extended survival with children who have this since it prevents the protein from absorbing the cellular wall. The cellular wall is where most of the damage happens. Preventing this would mean reversing the instability of the structure. The capsules are known to treat certain processing-deficient progeroid laminopathies in patients older than a year old. Calcitriol, a type of Vitamin D, rescues the cells that experience glitches in DNA replication from progerin. This shows how significant of a role Calcitriol plays when it comes to DNA damage.
Research on this medicine has been done in 30 countries, and Brown University along with other research centres have tracked more than 250 children, demonstrating an evident link between using this medicine and having prolonged survival. Upon using Lonafarnib, which was a failed pediatric brain cancer drug, researchers found a moderate weight gain and a reduction in artery hardening, as well as improved nuclear shape. These positive results have led to this drug being used universally since copious amounts of evidence gathered indicates its success. Before this treatment, the only options available were supportive care and therapies directed towards the complications of this progressive disease.
In conclusion, while progeria does have a high number of risk factors, the medical procedures involved in treating this disease have proven successful. With decades of research on this topic, the data gathered has effectively helped treat this and evidenced to have an optimistic lead on eradication. Hopefully, our research has aided readers to see the dire situation of patients with Hutchinson Gilford Progeria and bring more importance and attention to the subject.
An organisation that aims to carry out effective research on Hutchinson Gilford Progeria Syndrome is progeriaresearch.org
You can donate to show your support towards distributing treatment to patients with HGPS.


