
9 minute read
Nitrosamine: a public health issue - Dra. Marleby García González


The document published by the Food and Drug Administration (FDA) entitled "Control of nitrosamine impurities in human drugs; Guidance for Industry; Availability", updated February 24, 2021, is an international guidance that recommends steps that manufacturers of active substances and pharmaceutical products (drugs) should take to detect and prevent unacceptable levels of nitrosamine impurities in drug products.
The guide also describes conditions that can introduce nitrosamine impurities into drug production processes. The unexpected and fairly recent finding of nitrosamine impurities (which are potential human carcinogens) in drugs such as angiotensin II receptor blockers (ARBs or A2RBs), or sartans, ranitidine, nizatidine, and metformin, has made clear the need for a risk assessment strategy for the potential formation of nitrosamines in any pharmaceutical product.
Advertisement
According to this guidance, pharmaceutical manufacturers should take the following steps in order to mitigate nitrosamine in their marketed products:
• Evaluate nitrosamine impurities in drug substances, marketed and approved products, and pending applications. Risk assessments should be conducted promptly based on drug prioritization. Manufacturers must submit the risk assessment documentation upon request.
• Perform confirmation testing when nitrosamine impurities are identified. Due to the physical-chemical properties of nitrosamines, analytical methods must include specificity, excellent chromatographic separation, and high-detection capability.
• Changes implemented to prevent or reduce impurities in active substances and drug products must be reported to agencies, which includes the submission of the Drug Master File (DMF) under 21 CFR 314.420 (c) standard, changes to approved applications under 21 CFR 314.70 and 314.97 standards and pending applications under 21 CFR 314.60 and 314.96 standards.
In the United States, to ensure the safety of drug supply, confirmatory testing, and submission of required changes to drug applications must be completed within 3 years of the publication date of this guidance. The implementation schedule includes examining the root cause of contamination/formation of these compounds, identifying changes to eliminate the root cause, and confirming that the proposed changes will minimize the risk of contamination or formation with no adverse effects.
From now on, drug product manufacturers must comply with the guidance to potentially eliminate the risks of nitrosamine impurities in their marketed products.
WHAT ARE NITROSAMINES?
Nitrosamines, or more correctly, N-nitrosamines, refer to any molecule containing the nitroso functional group. These molecules are of concern because nitrosamine impurities are human carcinogens. Although these impurities are also present in some foods and potentially in drinking water supplies, their presence in medicines is considered unacceptable.
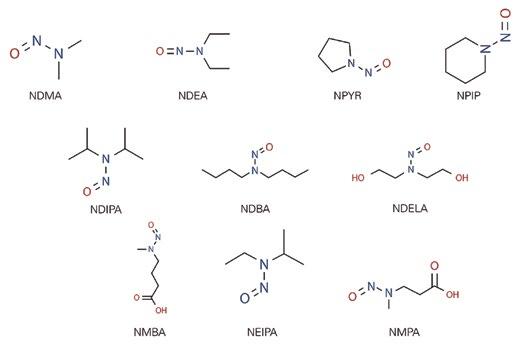
The presence of nitrosamines remains an issue in the human environment. Some of the most common sources of nitrosamines are tobacco (smoked or chewed), rubber products, cosmetics (creams, lotions, shampoos), metal cutting liquids, pesticides, and certain and well-identified pharmaceutical products. Nitrosamines are also present in food like bacon, beer, and canned fish. They can be formed when meat is heated at high temperatures. Nitrosamines, including NDMA, NDEA, and NPYR, among others, can also be found in drinking water.
WHY ARE THEY PRESENT?
The formation of nitrosamines is generally only possible when secondary or tertiary amines react with nitrous acid. By itself, nitrous acid is unstable, however, it can be formed in situ from nitrites (NO2) under acidic conditions.
In terms of sartan compounds, most contain a tetrazole ring and the formation of this ring involves the use of sodium nitrite. Coincidently, the solvents used are amines or contain traces of amines, and this probably causes the NDMA and NDEA nitrosamines, observed in them. The origins of the NDMA content in ranitidine batches are currently unclear.
However, during investigations, it was also concluded that the potential of nitrosamine impurity content was wider than just the simultaneous presence of nitrites and amines in the synthesis of the active pharmaceutical ingredient (API).
Evidence suggests that sources of nitrites or amines, as unintended contaminants from starting materials, reagents, and solvents, such as dimethylamine (DMF), may also provide circumstances in which nitrosamines can be formed. Carryover of nitrites or amines from later stages can also provide opportunities for the formation of these impurity compounds. Particularly, contamination from external sources has been identified as a highly likely source of nitrosamine input. Especially, contamination from the use of recycled materials and solvents that already contain levels of nitrosamines from downstream processes. An example is the use of recycled DMF, which is quenched with sodium nitrite to remove residual azide as part of the recovery process in the synthesis of some APIs. In addition, the recycling materials and solvents are often outsourced to third parties who may not have implemented adequate controls. Materials and solvents can be crossed-contaminated with nitrosamines or impurities that could react to form nitrosamines if the equipment is not thoroughly cleaned between customer batches.
Importantly, these additional mechanisms, in particular, cross-contamination, may affect several products that would not otherwise be expected to be at risk of nitrosamine formation by their natural synthesis route. These broader concerns have led regulatory agencies such as the EMA, FDA, and recently, COFEPRIS, to request that finished pharmaceutical product registrants conduct a risk assessment to determine the risk of nitrosamine content and verify that their drugs are free of these harmful impurities.
TOXICITY
Both NDMA and NDEA belong to the so-called “cohort of concern”, a group of highly potent mutagenic carcinogens that have been classified by the International Agency of Research on Cancer of the World Health Organization (WHO) as probable human carcinogens. Only limited impurity-specific toxicity data are available for NDMA and NDEA. Based on this information, most major regulators have adopted acceptable intermediate inputs for these specific impurities to provide a basis of safety for the consumption of drugs for the treatment of chronic diseases such as hypertension. Due to their structural similarity, international regulators consider that nitrosamines NDIPA, NEIPA, and NMBA should exhibit a similar toxicological profile to that of the NDMA and NDEA.
MOST RECOMMENDED METHODS OF ANALYSIS FOR THE DETECTION AND QUANTIFICATION OF NITROSAMINES
The low levels at which nitrosamine impurities occur create challenges for laboratory analysis testing, with few having access to adequate technologies. Several analytical methods that can be considered when determining the nitrosamine content in APIs and drugs have been published, primarily by the FDA and the European Medicines Agency (EMA).
The use of high-performance liquid chromatography-mass spectrometry (LCHRMS) technology has been recommended for its advantages of low detection limits and acceptable quantification. Other technologies also recommended, however, less used are the gas chromatography-mass spectrometry coupling (GC-MS) and the high-performance liquid chromatography (HPLC). In this order, the technologies are losing analytical sensitiveness in the methods described so far.
In Mexico, an analytical monograph for the quantification of these impurities has been evaluated in consultation with the Mexican Pharmacopoeia, which is expected to be published soon and become mandatory.
The CENEBA (National Center of Applied Bioprocesses Studies) is one of the few laboratories in the region with a validated analytical method capable of quantifying six different nitrosamines within the standards accepted by the most prominent and strict regulatory agencies.
In this Mexican laboratory, both technologies (LC-HRMS and GC-MS) are applied for this purpose and, depending on the nature of the sample to be analyzed, meet the expected detection and quantification limits requirements. The technology implemented at CENEBA reproduces the principles of international methods and with it, the methodological suitability of the matrices of regional medicines in Mexico and Latin America.

CORRECTIVE ACTION TO PROTECT PUBLIC HEALTH
Since 2018, WHO, EMA, and FDA issued public health alerts followed by guidance documents for manufacturers of APIs and pharmaceutical products on acceptable levels of nitrosamine impurities in different drugs.
An effective strategy will support manufacturers to identify points in their processes with the potential to introduce impurities, along with appropriate methods to detect, measure, and control them. Both manufacturers and global regulators play a role in ensuring that the levels of impurities remain within acceptable limits. To do so, they require tools to test and evaluate the risks related to nitrosamine impurities.
The FDA and EMA have already established deadlines to assess the manufacturing processes of all approved or marketed pharmaceutical products. The FDA establishes that manufacturers must complete a risk assessment by October 2021. If they identify one or more risk points where nitrosamines are potentially being introduced, the impurities should be confirmed by specific and sensitive analytical test methods. Then, manufacturers must report the changes carried out to mitigate the risk before 2023.
RISK TO GLOBAL DRUG SUPPLY
Beginning in 2018, the FDA and the manufacturers of the medicines worked together to protect patients by quickly removing ARBs from the market containing unacceptable limits of nitrosamines. However, the recalls resulted in a chronic drug shortage situation, where patients were at risk of not getting the medications, they needed to control their high blood pressure. In 2019, the FDA responded by prioritizing the review of a new generic product of valsartan. Its approval helped to alleviate the shortage of this critical drug resulting from multiple recalls of generic valsartan products.
Companies are responsible for understanding their manufacturing processes, including identifying and preventing the presence of unacceptable levels of impurities. Nevertheless, the global nature and complexity of the pharmaceutical supply chain make this an overwhelming task. Raw materials, excipients, substances, and finished drug products come from different countries around the world. The complexity of the manufacturing process for some drugs compounds the challenge. An inadequate risk assessment and control strategy can have dire consequences in the short and long term.
RISK OF NOT TRUSTING IN MEDICINES
ARBs and other drugs containing varying levels of nitrosamines are used to treat a wide range of common medical conditions. Valsartan, losartan, and irbesartan are anti-hypertensives drugs. Ranitidine is used to treat ulcers and acid indigestion. Nizatidine is also a treatment for stomach and intestinal ulcers. Metformin is used to treat high levels of sugar in the blood. Rifampicin and rifapentine are antibiotics used to treat tuberculosis. Given the prevalence of the conditions for which these drugs are used, analytical detection and monitoring of nitrosamine levels in processes and final products quickly rose to the level of public health imperative.
With each news headline about affected drugs, the public’s confidence in the safety of their medicines deteriorated. Patients debated whether they were more at risk by taking their medications than if they discontinued them. The FDA and healthcare professionals urged patients not to change their medications regimes unless instructed by their physicians because the interruption was potentially more dangerous to their health than exposure to unacceptable levels of nitrosamine exposure. When the list expanded to include non-ARB drugs, concerns grew exponentially among consumers. The drugs in question are found in the daily medication regimens of hundreds of millions of people worldwide.
Trust in medicine is difficult to measure, but the impact of the lack of it has been seen over and over around the world: the length and quality of people’s lives are decreased because, out of fear, they choose to quit life-saving treatments. For that reason, this is an issue of high national and international priority.
Dra. Marleby García González

CENEBA's General Director. A biochemist, expert in manufacturing processes of biotechnological drugs and the field of bio-analytics. She has 30 years of experience in the pharmaceutical industry in the areas of product development, regulatory affairs, and process validation.








