
24 minute read
LA BIOINFORMÁTICA EN LA INTERPRETACIÓN DE LA SECUENCIACIÓN MASIVA. Autores: José Miguel Lezana Rosales, Juan Francisco Quesada Espinosa, María José Gómez Rodríguez

from CLIN12LAB. Libro de sesiones de los Laboratorios Clínicos 2021
by Bioquímica y Análisis Clínicos. Hospital Universitario 12 de Octubre

Autores: José Miguel Lezana Rosales, Juan Francisco Quesada Espinosa, María José Gómez Rodríguez Servicio de Genética. Hospital 12 de Octubre. Madrid Palabras clave: Secuenciación masiva, Bioinformática, HPO
INTRODUCCIÓN
Advertisement
El genoma humano contiene un total de 60656 genes, de los cuales 19954 son codificantes de proteínas según GENCODE1. La alteración de las regiones codificantes u otras regiones de los genes (intrónicas flanqueantes, regiones de unión de factores de transcripción, regiones de metilación, etc.) puede dar lugar a enfermedad. Aunque los trastornos genéticos son poco comunes, en conjunto representan aproximadamente el 70% de las más de 6000 de enfermedades raras descritas2. En términos de incidencia, esto se traduce en que aproximadamente 1 de cada 17 personas se encuentra afecta por una enfermedad de etiología genética no oncológica. Además, nuestra constitución genética juega un papel, en mayor o menor medida, en todos los procesos fisiopatológicos, incluidos los trastornos comunes multifactoriales, como la diabetes, hipertensión, obesidad, etc. En relación a las enfermedades raras, existen actualmente varios interrogantes. Al revisar la literatura científica, se observa una gran disparidad en el número de enfermedades raras descritas. Uno de los motivos de esta variabilidad deriva de la distinta definición de enfermedad rara adoptada por las instituciones sanitarias. Más imprecisa aún resulta la cifra de prevalencia global para cada enfermedad, muy variable en función de la fuente bibliográfica consultada. Además, hay que tener en cuenta que las enfermedades raras se encuentran infradiagnosticadas, especialmente en aquellos países con recursos sanitarios limitados e inaccesibles que, en muchos casos por motivos socioculturales (consanguinidad, aislamiento genético, etc.), representan poblaciones de riesgo para este tipo de enfermedades. Qué número de enfermedades raras tiene una base genética mendeliana, es una cifra aún por dilucidar. Por ello, en muchos casos, no hay acuerdo sobre el número exacto, o no hay fundamentos sólidos en los artículos científicos que respalden los números citados3 . Por otra parte, la introducción de las técnicas de análisis genómico que ha tenido lugar durante la primera década del siglo XXI ha contribuido enormemente al diagnóstico de las enfermedades de etiología genética en los países con mayores recursos sanitarios. Una de estas tecnologías, la secuenciación masiva de ADN (NGS) o de próxima generación, permite el estudio de un elevado número de secuencias de ADN de un paciente en un único ensayo. Sin embargo, aumenta proporcionalmente el volumen de datos generados y la complejidad de su procesamiento. Por ello, el desarrollo de las técnicas de secuenciación masiva ha fomentado el surgimiento y la consolidación de una nueva disciplina biosanitaria, la bioinformática clínica. La bioinformática clínica es, por tanto, la disciplina sanitaria cuyo objetivo es desarrollar y mejorar las herramientas necesarias para la adquisición, el procesamiento, la organización, el almacenamiento y la explotación del dato genómico con fines diagnósticos.
QUÉ ES LA SECUENCIACIÓN MASIVA
La NGS es una tecnología que emplea la secuenciación en paralelo de millones de pequeños fragmentos de ADN para determinar su secuencia. En contraste con la secuenciación Sanger4, la velocidad de secuenciación y cantidad secuencias de ADN generadas con la NGS son exponencialmente mayores, llevándose a cabo con costes reducidos5 .
Dentro de la NGS existen diferentes estrategias de abordaje: la secuenciación del genoma completo (WGS), la secuenciación de las regiones codificantes e intrónicas flanqueantes (exoma, WES) y la secuenciación dirigida a paneles de genes. Con la disminución del coste de secuenciación por base, el WES y la WGS han demostrado ser los estudios de primera elección en términos de costeefectividad y de rendimiento diagnóstico para los pacientes afectos de enfermedades con sospecha de base genética6 . En este sentido, varios estudios han investigado los beneficios derivados de la aplicación de WES y WGS en la clínica, centrados principalmente en la discapacidad intelectual o las enfermedades mendelianas en general7–12 . Teniendo en cuenta que en estudios genéticos de WES el número de variantes de ADN obtenidas es muy elevado (alrededor de 20000-25000 por paciente, en la experiencia en nuestro Servicio de Genética), el reto es determinar cuál o cuáles serían compatibles con el fenotipo clínico del paciente. Para ello es crítico el diseño de una estrategia de filtrado de todas las variantes obtenidas, apoyado en la información de diversas bases de datos genéticas y analizar qué variantes candidatas pueden estar relacionadas con el fenotipo del paciente. Para ello resulta fundamental desarrollar un pipeline bioinformático que tenga en cuenta todos estos aspectos.
QUÉ ES LA BIOINFORMÁTICA
Uno de los puntos de inflexión en la explosión de la bioinformática, fue el Proyecto Genoma Humano13 donde convergieron la Biología y Genética Molecular con la Computación ante la necesidad de una interpretación de datos biológicos de una magnitud desconocida hasta la época.
KARMA
Figura 1. Flujo de procesos del pipeline de análisis Karma. Elaboración propia
A nivel formal, la Bioinformática se podría definir como la disciplina encargada de la resolución de problemas que provienen de la biología mediante metodologías aportadas por la ciencia de la computación14, o como un conjunto de métodos cuya misión es la extracción de conocimiento biológico a partir de la secuenciación, la expresión, la proteómica y los datos distribuidos del trazado isotópico15 . En un plano más práctico, la Bioinformática tendría como objetivos el almacenamiento de la información obtenida a partir de los resultados experimentales, el análisis computacional de los datos biológicos y la extracción de toda la información y conocimiento posible. Para ello, resulta imprescindible la creación de bases de datos, el desarrollo de algoritmos y herramientas estadísticas para establecer relaciones entre datos y la implementación de herramientas informáticas que permitan analizar e interpretar estos datos.
QUÉ ES UN PIPELINE DE ANÁLISIS
En el campo que nos ocupa, la Bioinformática resulta crucial en cuanto a que un óptimo diseño de pipeline de análisis permite una reducción fundamental del volumen de información derivada de las secuencias (variantes de ADN) y la interpretación de estas. Para ello se han de tener en cuenta bases de datos diversas y se han de emplear distintos programas, scripts y algoritmos de diseño ad-hoc que nos ayudan, en último término, a desentrañar la causa molecular que subyace en una determinada patología con origen genético. En nuestro Servicio de Genética, se ha diseñado un pipeline de análisis ad-hoc (en adelante, Karma), que cumple las recomendaciones de validación establecidas por la Association of Molecular Pathology16. El ámbito de aplicación del pipeline, será, principalmente, el análisis de WES a partir de archivos FASTQ, provenientes del secuenciador masivo (Figura 1). Karma se encarga, de una manera totalmente automatizada, del procedimiento de alineamiento de estos archivos al genoma de referencia (2 alineadores), a determinar las variantes con respecto a este genoma (2 genotipadores) para, más adelante, ofrecer un listado de variantes genéticas anotadas (2 anotadores), un análisis de variación en el número de copias (CNVs), determinar presencia de regiones de pérdida de heterocigosidad (ROH), informes de calidad, de cobertura, etc. Los resultados se gestionan y se analizan con un programa de análisis genómico (JNOMICS).
ARCHIVOS FASTQ
En el área de la secuenciación de ADN, el formato de archivo FASTQ17 se ha convertido un formato común de facto para el intercambio de datos entre las diferentes herramientas. Proporciona una extensión simple del formato FASTA18 ya que tiene la capacidad de almacenar una puntuación de calidad numérica asociada con cada nucleótido en una secuencia.
Los archivos FASTQ se generan a partir de las lecturas de los fragmentos de ADN que hace el secuenciador. En detalle, este fichero está compuesto por cuatro tipos de línea que representan a cada una de las secuencias: - Identificador de la secuencia.
- Secuencia de nucleótidos: Adenina (A), Guanina (G), Citosina (C) y Timina (T). Cuando el secuenciador no es capaz de asignar un nucleótido, se introduce el carácter ‘N’. - Indicador de fin de secuencia e inicio de calidades asociadas: carácter ‘+’. El contenido de este campo es opcional, la línea puede constar únicamente de dicho carácter. - Calidades Phred19, codificadas por un conjunto de caracteres ASCII.
Un paso clave en el análisis de secuencias es el alineamiento de estas a un genoma de referencia. Para ello existen diversos programas que gestionan el alineamiento, como BWA20 y Bowtie 221, generando como resultado un archivo SAM22 o su correspondiente archivo binario BAM. El papel trascendental de los alineadores es que reducen muy significativamente, mediante diferentes heurísticas como los algoritmos de Burrows-Weeler23 o NeedlemanWunsch24, el tiempo requerido de alinear millones de secuencias a un genoma de referencia. Si se empleara una estrategia de “fuerza bruta”, el procedimiento simplemente no sería abordable.
Un aspecto crítico en este paso es que los programas han de permitir el alineamiento imperfecto de las secuencias al genoma de referencia, con el fin de que secuencias con algún cambio con respecto a la referencia se alineen a ella y posteriormente se puedan detectar dicho(s) cambio(s). Los alineadores también han de tener la capacidad de discriminar entre posibles alineamientos múltiples, llevando a cabo el más específico posible. Hay que tener en cuenta que el proceso de alineamiento de secuencias cortas, como las empleadas en WES, tiene limitaciones. Por ejemplo las que se dan en regiones con elevada homología en el genoma (como pseudogenes con identidad cercana a 100%): en ellas los alineadores no tienen especificidad suficiente para discriminar si la lectura pertenece a una u otra región. Cabe decir que existen repositorios en la red donde se pueden identificar las regiones cuya secuenciación es dificultosa25 .
ARCHIVO DE VARIANTES. VCF
El archivo VCF26 es un formato de archivo de texto que contiene líneas de metainformación, un encabezado y a continuación líneas de datos, cada una con información sobre una posición en el genoma en donde ocurre un evento de variación.
Para la generación de estos archivos se emplean genotipadores como GATK27–29 o VarDict30 (son los utilizados en el Servicio de Genética). Estas herramientas tienen en cuenta, entre otros aspectos, en cuántas lecturas está presente la variante respecto a la referencia y con el grado de fiabilidad de estas. Si se supera cierto umbral en los parámetros de verificación internos de los programas, se genera una línea con la variante en el archivo VCF.
FILTRADO DE VARIANTES
El siguiente paso es poner en contexto el archivo VCF, que sólo caracteriza posicionalmente las variantes en el genoma, y determinar cuáles de ellas serán potenciales candidatas a la hora de diagnosticar un paciente. En primer lugar se enriquecerá este archivo mediante la adición a cada variante de su nomenclatura HGVS31, en qué gen se encuentra y otra información proveniente de bases de datos diversas. Por ejemplo: - Frecuencia según bases de datos poblacionales: 1000 genomas32, ExAC33, GnomAD34 . - Identificadores de la variante según repositorios clínicos: ClinVar35, COSMIC36, HGMD37 . - Modo de herencia del gen al que se asocian según
OMIM38 .
- Predicciones de patogenicidad. - Etc.
El Servicio de Genética cuenta con una base de datos de desarrollo propio (12OVar) en la que se alojan variantes de más de 2000 exomas (más de 1 millón de variantes únicas). De esta manera se está en disposición de correlacionar variantes entre diferentes pacientes, hacer estudios retrospectivos, de cohortes, etc. Esto representa una herramienta crucial en el reanálisis a posteriori de casos no diagnosticados. Finalmente, mediante la aplicación JNOMICS se interpretan las variantes provenientes de Karma (Figura 2). Una vez interpretadas, se generan de manera automatizada los informes diagnósticos. Gracias a JNOMICS, además, es posible seguir la traza de los estudios en todo momento pudiéndose gestionar y consultar las diferentes bases de datos que componen su núcleo (paneles virtuales de genes 12OVar, tiempos de respuesta, etc.).
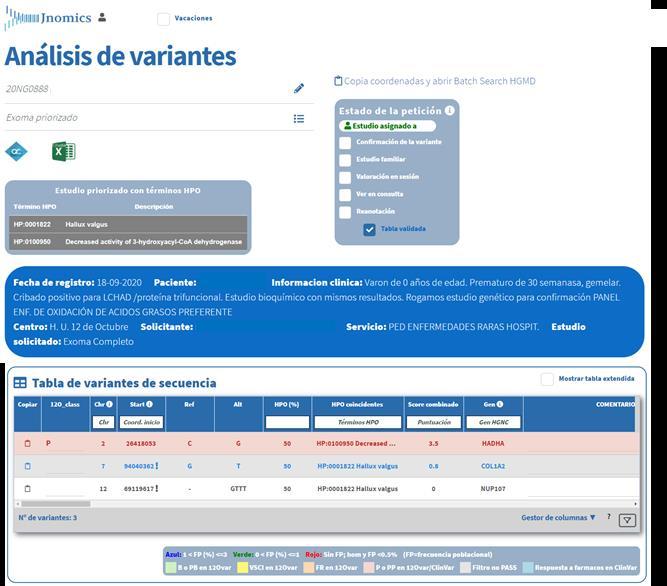
Figura 2. Módulo de análisis de variantes de JNOMICS. Elaboración propia
FILTRADO DE VARIANTES EMPLEANDO TÉRMINOS HPO
La ontología del fenotipo humano HPO39 representa una forma de estandarizar el vocabulario médico utilizado para designar las anomalías fenotípicas y los signos clínicos de la enfermedad humana. Su desarrollo se apoya en la literatura médica y en las bases de datos Orphanet40, DECIPHER41 y OMIM38. Actualmente, contiene más de 13,000 términos y más de 156,000 anotaciones a enfermedades hereditarias. Cada término en el HPO describe una anomalía fenotípica y está relacionado con todos aquellos genes para los que la enfermedad asociada puede cursar con la anomalía descrita. En el nuestro Servicio se ha integrado la priorización de variantes mediante el uso de términos HPO y características inherentes a ellas mismas: frecuencia poblacional, tipo de variante y predicciones de patogenicidad. Para esta integración, se ha desarrollado un script que ordena las variantes filtradas asignando una puntuación a cada variante, teniendo en cuenta lo siguiente: - Número de los términos HPO utilizados que están relacionados con el gen en el que se localiza la variante (Figura 3). - Tipo de variante. Mayor puntuación para las variantes radicales y canónicas de splice, seguido de las variantes de cambio de sentido y, por último, las sinónimas e intrónicas profundas. - Predicciones de patogenicidad. Mayor puntuación cuanto mayor es el número de predictores de patogenicidad in silico que apoya un efecto deletéreo de la variante genética. - Frecuencia de la variante en la base de datos poblacional según ExAC33 .
En un estudio publicado por nuestro grupo42 se reanalizaron los exomas completos de 33 pacientes con diferentes patologías y con diagnóstico genético molecular positivo (portadores de variantes clasificadas como patogénicas o probablemente patogénicas que explicaban su cuadro clínico y con datos de segregación compatibles). Como parte de este reanálisis, previamente a la priorización, se filtraron las variantes de potencial bajo impacto según diferentes criterios: frecuencia poblacional, tipo de variante, clasificación de patogenicidad según ClinVar, etc. A continuación se llevó a cabo la priorización puntuando las variantes como se detalló en el anterior punto. Se obtuvieron los siguientes resultados, detallados en la Tabla 1. Figura 3. Extracto de Relación término HPO/genes. Elaboración propia

Score Absoluto1
Score absoluto filtrado2
Score combinado3
Score combinado filtrado4

Media 7,81 4,47 3,125 1,43
SD 11,86 7,47 5,42 0,87
Mediana 2 1 1 1
Tabla 1. Resultados posicionales de las variantes según diferentes scores empleados en la priorización. 1Generado sólo teniendo en cuenta el porcentaje de términos HPO que se relaciona con cada gen asociado a la variante. 2Generado sólo teniendo en cuenta el porcentaje de términos HPO que se relaciona con cada gen asociado a la variante, eliminando variantes artefactuales manualmente. 3Generado según lo descrito anteriormente 4Generado según lo descrito anteriormente, eliminando variantes artefactuales manualmente. Elaboración propia

Figura 4. Distribución de posiciones en la tabla de las variantes causales tras aplicar priorizaciones según los diferentes scores. Elaboración propia


Figura 5. Visualización en JNOMICS de la una variante en la base de datos 12OVar enriquecida con términos HPO. Elaboración propia
Como queda demostrado en este estudio, la integración de los términos HPO en el análisis de datos de exoma permite detectar variantes asociadas a las patologías de forma más eficiente frente al uso de paneles virtuales, ya que las variantes potencialmente candidatas se encuentran priorizadas en las primeras posiciones de la tabla de variantes.
Este sistema de priorización representaría, por tanto, el primer abordaje para el análisis de variantes genéticas procedentes de secuenciación del exoma completo y un complemento al análisis mediante el uso de paneles virtuales de genes o al análisis de exoma completo.
OTROS ANÁLISIS POSIBLES A PARTIR DE DATOS DE EXOMA
Además del análisis de variantes de pequeño tamaño, a partir de Karma es posible realizar diferentes análisis complementarios para enriquecer al máximo la información y completar el diagnóstico: • QC:

Como en todo procedimiento analítico, es necesario llevar a cabo un control de calidad de los datos. En este caso, se analiza el estado de diferentes ficheros que comprenden el análisis o FASTQ: Mediante la adaptación del programa FastQC43 . o BAM: Scripts propios que evalúan cómo ha transcurrido el alineamiento (% off-target, uniformidad, % de duplicados…). o VCF: Determinación del número total de variantes, ratio variantes heterocigotas/homocigotas, determinación de contaminaciones cruzadas… • Cobertura:
Se evalúa la cobertura vertical media correspondiente al conjunto de genes, a cada gen en particular y se determina el % de bases y qué regiones que han superado o no determinados umbrales de cobertura. • CNVs (Figura 6): A partir de una línea base de alineamientos de muestras previamente secuenciadas, se compara exón a exón la cobertura y se determina en qué ventanas de exones hay una discrepancia entre la cobertura esperada y la observada, lo cual se correlaciona con deleciones o duplicaciones44 .
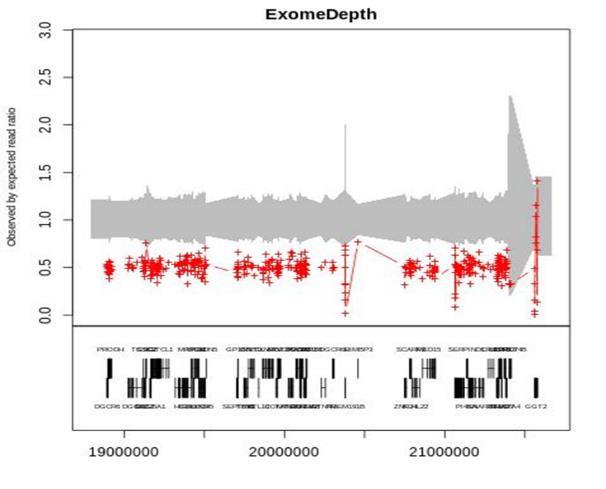
Figura 6. Deleción en heterocigosis que comprende varios genes detectada por Karma. Elaboración propia
Figura 7. Regiones cromosómicas con pérdida de heterocigosidad detectadas por Karma. Elaboración propia

• ROH (Figura 7): Se detectan regiones con pérdida de heterocigosidad a lo largo del genoma, empleando el perfil de variantes del archivo VCF.
RENDIMIENTOS DIAGNÓSTICOS EN WES EN EL SERVICIO DE GENÉTICA
En nuestra institución, y empleando Karma como pipeline de análisis y JNOMICS como herramienta de gestión e interpretación de variantes, se han completado 2072 estudios (noviembre 2018 – septiembre 2020). Los resultados han sido los siguientes: - 574 casos diagnosticados. - 312 casos no concluyentes. - 1128 casos no informativos.
Esto supone un rendimiento diagnóstico global del 27,7%, en concordancia con otras instituciones que han publicado sus datos: 25%, 26%, 28%, 30%, 52% y 52%8,12,45–48. Cabe decir, que los rendimientos más elevados tienen un sesgo de tipo de paciente y de patología. Si se desglosan los datos los rendimientos son los siguientes: - Pediatría: 34%
- Adultos: 24%
- Por enfermedad:
o Síndrome de Noonan y Rasopatías: 60% (15 de 25). o Síndrome de Marfan y Aortopatías: 50% (22 de 44). o Discapacidad intelectual: 43,5% (71 de 163). o Hipoacusia: 41% (52 de 127). o Hipercolesterolemia familiar: 31% (34 de 110). o Autismo: 14,5% (7 de 48). Del total de estudios diagnosticados, 38 del ellos (6,6%) contienen una CNV patogénica. Como acciones de mejora a futuro, para mejorar los rendimientos diagnósticos está el reanálisis de los estudios no informativos, empleando nuevos abordajes, ampliando el espectro de análisis a otros genes que se hayan descrito recientemente, etc49 .
LIMITACIONES EN EL ANÁLISIS DE WES
Las limitaciones principales en el análisis de WES son: - Sólo aborda las regiones codificantes e intrónicas flanqueantes, que suponen el 1,5-2% del genoma. - Técnica dependiente de captura: existen regiones, como las de baja complejidad o ricas en nucleótidos GCs que no se capturan (y por ende se secuencian) con eficiencia.
- Identificación de grandes deleciones e inserciones (>21 nucleótidos): en abordajes de NGS con lecturas cortas identificar este tipo de variantes es difícil. Sin embargo, mediante Karma se han detectado eventos con más de 50 nucleótidos de longitud. - Identificación de CNVs: aunque es posible, está limitada si la captura no ha sido uniforme y también es poco sensible si el número de exones afectados es bajo. - Reordenamientos genómicos. - Disomías uniparentales. - Variaciones epigenéticas. - Mutaciones en mosaico: debido a que la profundidad de lectura no resulta suficiente en muchos casos para detectar este tipo de eventos donde la frecuencia de alelo alternativo es baja. - Regiones repetitivas, pseudogenes. Se comentó como una de las limitaciones derivadas del alineamiento de secuencias.
1. Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis
I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res [Internet]. 2019 Jan 8 [consultado 15 octubre 2020];47(D1):D766–73. Disponible en: https://doi.org/10.1093/nar/gky955 2. Nguengang Wakap S, Lambert DM, Olry A, Rodwell C,
Gueydan C, Lanneau V, et al. European Journal of
Human Genetics Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet [Internet]. 2020 [consultado 29 octubre 2020];28(2):165-73. Disponible en: https://doi.org/10.1038/s41431-019-0508-0 3. Ferreira CR. The burden of rare diseases. Am J Med
Genet A [Internet]. 2019 [consultado 26 octubre 2020];179(6):885–92. Disponible en: https://pubmed.ncbi.nlm.nih.gov/30883013/ 4. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A [Internet]. 1977 [consultado 26 octubre 2020];74(12):5463–7. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/271968 /
5. Voelkerding K V., Dames SA, Durtschi JD. Nextgeneration sequencing:from basic research to diagnostics. Clin Chem [Internet]. 2009 [consultado 15 octubre 2020];55(4):641–58. Disponible en: from: https://doi.org/10.1373/clinchem.2008.112789 6. Chief Medical Officer. Chief Medical Officer annual report 2016: generation genome [Internet]. [consultado 15 octubre 2020]. London: Department of Health &
Social Care; 2017. Disponible en: https://www.gov.uk/government/publications/chiefmedical-officer-annual-report-2016-generation-genome 7. de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T,
Yntema HG, Kroes T, et al. Diagnostic Exome
Sequencing in Persons with Severe Intellectual
Disability. N Engl J Med [Internet]. 2012 Nov 15 [consultado 15 octubre 2020];367(20):1921–9.
Disponible en: http://www.nejm.org/doi/abs/10.1056/NEJMoa1206524 8. Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S,
Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA [Internet]. 2014 Nov 12 [consultado 15 octubre 2020];312(18):1880–7. Disponible en: https://doi.org/10.1001/jama.2014.14604 9. Sawyer SL, Hartley T, Dyment DA, Beaulieu CL,
Schwartzentruber J, Smith A, et al. Utility of wholeexome sequencing for those near the end of the diagnostic odyssey: Time to address gaps in care. Clin
Genet [Internet]. 2016 [cionsultado 15 octubre 2020];89(3):275–84. Disponible en: https://doi.org/10.1111/cge.12654 10. Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB,
Rimmer A, et al. Factors influencing success of clinical
genome sequencing across a broad spectrum of disorders. Nat Genet [Internet]. 2015 Jun 26 [consultado 15 octubre 2020];47(7):717–26. Disponible en: https://www.nature.com/articles/ng.3304 11. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A,
Ward PA, et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N Engl J Med [Internet]. 2013 Oct 17 [consultado 15 octubre 2020];369(16):1502–11. Disponible en: http://www.nejm.org/doi/10.1056/NEJMoa1306555 12. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA [Internet]. 2014 Nov 12 [consultado 15 octubre 2020];312(18):1870–9. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC432624 9/pdf/nihms661397.pdf 13. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC,
Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature [Internet]. 2001 Feb 15 [consultado 15 octubre 2020];409(6822):860–921.
Disponible en: https://doi.org/10.1038/35057062 14. Backofen R, Gilbert D. Bioinformatics and constraints.
Constraints. 2001 Jun;6(2–3):141–56. 15. Stephanopoulos G. Bioinformatics and metabolic engineering. Metab Eng [Internet]. 2000 [consultado 15 octubre 2020];2(3):157–8. Disponible en: https://doi.org/10.1006/mben.2000.0157 16. Roy S, Coldren C, Karunamurthy A, Kip NS, Klee EW,
Lincoln SE, et al. Standards and Guidelines for
Validating Next-Generation Sequencing Bioinformatics
Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American
Pathologists. J Mol Diagn [Internet]. 2018 [consultado 15 octubre 2020];20(1):4–27. Disponible en: https://doi.org/10.1016/j.jmoldx.2017.11.003 17. Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The
Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants.
Nucleic Acids Res [Internet]. 2010 Apr [consultado 22 febrero 2017];38(6):1767–71. Disponible en: https://doi.org/10.1093/nar/gkp1137 18. Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A [Internet]. 1988 [consultado 15 octubre 2020];85(8):2444–8. Disponible en: https://doi.org/10.1073/pnas.85.8.2444 19. GATK Team. Phred-scaled quality scores – GATK [Internet]. Cambridge, MA: Broad Institute; 2021 [consultado 15 octubre 2020]. Disponible en: https://gatk.broadinstitute.org/hc/enus/articles/360035531872-Phred-scaled-quality-scores 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics [Internet]. 2009 Jul 15 [consultado 12 febrero 2017];25(14):1754–60. Disponible en: http://doi.org/10.1093/bioinformatics/btp324
21. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods [Internet]. 2012 Apr [consultado 10 julio 2014];9(4):357–9. Disponible en: http://dx.doi.org/10.1038/nmeth.1923 22. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J,
Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics [Internet]. 2009 Aug 15 [consultado 14 febrero 2017];25(16):2078–9.
Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19505943 23. Burrows M, Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. 1994 [consuktado 15 octubre 2020]; Disponible en: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1 .1.121.6177
24. Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970 Mar 28;48(3):443–53. 25. GitHub. Genome-stratifications/GRCh37. Genome-ina-bottle. Genome-stratifications [Internet]. GitHUB; 2021 [consultado 15 octubre 2020]. Disponible en: https://github.com/genome-in-a-bottle/genomestratifications/tree/master/GRCh37
26. The Variant Call Format (VCF) Version 4.2
Specification. 2020. 27. DePristo MA, Banks E, Poplin R, Garimella K V,
Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet [Internet]. 2011 Apr 10 [consultado 12 febrero 2017];43(5):491–8. Disponible en: http://www.nature.com/doifinder/10.1038/ng.806 28. McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A MapReduce framework for analyzing nextgeneration DNA sequencing data. Genome Res. 2010
Sep;20(9):1297–303. 29. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43(1110):11.10.1-11.10.33 30. Lai Z, Markovets A, Ahdesmaki M, Chapman B,
Hofmann O, Mcewen R, et al. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res [Internet]. 2016 Jun 20 [consultado 15 octubre 2020];44(11):108. Disponible en: https://doi.org/10.1093/nar/gkw227 31. Den Dunnen JT, Antonarakis E. Nomenclature for the description of human sequence variations [Internet].
Human Genetics. Hum Genet; 2001 [consultado 15 octubre 2020];109(1):121–4. Disponible en: https://doi.org/10.1007/s004390100505 32. Auton A, Abecasis GR, Altshuler DM, Durbin RM,
Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature [Internet]. 2015 Sep 30 [consultado 30 septiembre 2015];526(7571):68–74.
Disponible en:http:/dx.doi.org/10.1038/nature15393 33. Consortium EA, Lek M, Karczewski K, Minikel E,
Samocha K, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans [Internet]. bioRxiv.
Cold Spring Harbor Labs Journals; 2015 Oct [consultado 31 octubre 2015]. Disponible en: http://biorxiv.org/content/early/2015/10/30/030338.abst ract
34. Karczewski KJ, Francioli LC, Tiao G, Cummings BB,
Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans.
Nature [Internet]. 2020 May 28 [consultado 15 octubre 2020];581(7809):434–43. Disponible en: https://doi.org/10.1038/s41586-020-2308-7 35. Landrum MJ, Lee JM, Benson M, Brown G, Chao C,
Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic
Acids Res [Internet]. 2015 Nov 17 [consultado 6 enero 2016];44(D1):D862-8. Disponible en: https://doi.org/10.1093/nar/gkv1222 36. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM,
Bindal N, et al. COSMIC: The Catalogue Of Somatic
Mutations In Cancer. Nucleic Acids Res [Internet]. 2019
Jan 8 [consultado 15 octubre 2020];47(D1):D941–7.
Disponible en: https://cancer.sanger.ac.uk 37. Stenson PD, Ball E V, Mort M, Phillips AD, Shiel JA,
Thomas NST, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat [Internet]. 2003 Jun [consultado 7 marzo 2016];21(6):577–81. Disponible en:https:/doi.org/10.1002/humu.10212 38. Hamosh A, Scott AF, Amberger J, Valle D, McKusick
VA. Online Mendelian Inheritance in Man (OMIM). Hum
Mutat [Internet]. 2000 [consultado 15 octubre 2020];15(1):57–61. Disponible en: https://doi.org/10.1002/(SICI)10981004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G 39. Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB,
Danis D, Gourdine JP, et al. Expansion of the Human
Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res [Internet]. 2019 Jan 8 [consutado 15 octubre 2020];47(D1):D1018–27.
Disponible en: https://doi.org/10.1093/nar/gky1105 40. Weinreich SS, Mangon R, Sikkens JJ, Teeuw ME,
Cornel MC. Orphanet: Een Europese database over zeldzame ziekten. Ned Tijdschr Geneeskd [Internet]. 2008 Mar 1 [consultado 15 octubre 2020];152(9):518–9. Disponible en: https://europepmc.org/article/med/18389888 41. Firth H V., Richards SM, Bevan AP, Clayton S, Corpas
M, Rajan D, et al. DECIPHER: Database of
Chromosomal Imbalance and Phenotype in Humans
Using Ensembl Resources. Am J Hum Genet [Internet]. 2009 Apr 10 [consultado 15 octubre 2020];84(4):524–33. Disponible en: https://doi.org/10.1016/j.ajhg.2009.03.010
42. Tuñón Le Poultel D, Lezana Rosales JM, Soengas
Gonda EM, Eguiburu Jaime JL, Sánchez Calvín MT,
Quesada Espinosa JF. Construcción y evaluación de un algoritmo de priorización de variantes de exoma empleando términos HPO [Internet]. Congreso Virtual
LabClin; 2020. Disponible en: https://www.labclin2020.es/index.php/bienvenida 43. Andrews S. FASTQC. A quality control tool for high throughput sequence data. BibSonomy [Internet]. [consultado 15 octubre 2020]. Disponible en: https://www.bibsonomy.org/bibtex/f230a919c34360709 aa298734d63dca3
44. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E,
Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics [Internet]. 2012 Nov 1 [consultado 12 febrero 2017];28(21):2747–54. Disponible en: https://academic.oup.com/bioinformatics/articlelookup/doi/10.1093/bioinformatics/bts526 45. Ji J, Shen L, Bootwalla M, Quindipan C, Tatarinova T,
Maglinte DT, et al. A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold
Spring Harb Mol Case Stud [Internet]. 2019 [consultado 15 octubre 2020];5(2). Disponible en: https://doi.org/10.1101/mcs.a003756 46. Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z,
Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genet Med [Internet]. 2015 Jul 2 [consultado 15 octubre 2020];17(7):578–86. Disponible en: https://doi.org/10.1038/gim.2014.154 47. Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K,
Shrestha R, et al. Diagnostic impact and costeffectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA
Pediatr [Internet]. 2017 Sep 1 [consultado 15 octubre 2020];171(9):855–62. Disponible en: https://doi.org/10.1001/jamapediatrics.2017.1755 48. Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Gaff
C, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J
Hum Genet [Internet]. 2018 May 1 [consulltado 15 octubre 2020];26(5):644–51. Disponible en: https://doi.org/10.1038/s41431-018-0099-1 49. Baker SW, Murrell JR, Nesbitt AI, Pechter KB,
Balciuniene J, Zhao X, et al. Automated Clinical Exome
Reanalysis Reveals Novel Diagnoses. J Mol
Diagnostics [Internet]. 2019 Jan 1 [consultado 15 octubre 2020];21(1):38–48. Disponible en: https://doi.org/10.1016/j.jmoldx.2018.07.008







