
Post




 Foto: Renate Fossum
Foto: Renate Fossum
Kjære leser,
11. januar 2024 var vi igjen samlet i Oslo for hematologisk Norsk aften hvor fjorårets ASH kongress ble oppsummet. Vi hadde nærmere 90 påmeldte leger, industri representanter og representanter fra relaterte organisasjoner. Det var veldig gøy at vi denne gang også hadde deltakere som til daglig jobber utenfor Oslo regionen.
Denne ettermiddagen og kvelden fikk vi høre gode presentasjoner med oppsummeringer fra ASH 2023, god faglig diskusjon i pauser, og ikke minst den sosiale delen avslutningsvis med felles bespisning. Intet unntak, det ble en fin sosial ramme under middagen denne gangen også.
Vi ønsker å rette en stor takk til vårt faglige panel som her har beskrevet deres mest interessante «funn» fra ASH 2023.
Neste hematologisk Norsk aften vil i samarbeid med Dagens Medisin bli arrangert i Madrid, lørdag 15. juni 2024, hvor vi får presentert nyhetene fra EHA 2024.
Følg med på www.norskaften.no for mer informasjon og påmelding.
Vennlig hilsen,
Sindre Hauge
Arrangør av Hematologisk Norsk Aften Apriil Event & Congress AS

norskaften.no
Post

6/ Hemolytiske anemier og Waldenströms makroglobulinemi
Sigbjørn Berentsen
11/ Lymfom: litt av det nye som kan brukes i hverdagen
Eirik Tjønnfjord
14/ Myelomatose: The return of conventional therapy
Fredrik Schjesvold
18/ Oppsummering KLL
Hoa Tran
20/ Akutt myelogen leukemi (AML)
Pål Tore Bentsen
INNHOLD Post
Sigbjørn Berentsen, seniorforsker, dr.med.
Seksjon for forskning og innovasjon, Haugesund sjukehus
Hemolytiske anemier og Waldenströms makroglobulinemi
På ASH 2023 var det en rekke presentasjoner innen emnet hemolytiske anemier, bl.a. om paroksysmal nattlig hemoglobinuri (PNH) og kuldeagglutininsykdom (CAD). Det ble også lagt fram nye data om Waldenströms makroglobulinemi (WM).
Paroksysmal nattlig hemoglobinuri (PNH)
Bakgrunn: Komplementhemming ved PNH
Flere presentasjoner om PNH handlet om forbedret behandlingseffekt ved bruk av nyere komplementhemmere. Figur 1 viser en skjematisk oversikt over komplementsystemet og virkningen av aktuelle inhibitorer.
Langtidseffekt av pegcetacoplan
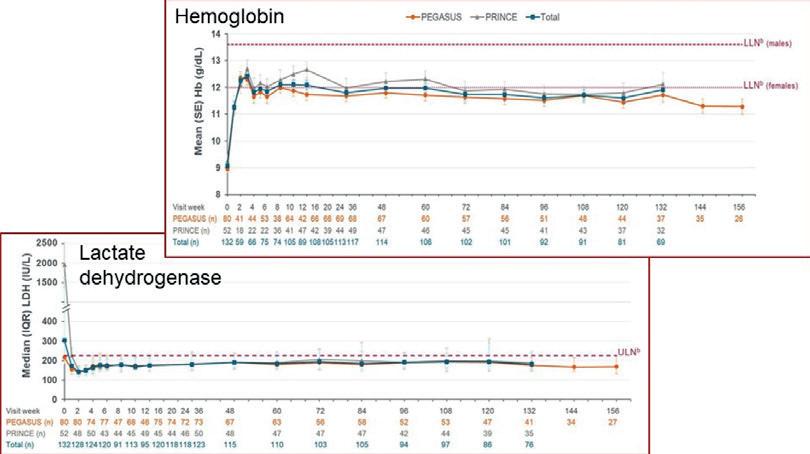
C. M. de Castro fra Durham, North Carolina, presenterte resultater fra 3 års oppfølging av PNH-pasienter behandlet med C3-inhibitoren pegcetacoplan. Studien var en integrert oppfølging av pasienter i PEGASUS-studien (tidligere behandlet med C5-hemmer) og PRINCE-studien (behandlingsnaïve pasienter), i alt 114. I begge gruppene inntrådte normalisering av hemoglobin (Hb), laktatdehydrogenase (LD), absolutt retikulocyttall og FACIT fatigue-skår hurtig og vedvarte i hele studieperioden på 3 år. Transfusjonsbehov ble ytterligere signifikant redusert ved overgang fra C5- til C3-hemming. Nye sikkerhetsproblemer ble ikke påvist. Noen av resultatene er vist i Figur 2
De Castro CM, Mulherin B, Patriquin CJ, et al. Efficacy and Safety Is Maintained in Adult Patients with Paroxysmal Nocturnal Hemoglobinuria Receiving Pegcetacoplan for up to 3 Years. Blood 2023; 142 (Suppl 1): 574.
Iptacopan ved PNH
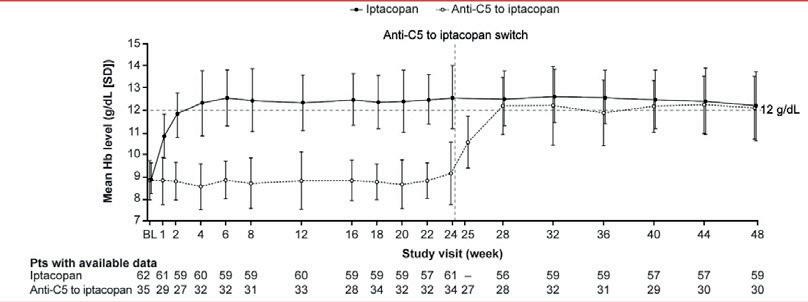
En fase 3-studie av behandling med den perorale faktor B-hemmeren iptacopan ved PNH («the APPLY-PNH trial») ble presentert av Antonio Risitano, Napoli, Italia. Studien omfattet både pasienter som hadde fått C5-inhibitorer og tidligere behandlingsnaïve pasienter. Pasienter i kontrollarmen fikk de første 24 ukene standardbehandling med C5-hemmer. Ved iptacopan monoterapi oppnådde pasientene klinisk betydelig bedring av Hb-nivå og hemolysemarkører, full normalisering av Hb hos et flertall, transfusjons-uavhengighet og bedring av pasientrapportert fatigue, med signifikant bedre effekt av iptacopan sammenliknet med C5-hemming. Figur 3 viser resultatene for Hb-nivå. Hos pasienter som var randomisert til C5-hemming, ble behandlingen endret til iptacopan etter 24 uker, og de oppnådde da samme effekt som pasientene i iptacopan-armen.
Risitano A, Kulasekararaj A, Roeth A, et al. Categorization of Hematological Responses to Oral Iptacopan Monotherapy in Anti-C5-Treated Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) and Persistent Anemia in the Phase III APPLY-PNH Trial and Complement Inhibitor-Naïve Patients in the Phase III APPOINT-PNH Trial. Blood 2023; 142 (Suppl 1): 4084.
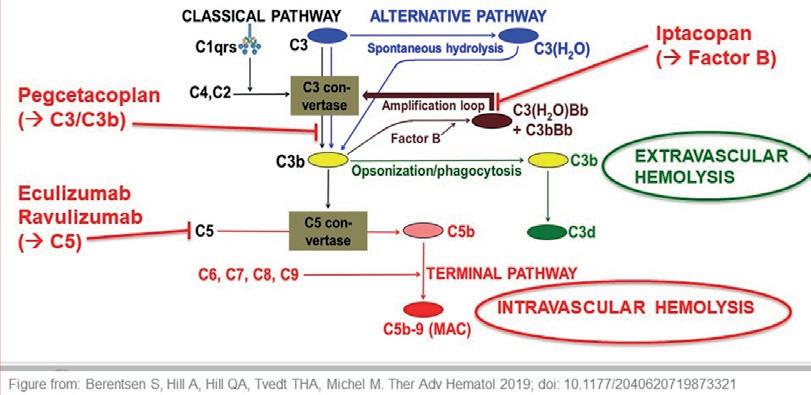
Figur 1. Komplementhemming ved PNH. Den perorale, lavmolekylære inhibitoren iptacopan virker på faktor B og hemmer derfor den alternative aktiveringsveien spesifikt. Pegcetacoplan, et pegylert syklisk peptid som tilføres subkutant, hemmer komplementproteinene C3 og C3b og kan på den måten blokkere hele komplementsystemet. Begge disse medikamentene har dermed potensiale til å hindre både ekstravaskulær og intravaskulær hemolyse. De etablerte monoklonale antistoffene eculizumab og ravulizumab virker på C5-nivå, hemmer derfor den terminale komplementkaskaden spesifikt og blokkerer intravaskulær men ikke ekstravaskulær hemolyse.
6
7
Figur 2. Hb- og LD-nivå hos pasienter prospektivt behandlet med pegcetacoplan for PNH. Grå kurver: tidligere ubehandlede pasienter; oransje kurver: pasienter som tidligere ved behandlet med C5-hemmer; grønne kurver: alle pasienter.
Figur 3. Iptacopan ved PNH i APPLY-PNH-studien. Kurvene viser Hb-nivå hos pasienter i iptacopan-armen (heltrukket kurve) og pasienter i C5-hemmer armen (stiplet kurve), som skiftet over til iptacopan etter 24 uker.
Kuldeagglutininsykdom (CAD)
Riliprubart – langtidsvirkende C1s-hemmer ved CAD Shirley D’Sa fra London, UK, la fram resultatene av en multinasjonal fase 1B-studie av riliprubart (BIVV020, SAR445088) for hemolytisk anemi ved CAD. Riliprubart er et humanisert monoklonalt antistoff som selektivt binder seg til den aktiverte formen av komplementprotein C1s. Molekylet inneholder mutasjoner som beskytter mot lysosomal degradering og har derfor lang biologisk halveringstid. Man ønsker å se om riliprubart kan gi like god effekt som den eksisterende C1s-hemmeren sutimlimab, men med dosering bare 4 ganger per år.
Tolv pasienter med verifisert CAD med hemolytisk anemi fikk riliprubart, enten 15 mg/kg eller 30 mg/kg intravenøst som én enkelt dose. Resultatene viser at bilirubinnivået (som generelt er den beste hemolyseparameteren ved CAD) ble normalt i løpet av få dager etter administrering av riliprubart, og Hb steg til normale eller lett subnormale nivåer innen 4 uker. Bedringen holdt seg deretter konstant i hele observasjonsperioden på 15 uker. Det ble ikke registrert bivirkninger med sannsynlig relasjon til studiemedikamentet og ingen alvorlige uønskede hendelser. Det planlegges en fase 3-studie.
Referentens merknad: Fra norsk side deltok Haukeland Universitetssjukehus i denne studien
D’Sa S, Vos JMI, Barcelini W, et al. Classical Complement Inhibition By SAR445088 (BIVV020) in Adults with Cold Agglutinin Disease: Safety, Tolerability and Activity Results from the Open-Label, Non-Randomized, Single-Dose Phase 1b Study. Blood 2023; 142 (Suppl 1): 3833.
Waldenströms makroglobulinemi (WM)
Kjemoimmunoterapi er fortsatt god behandling i første linje Både moderne kjemoimmunoterapi og nyere behandling med Bruton tyrosinkinase-hemmere (BTKi) har i tidligere prospektive studier vist god effekt og tilfredsstillende toleranse ved WM. Internasjonalt har man de siste årene i USA mest anbefalt BTKi i første linje, mens kjemoimmunoterapi fortsatt ofte foretrekkes i Europa. Anbefalinger fra International Workshop for WM (IWWM) sidestiller de to modalitetene, og det er ikke gjort direkte sammenliknende, prospektive studier.
På ASH 2023 presenterte Francisco Autore fra Roma, Italia, en retrospektiv “real life”-multisenterstudie av kjemoimmunoterapi hos 547 pasienter med behandlingstrengende WM fra Italia i tidsrommet 2008-22. De mest brukte regimene var bendamustin pluss rituksimab (BR, 245 pasienter) og deksametason-rituksimab-syklofosfamid (DRC, 116 pasienter), og studien fokuserte på sammenlikning av disse to regimene. Gruppene var ikke helt sammenliknbare med hensyn til komorbiditet (noe mer i DRCgruppen) og alvorlighetsgrad av WM (noe verre i BR-gruppen). Progresjonsfri overlevelse (PFS) var lengre i BR-gruppen (79% 5-års PFS ved BR; 55% ved DRC; p<0.001; Figur 4), men det var ingen signifikant forskjell i totaloverlevelse. Dosereduksjon av BR påvirket ikke resultatene. Begge regimene ble generelt godt tolerert. Forfatterne konkluderer med at kjemoimmunoterapi med BR eller DRC er svært effektiv førstelinjebehandling for uselekterte WM-pasienter. PFS er noe lengre ved BR enn ved DRC, men totaloverlevelsen er ikke signifikant ulik. Resultatene er gode også når bendamustindosen må reduseres pga. cytopeni.
Autore F, Tedeschi A, Benevolo G, et al. Real-Life Multicentre Study on 547 Patients Affected By Waldenstrom Macroglobulinemia Treated with ChemoImmunotherapy: Which Is the Best and Most Used First-Line Treatment?
Blood 2023; 142 (Suppl 1): 1667.
Ibrutinib pluss venetoklaks ved tidligere ubehandlet, symptomgivende WM
J. J. Castillo fra Boston, USA, presenterte en prospektiv fase 2-multisenterstudie av behandling med ibrutinib pluss venetoklaks ved tidligere ubehandlet, symptomgivende WM. Førtifem pasienter (30 menn og 15 kvinner) ble inkludert. Medianverdier ved behandlingsstart var alder 67 år (40-82 år), IgM-nivå 43 g/L (5,7-92 g/L), hemoglobin 10,2 g/dL (7,8-15,3 g/dL), og beinmargsaffeksjon av lymfoplasmacytisk lymfom 60% (5-90%). Alle pasientene hadde påvisbar MYD88 L265P-mutasjon, mens CXCR4-mutasjon forelå hos 17 pasienter (38%). Planlagt behandling ble gitt i 4-ukers sykluser og besto av ibrutinib 420 mg daglig i syklus 1. Venetoklaks ble lagt til f.o.m. syklus 2 i dosering 100 mg daglig i en uke, 200 mg daglig neste uke, og 400 mg daglig i 2 uker. Deretter var intensjonen en dosering av ibrutinib 420 mg daglig og venetoklaks 400 mg daglig i syklus 3-24.
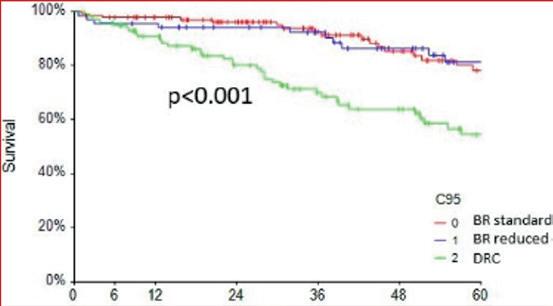
Figur 4. Retrospektiv «real life»-studie av bendamustin-rituksimab (BR) versus deksametason-rituksimab-syklofosfamid (DRC) ved WM. Kurvene viser progresjonsfri overlevelse i måneder ved BR i full dose (rød), BR i redusert dose (blå) og DRC (grønn kurve).
8
Post

Foto: Renate Fossum
•


BRUKINSA® ER INNFØRT SOM:
•
*Monoterapi til behandling av voksne pasienter med KLL som har mottatt minst én tidligere behandling (R/R KLL), eller voksne pasienter med tidligere ubehandlet kronisk lymfatisk leukemi (KLL) med 17p-delesjon/TP53-mutasjon og/eller 11q-delesjon.1–2
**Monoterapi til behandling av voksne pasienter med WM som har fått minst én tidligere behandling, eller som førstelinjebehandling for pasienter som er uegnet for kjemoimmunterapi.3
BRUKINSA® SIKKERHETSPROFIL:
BRUKINSA® har vist lavere forekomst av alle grader diaré, muskelspasmer og atrieflimmer sammenlignet med ibrutinib.4–5 Forekomst av nøytropeni grad ≥3 var lik for pasienter med KLL/SLL behandlet med BRUKINSA® og ibrutinib4 men høyere for pasienter med WM som ble behandlet med BRUKINSA® sammenlignet med ibrutinib5 selv om infeksjonsfrekvensen i begge studiene var lik for begge grupper.4–5 Blødningshendelser (alle grader) var lik mellom BRUKINSA® og ibrutinib hos pasienter med KLL4 og mindre hyppig hos WM-pasienter som ble behandlet med BRUKINSA®.5
† Ved median oppfølging på 29,6 måneder viste Brukinsa statistisk signifikant lengre PFS i ITT-populasjonen sammenlignet med ibrutinib (87 vs. 118 mht. sykdomsprogresjon/død); HR: 0,65, p=0,002).4
†† Brukinsa 28 % vs ibrutinib 19 %, p=0,095
BTK = Brutons tyrosinkinasehemmer; PFS = progresjonsfri overlevelse; R/R = relapserende/refraktær; KLL = kronisk lymfatisk leukemi; CR = komplett respons; VGPR = svært god delvis respons; ITT = intensjon om å behandle;
Referanser:
1. nyemetoder.no/metoder/zanubrutinib-brukinsa-indikasjon-iii (lest 22082023)
2. nyemetoder.no/metoder/zanubrutinib-brukinsa-indikasjon-v (lest 22082023)
3. nyemetoder.no/metoder/zanubrutinib-brukinsa (lest 22082023)
4. Brown JR, Eichhorst B, Hillmen P et al. Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med. 2023;26;388(4):319-332.
5. Tam CS, Opat S, D’Sa S, Jurczak W, Lee H-P, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The Aspen Study. Blood. 2020;136(18):2038–50. doi:10.1182/blood.2020006844
BeiGene Norway AS, email: nordics@beigene.com
0723-BRU-PRC-058
Utarbeidet december 2023
Brukinsa (zanubrutinib)
Antineoplastisk middel, proteinkinasehemmer. ATC-nr.: L01E L03
Utleveringsgruppe C. Reseptbelagt legemiddel. Kan forskrives på H-resept.
KAPSLER, harde 80 mg
Indikasjoner: BRUKINSA som monoterapi er indisert til 1) behandling av voksne med Waldenströms makroglobulinemi (WM) som har fått minst 1 tidligere behandling, eller som førstelinjebehandling for pasienter som er uegnet for kjemoimmunterapi, 2) behandling av voksne pasienter med marginalsonelymfom (MSL) som har mottatt minst én tidligere anti-CD20-basert behandling, 3) behandling av voksne pasienter med kronisk lymfatisk leukemi (KLL). 4) BRUKINSA i kombinasjon med obinutuzumab er indisert for behandling av voksne pasienter med refraktær eller tilbakefallende follikulært lymfom (FL) som har mottatt minst to tidligere systemiske behandlinger. Dosering: Monoterapi: Behandling skal innledes og overvåkes av lege med erfaring innen bruk av kreftlegemidler. Voksne: Anbefalt total daglig dose er 320 mg. Kan tas 1 gang daglig (4 kapsler à 80 mg), eller deles i 2 doser daglig (2 kapsler à 80 mg 2 ganger daglig). Behandling med Brukinsa bør fortsette til sykdomsprogresjon eller uakseptabel toksisitet. Kombinasjon med obinutuzumab: Zanubrutinib må administreres oralt før infusjon av obinutuzumab. Den anbefalte dosen er obinutuzumab 1000 mg intravenøst på dag 1, 8 og 15 av syklus 1, og på dag 1 i hver 28-dagers syklus fra syklus 2 til 6. Dosejustering ved bivirkninger: Se SPC. Asymptomatisk lymfocytose skal ikke anses som en bivirkning, og behandlingen skal fortsettes. Dosejustering ved samtidig bruk av CYP3A-hemmer/-induktor: Se SPC. Glemt dose: Det skal ikke tas dobbel dose som erstatning for glemt dose. Neste dose tas iht. opprinnelig plan. Spesielle pasientgrupper: Se SPC. Administrering: Tas med et glass vann til omtrent samme tid hver dag. Kan tas med eller uten mat. Skal svelges hele. Skal ikke tygges. Skal ikke knuses, deles eller åpnes. Kontraindikasjoner: Overfølsomhet for innholdsstoffene. Forsiktighetsregler: Blødninger: Alvorlige og fatale blødningstilfeller er sett ved behandling med zanubrutinib. Dosejustering kan være nødvendig ved bivirkninger grad ≥3. Warfarin eller andre vitamin K-antagonister bør ikke administreres samtidig. Pasienten bør overvåkes for tegn/symptomer på blødning og fullstendig blodtelling må også følges opp. Vurder nytte-risiko ved å holde tilbake zanubrutinib i 3 til 7 dager før og etter operasjonen, avhengig av type operasjon og risikoen for blødning. Infeksjoner: Fatale og ikke-fatale infeksjoner (inkl. bakterielle-, virale-, soppinfeksjon eller sepsis) og opportunistiske infeksjoner (f.eks. herpes viral-, cryptococcal aspergillus- og pneumocystis jiroveci-infeksjoner) er sett ved behandling med zanubrutinib. Infeksjoner grad ≥3 er sett, vanligst var pneumoni. Infeksjoner pga. hepatitt B-virus (HBV)-reaktivering er også sett. Før behandlingsstart bør HBV-status fastslås. Ved positiv HBV-test eller positiv hepatitt B-serologi, anbefales konsultasjon med ekspert på leversykdom før behandlingsstart. Pasienten bør overvåkes og behandles iht. retningslinjer for å forhindre reaktivering av hepatitt B. Vurder profylakse iht. retningslinjer ved økt infeksjonsrisiko. Pasienten bør overvåkes for infeksjonstegn og behandles deretter. Cytopeni: Grad 3/4 cytopeni, inkl. nøytropeni, trombocytopeni og anemi, er rapportert ved behandling med zanubrutinib. Fullstendig blodtelling skal utføres månedlig under behandlingen. Andre primære maligniteter, inkl. ikke-hud karsinom: Er sett hos pasienter med hematologisk malignitet behandlet med zanubrutinib. Hyppigste sekundære maligniteten var hudkreft (basalcellekarsinom og plateepitelkarsinom hud). Pasienten skal rådes til å bruke solbe-skyttelse. Atrieflimmer og -flutter: Er sett hos pasienter med hematologisk malignitet behandlet med zanubrutinib monoterapi, spesielt hos pasienter med hjerterisikofaktorer, hypertensjon, akutte infeksjoner og hos eldre (> 65 år). Pasienten skal overvåkes for tegn/symptomer på atrieflimmer og -flutter, og behandles etter behov. Tumorlysesyndrom: Tumorlysesyndrom har sjeldent blitt rapportert med zanubrutinib-monobehandling, spesielt hos pasienter som ble behandlet for KLL. Vurder relevante risikoer (f.eks. høy tumorbyrde eller urinsyrenivå i blodet og ta nødvendige forholdsregler. Overvåk pasienter nøye og gi passende behandling, Bilkjøring og bruk av maskiner:. Fatigue, svimmelhet og asteni er rapportert, og bør tas hensyn til ved vurdering av pasientens evne til å kjøre bil eller bruke maskiner. Interaksjoner: For utfyllende informasjon om relevante interaksjoner, bruk interaksjonsanalyse. Legemidler som kan øke plasmakonsentrasjonen av zanubrutinib: Samtidig bruk av BRUKINSA og legemidler som for eksempel Cytokrom P450-hemmere/induktorer kan øke/redusere plasmakonsentrasjonen av varandra. Graviditet, amming og fertilitet: Graviditetstesting anbefales for kvinner i fertil alder før behandling igangsettes. Graviditet: Skal ikke brukes under graviditet. Kvinner skal unngå å bli gravide under og i opptil 1 måned etter behandling. Fertile kvinner må bruke svært effektiv prevensjon under og i opptil 1 måned etter behandling. Ved bruk av hormonelle prevensjonsmidler må ekstra barrieremetode benyttes. Amming: Utskillelse i morsmelk er ukjent. Risiko for spedbarn som ammes kan ikke utelukkes. Amming bør avbrytes under behandling. Fertilitet: Ingen påvirkning av fertilitet eller reproduksjonsevne hos hann- eller hunnrotter ved doser opptil 300 mg/kg/døgn. Bivirkninger: Vanligste bivirkninger grad ≥3 (>3 %) er nøytropeni, lungebetennelse, hypertensjon*, trombocytopeni, blodplater redusert*, hemoglobin redusert*, anemi, blødning/hematom* og pneumoni (kombinasjon med obinutuzumab). *bare monoterapi. Overdosering/Forgiftning: Behandling: Nøye overvåkning og passende støttebehandling. Se Giftinformasjonens anbefalinger for proteinkinasehemmere L01E på www.felleskatalogen.no. Pakninger og priser: 120 stk. (flaske). 62835,40 kr. Refusjon: H-resept: L01E L03_1 Zanubrutinib. Refusjonsberettiget bruk: Der det er utarbeidet nasjonale handlingsprogrammer/nasjonal faglig retningslinje og/eller anbefalinger fra RHF/LIS spesialistgruppe skal rekvirering gjøres

10
INNFØRT TIL BEHANDLING AV KLL* & WM**
i tråd med disse. Vilkår: (216) Refusjon ytes kun etter resept fra sykehuslege eller avtalespesialist. Basert på SPC godkjent av SLV/EMA: 13.12.2023 Innehaver av markedsføringstillatelsen: BeiGene Ireland Limited. 10 Earlsfort Terrace Dublin 2 D02 T380, Irland, tlf. +353 1 566 766, e-post bg.ireland@beigene.com Les felleskatalogtekst eller preparatomtalen (SPC) for mer informasjon, se www.felleskatalogen.no Sist endret: 13.12.2023 BRUKINSA® (zanubrutinib) BTK-HEMMER TIL BEHANDLING AV KLL OG WM1-3
Signifikant lengre PFS sammenlignet med Ibrutinib ved R/R KLL† 4
Numerisk høyere rate for CR/VGPR med zanubrutinib sammenlignet med ibrutinib hos pasienter med WM†† 5 Dette legemidlet er underlagt særlig overvåking for å oppdage ny sikkerhetsinformasjon så raskt som mulig
Median oppfølgingstid ved rapportering av data var 24 måneder. Av uønskede hendelser var det 12 “progression events”, herunder 2 dødsfall under behandling og én transformasjon til diffust storcellet B-cellelymfom. Studien ble avsluttet før planlagt tid pga. forekomst av ventrikulær arytmi, men foredragsholder opplyste ikke hvor mange pasienter som fikk arytmi. Median behandlingstid var 10 måneder (1,9-21 måneder) før studien ble stoppet. Ved 24 måneder var raten av progresjonsfri overlevelse (PFS) 76%. Åttini prosent av pasientene hadde ikke trengt ny behandling, og totaloverlevelsen var 96%. Raten av svært god partiell respons (VGPR) var 42%.
CXCR4-mutasjoner så ikke ut til å påvirke PFS. Etter avsluttet behandling ble det ikke observert nye uønskede hendelser, heller ikke arytmi. Responsdata er vist i Figur 5
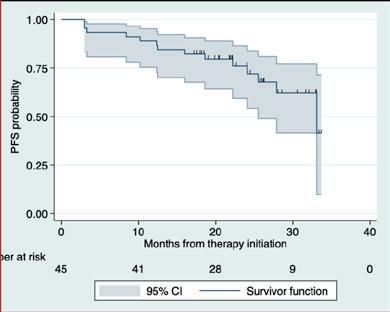
Forfatterne konkluderer med at ibrutinib pluss venetoklaks er høyeffektiv behandling ved WM, men pga. forekomst av uventet ventrikulær arytmi med ukjent genese vil de foreløpig ikke anbefale behandlingen.
Castillo JJ, Sarosiek SR, Branagan AR, et al. Ibrutinib and Venetoclax in Symptomatic, Treatment-Naive Patients with Waldenström Macroglobulinemia. Blood 2023; 142 (Suppl 1): 1661.
CAR-T-celler ved høymalign transformasjon av WM
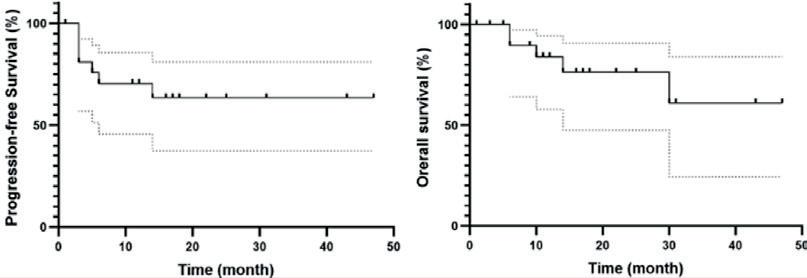
Histologisk transformert WM (tWM) har dårlig prognose, og responsen på kjemoimmunoterapi er vesentlig dårligere enn ved de novo diffust storcellet B-cellelymfom. En fransk-amerikansk retrospektiv multisenterstudie ved E. Durot (Reimes, Frankrike) og kolleger har undersøkt resultatene av behandling med anti-CD19 CAR-T-celler hos 22 pasienter med behandlingsresistent tWM eller residiv etter behandling (R/R-tWM). Materialet inkluderte også tilfeller av transformert nonsekretorisk eller ekstramedullært lymfoplasmacytisk lymfom. Overlevelseskurvene er vist i Figur 6.
Studien viser at anti-CD19 CAR-T-celleterapi er høyeffektiv ved R/R-tWM. Observerte bivirkninger omfattet som ventet cytokinfrigjøringssyndrom (CRS) og nevrologisk toksisitet (ICANS), men uventede bivirkinger ble ikke registrert. Lengre oppfølging er nødvendig for å bekrefte langtidsresultatene.
Durot E, Roos-Weil D, Chauchet A, et al. CD19-Targeting CAR T-Cell Therapy in Transformed Waldenström Macroglobulinemia/Lymphoplasmacytic Lymphoma: A Descar-T and US Collaborative Study. Blood 2023; 142 (Suppl 1): 616.
11
Figur 5. Responsdata ved behandling av Waldenströms makroglobulinemi med ibrutinib pluss venetoklaks.
Figur 6. Progresjonsfri og total overlevelse hos pasienter som har fått behandling med anti-CD19 CAR-T-celler for R/R-tWM.

VIKTIG SIKKERHETS-OG FORSKRIVNINGSINFORMASJON
REBLOZYL® (luspatercept)
25 mg pulver til injeksjonsvæske, oppløsning, 75 mg pulver til injeksjonsvæske, oppløsning. Reseptpliktigt legemiddel, reseptgruppe C.
Dette legemidlet er underlagt særlig overvåking for å oppdage ny sikkerhetsinformasjon så raskt som mulig. Helsepersonell oppfordres til å melde enhver mistenkt bivirkning til Statens Legemiddelverk på www.legemiddelverket.no/meldeskjema.
Indikasjoner:
Myelodysplastiske syndromer (MDS): Voksne med transfusjonsavhengig anemi pga. svært lav, lav og middels risiko MDS med ringsideroblaster, som har hatt utilfredsstillende respons til, eller ikke er kvalifisert for erytropoietinbasert behandling. β-talassemi: Voksne med transfusjonsavhengig og ikke-transfusjonsavhengig anemi knyttet til β-talassemi.
Dosering:
Før hver Reblozyl-administrasjon, skal Hb-nivået til pasienter vurderes. I tilfelle av en RBCtransfusjon som fant sted før dosering, må Hb-nivået før transfusjonen vurderes for doseringsformål. Anbefalt startdose er 1,0 mg/kg administrert én gang hver 3. uke. Dosen må kanskje justeres dersom pasienten ikke responderer på behandlingen innen en viss tid, dersom pasientens respons avtar helt eller hvis Hb-verdien øker. Se preparatomtalen for detaljert informasjon. Dersom pasienter opplever tap av respons, skal årsaksfaktorer (f.eks. en blødningshendelse) vurderes. Se preparatomtalen for instruksjoner om doseavbrudd eller dosereduksjoner på grunn av bivirkninger relatert til behandling med Reblozyl. Reblozyl bør avbrytes hvis pasienten ikke opplever reduksjon i transfusjonsbyrde (for pasienter med transfusjonsavhengig MDS eller β-talassemi) eller en økning fra baseline Hb for pasienter med ikke-transfusjonsavhengig β-talassemi etter 9 ukers behandling på maksimalt doseringsnivå hvis ingen andre forklaringer for responssvikt blir funnet (f.eks. blødning, kirurgi, andre samtidige sykdommer) eller hvis uakseptabel toksisitet oppstår under behandling.
Bivirkninger: Myelodysplastiske syndromer: Vanligste bivirkninger: Fatigue, diaré, asteni, kvalme, svimmelhet, ryggsmerter og hodepine. Alvorlige bivirkninger: Urinveisinfeksjon, ryggsmerter og synkope. Transfusjonsavhengig β-talassemi: Vanligste bivirkninger: Hodepine, skjelettsmerter og artralgi. Alvorlige bivirkninger: Tromboemboliske hendelser for dyp venetrombose, iskemisk slag portal venetrombose og lungeemboli. Ikke-transfusjonsavhengig β-talassemi: Vanligste bivirkninger: Skjelettsmerter, hodepine, artralgi, ryggsmerter, prehypertensjon og hypertensjon. Alvorlige bivirkninger: Traumatisk fraktur og ryggmargskompresjon på grunn av EMH-vev.
Kontraindikasjoner: Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene, graviditet og pasienter som trenger behandling for å kontrollere veksten av EMH-vev.
Advarsler og forsiktighetsregler:
Eldre: Begrensete data er tilgjengelig hos pasienter med β-talassemi ≥ 60 år. Nedsatt nyrefunksjon: Slike pasienter bør overvåkes nøye for bivirkninger, og dosejusteringer bør bli fulgt opp siden nedsatt nyrefunksjon kan føre til høyere eksponering av luspatercept. Pediatrisk populasjon: Reblozyl i MDS er ikke relevant. Reblozyl i β-talassemi er ikke relevant i hos pasienter yngre enn 6 år gamle og sikkerhet og effekt hos barn i alderen fra 6 år til under 18 år har ennå ikke blitt fastslått. Tromboemboliske hendelser (TEE) er rapportert i β-talassemi-pasienter. Den potensielle nytten av behandlingen skal veies opp mot den potensielle risikoen for TEE-er i β-talassemi-pasienter med splenektomi, og andre risikofaktorer for å utvikle TEE. EMH-vev og ryggmargskompresjon på grunn av EMH-vev er rapportert i β-talassemi-pasienter. Pasienter bør overvåkes ved oppstart og under behandling for symptomer og tegn eller komplikasjoner som følge av EMH-vev, og behandles i henhold til kliniske retningslinjer. EMH kan også oppstå etter langtidsbehandling med Reblozyl (dvs. etter 96 uker). Behandling med Reblozyl må seponeres ved alvorlige komplikasjoner på grunn av EMH-vev. Økt blodtrykk (systolisk og diastolisk) på i gjennomsnitt 5 mmHg fra baseline er rapportert hos MDS- og β-talassemipatienter. Behandlingen må kun startes dersom blodtrykket er tilstrekkelig kontrollert. Blodtrykket bør overvåkes før hver luspatercept-administrasjon. Dosen kan kreve justering. Den potensielle nytten av behandlingen bør reevalueres i tilfelle vedvarende hypertensjon eller forverring av preeksisterende hypertensjon. Traumatiske frakturer er observert hos ikke-transfusjonsavhengige β-talassemipatienter. Pasienter bør informeres om risikoen for traumatisk fraktur. Evnen til å kjøre bil og bruke maskiner påvirkes i liten grad fordi evnen til å reagere når du utfører disse oppgavene kan bli svekket pga. risiko for fatigue, vertigo, svimmelhet eller synkope. Overdosering kan forårsake en økning av Hb-verdier over ønsket nivå. I tilfelle overdosering skal behandlingen utsettes til Hb er ≤ 11 g/dl.
Interaksjoner: Ingen formelle kliniske interaksjonsstudier er utført. Samtidig bruk av jernchelerende midler har ingen effekt på farmakokinetikken. For utfyllende informasjon om relevante interaksjoner, bruk interaksjonsanalyse.
Pakninger og priser (pr. 24.08.2023): 25 mg: 1 stk. (hettegl.) kr 17 677,80. 75 mg: 1 stk. (hettegl.) kr 52 462,90.
Blå resept: Nei. Byttbar: Nei.
Refusjon: H-resept: B03X A06_1 Luspatercept; Refusjonsberettiget bruk: Der det er ut arbeidet nasjonale handlingsprogrammer/nasjonal faglig retningslinje og/eller anbefalinger fra RHF/LIS spesialistgruppe skal rekvirering gjøres i tråd med disse. Vilkår: (216) Refusjon ytes kun etter resept fra sykehuslege eller avtalespesialist.Reblozyl er under metodevurdering og er foreløpig ikke innført av Beslutningsforum for Nye Metoder.
Se felleskatalogen/preparatomtalen for fullstendig forskrivningsinformasjon og opplæringsmateriell/sjekkliste før forskrivning. Innehaver av markedsføringstillatelsen: Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Irland. Mer informasjon: Telefon: 23 12 06 37, www.bms.com/no
Versjon 1 Bristol
Myers Squibb
Torg 35 1366 Lysaker www.bms.com/no 2007-NO-2300002 Desember 2023
Lysaker
Lymfom: litt av det nye som kan brukes i hverdagen
Påpost-ASH setter vi fokus på noe av det nye som ble presentert på det 65. ASH møtet som gikk av stabelen fra 09-12. desember 2023 i San Diego, California, USA.
Som alltid var det mye nytt og spennende innen hematologiske sykdommer inkludert Lymfomer. Det er alt for mye til at man skal gå i detaljer, men jeg har plukket ut et par ting som jeg fant interessant og har laget et kort sammendrag her.
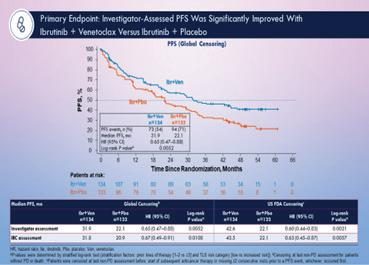
1. Ibrutinib Combined With Venetoclax in Patients With Relapsed/ Refractory Mantle Cell Lymphoma: Primary Analysis Results From the Randomized Phase 3 SYMPATICO Study – Wang M. et al
Kombinasjonen ibrutinib og venetoclax er vist å gi komplimenterende virkemøter og har vist synergistisk antitumor effect ved behnadling av KLL. Det samme er sett i prekliniske studier på MCL. I dette studiet kalt SYMPATICO, som er et fase 3 studie, har man evaluert effekten og tolerabiliteten av kombinasjonsbehandling med Ibrutinib (Brutons tyrosin kinase hemmer) og venetoclax (bcl-2 hemmer). Kombinasjonsbehandlingen er vurdert opp imot ibrutinib sammen med placebo (dvs ibrutinib monoterapi) til pasienter med refraktær og tilbakefall av MCL (R/R MCL).
I studiet var 267 pasienter inkludert, 134 i kombinasjonsarmen og 133 i kontroll armen og gruppene var sammenlignbare. Median follow-up var 51,2 måneder og en open-label sikkerhets run-in viste at kombinasjonen var sikker og veltolerert, og det ble ikke sett bivirkninger utover det som allerede var kjent for hver av medikamentene alene.
Etter 51.2 måneder så man at behandlingsstopp grunnet progresjon var hyppigst i kontrollarmen, mens behandlingsstopp grunnet adverse events var helt lik. Man så også at de ulike bivirkningene var som ved behandling med ibrutinib alene, nemlig neutropeni og diare, som også var de hyppigste grad 3 bivirkningene. Ut fra disse foreløpige funn og konklusjoner kan det virke som at kombinasjonsbehandling med ibrutinib og venetoclax er både mere effektivt og like trygt som monoterapi og dermed ville være et godt førstevalg til R/R MCL. Dette gjelder alle undergrupper inkludert i studiet (figur 3). Kombinasjonen gir en signifikant bedret PFS (progressjonfri survival) i alle undergrupper (figur 2). Både CR (komplett remissjon) og TTNT (time to next treatment) var signifikant bedre i intervensjonsgruppen. Overall survival ble ikke nådd, men var numerisk bedre og vi ser frem til videre resultater og vurdering av beslutningsforum.
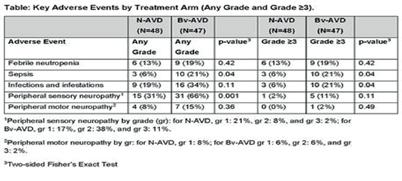
2. Nivolumab-AVD Is Better Tolerated and Improves Progression-Free Survival Compared to Bv-AVD in Older Patients (Aged ≥60 Years) with Advanced Stage Hodgkin Lymphoma Enrolled on SWOG S1826 – Rutherford S.C et al.
SWOG S1826, et randomisert studie av nivolumab(N)-AVD versus brentuximab vedotin(BV)- AVD ved advanced stage (AS) klassisk Hodgkin lymfom (HL) av Herrera A.F. et al ble presentert tidligere i 2023. Dette var et stort studie med 976 HL-pasienter hvor 489 pasienter ble randomisert til N-AVD og 487 pasienter havnet I kontrollarmen med B-AVD. Ved andre interim analyse, dvs ved oppnåelse av 50% av totalt PFS var nådd ble resultatene frigjort og
13
Eirik Tjønnfjord

disse favoriserte klart N-AVD (30 PFS i N-AVD vs 58 PFS i B-AVD armen). Ett års PFS var hhv 94% vs 86% og totalt så man 11 dødsfall med nesten dobbelt så mange i B-AVD armen (7 pga AE i B-AVD, mot 4 (3 pga AE) i N-AVD). Med henhold til adverse events grad 3 var det noen flere i N-AVD armen på 48,4% (45,1% var grad 3 nøytropeni), mens det i B-AVD armen var 30,5% AE, hvor 23,9% var grad 3 nøytropeni. De hyppigste komplikasjonene var stoffskifte problemer ved N-AVD, mens det var perifer nevropati i B-AVD armen.
Samlet vurdering var at N-AVD var bedre på PFS, og at AE var sjelden og kun 1% trengte rescue behandling i N-AVD og dermed er dette anbefalt som ny førstevalgs-behandling ved avansert HL. OS og PROs (pasient rapportert utkomme) var dog ikke nådd.
Ved ASH presenterte Rutherford S.C et al resultatene hos pasienter over 60 år. Også her så man at intervensjonsarmen, dvs N-AVD viste klart forbedret progresjonsfri overlevelse og EFS (event-fri overlevelse), samtidig som N-AVD var bedre tolerert enn B-AVD hos pasienter over 60 år med avansert Hodgkin lymfom. Dette så vi blant annet ved at det var en klart større gruppe i kontrollarmen, B-AVD, som måtte avslutte behandlingen grunnet toksisitet, sammen med at det var flere dødsfall i B-AVD gruppen tross helt sammenlignbare grupper basert på demografi.
N-AVD er derfor anbefalt å bli den nye standard behandlingen også til elder pasienter med avansert HL som er friske nok til å tale anthracycline-baserte kombinasjonsbehandlinger.
3. Abstrakt 104: Lisocabtagene Maraleucel in Relapsed/Refractory Large B-Cell Lymphoma: Real World Analysis from the Cell Therapy
Consortium – Riedell P.A. et al.
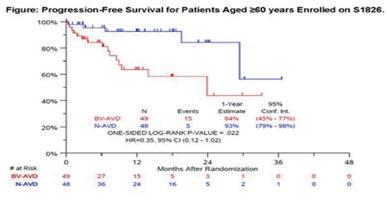
I dette abstraktet ble de første real-world data for lisocabtagene maraleucel (liso-cel) ved R/R DLBCL presentert. Lisocabtagene maraleucel (liso-cel) er en autolog CD19-basert CAR T celle behandling som er godkjent av FDA for relapsed/refractory (R/R) large B-celle lymfom (LBCL) basert på TRANSCEND (Abramson et al, Lancet 2020), TRANSFORM (Kamdar et al, Lancet 2022), og PILOT studiene (Sehgal et al, Lancet Oncol 2022). På ASH presenterte man så de første resultater fra oppfølging på behandling til pasienter utenfor studiene (real-world data). Totalt 101 pasienter mottok behandling (data er vist i figur 1). 68% hadde Charlston Comorbiditets score på >3 og gjennomsnittlig hadde de gjennomgått 3 behandlinger tidligere, mange var enda tyngre behandlet. Aldersmessig var medianalderen 71 år og over halvparten var menn. Bivirkninger var håndterbare og 6 måneders dødligheten ikke relatert til tilbakefall var på 8%. Resultatene var favorable (også ved høy risiko pasienter) med CR (komplett remissjon) rate på 65% og 6-mdr PFS rate på 64%. Det er fortsatt tidlig og mere data trengs før man kan konkludere, men det er lovende resultater til en svært vanskelig og tungt behandlet pasientgruppe. Lignende studier med lisocabtagene maraleucel (liso-cel) pågår også ved KLL og vi ser frem til ASH 2024 med oppdatering på resultatene innen begge pasient grupper.
14
Post

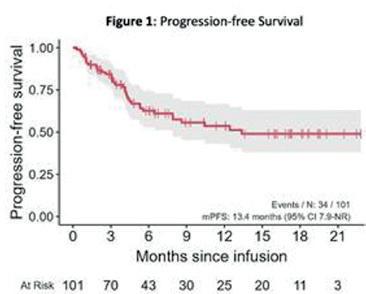
4. Pirtobrutinib in Richter Transformation: Updated Efficacy and Safety Results with 18-Month Median Survival Follow-up from the Phase 1/2 BRUIN Study – Wierda et al.
Richter transformasjon (RT) er en komplisert forverring til KLL som ses i ca 10% av pasientene og viser seg oftest som DLBCL (diffust storcellet B-celle lymfom), og har ekstremt dårlig prognose. Det er i dag ingen standard behandling til RT, men ofte forsøker man å inkludere dem i studier. De siste årene har det kommet mange nye behandlinger til KLL inkludert flere BTK-hemmer som blant annet Pirtobrutinib med håp om bedre effekt og resultater ved KLL. Pirtobrutinib er en høyt selektiv, ikke-kovalent BTK hemmer som tidligere har vist varig respons og har vært godt tolerert til B-celle maligniteter med dårlig prognose uavhengig av tidligere behandlinger. I BRUIN studiet som her er oppdatert har man sett lovende resultater også hos pasienter med RT. 82 pasienter var inkludert og man har sett en ORR (overall response rate) på 50%, hvorav 13.4 gikk i komplett remissjon, mens 36.6 oppnådde PR (delvis respons). Dette inkluderte også pasienter med transformert-KLL som tidligere hadde mottatt annen BTKhemmer. Som tidligere demonstrert er pirtobrutinib veltolerert og få pasienter trenger å stoppe behandlingen grunnet toksisitet. Richter transformasjon er stadig en svært krevende og alvorlig diagnose med svært dårlig prognose, men pirtobrutinib kan være
15
en potensiell behandlingsmulighet som alene eller i kombinasjon bør utprøves i ytterligere studier med håp om å bedre overlevelsen til en vanskelig behandlingsgruppe som RT.
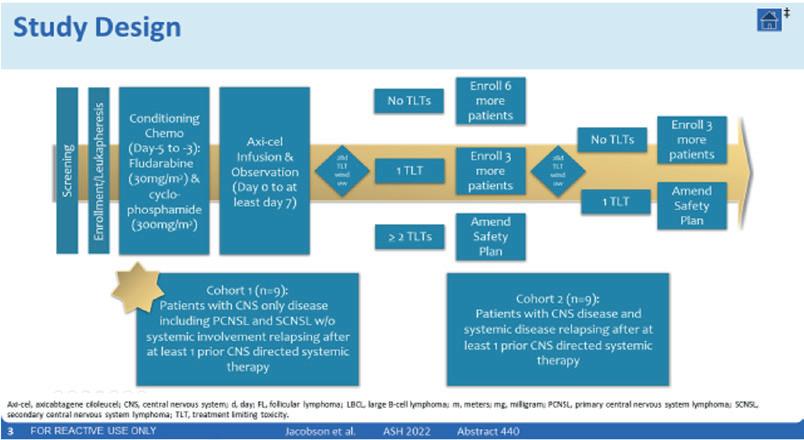
5. A Pilot Study of Axicabtagene Ciloleucel (Axi-cel) for the Treatment of Relapsed/Refractory Primary and Secondary CNS Lymphoma –Jacobsen C.A et al.
Axicabtagene ciloleucel (axi-cel) er en anti-CD19-CD28 annen generasjons autolog CAR T-celle behandling som av FDA er godkjent til behandling av LBCL i tredje linje (basert på ZUMA-1 studiet), men også ved primær R/tidlig-R LBCL i andre linje (ZUMA-7 studiet) og til follikulært lymfom som tredje linje behandling (ZUMA-5 studiet).
Disse studiene har dog ekskludert pasienter med tidligere eller aktiv CNS involvering av deres lymfom, samt at alle primær CNSlymfomer var ekskludert (absolutt eksklusjon). CD19 CAR-T celler har dog tidligere vist at de kan krysse over blod-hjerne barrieren og
at de er trygge til pasienter med sekundær CNS involvering (dette basererer seg på enkelt kasustikker). Dette pilot-studiet presentert her, ønsket å undersøke sikkerheten og effektiviten av Axi-cel ved både primær- og sekundær CNS-lymfom.
15 pasienter ble screenet og 9 ble inkludert. Så langt har det ikke blitt sett noen toksisitets begrensninger, kun 1 alvorlig AE i form av stafylokokk meningitt og 2 dødsfall pga progresjon.
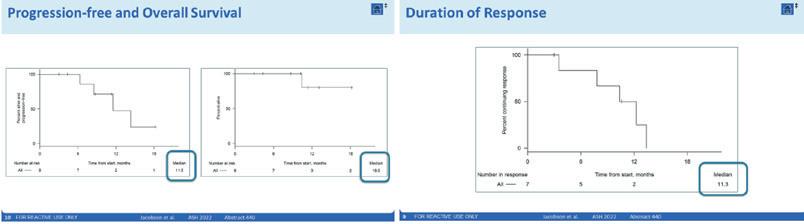
Axi-cel har foreløpig vist akseptabel toleranse til en svært vanskelig behandlet pasientgruppe med ekstremt dårlig prognose, sammen med lovende effekt med en CR rate på 67% og en mPFS på 11.5 måneder.
Ytterligere studier kreves og pågår, inkludert MRD analyse og fase 2 studier. Men de begrensede resultatene som foreligger gir håp til en spesielt vanskelig behandlingsgruppe.
16
Post
DARZALEX® + Rd FORLENGER
TOTALOVERLEVELSE1–3
DARZALEX® + Rd FORLENGER TOTALOVERLEVELSE1–3
Lenger oppfølgingstid (>5år) bekrefter signifikant OS fordel med DRd sammenlignet med Rd alene hos pasienter med nydiagnostisert myelomatose hvor HMAS ikke er aktuelt.*1,3
Lenger oppfølgingstid (>5år) bekrefter signifikant OS fordel med DRd sammenlignet med Rd alene hos pasienter med nydiagnostisert myelomatose hvor HMAS ikke er aktuelt.*1,3
Overall survival (median follow-up 64 months)1
Overall survival (median follow-up 64 months)1
DRd median: NR months
DRd median: NR months
Rd median: 65.5 months
Rd median: 65.5 months
34%
34%
reduksjon i risiko for død
Adapted from DARZALEX® SC summary of product characteristics.1
Adapted from DARZALEX® SC summary of product characteristics.1
*Median oppfølging på 64 måneder
DRd, DARZALEX® + lenalidomide + dexamethasone; mOS, median overall survival; HR, hazard ratio; CI, confidence interval; HMAS, høydosebehandling med autolog stamcellestøtte.
*Median oppfølging på 64 måneder1
DRd, DARZALEX® + lenalidomide + dexamethasone; mOS, median overall survival; HR, hazard ratio; CI, confidence interval; HMAS, høydosebehandling med autolog stamcellestøtte.
reduksjon i risiko for død
HR: 0.66 (95% CI: 0.53-0.83)
HR: 0.66 (95% CI: 0.53-0.83)
Ingen nye bivirkninger ble observert for DRd etter lenger oppfølgingstid1,3
Ingen nye bivirkninger ble observert for DRd etter lenger oppfølgingstid1,3
1. Darzalex SmPC, 11/2023, avsnitt 4.1, 4.8, 5.1. 2. Facon T, Kumar SK, Plesner T et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial Lancet Oncol. 2021;22:1582–1596. 3. Kumar SH, Moreau P, Bahlis NJ et al. Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) Alone in Transplant-Ineligible Patients with Newly Diagnosed Multiple Myeloma (NDMM): Updated Analysis of the Phase 3 Maia Study. Blood (2022) 140 (Supplement 1): 10150–10153.
1. Darzalex SmPC, 11/2023, avsnitt 4.1, 4.8, 5.1. 2. Facon T, Kumar SK, Plesner T et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial Lancet Oncol. 2021;22:1582–1596. 3. Kumar SH, Moreau P, Bahlis NJ et al. Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) Alone in Transplant-Ineligible Patients with Newly Diagnosed Multiple Myeloma (NDMM): Updated Analysis of the Phase 3 Maia Study. Blood (2022) 140 (Supplement 1): 10150–10153.
UTVALGT PRODUKT OG SIKKERHETSINFORMASJON FOR DARZALEX® INTRAVENØS (IV) OG SUBKUTAN (SC)
UTVALGT PRODUKT OG SIKKERHETSINFORMASJON FOR DARZALEX® INTRAVENØS (IV) OG SUBKUTAN (SC)
INDIKASJONER
Myelomatose (Darzalex® IV og SC)
INDIKASJONER
Myelomatose (Darzalex® IV og SC)
• I kombinasjon med lenalidomid og deksametason eller med bortezomib, melfalan og prednison til behandling av voksne pasienter med nydiagnostisert myelomatose, hvor autolog stamcelletransplantasjon ikke er aktuelt.
• Se SPC for fullstendig oversikt over godkjente indikasjoner.
• I kombinasjon med lenalidomid og deksametason eller med bortezomib, melfalan og prednison til behandling av voksne pasienter med nydiagnostisert myelomatose, hvor autolog stamcelletransplantasjon ikke er aktuelt.
DOSERING OG ADMINISTRASJONSMÅTE
• Se SPC for fullstendig oversikt over godkjente indikasjoner.
DOSERING OG ADMINISTRASJONSMÅTE
Gis som intravenøs infusjon eller subkutan injeksjon og skal administreres av helsepersonell med tilgjengelige ressurser for resuscitering. Subkutan formulering er ikke beregnet til intravenøs administrering, og skal kun gis ved subkutan injeksjon med angitt dosering. Det skal gis pre og post infusjonsbehandling/injeksjonsbehandling for å redusere risikoen for infusjonsrelaterte reaksjoner (IRR).
Gis som intravenøs infusjon eller subkutan injeksjon og skal administreres av helsepersonell med tilgjengelige ressurser for resuscitering. Subkutan formulering er ikke beregnet til intravenøs administrering, og skal kun gis ved subkutan injeksjon med angitt dosering. Det skal gis pre og post infusjonsbehandling/injeksjonsbehandling for å redusere risikoen for infusjonsrelaterte reaksjoner (IRR).
Myelomatose: Anbefalt dose er 16 mg/kg kroppsvekt gitt som IV. infusjon eller 1800 mg oppløsning til s.c. injeksjon gitt over ca. 3–5 minutter. For doseringsplan for daratumumab per indikasjon se SPC. For dose og doseringsplan for legemidler som gis sammen med daratumumab gitt som en i.v. infusjon eller s.c. injeksjon, se SPC og tilhørende preparatomtaler.
Myelomatose: Anbefalt dose er 16 mg/kg kroppsvekt gitt som IV. infusjon eller 1800 mg oppløsning til s.c. injeksjon gitt over ca. 3–5 minutter. For doseringsplan for daratumumab per indikasjon se SPC. For dose og doseringsplan for legemidler som gis sammen med daratumumab gitt som en i.v. infusjon eller s.c. injeksjon, se SPC og tilhørende preparatomtaler.
for virkestoffet eller noen av hjelpestoffene.
KONTRAINDIKASJONER
Overfølsomhet for virkestoffet eller noen av hjelpestoffene.
ADVARSLER OG FORSIKTIGHETSREGLER
Infusjonsrelaterte reaksjoner (IRR): Darzalex® kan forårsake kraftige og/eller alvorlige IRR. Det skal gis pre- og post-infusjonsbehandling/injeksjonsbehandling for å redusere risikoen for IRR, som ble rapportert i kliniske studier hos henholdsvis ca. 50 % av pasientene ved IV og 9 % ved SC. Ved enhver IRR, skal behandlingen avbrytes umiddelbart og symptomer håndteres. Dersom øyesymptomer oppstår, skal DARZALEX® avbrytes og umiddelbar oftalmologisk evaluering foretas før Darzalex® gjenopptas.
ADVARSLER OG FORSIKTIGHETSREGLER Infusjonsrelaterte reaksjoner (IRR): Darzalex® kan forårsake kraftige og/eller alvorlige IRR. Det skal gis pre- og post-infusjonsbehandling/injeksjonsbehandling for å redusere risikoen for IRR, som ble rapportert i kliniske studier hos henholdsvis ca. 50 % av pasientene ved IV og 9 % ved SC. Ved enhver IRR, skal behandlingen avbrytes umiddelbart og symptomer håndteres. Dersom øyesymptomer oppstår, skal DARZALEX® avbrytes og umiddelbar oftalmologisk evaluering foretas før Darzalex® gjenopptas.
Interferens med indirekte antiglobulintest (indirekte Coombs test): Darzalex® kan medføre positiv indirekte Coombs test. Pasienter skal typebestemmes og screenes før oppstart. Ved planlagt transfusjon skal blodtransfusjonssenteret gjøres oppmerksom på denne interferensen med indirekte antiglobulintester. Ved akutt transfusjon kan det gis ikke-kryssmatchede ABO/RhD-kompatible erytrocytter i henhold til lokal blodbankpraksis. Reaktivering av hepatitt B-virus (HBV): Hepatitt B-virusreaktivering er rapportert, og HBV-screening skal foretas hos alle pasienter før oppstart av behandling.
Interferens med indirekte antiglobulintest (indirekte Coombs test): Darzalex® kan medføre positiv indirekte Coombs test. Pasienter skal typebestemmes og screenes før oppstart. Ved planlagt transfusjon skal blodtransfusjonssenteret gjøres oppmerksom på denne interferensen med indirekte antiglobulintester. Ved akutt transfusjon kan det gis ikke-kryssmatchede ABO/RhD-kompatible erytrocytter i henhold til lokal blodbankpraksis. Reaktivering av hepatitt B-virus (HBV): Hepatitt B-virusreaktivering er rapportert, og HBV-screening skal foretas hos alle pasienter før oppstart av behandling.
Bivirkninger: De hyppigste bivirkningene uavhengig av grad (≥ 20 % av pasientene) med daratumumab (intravenøs eller subkutan formulering) gitt som monoterapi eller kombinasjonsbehandling var IRR, fatigue, kvalme, diaré, forstoppelse, feber, hoste, nøytropeni, trombocytopeni, anemi, perifert ødem, perifer sensorisk nevropati og øvre luftveisinfeksjon. Alvorlige bivirkninger var pneumoni, bronkitt, øvre luftveisinfeksjon, sepsis, lungeødem, influensa, feber, dehydrering, diaré, atrieflimmer og synkope. For fullstendig oversikt over bivikningene, se SPC
PAKNINGER, PRISER OG REFUSJON
Bivirkninger: De hyppigste bivirkningene uavhengig av grad (≥ 20 % av pasientene) med daratumumab (intravenøs eller subkutan formulering) gitt som monoterapi eller kombinasjonsbehandling var IRR, fatigue, kvalme, diaré, forstoppelse, feber, hoste, nøytropeni, trombocytopeni, anemi, perifert ødem, perifer sensorisk nevropati og øvre luftveisinfeksjon. Alvorlige bivirkninger var pneumoni, bronkitt, øvre luftveisinfeksjon, sepsis, lungeødem, influensa, feber, dehydrering, diaré, atrieflimmer og synkope. For fullstendig oversikt over bivikningene, se SPC
PAKNINGER, PRISER OG REFUSJON
Pakninger og priser: Injeksjonsvæske:15 ml (hettegl.) 72770,60 kr. Konsentrat til infusjonsvæske: 5 ml (hettegl.) 6097,40 kr. 20 ml (hettegl.) 24281,00 kr. Basert på SPC 11/2023.
Les fullstendig preparatomtale før foreskrivning av Darzalex®
Pakninger og priser: Injeksjonsvæske:15 ml (hettegl.) 72770,60 kr. Konsentrat til infusjonsvæske: 5 ml (hettegl.) 6097,40 kr. 20 ml (hettegl.) 24281,00 kr. Basert på SPC 11/2023.
Les fullstendig preparatomtale før foreskrivning av Darzalex®
KONTRAINDIKASJONER Overfølsomhet
CP-439133
351 336 317 300 281 258 241 223 204 157 117 65 26 8 4 0 369 343 324 308 294 270 251 232 214 195 186 350 344 334 316 302 286 273 255 248 200 148 103 42 16 5 0 368 346 338 328 305 297 280 266 249 246 240 DRd Rd No. at risk 100 80 60 40 20 0 % surviving Months HR: 0.66 (95% CI: 0.53–0.83) 3 9 15 21 27 33 39 45 51 60 63 66 69 72 75 78 0 6 12 18 24 30 36 42 48 54 57
CP-439133
351 336 317 300 281 258 241 223 204 157 117 65 26 8 4 0 369 343 324 308 294 270 251 232 214 195 186 350 344 334 316 302 286 273 255 248 200 148 103 42 16 5 0 368 346 338 328 305 297 280 266 249 246 240 DRd Rd No. at risk 100 80 60 40 20 0 % surviving Months HR: 0.66 (95% CI: 0.53–0.83) 3 9 15 21 27 33 39 45 51 60 63 66 69 72 75 78 0 6 12 18 24 30 36 42 48 54 57
Myelomatose: The return of conventional therapy
For en gangs skyld var det ikke immunterapi som skapte mest interesse av myelomatosepresentasjonene på ASH, selv om det selvfølgelig også var noe på denne fronten. For Norge var nok det morsomste at vi fikk 6 personer inn i den internasjonale myelomatosegruppen (IMWG), at vi hadde rekordmange presentasjoner (9), og at både Frida Askeland og Nita Moore ble gjort stas på med pris og høyrangerte postere.
Det aller viktigste foredraget var Perseus-studien, som ble presentert som late-breaking abstract den siste dagen. Perseus er en sammenligning av daratumumab som tillegg til VRd-induksjon, VRd-konsolidering, og lenalidomid-vedlikedhold, sammenlignet med standard, i pasienter som også fikk HMAS. Studien viste en betydelig økt PFS ved 48 måneder (84% vs 68%), med 58 reduksjon av progresjon eller død i intervensjonsarmen (HR 0.42). Den eneste subgruppen som ikke viste effekt, var pasienter over 65 år. Dette skyldes nok at det tilfeldigvis var en større andel med høyrisikosykdom og høy ISS-grad i denne armen, og får nok lite betydning for godkjenning av dette regimet. Man forventer en godkjenning av EMA i løpet av året, og den største forskjellen her vil være at daratumumab blir godkjent som vedlikehold. I studien stanset daratumumab etter 2 års MRD-negativitet.
REST-studien fra Norge, presentert av Frida Askeland, fikk en pris for høyest rangerte myelomatosepresentasjon av en lege i «training» innenfor prospektive kliniske studier. Studien gir isatuximab sammen med bortezomib og lenalidomid, med steroider bare i de 2 første syklusene, til nydiagnostiserte pasienter som ikke er aktuelle for HMAS. Resultatene må mest relevant sammenlignes med MAIA, dvs Dara-LenDex, i den samme populasjonen, og selv med en betydelig høyere median alder på 77 år, og 31% av pasientene over 80 år, ser vi at MRD-negativiteten er høyere i denne studien enn i MAIA, tydende på at tillegget av bortezomib med fraværet til dexametason, er fordelaktig for pasientene. Responsraten er 100%, noe som også er høyere enn MAIA. Lancet Haematology har allerede forespurt om vi kan submitte manuskriptet dit, noe som vil bli gjort etter sommeren.
Men noe immunterapi var det selvfølgelig. KarMMa-3, som sammenlignet et utvalg av standardbehandlinger (alle godkjente egentlig) med Ide-Cel, viste oppdaterte resultater. På PFS er det fortsatt en veldig overbevisende gevinst, med 13,8måneder vs 4,4måneder (HR 0.49), men OS-dataene var noe mer forvirrende. På totalpopulasjonen er HR 1.01, dvs ingen gevinst, noe som jo skaper litt diskusjon. Det som er kompliserende i studien er at studien inneholdt cross-over, dvs at de som progredierte på standard behandling, kunne få, og de fleste har fått, ide-cel ved tilbakefall. Ved sensitivitetsanalyser har man kunnet påvise at når man justerer for crossover, er det en sterk trend mot OS-gevinst for ide-cel, men dette er jo litt vanskeligere å tolke. Det er også kompliserende at en del pasienter progredierte eller av andre grunner ikke fikk ide-cel, og disse pasientene sier jo ikke noe om ide-cel-effekten, selv om de analyseres i den armen. Pasientene som skulle få ide-cel har en lengre periode uten behandling enn kontrollarmen, og fikk heller ikke den samme bridging-behandlingen som de i standard fikk fra start. Konklusjonen er vel, som vi allerede vet, at CAR-T ikke er det lureste å gi til pasienter som har en aggressivt utviklende myelomatose.
Spennende var det også med KarMMa-2, som ga ide-cel til pasienter som hadde dårlig respons etter HMAS. Og mest spennende var det som skjedde etterpå. I studien var det opp til investigator om man ga lenalidomid vedlikehold etterpå, som ble gitt til ca. en tredel av pasientene. I de pasientene som fikk lenalidomid vedlikehold, som bare var 8 pasienter, er det fortsatt ingen som har fått tilbakefall, med oppfølgingstid mellom 38 og 46 måneder, og dette i pasienter med dårlig prognose. Dette lover veldig godt for KarMMa-9-studien vi har, der pasienter med VGPR eller dårligere, randomiseres mellom ide-cel + lenalidomid vedlikehold, vs kun lenalidomid vedlikehold. Vi har inkludert 2 pasienter så langt, og begge er randomisert til ide-cel.
Noen ganger kommer det randomiserte hel-akademiske, enkle studier, som får umiddelbar impact. En slik var den asiatiske studien som randomiserte Pom-CyDex vs PomDex. Primærendepunktet for median PFS, viste en klar økning fra 5,8måneder til 10,9 måneder, med en høy-signifikant HR på 0.43. Dette kan og bør vi umiddelbart starte bruken av, i pasienter vi ellers hadde gitt PomDex til. Dosen av cyklofosfamid var 400mg ukentlig i 3 uker, og fri siste uken. Pom som vanlig.
En annen randomisert studie, som bekrefter det vi har trodd fra før, men som ikke tidligere har vært vist i en randomisert studie, var kombinasjonen av venetoclax med daratumumab, i pasienter med translokasjon 11;14. Regimet var sammenlignet med DVd, som jo fortsatt er en slags standard i 2.linje, selv om vi i Norge har gått over til å bruke IsaKd. Venetoclax ble gitt i doser både på 400mg daglig og 800mg daglig, og det ble ikke vist frem noen stor forskjell på disse. Median PFS økte fra 15,5 for DVd, som jo er som forventet, til 46,1 med venetoclax med daratumumab-dex. Hvordan dette hadde vært mot IsaKd er jo vanskelig å vite, men at venetoclax er gunstig for denne pasientgruppen virker klart.
Spørsmålet om vi skal gjøre tilbakefalls-HMAS har aldri blitt helt avklart. Det er det kanskje heller ikke nå, men en studie fra Tyskland (GMMG ReLApsE) ga oss i hvert fall noe mer å tygge på. Studien randomiserte mellom Rd og Rd=>HMAS=>Len vedlikehold, i tilbakefallspasienter som i hovedsak hadde fått 1 tidligere behandlingslinje med HMAS. Rd-arm-pasientene kunne få HMAS ved tilbakefall, som noen gjorde, og det kan forklare manglende OS-forskjell. Men PFS-resultatene var helt identiske, og dette tyder på at rollen for HMAS ved tilbakefall ikke er så viktig. Det skal legges til at de som fikk HMAS i studien, ikke fikk Rd fulldose etterpå, men fortsatte «bare» med vedlikehold. So, it’s complicated. Men studien har nok likevel svekket HMAS ved tilbakefall noe.
Noe av det aller mest spennende som ryktes, men ikke ble vist frem, er belantamab-VelDex mot DVd. I løpet av de neste ukene vil dette bli offentlig, og ryktene sier at daratumumab for første gang har blitt slått i en randomisert studie, og det til gangs. Så det er bare å følge med.
18
Fredrik Schjesvold
Godkjent av beslutningsforum, i kombinasjon med lenalidomid og deksametason, hos voksne myelomatosepasienter som har fått minst 3 tidligere behandlingslinjer1.
UTVALGT PRODUKT OG SIKKERHETSINFORMASJON FOR EMPLICITI®
VIKTIG SIKKERHETS-OG FORSKRIVNINGSINFORMASJON
Empliciti® (elotuzumab)
Utleveringsgruppe C, Reseptbelagt legemiddel.
Indikasjoner: I kombinasjon med lenalidomid og deksametason til behandling av myelomatose hos voksne som har fått minst 1 tidligere behandling. I kombinasjon med pomalidomid og deksametason til behandling av voksne med tilbakevendende og refraktær myelomatose som har fått minst 2 tidligere behandlinger, inkl. lenalidomid og en proteasomhemmer, og sykdomsprogresjon er vist med siste behandling.
Dosering: Voksne inkl. eldre: Premedisinering for forebygging av infusjonsrelaterte reaksjoner (IRR) skal gis. Empliciti i kombinasjon med lenalidomid og deksametason gis som IV, 10 mg/kg kroppsvekt ukentlig de 2 første syklusene, deretter 20 mg/kg kroppsvekt dag 1 i de påfølgende syklusene. Empliciti i kombinasjon med pomalidomid og deksametason gis som IV, 10 mg/kg kroppsvekt ukentlig de 2 første syklusene, deretter 20 mg/ kg kroppsvekt dag 1 i de påfølgende syklusene. En syklus er 28 dager. For detaljert doseringsskjema se www.felleskatalogen.no.
Vanligste bivirkninger: De vanligste bivirkningene (oppstod hos > 10 % av pasientene) ved elotuzumab behandling var IRR, diaré, herpes zoster, nasofaryngitt, hoste, pneumoni, øvre luftveisinfeksjon, lymfopeni og vektreduksjon.
Alvorlige biverkninger: Den alvorligste bivirkningen som kan oppstå under elotuzumab behandling er pneumoni.
Kontraindikasjoner*: Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene. Preparatomtalene for lenalidomid, pomalidomid og deksametason som brukes i kombinasjon med Empliciti skal gjennomgås før oppstart av behandling.
Advarsler og Forsiktighetsregler: IRR er rapportert hos pasienter som har fått elotuzumab. Premedisinering skal gis før infusjon. Pasienten bør overvåkes for utvikling av Sekundære primære maligniteter (SPM). I kliniske studier hos pasienter med myelomatose var forekomsten av alle infeksjoner, inkludert pneumoni, høyere hos pasienter behandlet med Empliciti. Pasienter bør overvåkes og infeksjoner bør håndteres med standardbehandling. For fullstendig informasjon, se preparatomtale og SPC tilgjengelig på www.felleskatalogen.no.
Interaksjoner: Ingen farmakokinetiske interaksjonsstudier er utført. Metabolisme via CYP450 enzymer eller andre legemiddelmetaboliserende enzymer forventes ikke. Empliciti kan påvises i serumproteinelektroforese (SPEP) og ved immunfiksering i serum hos myelompasienter og kan interferere med riktig responsklassifisering. Tilstedeværelse av elotuzumab i pasientens serum kan forårsake en liten topp i begynnelsen av gamma regionen på SPEP som tilsvarer IgGƙ ved immunfiksering i serum. Denne interferensen kan påvirke vurderingen av fullstendig respons og mulig tilbakefall fra fullstendig respons hos pasienter med IgG kappa myelomprotein. I tilfeller hvor det påvises ekstra topper ved immunfiksering i serum bør en mulig biklonal gammopati utelukkes.
Pakninger, priser og refusjon: 300 mg: 1 stk. (hettegl.) 15190,30 kroner. 400 mg: 1 stk. (hettegl.) kr 20241,70 kroner. Empliciti er godkjent refundert i kombinasjon med lenalidomid og deksametason til pasienter som har fått 3 tidligere behandlingslinjer.
Se felleskatalogen for fullstendig forskrivningsinformasjon før forskrivning.
Bristol Myers Squibb Norge, Lysaker Torg 35 1366 Lysaker, telefon: 23 12 06 37 , www.bms.com/no
Versjon 1
Bristol
Myers Squibb Lysaker Torg 35 1366 Lysaker www.bms.com/no
Beslutningsforum
23.05.2022 sak 064-2022 689-NO-2300001 / Utarbeidet feb 2023
1.
for nye metoder


Helseforetakene anbefaler Calquence som førstevalg BTKi i anskaffelsen fra 1/10-20231
CalquenCe ® (akalabrutinib)
Helseforetakene anbefaler Calquence i anskaffelsen fra 1/10-2023. Førstevalg BTK hemmer ved KLL i førstelinje for høyrisiko pasienter (del17p, mutTP53 eller del11q) og for pasienter med tilbakefall1
CALQUENCE er indisert for behandling av voksne med tidligere ubehandlet kronisk lymfatisk leukemi (KLL) som monoterapi eller i kombinasjon med obinutuzumab, eller som monoterapi for behandling av voksne med kronisk lymfatisk leukemi (KLL) som har fått minst én tidligere behandling.2
Viktig sikkerhetsinformasjon
Alvorlige blødninger er sett. Pasienter som bruker antitrombotiske midler kan ha økt risiko for blødning og forsiktighet skal utvises ved bruk. Atrieflimmer/flutter forekom hos pasienter med hematologiske maligniteter både ved monoterapi og i kombinasjon med obinutuzumab.2
Viktig informasjon om Calquence (akalabrutinib)
Indikasjoner: Som monoterapi eller i kombinasjon med obinutuzumab, til behandling av voksne med tidligere ubehandlet kronisk lymfatisk leukemi (KLL). Som monoterapi til behandling av voksne med KLL som har fått minst 1 tidligere behandling. Dosering: Behandling skal igangsettes og følges opp av lege med erfaring i bruk av legemidler mot kreft. Voksne: Anbefalt dose er 100 mg 2 ganger daglig. Behandling bør fortsette inntil sykdomsprogresjon eller uakseptabel toksisitet. Forsiktighetsregler: Alvorlige blødninger, inkl. blødning i CNS og gastrointestinal blødning er sett. Pasienter som bruker antitrombotiske midler kan ha økt risiko for blødning og forsiktighet skal utvises ved bruk. Warfarin eller an-dre vitamin K-antagonister skal ikke gis samtidig med akalabrutinib. Atrieflimmer/flutter forekom hos pasienter med hematologiske maligniteter både ved monoterapi og i kombinasjon med obinutuzumab. Overvåk for symptomer på atrieflimmer og atrieflutter og foreta EKG om nødvendig. Ved høy risiko for tromboembolisk sykdom, skal nøye kontrollert behandling med antikoagulanter og andre behandlingsalternativer vurderes. Interaksjoner: Samtidig bruk av sterke CYP3A-hemmere skal unngås; kan gi økt eksponering for akalabrutinib og dermed økt risiko for toksisitet. Dersom slike hemmere skal brukes i en kort periode (f.eks. antiinfektiva i opptil 7 dager), skal behandlingen avbrytes. Pasienten skal overvåkes nøye hvis en moderat CYP3A-hemmer brukes. Samtidig bruk av en CYP3A-induktor og sterke CYP3A4-induktorer skal unngås; vil kunne gi redusert eksponering og risiko for manglende effekt. Akalabrutinib kan øke eksponeringen for samtidig administrerte BCRP-substrater (f.eks. metotreksat) ved å hemme BCRP i tarm. Metabolitten ACP-5862 kan øke eksponeringen for samtidig administrerte MATE1-substrater (f.eks. metformin). Bivirkninger: Svært vanlige (≥1/10): Anemi, nøytropeni, trombocytopeni. Abdominalsmerter, diaré, forstoppelse, kvalme, oppkast. Fatigue. Hud: Utslett. Infeksiøse: Nasofaryngitt, pneumoni, sinusitt, urinveisinfeksjon, øvre luftveisinfeksjon. Blåmerke, blødning/hematom, kontusjon, petekkier. Artralgi, muskel-skjelettsmerter. Hodepine, svimmelhet. Ny primær malignitet. Undersøkelser: Redusert Hb, redusert absolutt nøytrofiltall, redusert trombocyttall. Vanlige (≥1/100 til <1/10): Trombocytopeni. Asteni. Atrieflimmer/-flutter. Bronkitt, herpesvirusinfeksjon. Kar: Ekkymose, epistakse, gastrointestinal blødning, intrakraniell blødning, hypertension. Ikke-melanom hudkreft, sekundær primær malignitet. Pakninger og priser: 60 stk. (blister) kr 76267,60. Reseptgruppe C. Refusjon: H-resept. Rekvirering skal gjøres i tråd med anbefalinger fra RHF, og refusjon ytes kun etter resept fra sykehuslege eller avtalespesialist. Calquence er godkjent refundert som monoterapi eller i kombinasjon med et anti CD20-antistoff, til behandling av voksne pasienter med tidligere ubehandlet kronisk lymfatisk leukemi (KLL) med 17p-delesjon/TP53-mutasjon og/eller 11q-delesjon, eller som monoterapi hos KLL pasienter som har fått minst én tidligere behandling. Calquence inngår i Helseforetakenes anbefaling for KLL (Onkologi 2307, gjeldende fra 1.10.2023): - Som monoterapi eller i kombinasjon med et anti CD20-antistoff, til behandling av voksne pasienter med tidligere ubehandlet kronisk lymfatisk leukemi (KLL) med 17p-delesjon/TP53-mutasjon og/eller 11q-delesjon (førstevalg BTK hemmer). - Til behandling av voksne pasienter med kronisk lymfatisk leukemi (KLL) som har fått minst én tidligere behandling (førstevalg BTK-hemmer).
For mer info. om Calquence, les FK tekst på www.felleskatalogen.no eller godkjent SPC. Beslutningsforum for Nye Metoder, sak 153-20221, og sak 167-20221, www.nyemetoder.no
ID: NO-11408-11-23-ONC
Referanser: 1. https://www.sykehusinnkjop.no/avtaler-legemidler/onkologi 2. Calquence SPC. section 4.1, 4.4, 5.1
20
AstraZeneca Nordic • Postboks 6050 Etterstad 0601 Oslo • www.astrazeneca.no
NO-11384-11-23-ONC
Hoa Tran
Overlege PhD, Hematologisk avdeling, Ahus
Oppsummering KLL
For KLL pasienter viser oppfølgende data fra kliniske studier (CLL13, CAPTIVATE, FLAIR og GLOW) at målrettet tidsbegrenset behandling med I + V eller V+ 0 eller R + V eller IVO resulterer i signifikant bedre progresjonsfri overlevelse (PFS) og overlevelse (der data foreligger) med akseptable bivirkninger enn tradisjonelle kjemoterapi (FCR/BR/Chl-O). Studiene viser også at man oppnår høy respons rate ved rebehandling ved residiv etter målrettede behandling (CAPTIVATE og MURANO). Studie på CAR-T (TRANSCEND) og non-kovalent BTK-hemmer pirtubrutinib (BRUIN) hos pasienter som har progredierte på BCL2 – og eller BTK-hemmere viser lovende resultater. Pirtubrutinib viser en overall responsrate på 79 -83 % hos BCL2 og BTK-hemmer resistente pasienter i BRUIN studien. Den viser også median PFS 16 – 23 måneder etter en oppfølgingstid på 27 måneder.
Studier på trippel kombinasjon pågår. For KLL ubehandlet pasienter pågår det fase 2 studie på tidsbegrenset behandling med Zanubrutinib + Ven-Obinutuzumab samt studier på acalabrutinib + VenObi (AVO). I tillegg er det CLL16 fase III studien på AVO vs VenO hos høyrisk KLL pasienter og AMPLIFY studien der AVO vs kjemoterapi hos pasienter uten Tp53 mutasjoner/del17p.
I-ibrutinib, V-venetoclax, R – Rituximab, O- obinutuzumab, CIT –Chemoimmunotherapy – FCR – Fludarabin. Cyclophosphamide- Rituximab eller BR – Bendamustin Rituximab. Chl – Chlorambucil.
Tabellen viser oversikt over studier på KLL pasienter som ikke har fått behandling tidligere.
Studie
CLL 13 / GAIA (n= 230 i hver arm).
4 års Oppfølging.
Moritz Furstenau et al ASH 2023. Abstract 635
FLAIR N = 274 (n=523)
43 måneders oppfølging.
Hillmen B. Et al ASH 2023, Abstract 631
CAPTIVATE
N = 153
5 års oppfølging
Gia paolo et al ASH 2023
Abstract 633
GLOW
N= 211 57 måneders oppfølging
Carol Moreno et al, ASH 2023
III CIT vs RV vs VO vs IVO
Totalt 4 arm.
III I+ V i minimum 24 måneder avh MRD vs FCR
Ubehandet, spreke (FIT). Pas med Tp53 mutasjon eller Del17q mutasjon er ikke inkludert
Ubehandlet <75 år og del 17q < 20%
II I + V i 12 måneder eller lenger. Randomiseres avh MRD status.
Ubehandlet < 70 år Del 11q, del 17q , Tp53 mutasjon er inkludert
III I + V vs Chl + O
Behandling i 12 måneder
Ubehandlet, CIRS > 6 /Cr < 70 eller > 65 år ECOG 0-2. Ikke inkludert Tp53 eller del17q
I-ibrutinib, V-venetoclax, R – Rituximab, O- obinutuzumab, CIT – Chemoimmunotherapy – FCR eller BR
62 % - CIT (BR eller FCR)
70 % - RV
82 % - VO
85,5 % - IVO
PFS ed 4 års oppfølging
93,5 % - I + V OS – 95 %
64,8 % - FCR OS – 87 %
PFS ved 3 års oppfølging
97,2 % - I + V OS – 98 %
76,8 % - FCR OS – 93 %
PFS ved 5 års oppfølging
70 % - for alle pasienter.
68 % -umutert
64 %- del 11q
60 % - kompleks kariotype.
45 % -Tp53 pos/del17q
66,5 % - I+V
19,5 % - Chl + O
Behandlingsfri OS
76 % - I + V vs 35 % - Chl -O
21
Fase Behandling Pasientgruppe PFS
Tabellen viser oversikt over studier på KLL pasienter som har fått behandling tidligere.
Studie
MURANO -rebehandling
N = 389
Ved 102 mnd oppfølging
Seymour J et al, ASH 2023
Abstract 1898
BRUIN
N = 282 hvorav 170 KLL/SLL pasienter.
Woyach J et al, ASH 2023
Abstract 325
CAPTIVATE
Rebehandling
Gia paolo et al ASH 2023
Abstract 633
TRANSCEND (n= 118)
Tanya Siddiqqi, ASH 2023
Abstract 330
Fase Behandling Pasientgruppe
III Ven + R Eller BTK-hemmer
Residiv etter å ha fått VenR eller BR.
Best overall respons (BOR) for pas som har fått Ven + R er 80 % og BR er 79 %.
BOR blant pasienter som ble rebehandlet med VenR er 76 – 88% og 78 -86 % for de som ble rebehandlet med BTKi.
Konklusjon: Det er høy responsrate ved rebehandling, enten med VenR eller med BTK-hemmer.
I/II Pirtobrutinib (non-kovalent BTK hemmer)
Residiv KLL, 89 % har tidligere fått BKTi.
Ved 27 måneders oppfølging.
ORR – 83 % for pas som ikke har fått BCL2-hemmer vs 79% hos de som har fått BCL2-hemmer.
Median PFS var 23 vs 16 måneder for de 2 gruppene.
II I + V eller Ibrutinib Residiv etter I + V Responsrate ved rebehandling med Ibrutinib - 86 % (n= 21) I + V – 83 % ( n = 6)
CAR-T KLL pasienter som har progrediert etter BCL2 eller BTK hemmere.
ORR 44 %, CR/CR1 18 % og PFS 12 måneder, median OS er 30 måneder med uMRD 60 % i blod.
I-ibrutinib, V-venetoclax, R – Rituximab, O- obinutuzumab, CIT – Chemoimmunotherapy – FCR eller BR- Bendamustin-Rituximab
22 Post
15. juni Madrid
Save the date



IMBRUVICA ® + VENETOCLAX
deliver deep, durable responses as the first all-oral fixed duration and chemo free combination to demonstrate an OS advantage in previously untreated CLL 1,2
STUDY BACKGROUND: GLOW is an international phase 3 trial evaluating the efficacy and safety of ibrutinib-venetoclax in older patients and/or those with previously untreated CLL after a 15 months fixed duration treatment period. Patients assigned to ibrutinib-venetoclax received three cycles of ibrutinib lead-in at 420 mg OD followed by 12 cycles of the combination of ibrutinib + venetoclax.




OS advantage vs CIb+O of patients on IMBRUVICA® + venetoclax were alive at 3.5 years1,2 (HR: 0.487 vs CIb+O; nominal p=0.0205†)
Superior PFS vs CIb+O regardless of genetic profile of patients on IMBRUVICA® + venetoclax were progression-free at 3.5 years1,2
Sustained uMRD efficacy vs CIb+O of patients on IMBRUVICA® + venetoclax did not require subsequent treatment at 3.5 years1,2
Utvalgt produkt-og sikkerhetsinformasjon for IMBRUVICA ® (Ibrutinib)
Indikasjoner: Som monoterapi til behandling av voksne med residiverende eller refraktær mantelcellelymfom (MCL). Som monoterapi eller i kombinasjon med rituksimab eller obinutuzumab eller venetoklaks til behandling av voksne med tidligere ubehandlet kronisk lymfatisk leukemi (KLL). Som monoterapi eller i kombinasjon med bendamustin og rituksimab (BR) til behandling av voksne med KLL som har fått minst én tidligere behandling. Som monoterapi til behandling av voksne med Waldenströms makroglobulinemi (WM) som har fått minst én tidligere behandling, eller som førstelinjebehandling hos pasienter som ikke er egnet for kjemoimmunterapi. I kombinasjon med rituksimab til behandling av voksne med WM.
Dosering og administrasjonsmåte
Behandlingen skal innledes av og gjennomføres under tilsyn av lege med erfaring i bruk av legemidler mot kreft. Behandlingen skal fortsette til sykdomsprogresjon eller til pasienten ikke lenger tolererer den. Mantelcellelymfom (MCL): Voksne: Anbefalt dose er 560 mg 1 gang daglig. Kronisk lymfatisk leukemi (KLL): Voksne: Anbefalt dose, enten som monoterapi eller i kombinasjon, er 420 mg 1 gang daglig. Waldenströms makroglobulinemi (WM): Voksne: Anbefalt dose er 420 mg 1 gang daglig. Ved kombinasjon med anti-CD20-behandling, anbefales det å gi ibrutinib før anti-CD20-behandling når de gis på samme dag.
Forsiktighetsregler:
Blødningsrelaterte hendelser:
Spesiell forsiktighet ved antikoagulasjonsbehandling.
Warfarin/vitamin K-antagonister skal ikke gis samtidig med IMBRUVICA, og fiskeolje og vitamin E-preparater skal unngås.
Atrieflimmer/-flutter, ventrikulær arytmi eller hjertesvikt:
Ved eksisterende atrieflimmer som krever antikoagulasjonsbehandling bør oppstart med andre behandlingsalternativer vurderes. Ved eksisternede relevante risikofatorer for hjertehendelser, eller utvikling under behandling, bør nytte/risiko evalueres, og andre alternativer eller samtidig antikoagulasjonsbehandling vurderes.
• Ved tegn/symptomer på ventrikulær takyarytmi skal behandling midlertidig seponeres.
• Ved grad 2 hjertesvikt skal IMBRUVICA holdes tilbake inntil hjertehendelser når grad 1 eller baseline verdier. Virusreaktivering: Hepatitt B-virus (HBV)-status bør fastslås før behandlingsoppstart. Ved positiv hepatitt B-serologi, bør pasienten overvåkes og behandles for å forebygge hepatitt B-reaktivering.
Bivirkninger:
Vanligste bivirkninger (≥ 20 %): Artralgi, blødninger, blåmerker, diaré, kvalme, muskel-skjelettsmerter, nøytropeni, trombocytopeni, øvre luftveisinfeksjon og utslett
• Vanligste grad 3/4 bivirkninger (≥5%): Hypertensjon, lymfocytose, nøytropeni, pneumoni og trombocytopeni.
Interaksjoner og dosejustering:
Samtidig bruk av moderate til sterke CYP 3A4-hemmere/-induktorer kan gi hhv. økt eller redusert eksponering for IMBRUVICA, og dosejustering eller midlertidig seponering kan være nødvendig.
Kontraindikasjoner: Preparater som inneholder johannesurt
Pakninger,priser og refusjon:
Pakninger og priser:
140 mg: 28 stk.1 (endose) 19 576,60. 280 mg: 28 stk.1 (endose) 39 117,00. 420 mg: 28 stk.1 (endose) 58 657,30.
560 mg: 28 stk.1 (endose)78 197,70.
Refusjon: 1H-resept: L01E L01_1 Ibrutinib
Refusjonsberettiget bruk: Der det er utarbeidet nasjonale handlingsprogrammer/nasjonal faglige retningslinjer og/eller anbefalinger fra RHF/LIS spesialistgruppe skal rekvirering gjøres i tråd med disse.
Vilkår: 216 Refusjon ytes kun etter resept fra sykehuslege eller avtalespesialist.
Basert på SPC godkjent av SLV/EMEA 09/2023
CP-410207
1. Niemann C, Munir T, Moreno C et al. Fixed-duration ibrutinib–venetoclax versus chlorambucil– obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. HYPERLINK “http://www.thelancet.com/oncology”www.thelancet.com/oncology Published online November 6, 2023 https://doi.org/10.1016/S1470-2045(23)00452-7
2. Kater AP, et al. Fixed-duration ibrutinib-venetoclax in patients with chronic lymphocytic leukemia and comorbidities. NEJM Evidence. 2022; doi:10.1056/EVIDoa2200006.
Pål Tore Bentsen
Haukeland Universitetssjukehus
Akutt Myelogen Leukemi (AML)
Sammen med endringer i diagnostiske klassifikasjoner og risikostratifisering har behandlingsmulighetene ved AML endret seg drastisk i løpet av det siste tiåret, særlig ved innføring av venetoclax (Ven) i kombinasjon med hypometylerende behandling (HMA), men også med stadig flere målrettede behandlingsmodaliteter, særlig signalveishemmere. De viktigste fremskrittene har man sett hos eldre og komorbide pasienter samt hos pasienter med behandlingsfølsomme mutasjoner, og samlet sett er langtidsoverlevelsen økende. Flere pågående studier addresserer nå hvordan ulike målrettede behandlinger best kan kombineres, sekvenseres og integreres i behandlingsalgoritmene ved AML, og forhåpentligvis vil dette bidra til å redusere resistensproblematikk og ytterligere bedre langtidsoverlevelsen. Allogen hematopoietisk stamcelletransplantasjon (allo-HSCT) er fortsatt en viktig kurativ behandlingsopsjon hos de fleste yngre pasienter med høy- eller intermediær-risiko sykdom, samt hos spreke eldre pasienter, særlig ved høyrisikosykdom. Likevel er fortsatt relaps den viktigste årsaken til behandlingssvikt etter allo-HSCT, og utvikling av effektive skreddersydde strategier for post-transplantasjons vedlikehold eller annen risikotilpasset intervensjon er et svært sentralt forskningsområde. Samtidig er mangel på målrettet behandling ved spesielt utfordrende høyrisikogenetikk, som ved RAS- og TP53-mutasjon samt inv(3), fortsatt en betydelig utfordring, og det er bred enighet om at særlig slike pasienter i størst mulig grad bør inkluderes i studieprotokoller. Uansett ser MRD-bestemmelser ut til å bli et stadig viktigere verktøy for å bestemme behandlingsintensitet og –varighet.
Det ble på møtet presentert totalt omkring 650 abstracts tilknyttet akutte myeloide maligniteter, hvorav et utvalg innen AML kort omtales her.
Oppdaterte behandlingsresultater
David Grinblatt presenterte (abstract #593) 10 års resultater fra en multisenter prospektiv observasjonsstudie gjennom Connect®registeret, hvor pasienter med de novo AML >55 år er gruppert i 2 tidsperioder ihht. tilgjengelighet av nyere diagnostikk, hhv. 2013-16 og 2017-22. Studiepopulasjonen på totalt 773 pasienter med median alder på hhv. 70 og 71 år, kom ut med median samlet overlevelse (´overall survival´- OS) på hhv. 11 og 16 måneder. Molekylær testing ble gjennomført hos 69% og 87% i første/ siste periode, og var assosiert med høyere median OS. Andelen behandlet med allo-HSCT økte fra 22% til 31% hos pasienter <75 år. Det ble ikke funnet signifikant forskjell i andelen som fikk intensiv kjemoterapi, men ´targeted therapy´ ble brukt hos en større andel av dem som ikke fikk dette i den siste tidsperioden. Forfatterne konkluderte med at overlevelsen har økt over tid, og angir økt grad av molekylær testing, mer bruk av allo-HSCT og nyere, målrettede behandlinger som mulige årsaksfaktorer.
I abstract #967 beskrev Mathilde Hunault-Berger og kollegaer resultater av den store franske multisenter BIG-1 studien, hvor voksne (18-60 år) pasienter med de novo-, s- eller tAML gjennomgikk randomisering mellom idarubicin eller daunorubicin sammen med cytarabin i induksjonsbehandling (R1), og mellom HDAC eller IDAC i konsolidering (R2). Tillegg av midostaurin kunne gies ved FLT3-mutert sykdom fra 2018. Ved intermediær- eller høyrisikosykdom ble pasientene tilbudt allo-HSCT i første remisjon. 1405 pasienter ble inkludert mellom 2015 og 2019 hvorav 1357 var evaluerbare, med median oppfølging i 5,4 år. Samlet oppnådde
84,6% CR/CRp/CRi, hvorav 41,6% fikk HSCT i første remisjon. 6 års OS var 53,4%, uten signifikante forskjeller ihht. R1 eller R2, men med positiv effekt av HSCT. Idarubicin gav noe økt hematologisk toksisitet. Det ble konkludert med at nytte-/risikoavveining går i favør av Daunorubicin i induksjon, og at IDAC bekreftes som standard konsolidering.
MRD
I NRCI AML 18-studien fra Storbritannia presentert av Nigel Russell (abstract #830), ble eldre pasienter (>60 år) som ikke var flowcytometrisk MRD-negative etter induksjonskur I (inkl. 0/1/2 doser gemtuzumab ozogamicin), randomisert til fortsatt standard kjemoterapi med daunorubicin og AraC (DA), eller til intensivert kjemoterapi med opptil 2 kurer enten FLAG-Ida eller DA + cladribin (DAC). Primært endepunkt var overlevelse (OS), sekundært MRDnegativitet. Totalt ble 523 pasienter randomisert, og samlet 5 års OS var 32% med intensivert kjemoterapi, 27% uten. Forskjellen var signifikant dersom pasienter med ukjent MRD-status ble tatt ut av analysen, tross økt tidlig toksisitet ved intensivering, særlig med FLAG-Ida. DAC var det regimet som fikk flest pasienter videre til allo-HSCT, og som gav mest MRD-negativitet. Forfatterne konkluderte med at eldre pasienter med MRD-positivitet etter første induksjonskur, har en overlevelsesgevinst av intensivert videre kjemoterapi.
MRD-bestemmelse av FLT3-ITD har så langt vært begrenset av dårlig sensitivitet både i PCR-baserte og NGS-baserte testplattformer. Nylig utviklede FLT3-ITD spesifikke kombinerte PCR-NGS fremstår mer lovende, men er så langt ikke testet ut i prospektive, randomiserte studier som inkluderer FLT3-hemmere. Alexander Perl presenterte MRD-fokuserte resultater fra QuANTUM-First (abstract #832). Denne fase 3-studien viste som kjent at tillegg av den selektive type II FLT3-hemmeren quizartinib ved intensiv kjemoterapi, fulgt av vedlikehold som monoterapi, gav en signifikant overlevelsesgevinst ved nydiagnostisert FLT3-ITD positiv AML. Det ble her benyttet en protokollspesifikk PCR-NGS MRD målt i perifert blod eller benmarg i remisjon etter induksjon og etter konsolidering. Analysen ble utført hos 87,2% av de 368 (av totalt 539) pasientene som oppnådde CRc etter induksjon, og hos 337 pasienter etter konsolidering. Der var ingen signifikant forskjell mellom studiearmene i andelen CRc-pasienter med MRD <10-4 etter induksjon, mens en større andel hadde udetekterbar MRD i quizartinib-armen. Mediant beste FLT3-ITD VAF var også lavere ved fullført konsolidering for quizartinib-behandlete pasienter. Blant pasienter med påvisbar MRD etter induksjon, hadde pasienter i quizartinib-armen lenger OS, og forfatterne konkluderte med at FLT-ITD spesifikk MRD har potensiell prognostisk nytte, samt at overlevelsesgevinsten ved quizartinib sannsynligvis delvis kan relateres til dyp og vedvarende reduksjon av leukemibyrden.
FLT3-ITD har av mange vært ansett som indikasjon for allo-HSCT i første remisjon også hos MRD-negative intermediær-risiko pasienter, men dette synet baserer seg på studier uten høysensitiv MRD-metodikk. Et av de mest omtalte arbeidene innen AML på ASH –23 ble presentert av Jad Othman (abstract #425). Dette tar utgangspunkt i data fra totalt 737 pasienter i de 2 sekvensielt randomiserte britiske studiene UK NRCI AML17 og –19, som begge inkorporerte høysensitiv qPCR-basert NPM1-MRD. Begge studiene inkluderte pasienter i aldersgruppen 18-60 år med nydiagnostisert AML. I 17-studien ble beslutning om allo-HSCT i første remisjon
25
(CR1-allo) basert på utgangsrisiko og induksjonsrespons, mens CR1-allo i 19-studien kun ble anbefalt pasienter som var MRDpositive i perifert blod etter kjemoterapi-syklus 2, uavhengig av øvrige risikofaktorer, som FLT3-ITD. Begge studiene var gjenomført før midostaurin og FLT3-ITD MRD ble tilgjengelig. I dette arbeidet ble resultatene for NPM1-muterte pasienter analysert mhp. nytte av CR1-allo avhengig av FLT3-ITD og molekylær MRD-status. Blant MRD-positive pasienter ble CR1-allo gjennomført hos hhv. 27% og 60% i de 2 studiene, og blant MRD-negative hos hhv. 18% og 16%. MRD-positive pasienter i 17-studien hadde en 3 års kumulativ relapsinsidens (CIR) på 84% og overlevelse (OS) på 25%, mens CIR i 19-studien var 50% og OS 51%. Samlet sett gav CR1-allo en signifikant overlevelsesgevinst hos MRD-positive (3 års OS 61% vs. 24%, HR 0,42; p<001). MRD-negative pasienter hadde derimot utmerkede resultater i begge studiene (3 års CIR 33% og 24%, OS 75% og 83% for hhv. -17 og -19), uten overlevelsesgevinst av CR1-allo (HR 0,82; 95%CI 0,51-1,34; p=0,4). Totalt inkluderte studiene 286 NPM1-muterte FLT3-ITD muterte pasienter med tilgjengelig MRD-status etter syklus 2, hvorav 28% MRD-positive, og av disse gikk 40% til CR1-allo, som var klart assosiert med bedre OS (45% vs. 18% etter 3 år, HR 0,52, p=0,03) i tråd med den generelle MRDpositive populasjonen. For de 70% av de NPM1-muterte FLT3-ITD muterte pasientene som oppnådde MRD-negativitet etter syklus 2, var samlede resultater lovende, med 3 års OS på hhv. 75% og 80% for AML17 og -19. CR1-allo ble gjennomført hos 20% av disse pasientene, uten at dette gav overlevelsesgevinst (HR 0,80; 95%CI 0,37-1,72; p=0,6), også etter at analysen ble begrenset til pasienter med høy (>0,5) allelratio, og der var heller ingen interaksjon mellom allelratio og transplantasjonsgevinst. Konklusjonen ble at pasienter som oppnår MRD-negativitet etter syklus 2 har lav relapsrisiko, uten gevinst av CR1-allo, og at dette også gjelder FLT3-ITD muterte pasienter, i motsetning til MRD-positive pasienter som har klar nytte av CR1-allo.
Kunstig Intelligens (KI)
KI er aktuelt som aldri før, og er ikke uventet også på vei inn i leukemi-feltet, selv om det nok vil gå noe tid før slike teknologier finner sin integrerte plass i behandlingsalgoritmene. Bl.a. la Alberto Hernández Sánchez frem et arbeid (abstract #62) om bruk av maskinlæring for individualisert prediksjon av resultater etter CR1 oppnådd med intensiv kjemoterapi hos voksne AMLpasienter, hvor data fra HARMONY-platformen bestående av 7467 AML-pasienter ble brukt som utgangspunkt for utvikling av en maskinlæringsmodell basert på nonparametrisk ‘Bayesian Additive Regression Trees (BART)’. Pasienter i aldersgruppen 18-70 år uten CR1-allo ble inkludert, og kjemorefraktære pasienter samt pasienter med relapsfri overlevelse på <2 mnd. ble ekskludert. Individualiserte estimater av RFS, CIR og OS ble basert på data fra 1863 pasienter hvor man også hadde NGS-data, og genetiske avvik tilstede hos minst 10 pasienter ble inkludert i modellen. Modellens treffsikkerhet ble testet ved kryssvalidering med ROC AUC etter 1, 2, 3, 4 og 5 år, og ekstern validering gjort mot en offentlig tilgjengelige database som inkluderte 722 pasienter basert på identiske inklusjonskriterier. Sammenlignet med prognostisk klassifikasjon ihht. ELN 2022, hadde modellen større treffsikkerhet for utfallsvariablene ved samtlige predefinerte tidspunkt, og dette ble også bekreftet i den eksterne kohorten, tross ulik distribusjon av risikokategorier. Forfatterne konkluderte med at man ved analyse av store veldefinerte pasientkohorter kan utvikle prediksjonsmodeller med økt presisjon, og at HARMONY-modellen kan brukes som klinisk beslutningsstøtte hos pasienter i aktuell aldersgruppe som ikke får CR1-allo. Det er behov for videre prospektiv validering samt inklusjon av MRD og pasienter som får målrettet behandling.
Respons etter avbrudd av Hypometylerende behandling i kombinasjon med Venetoclax
De sist oppdaterte resultatene fra Viale-A har vist en langtidsoverlevelse omkring 30% ved nydiagnostisert AML behandlet med azacitidin-venetoclax (Aza-Ven) (Pratz et al., ASH 2022), og behandlingsfrie remisjoner er også rapportert (Chua et al., Blood Adv. 2022). I abstract #161 presenterte Garciaz og kollegaer resultater fra en retrospektiv fransk-amerikansk studie av 83 pasienter med nydiagnostisert (nd, n=60)) eller relapsert/refraktær (R/R, N=23) AML i CR som stoppet behandling med azacitidin og/eller venetoclax i >3 mnd på annen indikasjon enn leukemiprogresjon, dvs. pga. toksisitet, pasientønske eller ukjent. Median alder for nd-gruppen var 76 år, og mediant antall sykluser 4 (1-17). 43% av pasientene stoppet begge medikamenter, mens 57% stoppet venetoclax. Denne kohorten hadde en median OS på 44 mnd., median behandlingsfri remisjon (TFR) på 15 mnd, og 27,7% oppnådde ny CR ved rebehandling med samme kombinasjon. Ved R/R AML var median alder 73, med mediant 1 tidligere behandlingslinje. 70% av disse stoppet begge medikamenter, 30% kun Ven. Her var median OS 19 mnd. og TFR 10 mnd. Ved rebehandling oppnådde 50% av disse ny CR. I multivariat analyse var R/R sykdom (HR 0,47) og IDH-mutasjoner (HR 2,1) signifikant mhp. OS, i motsetning til cytogenetikk, NPM1-mutasjoner eller type avbrutt behandling. Forfatternes konklusjon var at seponering av Ven-Aza i remisjon er assosiert med vedvarende responser og langtidsoverlevelse, og at rebehandling kan indusere nye remisjoner.
FLT3 og ekspresjonsprofilering
Harry Erba (abstract #972) presenterte oppdaterte sikkerhetsdata for ulike behandlingsfaser og aldersgrupper i QuANTUM-First studien, hvor det ble vist at quizartinib var assosiert med cytopenier og infeksjoner i alle behandlingsfaser, med flere fatale infeksjoner på quizartinib ifm. induksjons- og konsolideringsfasen, men ikke under vedlikeholdsbehandling. QTc-forlengelse var også assosiert med quizartinib, men QTcF>500 ms var uvanlig og forekom kun ifm. induksjon og konsolidering. Pasienter >65 år hadde mer TESAE i begge studiearmene. Likevel ble konklusjonen en positiv samlet nytte-risikoprofil for Quizartinib.
I den spanske randomiserte fase II multisenterstudien Pethema QUIWI, ble pasienter randomisert mellom Quizartinib og placebo i tillegg til standard kjemoterapi ved AML uten FLT3-mutasjon, dvs. FLT3 villtype (wt). Orgueira og medarbeidere beskrev i abstract #974 resultater fra RNA-sekvensering av benmargs- og blodprøver fra et utvalg på 161 wt-pasienter i studien sammenholdt med 55 FLT-ITD positive pasienter, basert på tidligere funn hos en undergruppe wt-pasienter av en såkalt FLT3-lignende ekspresjonssignatur tilsvarende det man ser ved FLT3-ITD og -TKD. Denne signaturen ble funnet hos 49,67% av pasientene i wt-gruppen, og disse pasientene hadde i QUIWI-studien signifikant lavere mortalitet og høyere EFS, RFS samt OS (HR 0,41; p=0,01) med quizartinib enn med placebo, mens det ikke ble funnet noen signifikante forskjeller blant wt-pasientene uten FLT3-lignende ekspresjonssignatur. Det ble heller ikke sett noen sammenheng mellom ekspresjonssignatur og ELN-17 klassifikasjon. Ekspresjonsdata ser dermed ut til å kunne fungere som prediktiv biomarkør ved behandling med quizartinib, og kan bidra til å danne utgangspunkt for persontilpasset behandling.
26
Post
Jakavi ved GvHD1
Den første godkjente andrelinje-behandlingen for akutt og kronisk transplantat-mot-vertsykdom (GvHD).1
Jakavi er for behandling av pasienter ≥12 år med akutt eller kronisk GvHD som har respondert utilstrekkelig på kortikosteroider eller andre systemiske behandlinger.1
For alle godkjente indikasjoner med Jakavi er de vanligste bivirkningene trombocytopeni, anemi, nøytropeni, cytomegalovirusinfeksjon (CMV), sepsis, urinveisinfeksjoner, blødninger, blåmerker, vektøkning, hyperkolesterolemi, hypertensjon, svimmelhet, hodepine og økt alanin- og aspartataminotransferase.1
Fullstendig blodcelletelling, inkl. differensialtelling av hvite blodceller, må utføres før behandling startes, og kontrolleres hver 2.-4. uke til dosering er stabilisert, og deretter etter klinisk behov.1

Skann QR-koden for å registrere ditt samtykke og motta elektronisk informasjon fra Novartis, eller gå inn på www.medhub.no/hold-deg-oppdatert
Sikkerhetsinformasjon: Indikasjon: Jakavi er indisert til behandling av sykdomsrelatert splenomegali eller symptomer hos voksne pasienter med primær myelofibrose (også kjent som kronisk idiopatisk myelofibrose), post-polycytemia vera myelofibrose eller post-essensiell trombocytemi myelofibrose. Jakavi er indisert til behandling av voksne pasienter med polycytemia vera (PV) som er resistente mot eller intolerante overfor hydroksyurea. Jakavi er indisert til behandling av pasienter som er 12 år og eldre med akutt transplantat-mot-vert-sykdom (graft versus host disease, GvHD) eller kronisk transplantat-mot-vert-sykdom (GvHD) som har respondert utilstrekkelig på kortikosteroider eller andre systemiske behandlinger. Dosering: Fullstendig blodcelletelling, inkludert differensialtelling av hvite blodceller, må utføres før behandling med Jakavi startes opp og bør kontrolleres hver 2. til 4. uke til dosering av Jakavi er stabilisert, og deretter etter klinisk behov. Den anbefalte startdosen med Jakavi ved myelofibrose (MF) er basert på blodplatetall.Ved blodplatetall 50 x 109/liter til <75 x 109/liter gis 5 mg oralt to ganger daglig.Ved blodplatetall 75 x 109/liter til <100 x 109/liter gis 10 mg oralt to ganger daglig. Ved blodplatetall 100 til 200 x 109/liter gis 15 mg oralt to ganger daglig.Ved blodplatetall > 200 x 109/liter gis 20 mg oralt to ganger daglig. Den anbefalte startdosen med Jakavi ved polycytemia vera (PV) er 10 mg gitt oralt to ganger daglig. Den anbefalte startdosen med Jakavi ved akutt og kronisk transplantat-mot-vert-sykdom (GvHD) er 10 mg gitt oralt to ganger daglig. Jakavi kan gis samtidig ved fortsatt bruk av kortikosteroider og/eller kalsineurinhemmere. Vanligste bivirkninger: Bivirkninger som ble rapportert for alle indikasjoner var trombocytopeni, anemi, nøytropeni, cytomegalovirus (CMV)- infeksjon, sepsis, blåmerker og blødninger, hyperkolesterolemi, økt alaninamino- og aspartataminotransferase, hodepine og hypertensjon. Kontraindikasjoner: Graviditet og amming. Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene. Advarsler og forsiktighetsregler: Behandling med Jakavi kan føre til hematologiske bivirkninger, inkludert trombocytopeni, anemi og nøytropeni. Risikoen for alvorlige bakterielle og mykobakterielle infeksjoner og sopp- og virusinfeksjoner, bør vurderes. Pasienten bør følges nøye for tegn/symptomer på infeksjoner og egnet behandling initieres straks. Behandling med Jakavi bør ikke startes før aktive, alvorlige infeksjoner er behandlet. Progressiv multifokal leukoencefalopati (PML) er rapportert ved behandling med Jakavi. Pasienter bør monitoreres og ved nyoppståtte eller forverrede symptomer på PML bør pasienten henvises til nevrolog og videre dosering stoppes inntil PML er utelukket. Anbefalt startdose for MF basert på antall blodplater ved enhver form for nedsatt leverfunksjon bør reduseres med ca. 50%, og administrering bør skje 2 ganger daglig.Ved PV er anbefalt startdose 5 mg 2 × daglig ved nedsatt leverfunksjon. Hos pasienter med GvHD og lett, moderat eller alvorlig nedsatt leverfunksjon som ikke er relatert til GvHD, bør startdosen av ruksolitinib reduseres med 50 %. Hos pasienter som har leverrelatert GvHD og en økning av total bilirubin til > 3 x ULN, bør blodtellinger overvåkes oftere for toksisitet, og en dosereduksjon med ett dosenivå er anbefalt. Startdosen ved alvorlig nedsatt nyrefunksjon bør være redusert til ca. 50% og gis 2 ganger daglig ved MF, og 5 mg 2 ganger daglig ved PV og GvHD. Reseptgruppe, refusjon & pris: Reseptgruppe C. Godkjent etter H-resept. Jakavi 20 mg tabletter, 56 stk, kr 42 601,40. Jakavi 15 mg tabletter, 56 stk, kr 42 601,40. Jakavi 10 mg tabletter, 56 stk, kr 42 329,60. Jakavi 5 mg tabletter, 56 stk, kr 21 540,70 per 23.09.2022.
Se Felleskatalogen for mer informasjon ved å skanne QR-koden eller gå inn på: https://www.felleskatalogen.no/medisin/jakavi-novartis-578770
Referanser: 1. JAKAVI SPC avsnitt 4.1, 4.2, 4.3, 4.4 & 4.8. 2. Felleskatalogen. Jakavi «Novartis» [Internett]. Oslo: Felleskatalogen AS; 2022 [hentet 08.07.2022].Tilgjengelig fra: https://www.felleskatalogen.no/medisin/jakavi-novartis-578770
NOVARTIS NORGE AS - PB. 4284, Nydalen, 0401 OSLO - Tlf: 23 05 20 00 - www.novartis.no
NO2402013595
Øk trombocyttantallet* uten matrestriksjoner1
Doptelet® (avatrombopag) er en oral TPO-RA for behandling av primær kronisk immunologisk trombocytopeni (ITP) hos voksne pasienter som er refraktære overfor for eksempel kortikosteroider og immunoglobuliner. Administreres regelmessig med mat.1

Anbudsvinner av LIS 2299I ITP peroral
Periode: 01/09.2022 - 31.08.2023 med forlengelse til 31.08.20242
Vanligste bivirkninger er hodepine og fatigue. Vær oppmerksom på at trombopoeitinreseptoragonister kan være forbundet med økt risiko for tromboemboliske hendelser.1
TPO-RA = trombopoietinreseptoragonist.
*Målet er å nå et stabilt blodplatenivå på ≥ 50 x 109/l og ≤ 150 x 109/l.
1. Doptelet EMA Summary of Product Characteristics. Available at: www.ema.europa.eu/en/medicines/human/EPAR/doptelet Last accessed: 09/Feb/2024.
2. LIS 2299I ITP – Meddelelse om valg av leverandør fra Sykehusinnkjøp, saksnummer: 2021/1226;03/2022.
Doptelet (avatrombopag) tabletter
Reseptgruppe C H
Indikasjon: Primær kronisk immunologisk trombocytopeni (ITP) hos voksne som er refraktære overfor andre behandlinger. Dosering: Laveste dose for å oppnå og opprettholde blodplatenivå på ≥ 50 × 10 9/liter. Startdose på 20 mg 1 gang daglig, maks 40 mg daglig. Overvåkning av blodplatetall påkrevet. Se SPC for detaljer rundt hyppighet av overvåkning og dosejustering basert på respons av blodplatetall, samt anbefalt startdose ved samtidig administrering av andre legemidler. Behandling skal innledes av lege med erfaring innen hematologiske sykdommer. Blodplatetall måles før behandlings oppstart og på prosedyredagen. Se Forsiktighetsregler og Interaksjoner i SPC. Kontraindikasjoner: Overfølsomhet for innholdsstoffene. Forsiktighetsregler: Økt risiko for trombotiske/trombo emboliske hendelser ved kronisk leversykdom. Bør ikke brukes ved kronisk immunologisk trombocytopeni i forsøk på å normalisere blodplatetallet. Bør kun brukes ved alvorlig nedsatt leverfunksjon dersom for ventet nytte overgår forventet risiko. Behandling i samsvar med klinisk praksis og nøye oppfølging mtp. tegn på forverring eller nyoppstått hepatisk encefalopati, ascites og trombose eller blødnings tendens. Trombocytopeni vil sannsynligvis komme tilbake etter avsluttet behandling hos pasienter med ITP, noe som kan føre til blødninger . Nøye overvåkning og gjenopptatt ITP-behandling anbefales. Risiko for økt retikulin benmarg . Undersøkelser for cellulære morfologiske abnormaliteter og fullstendig hematologisk status anbefales. Ved opprettholdt effekt og unormalt perifert blodutstryk bør nytte-risiko av avatrombopag og alternative ITP-behandlinger vurderes på nytt. Skal ikke brukes for behandling av trombocytopeni grunnet MDS, da det er fare for progresjon av eksisterende MDS Graviditet, amming og fertilitet: Ikke anbefalt under graviditet og hos fertile kvinner som ikke bruker prevensjon. Risiko for barn som ammes kan ikke utelukkes; nytte-/risiko må vurderes. Effekt på fertilitet kan ikke utelukkes. Bivirkninger: Svært vanlige: Fatigue, hodepine. Vanlige: Anemi, splenomegali, trombocytopeni, diaré, flatulens, kvalme, oppkast, øvre abdominalsmerte, asteni, akne, kløe, petekkier, utslett, hypertensjon, dyspné, epistakse, artralgi, muskel-skjelettsmerter, myalgi, ryggsmerter, smerte i ekstremitet, migrene, parestesi, svimmelhet, ubehag i hodet, hyperlipidemi, redusert appetitt, redusert blodglukose, redusert blodplatetall, økt ALAT, økt LDH, økt blodglukose, økt blodplatetall, økt gastrin, økte triglyserider. Mindre vanlige: Hjerteinfarkt, dyp venetrombose, trombose i halsvene, portvenetrombose, lungeembolisme, cerebrovaskulær hendelse, TIA, myelofibrose, retinal arterieokklusjon, synssvekkelse. For mer informasjon, se felleskatalog eller preparatomtalen (SPC). Innehaver av markedsføringstillatelsen: Swedish Orphan Biovitrum AB, SE-112 76 Stockholm, Sweden. Dato: 06.04.2021. Basert på SPC godkjent av SLV/EMA: 09.02.2024. Pakninger og priser: Blisterpakninger: 10 stk. kr. 7471,70; 15 stk. kr. 11189,40; 30 stk. kr. 22342,50 (priser og ev. refusjon oppdateres hver 14. dag). Refusjon: H-resept. Vilkår 216. Refusjon ytes kun etter resept fra sykehuslege eller avtale spesialist.
Swedish Orphan Biovitrum AS, Dronning Eufemias gate 16, 0191 Oslo PP-22186 FEB 2024 © 2024 Swedish Orphan Biovitrum AB (publ) – All rights reserved. Sobi ™ and Doptelet™ are trademarks of Swedish Orphan Biovitrum AB (publ).
FLT3-rettede tripletter
I abstract #158 (Yilmaz et al.) ble foreløpige resultat resultat fra en fase I/II-studie med quizartinib, venetoclax og decitabin i kombinasjon ved nd AML uaktuell for intensiv kjemoterapi (n=10) eller ved R/R FLT3-ITD mutert sykdom (n=40) med inntil 5 tidligere behandlingslinjer presentert, hvorav 78% av R/R-pasientene var tidligere eksponert for gilteritinib. Venetoclax ble gitt i 14-21 dager i syklus 1, deretter 14 dager i påfølgende sykluser. Decitabin ble gitt i 10 dager ved induksjon, deretter 5 dager. Quizartinib ble administrert kontinuerlig. Etter median oppfølging i 33 mnd. oppnådde man 68% CRc i R/R-populasjonen og median OS på 7,1 mnd. Hos nd-pasienter var median oppfølging 15 mnd., CRc 100% og median OS er ennå ikke nådd. Det er så langt ikke observert dosebegrensende toksisitet ved Quizartinib 30 mg, mens 40 mg resulterte i 2 tilfeller av langvarig grad 4 agranulocytose.
En annen pågående fase I/II-studie utgående fra MD Anderson benytter den perorale tripletten ASTX727(decitabin/cedazuridin), gilteritinib og venetoclax ved R/R FLT3-mutert AML eller R/R intermediær-2/høyrisiko MDS/KMML eller nd AML uaktuell for intensiv kjemoterapi. Briski og kollegaer presenterte (abstract#2910) foreløpige data fra n=15 pasienter i den tungt forbehandlede (opptil 9 linjer) R/R kohorten, hvorav 1 pasient med FLT3-TKD, behandlet i 28 dagers sykluser med ASTX727 dag 1-5, venetoclax i 14-28 dager og gilteritinib kontinuerlig. Median alder var 65 år og mediant antall sykluser 2 (1-8). Samlet responsrate (ORR) var 53%, og 6 av 12 tidligere gilteritinib-eksponerte pasienter responderte. Som forventet dominerte nøytropeni og infeksjoner innen toksisitet, med 2 sepsis-relaterte dødsfall.
Andre Venetoclax-kombinasjoner
Xiaohui Suo presenterte oppdaterte resultater fra en kinesisk fase II multisenter enarmet studie av venetoclax i kombinasjon med daunorubicin og cytarabin (2+6) (abstract #969), som inkluderte n=70 pasienter med nydiagnostisert AML i aldersgruppen 16-60 år (median 44), hvorav 46%/13%/41% med ‘favorable-’/’intermediate-’/’high risk’ ihht. ELN 2022. ORR etter 1 induksjonssyklus var 93%, CRc 90% og 89,5% oppnådde udetekterbar MRD. Median nøytrofil aplasitid var 13 dager (5-36), og estimert 12 mnd. OS/ EFS/DFS var hhv. 85/81/82%. 6 av pasientene har så langt gått videre til allo-HSCT.
I en annen kinesisk multisenterstudie hvorfra Jing Lu la frem foreløpige resultater for n=102 pasienter (abstract #970), blir venetoclax og decitabin (VEN-DAC) sammenlignet med konvensjonell kjemoterapi med idarubicin og AraC 3 + 7 (IA-12) som induksjonsbehandling hos yngre pasienter (18-59 år) med nydiagnostisert AML via 1:1-randomisering. Decitabin gies i 5 dager og venetoclax via ‘ramp-up’ i 28 dager. Pasienter uten respons kunne motta ytterligere 1 identisk syklus, og konsolidering ble gjennomført med intermediærdose AraC (IDAC). Primært endepunkt var CRc, som ble oppnådd hos 85,5% i VEN-DAC gruppen, mot 78,7% i IA-12 gruppen (ns), med hhv. 67,3% og 53,2% MRD-negativitet (ns). For intermediær- og høyrisiko-pasientene, var andelen CRc og MRD-negative signifikant høyere ved VEN-DAC. Pasientene i denne gruppen hadde også lavere transfusjonsbehov og kortere varighet av nøytropeni. Det ble konkludert med at VEN-DAC har sammenlignbar effektivitet og lavere toksisitet enn IA-12, og at særlig høyrisikogruppen oppnår flere og dypere remisjoner med VEN-DAC.
Den italienske multisenter singel-arm Ven DEC GITMO-studien (Russo et al., abstract #1513) har benyttet venetoclax i kombinasjon med decitabin som bro til allo-HSCT hos eldre (60-75 år) pasienter
med intermediær- eller høyrisiko AML, med andel CR1-allo som primært endepunkt. Det ble gitt 2 28-dagers sykluser (C1-2) med opptil 400 mg venetoclax i kombinasjon med 5 dager decitabin, og protokollbestemt allo-HSCT skulle utføres innen 2 mnd. ved remisjon. Ved manglende CR kunne ytterligere 2 sykluser (C3-4) gies. Blant n=93 behandlede pasienter kom 74% i CR etter C2, og 20% av de øvrige oppnådde CR etter C3-4, som også ble gitt til mange pasienter i påvente av allo av logistiske årsaker. C2-4 ble som hovedregel gitt poliklinisk. Median tid til respons var 65(49-175) dager. Totalt oppnådde 55% primært endepunkt, og blant pasientene i CR var andelen 82%. Protrahert nøytropeni opptrådte hos 24%, og 7 av pasientene avbrøt behandlingen i CR pga. toksisitet. Det ble konkludert med at VEN-DEC regimet gir høy remisjonsrate og økt mulighet for allo-HSCT, med i stor grad poliklinisk behandling og relativt lav toksisitet.
IDH-muterte
Særlig IDH2-muterte cellelinjer er i prekliniske studier særlig følsomme for BCL2-hemming, og dette har gitt betydelig interesse for nye venetoclax-baserte kombinasjoner ved IDH-mutert AML.
Himachandana Atluri presenterte foreløpige resultater fra en fase Ib/II-studie med en hel-oral triplett bestående av ASTX727 (decitabin/cedazuridine), venetoclax og ivosidenib eller enasidenib hos nydiagnostiserte (nd) AML-pasienter uaktuelle for intensiv terapi samt R/R pasienter, med mutasjon i IDH1 eller –2 (abstract #968). Ved R/R sykdom tillot protokollen tidligere bruk av HMA, venetoclax eller IDH-hemmer (IDHi). Blant n=50 evaluerbare pasienter var median alder 72(41-86), og 52% hadde R/R sykdom, med mediant 2 tidligere behandlingslinjer. For nd-pasientene var CRc 96% med 91% flowcytometrisk MRD-negativitet ved responderende sykdom, mens tilsvarende ble oppnådd hos hhv. 46% og 67% ved R/R AML. 30% av tidligere venetoclax-eksponerte og 71% av IDHi-eksponerte oppnådde ny CRc. Av toksisitet ble det spesielt bemerket hyperbilirubinemi v/ enasidenib samt differensieringssyndrom (DS) hos 10% av pasientene.
Sluttresultatene fra en canadisk enarmet fase Ib/II-studie med kombinasjon av enasidenib og venetoclax ved IDH2-mutert relapsert/refraktær myeloid malignitet (MDS, AML) ble presentert av Guillaume Richard-Carpentier (abstract #159). Blant n=27 inkluderte pasienter var median alder 70(23-84) år, og pasientene hadde mottatt 1-2 tidligere behandlingslinjer. Blant eksklusjonskriteriene var tidligere eksposisjon for IDH2i eller BCL2i. ORR var 70%, med 57% CR og median tid til respons på 1 mnd; 2,8 mnd til beste respons. Man fant også en assosiasjon mellom fallende IDH2-mut VAF og respons, med best effekt ved R172-mutasjoner. Etter median oppfølging på 17,1(0,9-31,4) mnd var median OS 9,4 mnd og 2 års OS 42%. 4 pas. gikk videre til allo-HSCT, hvorav 2 fortsatt er i live og i remisjon. Forekomst av hyperbilirubinemi var 48%, og det ble ikke sett tilfeller av DS. Av øvrig toksisitet dominerte nøytropeni, trombocytopeni og lungeinfeksjoner.
MULTIKINASE-Inhibisjon
Tuspetinib er en oral hemmer av SYK, FLT3, JAK1/2, RSK1/2, KIT og TAK1-TAB1. Naval Daver la frem foreløpige resultater av monoterapi med denne kinasehemmeren i en global multisenterstudie ved R/R AML, som også inkluderer kombinasjonsbehandling med Venetoclax (abstract #162). Blant n=100 pasienter var 35% FLT3-muterte hvorav 51% tidligere eksponert for FLT3i og 60% for venetoclax. I monoterapi så man en ORR hos 14,3% av venetoclax-eksponerte og hos 23,1% av FLT3i-eksponerte FLT3-muterte, mot 66,7% ved FLT3-mutasjon i kombinasjon med NPM1-mutasjon. Det ble også sett responser hos TP53- og
29
RAS-muterte, uten dosebegrensende toksisitet opp til 160 mg. Det ble konkludert med lovende respons- og tolerabilitetsdata, uten modne data for kombinasjonsbehandlingen.
En annen interessant nyere kinasehemmer, særlig ved FLT3-mutert sykdom, er emavusertib (CA-4948), som i tillegg til FLT3 også hemmer IRAK4, som typisk oppreguleres under behandling med FLT3i samt ved spleisefaktor-mutasjonene U2AF1 og SF3B1 via IRAK4-L, med resulterende økt TLR-signalering. Winer og medarbeidere presenterte i abstract #2924 foreløpige erfaringer fra monoterapi med denne hemmeren i den pågående TakeAim-studien (fase I/ IIa) for inkluderte FLT3-muterte R/R-pasienter. Av n=10 (9 ITD, 1 TKD) inkluderte pasienter, hvorav 7 Gilteritinib-eksponerte, oppnådde 5 av 9 evaluerbare >90% blastreduksjon uten DLT. Mutasjonsprofilering tyder også på en sykdomsmodifiserende effekt av emavusertib. Studien fortsetter å inkludere.
Hemming av E-Selectin
E-selectin er en markør for kjemoresistens og er høyt uttrykt i det leukemiske mikromiljøet, med økt uttrykk etter at blastene eksponeres for HMA. AML etter HMA-behandling av myeloide maligniteter, såkalt ‘treated-secondary AML (ts-AML)’ har dårlig prognose, og Huante og medarbeidere presenterte i abstract #2922 resultater fra en fase Ib/II-studie med bruk av den parenterale E-selectin antagonisten uproleselan i kombinasjon med cladribin og lavdose AraC (LDAC) ved ubehandlet ts-AML. Det ble benyttet 4-ukers sykluser med 2 dosenivåer av cladribin og LDAC. Blant n=20 pasienter var 55% tidligere eksponert for venetoclax og 25% stamcelletransplantert. Samtlige hadde ugunstig cytogenetikk. 72% av pasientene oppnådde blastreduksjon, og ORR var 50%, 20% ved tidligere venetoclax, med median OS på 5,3 mnd. 1 pasient som oppnådde MRD-negativitet gikk videre til allo-HSCT. 65% fikk febril nøytropeni, men det forekom ingen DLT. Man studerer også betydningen av plasma E-selectin som biomarkør på respons.
Meninhemmere
Menin er virkelig i vinden ved AML om dagen. Samspillet mellom dette viktige stillasproteinet og KMT2A er avgjørende for sykdomsprossessen ved leukemier med rearrangering av KMT2A (KMT2Ar) eller NUP98r, eller mutert Nucleophosmin 1 (NPM1m). Især KMT2Ar sykdom er forbundet med svært høy recidivrisiko og elendig prognose. Hemming av menin/KMT2A-interaksjonen med den selektive orale meninhemmeren revumenib har så langt vist klinisk effekt og tolerabilitet ved svært refraktær akutt leukemi (Issa, Nature 2023), med synergisme med BCL2-hemming ved KMT2Ar og NPM1m (Carter, Blood 2021). Kombinasjonsterapi har derfor vakt interesse, bl.a. hos Issa og kollegaer, som i abstract #58 beskrev foreløpige resultater fra n=8 pasienter i en fase I/II-studie på kombinasjon med decitabin/cedazuridin og venetoclax ved R/R AML og MPAL. Median alder var 27 år, og 63% var tidligere behandlet med HMA, venetoclax eller SCT. ORR er så langt 100%, med 43% MRD-negativitet. Av toksisitet bemerkes cytopenier, hyperfosfatemi og transaminitt.
Resultater fra interimanalysen av fase II Augment-101 studien presentert av Ibrahim Aldoss i ‘Late Breaking’-sesjonen (abstract #LBA-5) har fått mye oppmerksomhet. Man har her brukt revumenib i monoterapi ved R/R AML (83%), -ALL eller -MPAL, hvor middelet er gitt i 28-dagers sykluser kombinert med en CYP3A4-hemmer, og man har nå nådd den forhåndsspesifisert evaluerbare effekt-populasjonen på n=57 KMT2Ar pasienter med 6 mnd, oppfølging, med median alder 34(1,3-75). Populasjonen var tungt forbehandlet (opptil 11 linjer), og nær halvparten var allotransplantert. Man så her en ORR på 63,2%, CRc på 43,9%
hvorav 68,2% MRD-negative. 38,9% av responderne gikk videre til transplantasjon, med post-allo vedlikehold med revumenib hos halvparten av disse. I sikkerhetspopulasjon på n=94 pasienter opptrådte behandlingsrelaterte bivirkninger (TRAE) hos 81,9%, og kvalme, differensieringssyndrom og QTc-forlengelse var vanligst. Grad≥3 febril nøytropeni forekom hos 13,8%, og 6,4% av pasientene måtte avbryte behandlingen pga. TRAE. Studien nådde sitt primære endepunkt, og forfatterne konkluderte med klinisk meningsfull effekt av revumenib i denne tungt forbehandlede populasjonen. På effektgrunnlag ble studien stoppet tidlig for denne kohorten, men fortsetter å inkludere NPM1m pasienter.
Andre meninhemmere som studeres ved R/R AML er JNJ-75276617 og Ziftomenib (abstract #57 og #1553), hvor Jabbour og kollegaer viste ORR på 50% ved monoterapi med førstnevnte, mens KOMET008 studien (abstract #1553) undersøker effekt av sistnevnte i kombinasjon med FLAG-IDA eller LDAC etter lovende resultater i monoterapi i KOMET-001. Her foreligger foreløpig ingen modne data.
Antistoffer og celleterapi Både bi- og trispesifikke antistoffer samt CAR-T/NK er under utvikling for AML, foreløpig med lite modne data. Å finne passende målmolekyler for CAR ved AML har vært en betydelig utfordring, og så langt har CD123, CD33 og CLL-1 vært best studert. Det ble presentert foreløpige resultat bl.a. i abstract #34565, #771 og #2106, hvor man ser ORR opptil omkring 70% hos til dels tungt forbehandlede pasienter. CD123-CAR har vært assosiert med betydelig toksisitet (cytopenier, kapillærlekkasje, CRS, ICANS), men man ser nå lovende resultater ved å benytte såkalt adapter-CAR-T teknologi hvor et ‘targeting molecule’ med kort halveringstid fungerer som en “på/av-bryter” for CAR-T cellene. CD123 er også aktuelt for konjugater, og Daver og medarbeidere presenterte i abstract #2906 foreløpige resultat fra n=50 pasienter i en ublindet multisenter fase I/II studie av pivekimab sunirin, som er et anti-CD123 med påkoblet alkylator. I studien gies dette kombinert med Aza-Ven til voksne med nydiagnostisert CD123-positiv AML (median alder 74), så langt med akseptabel toleranse og tilsynelatende robuste responser, med cCR på 66%, hvorav 73% MRD-negative i en kohort med høy andel høyrisikopasienter, bl.a. 38% TP53-muterte.
TP53
TP53-mutasjoner er tilstede hos omlag 30% av R/R AML og er assosiert med høy grad av resistens både mot kjemoterapi og målrettet behandling, høy relapsrate og svært redusert overlevelse. Sykdomsbiologien er også ufullstendig kartlagt. Bl.a. har lite vært kjent omkring cellulære hierarkier hos slike pasienter. Man vet at der er en invers regulatorisk relasjon mellom MYC og TP53, og et økt uttrykk av CD34 ved TP53mut sykdom. Nishida og medarbeidere presenterte i abstract #583 et svært interessant arbeid, med funn av signifikant økt MYC mRNA i den leukemiske stamcellefraksjonen (LSC) ved TP53mut AML sammenlignet med TP53wt AML. Dette ble bekreftet på proteinnivå med massecytometri (CyTOF), og ved enkeltcelle RNA-sekvensering av benmarg fra friske, TP53mut og TP53wt, fant man en betydelig anrikning av stamcellelignende (HSC) celler hos TP53mut, med reduksjon av progenitor- og GMP-lignende celler. HSC-lignende celler fra TP53mut hadde også høyere uttrykk av MYC, og CD34+ AML celler, særlig de med samtidig uttrykk av CD38, hadde betydelig økt mengde c-MYC protein ved CyTOF enn CD34+ HSC fra friske. De var også mye mer sensitive for behandling med GT19715, en cereblon-modulator innrettet mot c-MYC proteinet, tydende på et terapeutisk vindu. GT19715 induserte betydelige metabolske svekkelser i sensitive celler, og medførte en dyp blastreduksjon i TP53mut prøver samt
30
Post
i en svært aggressiv TP53mut PDX-modell, uten holdepunkt for alvorlig toksisitet i musemodeller med operativ cereblon-mediert proteindegradering.
131I-Apamistamab (Apa) er et anti-CD45 radioimmunkonjugat som konseptuelt gir en målrettet myeloablasjon. I den randomiserte multisenter fase III-studien SIERRA kombineres Apa med fludarabin og TBI 2 Gy som induksjon og kondisjonering ved allo-HSCT for R/R AML, sammenlignet med konvensjonell behandling (CC) i et crossover-design. Choe og medarbeidere presenterte
i abstract #469 data for den TP53-muterte R/R populasjonen i studien (n=37, median alder 65), med funn av median OS på 5,72 mnd. i Apa-armen, hvilket ikke skiller seg signifikant fra TP53wt (6,37; p=0,16), mot 2,96 mnd i CC-armen. Ekskluderte man crossover-pasientene fra CC-gruppen sank median OS for TP53mut til 1,66 mnd (høysignifikant ift. Apa). Forfatterne konkluderte med at intervensjonsarmen gav signifikant økt overlevelse hos TP53-muterte, og dermed motvirket den negative effekten av mutasjonen. Det blir spennende å følge med på langtidsresultater av denne strategien.


norskaften.no
