
12 minute read
Expert Panel - Medical Device Regulation

from Health Business 22.6
by PSI Media

EXPERT PANEL
MEDICAL DEVICE REGULATION
In this issue’s Expert Panel, Benedict Wallner, general manager and Regina von Campe, quality assurance manager at preOx.RS GmbH discuss The European Union Medical Device Regulation - Regulation (EU) 2017/745 (EU MDR) and how it affects SMEs
Benedict Wallner, preOx.RS GmbH, general manager
Benedict Wallner has 35 years’ experience in the medical device industry, having started in 1987 as a sales representative for laparoscopic surgery at WISAP where he played a major role in developing minimal invasive surgery in the early nineties. In 1993, he made the switch to single-use medical devices as a sales manager. Since 2001, Benedict has been a partner at preOx.RS GmbH and since 2015, has been general manager. In his current role, he is responsible for sales marketing and product development.

Regina von Campe has 32 years’ experience in the medical device industry, having started as a product manager in 1990 at Sherwood Davis & Geck GmbH. Regina became the regulatory affairs manager at preOx. RS GmbH in 2006. In her current role, she is responsible for quality assurance and regulatory affairs.
Regulation (EU) 2017/745 is an EU regulation on the clinical investigation and sale of medical devices for human use. The regulation came into force on 25 May 2017, with previously approved medical devices having a transition time of three years to meet the new requirements (up to 26 May 2021).
The regulation replaced Directive 90/385/ EEC, Directive 93/42/EEC. In comparison to the previous Medical Device Directive (MDD), noticeable differences include changes in device classification and device scope, stricter oversight of manufacturers by Notified Bodies, the introduction of the “Person Responsible for Regulatory Compliance” (PRRC), and increased post-market surveillance activities..
Medical devices With the new regulation now in place, we discuss the effect it has had on the medical device industry. preOx.RS GmbH has been developing, manufacturing and marketing innovative medical products since 1998 with a focus on single-use medical devices
We know that medical devices are incredibly important and are indispensable in any health service, by improving patient care, reducing costs and saving time. We began by asking our panellists about the importance of medical devices. Benedict says: “Medical devices are used in many diverse settings, for example, by laypersons at home and clinicians in hospitals as well as by healthcare professionals in medical facilities. Medical devices play a very important role in the prevention of health problems with the screening, diagnosis and finally treatment of illness. Medical devices are irreplaceable tools to drive healthcare systems all over the world. They keep us alive, if necessary, and help us to recover. Without medical devices, the survival of E
humankind is challenged. Without medical devices, only the fittest will survive.”
SMEs and cost With the introduction of any new regulation, it can always seem more difficult for SMEs to get to grips with it, as they don’t have the workforce, money or legal teams that a large corporation has. Benedict says: “SMEs often cover niche markets with niche products. The MDR requirements for niche products are the same as for products that sell millions, and so the cost is also the same. The turnover with niche products is smaller so the challenge is to establish a price level which enables SMEs to invest in MDR transition.”
Benedict continued: “The biggest challenge is personnel resources. To support their own Regulatory Affairs, SMEs must look to consultancy. Consultants are rare and expensive, and everyone is looking for one. In the end, MDR is the highest financial risk for SMEs ever and many companies will close.”
On the topic of cost implications of MDR for SMEs, Benedict says: “The cost of MDR is higher than before. Additional regulatory work leads to a cost increase. Depending on the product portfolio of a company, the cost increase could cause a company to put a stop to products with a low margin or low turnover. SMEs often have low-turnover niche products. These products run the risk of discontinuation.”
Because of this, our experts warn that there will be a negative effect on health care and health services. Regina says: “Devices will vanish from the market, simply because it doesn’t pay off. The availability of products, especially of niche products for treatments which are not regularly used, will get worse.”
In order to prepare for MDR, SMEs face the same process and regulatory journey to approval. Benedict says: “It is very hard for small companies to cover all the necessary regulatory fields and to finance the expenditure. To prepare for MDR, small companies must follow the same ways as big players, so the effort is the same. The major difference is the manpower SMEs can put into the transition. In many SMEs, you have only one person responsible for Regulatory Affairs who must cover the transition to MDR. A regulatory affairs manager needs support from IT solutions. There are many on the market to help companies in the transition but the challenge for SMEs is to find a solution which covers their needs, with easy handling, and minimal cost.” Advice for SMEs However, hope is not lost for SMEs in the medical device field. Our experts have some advice for SMEs navigating MDR. Benedict says: “SMEs must build networks and cooperations with each other to bundle experience and know how. Now is the time to share and leave old competitiveness behind. If SMEs take this path, the MDR barrier can be overcome. We are already working in a network, but taking the step to share more for the benefit of all is difficult as there are too many concerns.”
On that note, is there anything that regulators could do to help SMEs with MDR? Of course, it would be in their best interest to do so, as this can only lead to the approval of more medical devices which could be beneficial to population health. Our experts believe that more could be done to help SMEs. Regina says: “The actual situation is that from the 80 former notified bodies, only 34 are newly certified and ready for MDR. If a company like us lose their notified body because they are not certified yet, there is no chance of getting a new notified body. If you have a certified notified body, the time for recertification of products takes more than 18 months. Regulators must accept that the timeframe for changing to MDR is not realistic. The argument that we

had a five-year transfer period and medical device companies did not use this time to prepare for MDR occludes the reality. The reality is that we have too few Notified Bodies for too many companies and medical products. This leads to an overload in the system of certification and finally to the collapse of medical device allocation.”
Regina says that taking all of this into account, there are several actions that regulators can take to help SMEs. This includes expanding the transition time from the previous MDD to the current MDR by a minimum of an additional three years and establishing a scope of action for how to transfer existing products to MDR.
Regina calls for the focus to be on high-risk products to be prioritised and for niche and existing products with a low-risk classification to be excluded from immediate re-certification.
Regina is also calling for the certification of new Notified Bodies to be sped up.
At the end of our conversation, we asked our panellists if there was anything they would like to add. Benedict cited Chapter 2 of the Medical Device Regulation, which reads: “This Regulation aims to ensure the smooth functioning of the internal market as regards medical devices, taking as a base a high level of protection of “This Regulation aims to ensure the smooth functioning of the internal market as regards medical devices, taking as a base a high level of protection of health for patients and users, and taking into account the small- and mediumsized enterprises that are active in this sector.”
health for patients and users, and taking into account the small- and medium-sized enterprises that are active in this sector. At the same time, this Regulation sets high standards of quality and safety for medical devices in order to meet common safety concerns as regards such products.”
However Benedict disagrees with this statement, saying: “This 2nd chapter of the MDR illustrates how unrealistic the claim of the new regulation is. It demonstrates an attitude against all medical device companies and implies that medical devices were not safe and had no high standards before MDR. It also implies that there are public concerns about the safety of medical devices and the reliability of medical device manufacturers.”
Benedict continues: “Based on chapter 2, SMEs must ask the question what the intention was with “taking into account the small- and medium-sized enterprises”. Was the intention to eliminate them?
“In the end, MDR overregulates the medical device market in Europe without achieving more safety and patient benefits than before.”
Though MDR has been in effect for a while now, it is clear there are still barriers for SMEs in achieving accreditation. The result of this may be a reduction in the production and invention of medical devices, the closure of medical device companies and therefore a negative effect on healthcare and health services. L

Does Scan4Safety hold the answer to reducing NHS Never Events?
With hundreds of preventable patient safety incidents occurring in the NHS every year, Glen Hodgson, head of healthcare at GS1 UK looks at how they can be avoided
Each year, NHS England releases a report highlighting the number and types of “Never Events” that have occurred across healthcare provider organisations in England.
Described as “patient safety incidents that are wholly preventable where guidance or safety recommendations that provide strong systemic protective barriers are available at a national level and have been implemented by healthcare providers”, hundreds of incidents arise across the NHS annually.
From 1 April 2021 to 31 March 2022, 407 Never Events were reported across the NHS, including a range of safety incidents including wrong implant/prosthesis and medication administration via the wrong route.
A national approach to investigating Never Events The Healthcare Safety Investigation Branch (HSIB) is responsible for conducting independent safety investigations of such incidents in NHS-funded care. Its purpose is “to improve patient safety by carrying out independent and effective investigations without apportioning blame or liability”.
Since launching in 2017, the HSIB has completed and published 76 reports based on national investigations and has made 215 improvement recommendations for improvement. Ten of these reports have concerned Never Events and the learning from these was aggregated in a National Learning Report on the subject1 .
National drivers for traceability Many Never Events do not result in significant patient harm, it is the repercussions that are often more wide-reaching. However, when such patient safety incidents are occurring and recurring, then the importance and effectively addressing them is clear.
Take the parallels of the 2011 PIP breast implant scandal versus the 2013 horsemeat scandal. In both cases, substandard products were identified as a risk to population health. However, due the widespread use of GS1 standards for product identification, contaminated horsemeat products were removed from retail shelves within two hours. In contrast, to this day, it is estimated that many of the 47,000 patients with faulty PIP implants, are still living with them2 .
Fast-forward to 2020, and the Independent Medicines and Medical Devices Safety Review (IMMDSR)3, chaired by Baroness Julia Cumberlege, reflects these same challenges.
The IMMDSR explained how the lack of standardised data capture and sharing, made it difficult to monitor trends for adverse events and to then trace these reports back to affected patients. This prompted a central strategy to develop a

national patient-identifiable database to improve traceability. The foundations of which would rely on the effective capture of accurate data, at the point of care, and associating it with the correct patient.
Used for the unique identification of people, products and places, GS1 standards would enable data to captured directly at the point of care via through the scanning of a GS1 barcode – a process known as Scan4Safety4 .
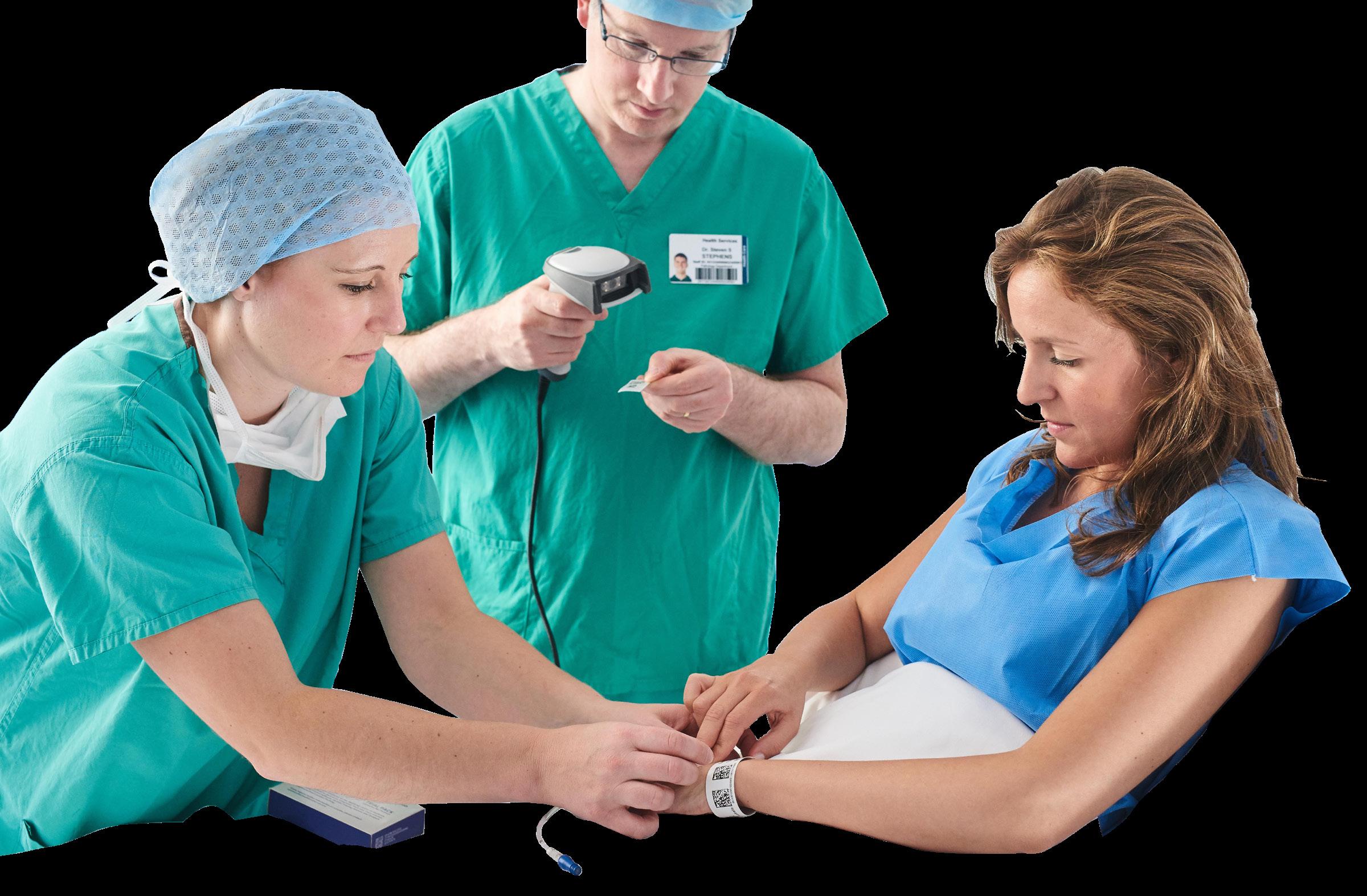
Learning from safety incidents The Cumberlege Review highlights the benefits of Scan4Safety implementation with reference also noted in some HSIB report findings.
In 2017, the HSIB investigated an incident where a patient received an incompatible hip prosthesis during surgery5. “The error was identified when data from the procedure was recorded in the National Joint Registry several days later.”
As part of the recommendations, it was suggested that “an interim basic scanning system [could be developed and implemented] to identify wrong prostheses prior to implantation”, and for “the Department of Health and Social Care to expand the remit of the working group which includes University Hospitals of Derby and Burton’s Scan4Safety programme.”
Dr Sean Weaver, deputy medical director at HSIB explains, “Different people do things differently. A barcode scanning system could standardise the selection and identification of equipment to prevent errors”. Scan4Safety driving traceability across the NHS So, how can technology be used to improve patient care? For Leeds Teaching Hospitals NHS Trust, implementing Scan4Safety has provided a wealth of data to help mitigate the risk of errors at the point of care.
Medical director of operations, Mr Stephen Bush explains, “Using Scan4Safety has helped us to track activity and inform our decisionmaking around patient care and flow. We now have better and more rapid access to data which has allowed us to improve our ability to care for patients across our organisation”. Leeds have implemented GS1 standards to uniquely identify patients, products, and places across multiple trust sites. Mr Bush continued, “Barcodes and GS1 standards are the key to enable clinicians to reduce patient harm”. The traceability benefits also go a long way. ‘‘Real-time scanning of devices used in patient care ensures that the right product is rapidly identified for that patient and assures us that we have the correct stock levels to care for future patients.’’

The power of Scan4Safety To improve patient safety across healthcare systems, introducing system-wide effective risk management strategies are crucial. Dr Weaver explains, “Investigations should be done to maximise learning to make it as safe as possible”. Scan4Safety has been proven to reduce errors and decrease the risk of Never Events.
To deliver better patient care, Scan4Safety should be made commonplace for all healthcare provider organisations. The tools exist. As Mr Stephen Bush said, “Make the easy thing the right thing and it will work.”L Credits to: Dr Sean Weaver, deputy medical director, HSIB, and Mr Stephen Bush, medical director operations, Leeds Teaching Hospitals NHS Trust
FURTHER INFORMATION
www.gs1uk.org/industries/healthcare healthcare@gs1uk.org
References:
1. Never events: analysis of HSIB’s national investigations 2. https://www.nhs.uk/ conditions/pip-implants/ 3. https://www.immdsreview.org.uk/ downloads/IMMDSReview_Web.pdf 4. https://healthcare.gs1uk. org/scan4safety/ 5. https://www.hsib.org.uk/ investigations-and-reports/ implantation-of-wrong-prosthesesduring-joint-replacement-surgery/









